Introduction
Hantaviruses are rodent-borne negative-stranded RNA
viruses, belonging to the genus Hantavirus, family
Bunyaviridae (1,2). Thus far, two severe human diseases
caused by Hantavirus have been identified, and they are
categorized by geographical distribution and target organ as Old
World hantaviruses, which cause hemorrhagic fever with renal
syndrome (HFRS) in Europe and Asia, and New World hantaviruses,
which cause hantavirus pulmonary syndrome (HPS) in America
(1–5). These two diseases are characterized
by fever, bleeding and shock (1–5).
Approximately 10,000 cases of HFRS are reported in mainland China
annually, with a mortality rate of 0.1–15% (5–7).
The pathogens responsible for HFRS in China are mainly Hantaan
virus (HTNV) and Seoul virus (6,8).
The clinical presentation of HFRS can be divided into five
sequential stages; febrile, hypotensive, oliguric, diuretic and
convalescent (9). High fever,
thrombocytopenia and capillary leak syndrome are typical symptoms
experienced throughout infection (5,9).
Although multiple hypothesizes have been developed
to explain the pathogenesis of Hantavirus, the precise mechanism
remains undefined (1,10–12). Numerous cytokines and chemokines,
such as vascular endothelial growth factor, tumor necrosis
factor-α, interleukin-6 (IL-6) and IL-8 are elevated in HFRS
patients, and the induction of pro-inflammatory cytokines and
chemokines is considered to play a pivotal role in the pathogenesis
of Hantavirus (13–15). These cytokines increase the
expression of adhesion factors on endothelial cells, enabling
transmigration of leukocytes to the sites of infection and re-set
the hypothalamus thermoregulatory center, causing fever. IL-1β,
which is considered to be an endogenous pyrogen, is elevated in the
plasma of HFRS patients (15).
Recently, Zhu et al (16)
demonstrated that IL-1β caused increased vascular permeability by
activation of IL receptors and activation of the MYD88-ARNO-ARF6
cascade to disrupt vascular stability. In addition, Hottz et
al (17) found that IL-1β
expression is elevated in the platelets and platelet-derived
microparticles of dengue virus-infected patients, and that the
dengue virus initiates the assembly of inflammasomes, activation of
caspase-1, and caspase-1-dependent IL-1β secretion. It appears that
IL-1β can contribute to the pathogenesis of viral infection by
inducing excessive hyperpermeability of the vascular system.
IL-1β induction involves the formation of an
inflammasome to generate bioactive caspase-1 by cleavage of
pro-IL-1β (18,19). Inflammasomes are initiated by
pattern-recognition receptors activated by viral or bacterial
components and toxins, or even crystalline structures (18,20,21). Thus far, retinoic acid-inducible
(RIG)-I-like receptors, nucleotide-binding oligomerization
domain-like receptors (NLRs) and absent in melanoma 2 (AIM2)-like
receptors have been found to initiate inflammasome formation, and
the majority of RNA viruses are capable of initiating the
nucleotide-binding domain, leucine-rich repeat containing protein 3
(NLRP3) inflammasome (22–25).
NLRP3, also known as cryopyrin, NALP3 or PYPAF1, contains an
N-terminal pyrin domain (PYD), a central nucleotide-binding domain
and C-terminal leucine-rich repeats (26). Following activation, NLRP3
oligomerizes to form a large protein complex, and recruits the
adaptor apoptosis-associated speck-like protein (ASC). ASC contains
a PYD and a C-terminal caspase-recruitment domain (CARD), allowing
it to interact with NLRP3 and pro-caspase-1. Pro-caspase-1 contains
a pro-domain (CARD domain), a 20-kDa subunit (p20) and a 10-kDa
subunit (p10), and is activated by proteolytic cleavage to generate
caspase-1, which subsequently cleaves the pro-IL-1β into IL-1β,
which is secreted (27,28).
The mechanisms preceding the formation of the
inflammasome are not completely understood, but are characterized
by three models. First, the formation of pores on the surface of
cells allows extracellular NLRP3 agonists to enter the cytosol and
directly activate NLRP3 (29).
Second, lysosome rupture results in the release of cathepsin B,
which activates NLRP3 (30).
Third, all NLRP3 agonists trigger the generation of reactive oxygen
species (ROS) by the mitochondria, which leads to the activation of
the NLRP3 inflammasome (31–35).
The present study examined the level of IL-1β during
HTNV infection of the human monocytic cell line, THP-1, and
demonstrated that induction of IL-1β by HTNV was dependent on the
NLRP3 inflammasome.
Materials and methods
Reagents and antibodies
Adenosine triphosphate (ATP) was obtained from Roche
Diagnostics (Basel, Switzerland); MG132, Z-VAD-FMK, pyrrolidine
dithiocarbamate (PDTC) and apocynin were purchased from
Merck-Calbiochem (Darmstadt, Germany); and lipopolysaccharide
(LPS), phorbol 12-myristate-13-acetate (PMA), Boc-D-CMK and H2DCFDA
were obtained from Sigma-Aldrich (St. Louis, MO, USA). The ATP
assay kit and radioimmunoprecipitation buffer (Beyotime, Shanghai,
China) were used for cell lysis. Antibodies directed towards human
IL-1β (#2021) were purchased from Cell Signaling Technology
(Danvers, MA, USA), caspase-1 (#PAB0811) was obtained from Abnova
(Taipei, Taiwan) [an additional cleaved caspase-1 antibody was
obtained from Santa Cruz Biotechnology, Santa Cruz, CA, USA
(sc-22163)], and actin from Beyotime. Monoclonal antibody (mAb) 1A8
[specific to the HTNV nucleocapdis protein (NP)] was prepared in
our laboratory as previously described (8,36).
Cells and viruses
THP-1 [TIB-202D; American Type Culture Collection
(ATCC), Manassas, VA, USA] were cultured in RPMI-1640 supplemented
with 10% fetal bovine serum (HyClone, Logan, UT, USA) (culture
medium), 100 μM non-essential amino acids, 100 U/ml
penicillin and 100 μg/ml streptomycin, and were incubated at
37°C with 5% CO2. THP-1 cells were differentiated with 1
μM PMA for 12 h, and subsequently incubated with
heat-inactivated HTNV, HTNV at indicated multiplicity of infection
(MOI) or mock (medium only) for 1 h before the supernatant was
removed and replaced with culture medium. PMA-THP-1 were cultured
for 6 h to 3 days before use.
HTNV 76-118 was propagated in the sucking mice
brain. All the animal experiments were approved by the Fourth
Military Medical University Medical Ethics Committee (Xi’an, China)
(approval no. XJYYLL-2012508) (37). Viral titer was determined by
TCID50 on Vero E6 cells (Vero C1008; ATCC CRL-1586), and
was converted to plague-forming units using the Reed-Muench method.
The virus was heat inactivated at 56°C for 30 min. Inactivation was
confirmed when infection of Vero E6 cells for 3 days yielded no
detectable viral nucleoprotein.
Immunofluorescence assay
For the detection of immunofluorescence, human
umbilical vein endothelial cells (HUVEC) and Vero E6 cells were
directly seeded in coverslips in a 24-well plate, and when adherent
and 70% confluent were infected with HTNV (MOI=1) for 90 min. The
THP-1 cells were differentiated with PMA for 12 h in a 24-well
plate supplement with coverslips. Seventy-two hours post-infection,
cells were washed three times with Dulbecco’s phosphate-buffered
saline (DPBS; HyClone) and fixed with 4% paraformaldehyde for 15
min at room temperature. Cells were subsequently permeabilized with
0.5% Triton X-100 (Sigma-Aldrich) for 10 min and washed again with
DPBS. HTNV NP present in cytosol was stained by mAb fluorescein
isothiocyanate-1A8 specific for HTNV nucleoprotein and Hoechst
33258 (100 ng/ml; Beyotime) was used to stain the cell nuclei. The
cells were observed using a BX60 fluorescence microscope (Olympus,
Tokyo, Japan).
Quantitative polymerase chain reaction
(qPCR)
The specific oligonucleotide primers were: Human
GAPDH forward, 5′-ACC CAC TCC TCC ACC TTT G-3′; and reverse,
5′-ATC TTG TGC TCT TGC TGG G-3′; NLRP3 forward, 5′-CTT CCT
TTC CAG TTT GCT GC-3′; and reverse, 5′-TCT CGC AGT CCA CTT CCT
TT-3′; human IL-1β forward, 5′-CAG CCA ATC TTC ATT GCT
CA-3′; and reverse, 5′-TCG GAG ATT CGT AGC TGG AT-3′; caspase-1
forward, 5′-GGA CTC TCA GCA GCT CCT CAG GCA-3′; and reverse, 5′-GCA
AAG CTT GAC ATT CCC TTC TGA GCC-3′; and ASC forward, 5′-CCT
ACG GCG CCG AGC TCA C-3′; and reverse, 5′-CTC CAG AGC CCT GGT GCG
T-3′; which were synthesized at Sangon Biotech (Shanghai, China).
For qPCR, total RNA was isolated using RNAiso and converted to cDNA
immediately using the PrimeScript™ RT Master mix (Perfect
Real-Time) (Takara, Dalian, China) following the manufacturer’s
instructions and stored at -20°C until use. Each cDNA was amplified
with the previously listed primers and SYBR Premix Ex Taq™ II (Tli
RNaseH Plus) (Takara) for 40 cycles, and results were analyzed
using the Mx3005P System Software (ABI Stratagene, La Jolla, CA,
USA).
Caspase-1 activity assays
The activity of caspase-1 was determined based on
the ability of caspase-1 to change acetyl-Tyr-Val-Ala-Asp
p-nitroanilide (Ac-YVAD-pNA) into the yellow formazan product
p-nitroaniline (pNA) using a Caspase-1 Activity kit (Beyotime).
Cell lysates were centrifuged at 13,000 x g for 10 min, and the
protein concentrations were determined by the Bradford protein
assay. Cellular extracts (30 μg of protein) were incubated
in a 96-well microtiter plate with 20 ng of Ac-DEVD-pNA overnight
at 37°C. The absorbance values of pNA at 405 nm, optical density
(OD)405, were measured using a 96-well plate reader
(BioTek, Santa Barbara, CA, USA). An increase in the
OD405 indicated activation of caspase-1.
RNA interference
Small hairpin (sh) RNA lentivirus against human
NLRP3 (target, 5′-GGA GAG ACC TTT ATG AGA AAG-3′), human
ASC (target, 5′-GCA AGA TGC GGA AGC TCT TCA-3′), human
caspase-1 (target, 5′-GCA CAC GTC TTG CTC TCA TTA-3′) and scrambled
sequence (5′-GTT CTC CGA ACG TGT CAC GT-3′) were synthesized at
GenePharma (Shanghai, China). Undifferentiated THP-1 cells were
infected with shRNA lentivirus (MOI=100) and selected over three
passages in the presence of puromycin (Sangon, Shanghai, China).
Specifically, THP-1 cells stably expressing indicated shRNAs were
selected with 1 μg/ml puromycin for 2 weeks, followed by 0.5
μg/ml puromycin for an additional 2 weeks, with the medium
changed every 3 days.
Enzyme-linked immunosorbent assay (ELISA)
and immunoblot
The supernatant and cell lysate of
PMA-differentiated THP-1 cells were collected at the given time
points between 6 h and 3 days post-infection, as indicated. The
concentration of the IL-1β level in the cell culture supernatant
was determined by ELISA according to the manufacturer’s
instructions (R&D Systems, Minneapolis, MN, USA). Samples were
tested in triplicate and data were analyzed using GraphPad software
version 5.0 (GraphPad Software, San Diego, CA, USA). The presence
of pro-caspase-1 and pro-IL-1β in cell lysate was measured by
immunoblot as described previously (38), and the bio-active form of IL-1β
and caspase-1 in the supernatant were detected with antibodies that
target IL-1β and caspase-1.
ATP assay
THP-1 culture supernatant was collected at the
indicated time points post-infection. The concentration of ATP was
determined immediately using the ATP assay kit (Beyotime) according
to the manufacturer’s instructions.
ROS release assay
The assay is based on the incorporation of
2′,7′-dichlorofluorescein diacetate into the cell. THP-1 cells were
infected with HTNV for 1 day, or were treated with 100 μM
H2O2 as a positive control or with cold DPBS
as a negative control for 10 min, stained with 10 μM H2DCFDA
(Sigma-Aldrich), and subsequently ROS release was evaluated by
Cytomics FC 500 (Beckman-Coulter, Fullerton, CA, USA).
Statistical analyses
All the data are expressed as mean ± standard
deviation. The statistical significance of the obtained data was
analyzed using a two-tailed unpaired t-test in GraphPad Prism 5.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Induction and secretion of IL-1β by HTNV
in THP-1 cells
The human monocytic cell line, THP-1, is widely used
in inflammasome research (19),
and it has previously been established that Hantavirus can infect
THP-1 cells (39,40). Vero E6, HUVEC and
PMA-differentiated THP-1 were infected with HTNV at an MOI of 1,
and intracellular nucleoprotein was detected at 3 days
post-infection by immunofluorescent staining. The fraction of THP-1
cells infected with HTNV was lower than the fraction of Vero E6 and
HUVEC cells (Fig. 1A), consistent
with previously published data (40). As the level of secreted IL-1β
indicates activation of inflammasome, ELISA was used to assess the
concentration of IL-1β in cell supernatants. Significantly high
levels of IL-1β (95.6±27.0 pg/ml) secretion from PMA-differentiated
THP-1 were detected 24 h after HTNV infection compared to the
control group (16.2±5.0 pg/ml) (P<0.05) (Fig. 1B). The concentration of IL-1β
corresponded with the MOI of HTNV (Fig. 1B), and the level of IL-1β in the
supernatant peaked at 24 h post-infection (Fig. 1D). PMA-differentiated THP-1 cells
incubated with heat-inactivated HTNV also secreted increased levels
of IL-1β (33.2±9.0 pg/ml), however, this was less than that of the
PMA-differentiated THP-1 cells incubated with infectious HTNV and
there was no statistical significance compared to the mock group
(Fig. 1B).
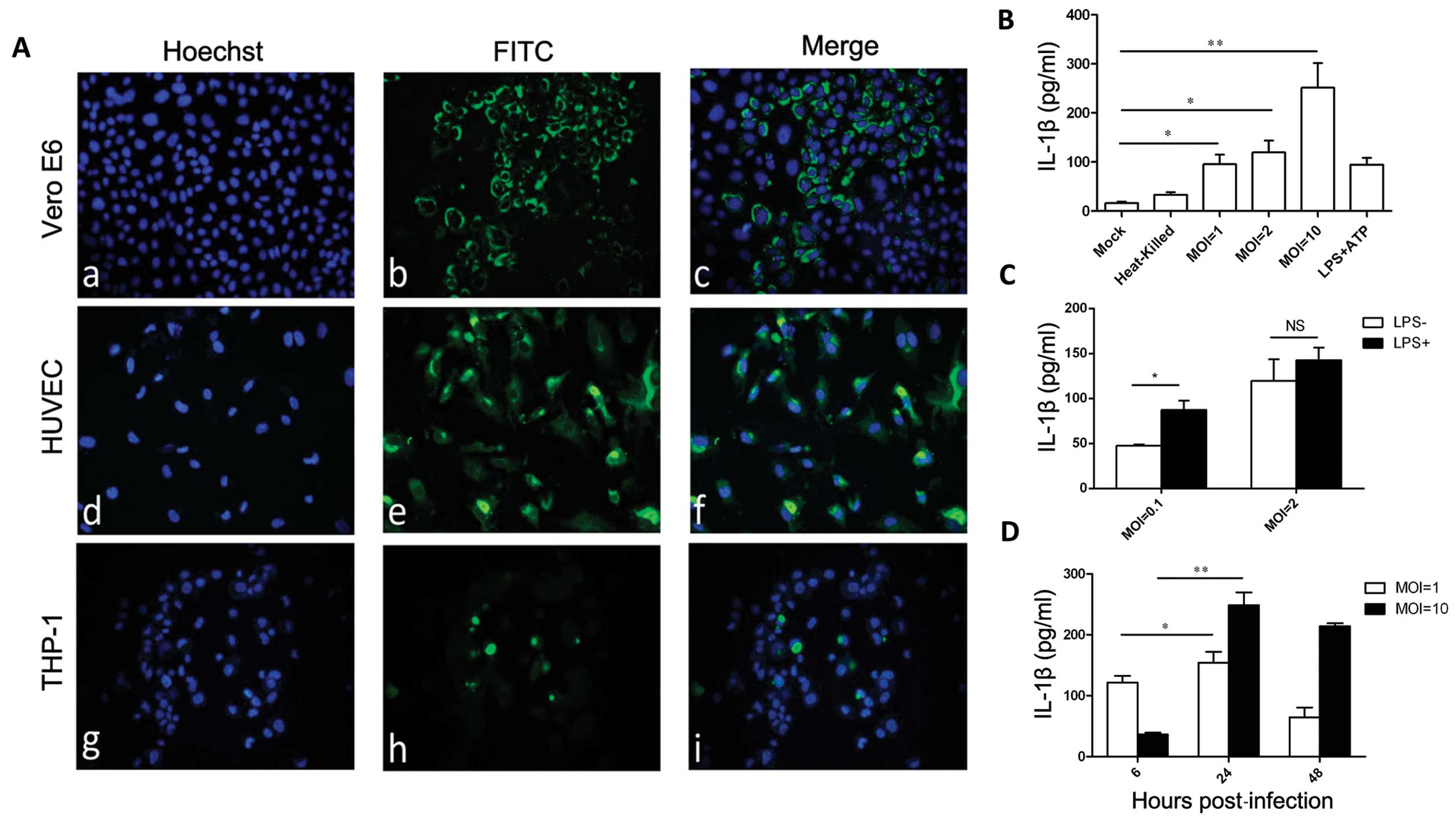 | Figure 1Hantaan virus (HTNV) induces the
production of interleukin-1β (IL-1β) in THP-1 cells. (A)
Immunofluorescent staining of HTNV nucleoprotein in infected Vero
E6, human umbilical vein endothelial cells (HUVEC) and phorbol
12-myristate-13-acetate (PMA) differentiated-THP-1. (A–C) Vero E6,
(D–F) HUVEC and (G–I) THP-1 were infected with HTNV and were fixed
72 h post-infection. (A, D and G) Nuclei were stained with Hoechst;
and (B, E and H) HTNV nucleoprotein was detected with fluorescein
isothiocyanate (FITC)-1A8. Data are representative of three
independent experiments. (B) Enzyme-linked immunosorbent assay
(ELISA) detection of IL-1β in the supernatant of HTNV-infected
THP-1. PMA-differentiated THP-1 cells were incubated with
infectious HTNV at a multiplicity of infection (MOI) of 1, 2 or 10,
or heat-inactivated HTNV, lipopolysaccharide (LPS) + adenosine
triphosphate (ATP) at 24 h post-infection. (C) ELISA detection of
IL-1β in the supernatant of HTNV-infected THP-1. PMA-differentiated
THP-1 cells were incubated with HTNV at an MOI of 2 or 0.1 in the
presence or absence of PMA, and 24 h post-infection the IL-1β level
in the supernatant was measured by ELISA. (D) ELISA detection of
IL-1β in the supernatant of HTNV-infected THP-1. PMA-differentiated
THP-1 cells were incubated with HTNV at an MOI of 1 or 10, and the
level of IL-1β in the supernatant was measured by ELISA at the
indicated times post-infection. (B–D) Data are representative of
three independent experiments each performed in triplicate (errors
bars represent standard error of the mean). *P<0.05,
**P<0.01; NS, not significant. |
Secretion of IL-1β requires two independent
processes. First is the expression of pro-IL-1β and, second is the
inflammasome cleavage of pro-IL-1β into IL-1β (19). LPS is routinely employed in
vitro to initiate the expression of pro-IL-1β (41). To examine whether LPS-enhanced
pro-IL-1β expression further increased HTNV-induced IL-1β
secretion, PMA-differentiated THP-1 cells were infected in the
presence or absence of LPS. The addition of LPS increased the level
of IL-1β secreted by HTNV-infected PMA-differentiated THP-1 cells
24 h post-infection, as illustrated in Fig. 1C; 87.3±14.8 pg/ml for MOI= 0.1 and
142.8±19.7 pg/ml for MOI=2 compared to 47.8±1.6 pg/ml for MOI= 0.1
and 119.7±33.8 pg/ml for MOI=2. There was no statistical
significance between these two groups, indicating that HTNV was
likely inducing pro-IL-1β expression independently.
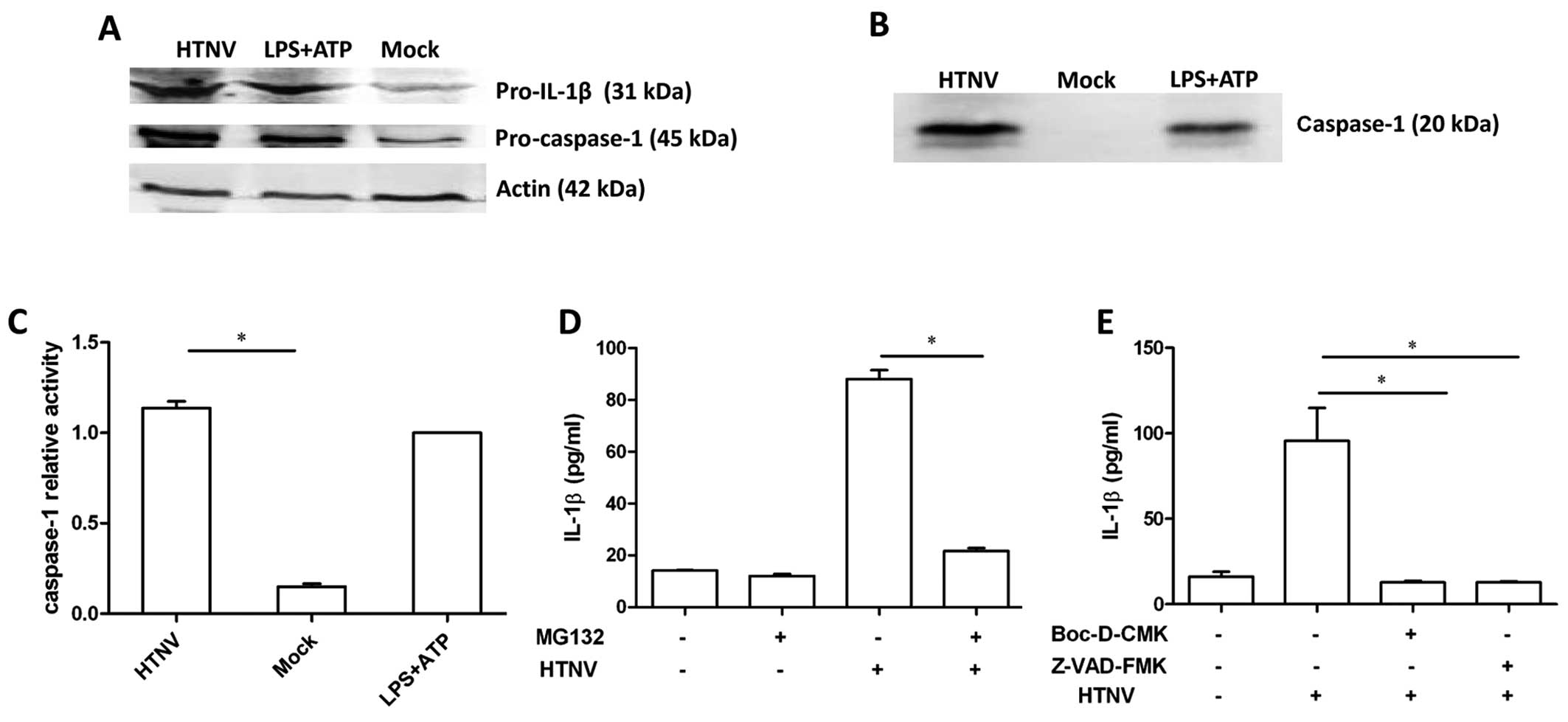
HTNV activates caspase-1 and pro-IL-1β in
THP-1 cells
As HTNV induced IL-1β secretion in the absence of
LPS, it is likely that HTNV induced pro-IL-1β expression in THP-1
cells. To evaluate this hypothesis, the total protein from
HTNV-infected THP-1 cells was isolated and the level of pro-IL-1β
and pro-caspase-1 protein was assessed by immunoblot. Increased
levels of pro-IL-1β and pro-caspase-1 were detected in cells
incubated with either HTNV or LPS and ATP (Fig. 2A), which is consistent with our
previous results (Fig. 1C).
Bioactive caspase-1 is required to cleave pro-IL-1β into IL-1β. In
order to investigate whether caspase-1 was activated during HTNV
infection, the culture supernatant of HTNV-infected THP-1 was
ultra-filtered and an increased concentration of secreted caspase-1
was detected post-infection (Fig.
2B). In addition, the activity of caspase-1 was detected in
THP-1 cell lysates, and the activation of caspase-1 was
significantly increased 12 h post-infection (1.137±0.064) compared
to the mock group (0.15±0.026) (P<0.0001) (Fig. 2C). These results indicate that
HTNV-infected THP-1 generated the bioactive form of caspase-1.
Incubation with the potent proteasome inhibitor
MG132 significantly reduced the concentration of IL-1β in the
culture supernatant of HTNV-infected cells, 88.1±4.8 pg/ml compared
to 21.6±1.6 pg/ml (P=0.047) (Fig.
2D), indicating that the proteasome may participate in the
process of HTNV IL-1β induction. To further investigate the role of
caspase-1 in HTNV-induced IL-1β secretion, cells were incubated
with the specific inhibitors of caspase-1, Z-VAD-FMK and Boc-D-CMK,
in advance of HTNV infection. Z-VAD-FMK and Boc-D-CMK significantly
reduced the level of IL-1β secretion (P<0.05) (Fig. 2E), indicating that caspase-1
participated in HTNV-induced IL-1β secretion.
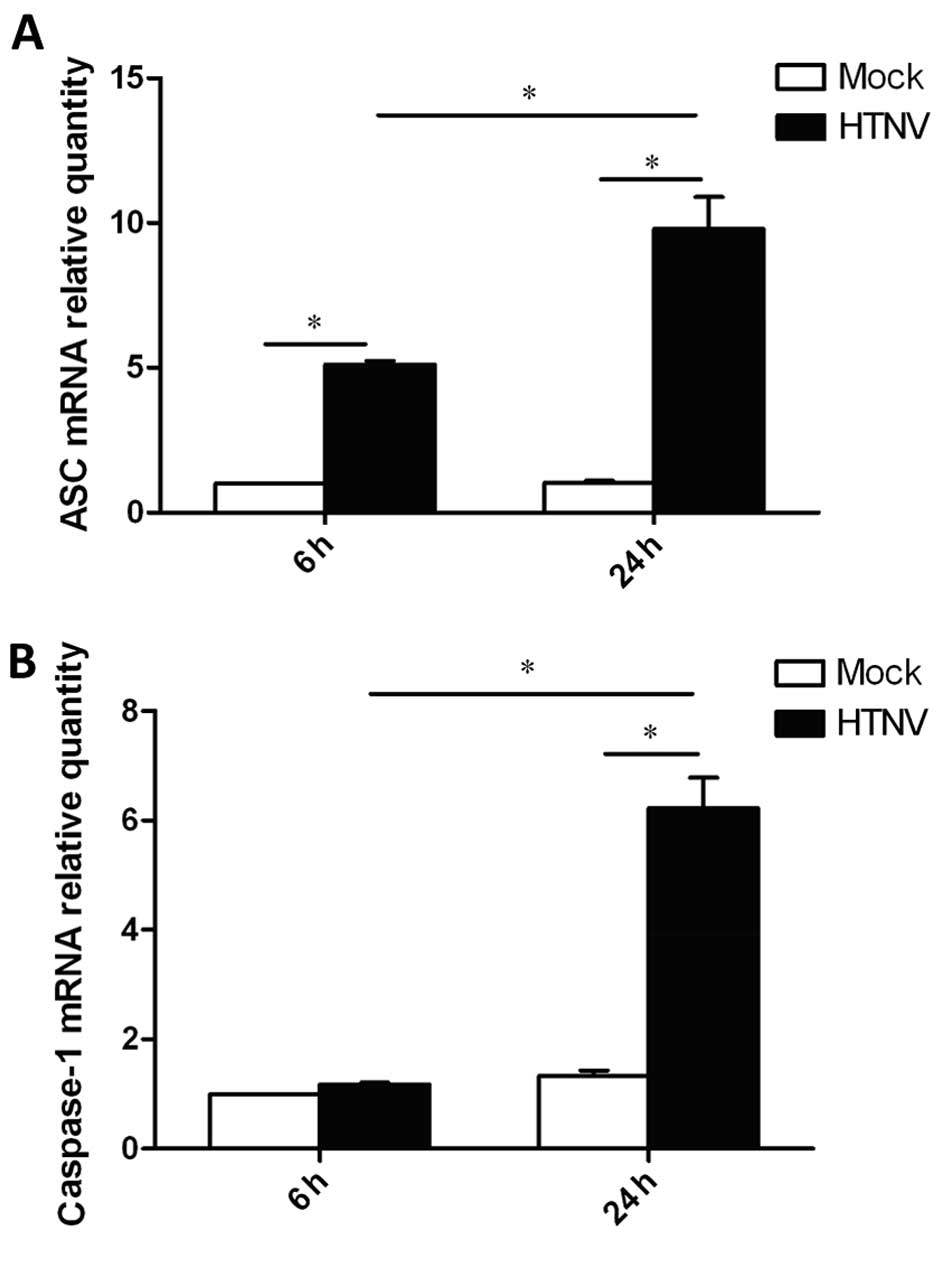
mRNA level of ASC and caspase-1 are
elevated in the HTNV-infected THP-1
To address the mechanism by which IL-1β secretion
was triggered, the total cellular RNA was extracted from
HTNV-infected THP-1 cells. Using qPCR, the mRNA levels of
ASC and caspase-1, key molecules forming the inflammasome,
were measured (Fig. 3). The level
of caspase-1 mRNA was increased by ~6-fold in HTNV-infected cells
24 h post-infection (Fig. 3B).
The level of ASC mRNA was increased by ~5-fold 6 h
post-infection, and reached an increase of 10-fold 24 h
post-infection (Fig. 3A). Thus,
these results indicate that HTNV-infected THP-1 induce expression
of ASC and caspase-1, which participate in the induction of
IL-1β secretion.
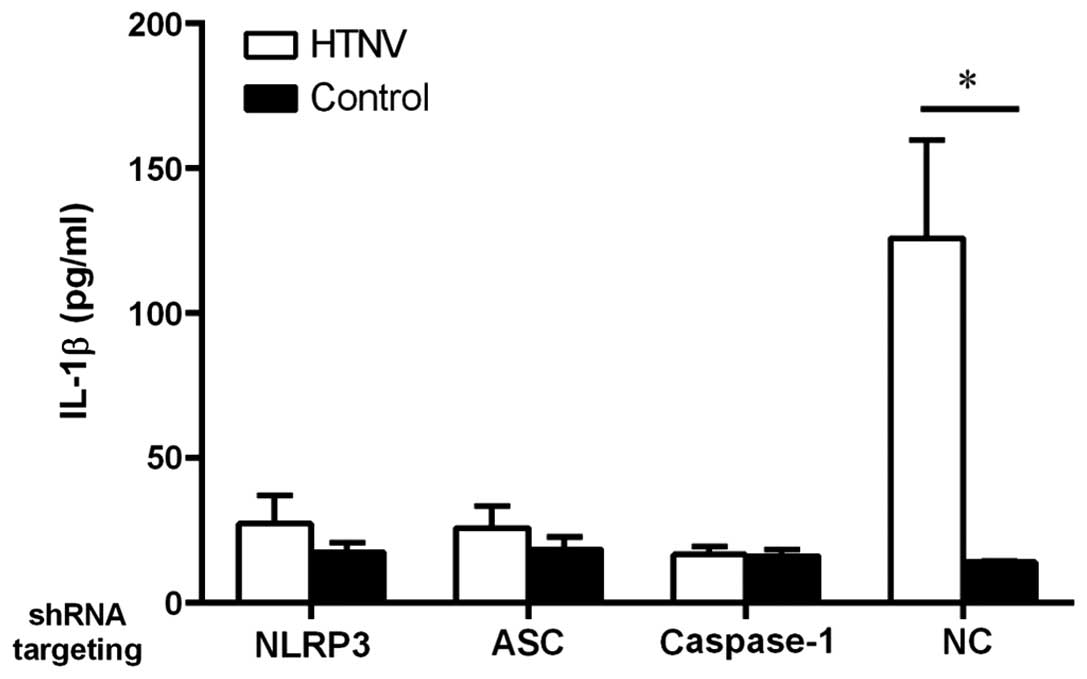
Effect of NLRP3, ASC and caspase-1 shRNA
on IL-1β secretion
As the NLRP3 inflammasome is activated by numerous
viruses (17,19,24,25,26,42–52), we suspect that NLRP3 may
participate in HTNV induction of IL-1β. To evaluate the role of
NLRP3 in the induction of IL-1β following HTNV infection, an shRNA
lentivirus was used to knockdown the expression of NLRP3. In
comparison to the non-targeting control shRNA (NC), the cells in
which NLRP3 was knocked down secreted less IL-1β (Fig. 4). As a control, the level of IL-1β
induced by knockdown of ASC and caspase-1 was also compared
to further examine the role of NLRP3 in HTNV infection and
this determined that, as expected, the cells in which ASC
and caspase-1 were knocked down also generated less IL-1β (Fig. 4). These results indicate that the
assembly of the NLRP3 inflammasome complex is responsible for the
increased secretion of IL-1β in HTNV-infected THP-1 cells.
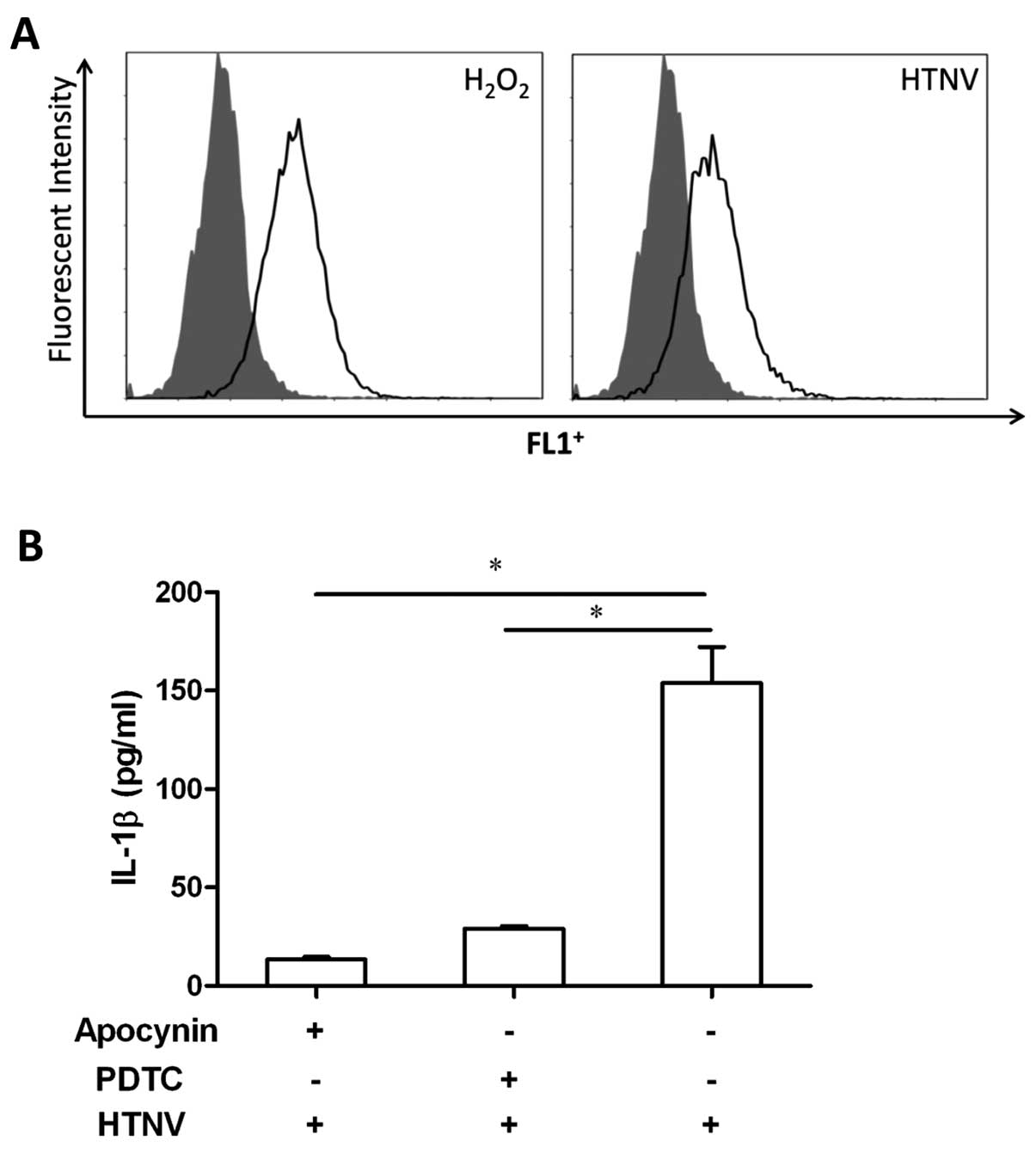
HTNV activation of NLRP3 correlates with
ROS release but not the extracellular ATP level
ATP is regarded as a damage signal that can induce
the formation of inflammasome (19,53). We speculated that extracellular
ATP may be involved in HTNV-induced IL-1β secretion. The
supernatant from HTNV-infected THP-1 cells was collected at
different time points, but no change was found in the level of ATP
correlating with HTNV infection (data not shown). However,
increased levels of ROS were detected in HTNV-infected THP-1 cells
(Fig. 5A). To further investigate
the role of ROS in inflammasome formation during HTNV infection,
THP-1 cells were treated with the ROS inhibitors apocynin and
ammonium PDTC, prior to HTNV infection. The level of IL-1β in the
supernatant of HTNV-infected cells was decreased by ROS inhibitors
(Fig. 5B); 153.9±36.7 pg/ml
compared to 13.5±2.5 pg/ml for apocynin (P<0.001) and 29.0±2.8
pg/ml for PDTC (P<0.001). Thus, during HTNV infection, ROS
induced by HTNV contributed to NLRP3 inflammasome formation in
THP-1 cells.
Discussion
Secreted cytokines produce an inflammatory
microenvironment aiding the elimination of pathogens; however,
excessive cytokine secretion can cause damage to host tissues and
produce a ‘cytokine storm’, which is speculated to be the main
cause of mortality resulting from SARS-CoV and pandemic influenza A
infection (54–57). Induction of pro-inflammatory
molecules may play an important role in the pathogenesis of HFRS
(13–15); however, little is known regarding
the mechanism by which hantavirus elicits an inflammatory
processes. As an important cytokine in the inflammation and
angiogenesis, IL-1β is critical for induction of fever and an
inflammatory microenvironment inhibiting pathogen invasion
(16,58,59). However, IL-1β is also considered
as a detrimental factor in certain cases, inducing
hyperpermeability of the endothelium. IL-1β has been found to
induce endothelial hyperperme-ability during dengue hemorrhagic
fever (17,24,25). In vivo and in vitro
studies have reported that the level of circulating IL-1β was
elevated in HFRS patients (15)
and was induced in hantavirus-infected cells (39,40). The present study reports that
secretion of IL-1β from HTNV-infected THP-1 cells requires the
assembly of the NLRP3 inflammasome and is ROS-signal dependent.
Not only pathogens, but also aluminum, asbestos,
silica crystals and monosodium urate crystals were identified as
‘danger’ signals inducing formation of the inflammasome (30,60). The present study identified that
HTNV efficiently induces the expression of pro-IL-1β in THP-1
cells, and the secretion of mature IL-1β. Previously, TLR3 and
RIG-I have been shown to sense hantavirus and induce expression of
type I interferon (61,62), and the NLRP3 inflammasome can be
induced by viral RNA via activation of TLR3 and RIG-I (51,63). HTNV induced less IL-1β in the
presence of the ROS and NF-κB inhibitor PDTC in the present study.
PDTC likely inhibited the translation of pro-IL-1β and inflammasome
formation. A live virus induces more mature IL-1β secretion
compared to a heat-inactivated virus, suggesting that
HTNV-induction of IL-1β secretion requires cytosolic viral
replication.
The outcome of the inflammasome signal is generation
of bioactive caspase-1 and subsequent cleavage of pro-IL-1β
(18,20). The mRNA level of ASC and
caspase-1 were increased 24 h post-infection in the present study,
indicating that HTNV may alter the transcriptional level
inflammasome components, as suggested by previous findings
(46,64). A significant increase was also
identified in the level of ASC and caspase proteins in
HTNV-infected THP-1, and increased caspase-1 activity. By knocking
down the level of caspase-1 in THP-1, the secretion of IL-1β was
reduced, consistent with the established mechanism by which RNA
viruses activate inflammasome in primary macrophages or THP-1 cells
(18,46,65–68).
As opposed to the AIM2 and IFI16 inflammasomes,
which can sense DNA (69–72), the molecular partners of NLRP3
remain unknown (19,42). Previous studies have indicated
that certain pathogens can directly activate the inflammasome. For
example, the M2 ion channel protein of influenza A has been
reported to activate the NLRP3 inflammasome (23). Dengue virus interacts with CLEC5A
to form the NLRP3 inflammasome (25). The component of the hantavirus
HTNV that is responsible for induction of the NLRP3 inflammasome
requires further investigation, but the present results support the
role of ROS in the induction of the NLRP3 inflammasome complex.
In conclusion, the present study indicates that the
NLRP3 inflammasome complex formation and the subsequent induction
of bioactive caspase-1 and the cleavage of pro-IL-1β are
responsible for HTNV-induced IL-1β secretion in THP-1 cells. These
results provide an important insight into the role of the NLRP3
inflammasome in the pathogenesis of HFRS, and may highlight
potential strategies for the treatment of HFRS.
Acknowledgments
The authors would like to thank Dr Huijie Bian for
providing the THP-1 cell line. The present study was supported in
part by grants from the Major State Basic Research Development
Program of China (973) (no. 2012CB518905), the National Major
Infectious Diseases Prevention and Control Special Issues (no.
2013ZX10004609-002) and the National Natural Science Foundation
General Program of China (nos. 30970148 and 30972590).
References
|
1
|
Vaheri A, Strandin T, Hepojoki J, et al:
Uncovering the mysteries of hantavirus infections. Nat Rev
Microbiol. 11:539–550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mir MA: Hantaviruses. Clin Lab Med.
30:67–91. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mustonen J, Mäkelä S, Outinen T, et al:
The pathogenesis of nephropathia epidemica: new knowledge and
unanswered questions. Antiviral Res. 100:589–604. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Macneil A, Nichol ST and Spiropoulou CF:
Hantavirus pulmonary syndrome. Virus Res. 162:138–147. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jonsson CB, Figueiredo LT and Vapalahti O:
A global perspective on hantavirus ecology, epidemiology, and
disease. Clin Microbiol Rev. 23:412–441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Watson DC, Sargianou M, Papa A, Chra P,
Starakis I and Panos G: Epidemiology of Hantavirus infections in
humans: a comprehensive, global overview. Crit Rev Microbiol.
40:261–272. 2014. View Article : Google Scholar
|
|
7
|
Yi J, Xu Z, Zhuang R, et al: Hantaan virus
RNA load in patients having hemorrhagic fever with renal syndrome:
correlation with disease severity. J Infect Dis. 207:1457–1461.
2013. View Article : Google Scholar
|
|
8
|
Yu L, Bai W, Wu X, et al: A recombinant
pseudotyped lentivirus expressing the envelope glycoprotein of
Hantaan virus induced protective immunity in mice. Virol J.
10:3012013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Linderholm M and Elgh F: Clinical
characteristics of hantavirus infections on the Eurasian continent.
Curr Top Microbiol Immunol. 256:135–151. 2001.PubMed/NCBI
|
|
10
|
Gupta S, Braun M, Tischler ND, et al:
Hantavirus-infection confers resistance to cytotoxic
lymphocyte-mediated apoptosis. PLoS Pathog. 9:e10032722013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krautkrämer E, Grouls S, Stein N, Reiser J
and Zeier M: Pathogenic old world hantaviruses infect renal
glomerular and tubular cells and induce disassembling of
cell-to-cell contacts. J Virol. 85:9811–9823. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rang A: Modulation of innate immune
responses by hantaviruses. Crit Rev Immunol. 30:515–527. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li M, Ji Y, Dong Y, Zhou Y, Ren H and Xie
M: The detection of vascular endothelial growth factor in serum of
patients with hemorrhagic fever with renal syndrome. Inflammation.
36:962–967. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saksida A, Wraber B and Avšič-Županc T:
Serum levels of inflammatory and regulatory cytokines in patients
with hemorrhagic fever with renal syndrome. BMC Infect Dis.
11:1422011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang L, Li XL, Dai Y, Qiu ZF and Li TS:
Change of plasma pro-inflammatory cytokines levels in patients with
hemorrhagic fever with renal syndrome. Zhongguo Yi Xue Ke Xue Yuan
Xue Bao. 30:607–609. 2008.In Chinese. PubMed/NCBI
|
|
16
|
Zhu W, London NR, Gibson CC, et al:
Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt
vascular stability. Nature. 492:252–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hottz ED, Lopes JF, Freitas C, et al:
Platelets mediate increased endothelium permeability in dengue
through NLRP3-inflammasome activation. Blood. 122:3405–3414. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gram AM, Frenkel J and Ressing ME:
Inflammasomes and viruses: cellular defence versus viral offence. J
Gen Virol. 93:2063–2075. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bauernfeind F, Ablasser A, Bartok E, et
al: Inflammasomes: current understanding and open questions. Cell
Mol Life Sci. 68:765–783. 2011. View Article : Google Scholar
|
|
20
|
Rathinam VA, Vanaja SK and Fitzgerald KA:
Regulation of inflammasome signaling. Nat Immunol. 13:333–342.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kanneganti TD: Central roles of NLRs and
inflammasomes in viral infection. Nat Rev Immunol. 10:688–698.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito M, Yanagi Y and Ichinohe T:
Encephalomyocarditis virus viroporin 2B activates NLRP3
inflammasome. PLoS Pathog. 8:e10028572012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ichinohe T, Pang IK and Iwasaki A:
Influenza virus activates inflammasomes via its intracellular M2
ion channel. Nat Immunol. 11:404–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tan TY and Chu JJ: Dengue virus-infected
human monocytes trigger late activation of caspase-1, which
mediates pro-inflammatory IL-1β secretion and pyroptosis. J Gen
Virol. 94:2215–2220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu MF, Chen ST, Yang AH, et al: CLEC5A is
critical for dengue virus-induced inflammasome activation in human
macrophages. Blood. 121:95–106. 2013. View Article : Google Scholar
|
|
26
|
Dowling JK and O’Neill LA: Biochemical
regulation of the inflammasome. Crit Rev Biochem Mol Biol.
47:424–443. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lupfer C and Kanneganti TD: The expanding
role of NLRs in antiviral immunity. Immunol Rev. 255:13–24. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Proell M, Gerlic M, Mace PD, Reed JC and
Riedl SJ: The CARD plays a critical role in ASC foci formation and
inflammasome signaling. Biochem J. 449:613–621. 2013. View Article : Google Scholar
|
|
29
|
Kanneganti TD, Lamkanfi M, Kim YG, et al:
Pannexin-1-mediated recognition of bacterial molecules activates
the cryopyrin inflammasome independent of Toll-like receptor
signaling. Immunity. 26:433–443. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hornung V, Bauernfeind F, Halle A, et al:
Silica crystals and aluminum salts activate the NALP3 inflammasome
through phagosomal destabilization. Nat Immunol. 9:847–856. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cruz CM, Rinna A, Forman HJ, Ventura AL,
Persechini PM and Ojcius DM: ATP activates a reactive oxygen
species-dependent oxidative stress response and secretion of
proinflammatory cytokines in macrophages. J Biol Chem.
282:2871–2879. 2007. View Article : Google Scholar
|
|
32
|
Cassel SL, Eisenbarth SC, Iyer SS, et al:
The Nalp3 inflammasome is essential for the development of
silicosis. Proc Natl Acad Sci USA. 105:9035–9040. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koshiba T, Bashiruddin N and Kawabata S:
Mitochondria and antiviral innate immunity. Int J Biochem Mol Biol.
2:257–262. 2011.PubMed/NCBI
|
|
34
|
Nakahira K, Haspel JA, Rathinam VA, et al:
Autophagy proteins regulate innate immune responses by inhibiting
the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011. View Article : Google Scholar :
|
|
35
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
36
|
Xu Z, Wei L, Wang L, Wang H and Jiang S:
The in vitro and in vivo protective activity of monoclonal
antibodies directed against Hantaan virus: potential application
for immunotherapy and passive immunization. Biochem Biophys Res
Commun. 298:552–558. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cheng L, Yu L, Wu X, et al: Induction of
specific humoral and cellular immune responses in a mouse model
following gene fusion of HSP70C and Hantaan virus Gn and S0.7 in an
adenoviral vector. PLoS One. 9:e881832014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Deng JX, Nie XJ, Lei YF, et al: The highly
conserved 5′ untranslated region as an effective target towards the
inhibition of Enterovirus 71 replication by unmodified and
appropriate 2′-modified siRNAs. J Biomed Sci. 19:732012. View Article : Google Scholar
|
|
39
|
Markotic A, Hensley L, Daddario K, Spik K,
Anderson K and Schmaljohn C: Pathogenic hantaviruses elicit
different immunoreactions in THP-1 cells and primary monocytes and
induce differentiation of human monocytes to dendritic-like cells.
Coll Antropol. 31:1159–1167. 2007.
|
|
40
|
Shin OS, Yanagihara R and Song JW:
Distinct innate immune responses in human macrophages and
endothelial cells infected with shrew-borne hantaviruses. Virology.
434:43–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Davis BK, Wen H and Ting JP: The
inflammasome NLRs in immunity, inflammation, and associated
diseases. Annu Rev Immunol. 29:707–735. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Leavy O: Inflammasome: Turning on and off
NLRP3. Nat Rev Immunol. 13:12013. View Article : Google Scholar
|
|
43
|
Bird L: Innate immunity: Linking
mitochondria and microbes to inflammasomes. Nat Rev Immunol.
12:2292012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Henao-Mejia J, Elinav E, Strowig T and
Flavell RA: Inflammasomes: far beyond inflammation. Nat Immunol.
13:321–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Leemans JC, Cassel SL and Sutterwala FS:
Sensing damage by the NLRP3 inflammasome. Immunol Rev. 243:152–162.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Burdette D, Haskett A, Presser L, McRae S,
Iqbal J and Waris G: Hepatitis C virus activates interleukin-1β via
caspase-1-inflammasome complex. J Gen Virol. 93:235–246. 2012.
View Article : Google Scholar :
|
|
47
|
Barlan AU, Griffin TM, McGuire KA and
Wiethoff CM: Adenovirus membrane penetration activates the NLRP3
inflammasome. J Virol. 85:146–155. 2011. View Article : Google Scholar :
|
|
48
|
Nour AM, Reichelt M, Ku CC, Ho MY,
Heineman TC and Arvin AM: Varicella-zoster virus infection triggers
formation of an interleukin-1β (IL-1β)-processing inflammasome
complex. J Biol Chem. 286:17921–17933. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rajan JV, Rodriguez D, Miao EA and Aderem
A: The NLRP3 inflammasome detects encephalomyocarditis virus and
vesicular stomatitis virus infection. J Virol. 85:4167–4172. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Komune N, Ichinohe T, Ito M and Yanagi Y:
Measles virus V protein inhibits NLRP3 inflammasome-mediated
interleukin-1β secretion. J Virol. 85:13019–13026. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Poeck H, Bscheider M, Gross O, et al:
Recognition of RNA virus by RIG-I results in activation of CARD9
and inflammasome signaling for interleukin 1 beta production. Nat
Immunol. 11:63–69. 2010. View Article : Google Scholar
|
|
52
|
Kaushik DK, Gupta M, Kumawat KL and Basu
A: NLRP3 inflammasome: key mediator of neuroinflammation in murine
Japanese encephalitis. PLoS One. 7:e322702012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lau SK, Lau CC, Chan KH, et al: Delayed
induction of proinflammatory cytokines and suppression of innate
antiviral response by the novel Middle East respiratory syndrome
coronavirus: implications for pathogenesis and treatment. J Gen
Virol. 94:2679–2690. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tisoncik JR, Korth MJ, Simmons CP, Farrar
J, Martin TR and Katze MG: Into the eye of the cytokine storm.
Microbiol Mol Biol Rev. 76:16–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cheng XW, Lu J, Wu CL, et al: Three fatal
cases of pandemic 2009 influenza A virus infection in Shenzhen are
associated with cytokine storm. Respir Physiol Neurobiol.
175:185–187. 2011. View Article : Google Scholar
|
|
57
|
Huang KJ, Su IJ, Theron M, et al: An
interferon-gamma-related cytokine storm in SARS patients. J Med
Virol. 75:185–194. 2005. View Article : Google Scholar
|
|
58
|
Hussain SP and Harris CC: Inflammation and
cancer: an ancient link with novel potentials. Int J Cancer.
121:2373–2380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sims JE and Smith DE: The IL-1 family:
regulators of immunity. Nat Rev Immunol. 10:89–102. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dostert C, Petrilli V, Van Bruggen R,
Steele C, Mossman BT and Tschopp J: Innate immune activation
through Nalp3 inflammasome sensing of asbestos and silica. Science.
320:674–677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Handke W, Oelschlegel R, Franke R, Kruger
DH and Rang A: Hantaan virus triggers TLR3-dependent innate immune
responses. J Immunol. 182:2849–2858. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lee MH, Lalwani P, Raftery MJ, et al: RNA
helicase retinoic acid-inducible gene I as a sensor of Hantaan
virus replication. J Gen Virol. 92:2191–2200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Guillot L, Le Goffic R, Bloch S, et al:
Involvement of toll-like receptor 3 in the immune response of lung
epithelial cells to double-stranded RNA and influenza A virus. J
Biol Chem. 280:5571–5580. 2005. View Article : Google Scholar
|
|
64
|
Kummer JA, Broekhuizen R, Everett H, et
al: Inflammasome components NALP 1 and 3 show distinct but separate
expression profiles in human tissues suggesting a site-specific
role in the inflammatory response. J Histochem Cytochem.
55:443–452. 2007. View Article : Google Scholar
|
|
65
|
Allen IC, Scull MA, Moore CB, et al: The
NLRP3 inflammasome mediates in vivo innate immunity to influenza A
virus through recognition of viral RNA. Immunity. 30:556–565. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ichinohe T, Lee HK, Ogura Y, Flavell R and
Iwasaki A: Inflammasome recognition of influenza virus is essential
for adaptive immune responses. J Exp Med. 206:79–87. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kanneganti TD, Body-Malapel M, Amer A, et
al: Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in
response to viral infection and double-stranded RNA. J Biol Chem.
281:36560–36568. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Thomas PG, Dash P, Aldridge JR Jr, et al:
The intracellular sensor NLRP3 mediates key innate and healing
responses to influenza A virus via the regulation of caspase-1.
Immunity. 30:566–575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kerur N, Veettil MV, Sharma-Walia N, et
al: IFI16 acts as a nuclear pathogen sensor to induce the
inflammasome in response to Kaposi sarcoma-associated herpesvirus
infection. Cell Host Microbe. 9:363–375. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Unterholzner L, Keating SE, Baran M, et
al: IFI16 is an innate immune sensor for intracellular DNA. Nat
Immunol. 11:997–1004. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rathinam VA, Jiang Z, Waggoner SN, et al:
The AIM2 inflammasome is essential for host defense against
cytosolic bacteria and DNA viruses. Nat Immunol. 11:395–402. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hornung V, Ablasser A, Charrel-Dennis M,
et al: AIM2 recognizes cytosolic dsDNA and forms a
caspase-1-activating inflammasome with ASC. Nature. 458:514–518.
2009. View Article : Google Scholar : PubMed/NCBI
|