1. Introduction
Interferons (IFNs) were first characterized for
their ability to ‘interfere’ with viral replication, and indeed one
of their major functions is the establishment of a robust antiviral
state in response to infection (1,2).
In addition, IFNs activate immune cells and facilitate the
recognition of virus-infected cells and tumor cells by the immune
system, as they stimulate antigen presentation to T lymphocytes
(3). IFNs are usually subdivided
into three classes: type I (including IFN-α, IFN-β and IFN-ω), type
II (in humans, IFN-γ) and type III [including interleukin (IL)-28
and IL-29] (3). Type I IFNs are
produced by virus-infected cells. Plasmacytoid dendritic cells
(DCs) and mononuclear phagocytes are the major sources of IFN-α
(4), while IFN-β is produced by a
number of cell types, including fibroblasts (5).
IFN-β is the principal antiviral factor secreted by
infected mammalian cells in response to the activation of retinoic
acid-inducible gene (RIG-I) following Newcastle disease virus (NDV)
infection. Type I IFNs bind to the IFN-α membrane receptor (IFNAR)
(2) on infected cells and through
Janus kinase (JAK) and signal transducer and activator of
transcription (STAT) signaling (6), induce the expression of genes whose
products enhance the susceptibility of cells to cytotoxic natural
killer (NK) cell- and T cell-mediated killing (7). In addition, type I IFNs induce
resistance to viral replication in all cells, thereby involving
autocrine and paracrine actions (7).
In this review, immune activation by NDV in humans
is compared to immune evasion by Ebola virus (EBOV). Such timely
comparison is justified as both phenomena are associated with the
activation of the same two signaling pathways.
Unlike NDV, which is a pathogen found in birds but
not in humans, EBOV, as a virus affecting primates, is a
devastating pathogen affecting humans. During approximately 200
million years of evolution, viruses from mammals, (derived from
therapsids), have had time to adapt to the immune systems of their
host. According to a recent whole-genome analysis, 95% of bird
species (derived from sauropsids) developed during a rapid
radiation following the Cretaceous-Paleogene mass extinction
approximately 66 million years ago (8). Thus, bird viruses have had a shorter
time period for adaptation to the host immune system than mammalian
viruses. The characteristics of the two RNA viruses, NDV and EBOV,
are reviewed herein with particular focus on the aspect of the
species-specificity of viral escape mechanisms antagonizing type I
IFN responses.
2. Newcastle disease virus
NDV in birds
NDV is one of the most important diseases affecting
poultry worldwide. NDV outbreaks were first reported in Indonesia,
and, subsequently, in Newcastle-upon-Tyne in the year 1926.
Infections by virulent NDV strains cause severe economic losses and
may have flock mortality rates of up to 100%. Therefore, NDV has a
significant impact on the world economy, possibly more so than any
other disease affecting animals (9).
NDV belongs to the avian paramyxovirus serotype
(APMV)-1 family and is the most characterized member among the nine
APMV serotypes. It is possible that all species of birds are
susceptible to NDV infection. However, the disease may vary greatly
depending upon the virus strain and the host species. Eighteen NDV
strains from four lineages have been identified and classified as
velogenic, mesogenic and lentogenic according to their pathotypes
(10). NDV attaches to
respiratory epithelial cells through the viral HN protein, which
binds to sialic acid containing cell surface receptors, such as
gangliosides and N-glycoproteins. This is followed by the
activation of the F protein, which leads to the fusion of the viral
and the host cell membranes. In the cytoplasm of the host cell, the
viral genome, a 15 kb non-segmented negative single-stranded RNA
(ssRNA), is transcribed into mRNAs and is translated into viral
proteins (11). Respiratory
disease can be mild in the case of lentogenic viruses, more severe
with mesogenic viruses and severe with a high mortality rate in the
ase of velogenic viruses. Velogenic viruses are further subdivided
into viscerotropic, which cause mortality with haemorrhagic lesions
in the intestines, and neurotropic, when neurological diseases
predominate without haemorrhagic lesions in the intestines
(12).
All NDV strains encode seven proteins: N, P, V, M,
F, HN and L. The V protein is not essential for viral replication
in vitro and serves as an accessory protein. The V protein
is a frameshift variant of the NDV phosphoprotein P. and, while the
P protein consists of 395 amino acids, the V protein consists of
only 239 amino acids. The incorporation of two G nucleotides at the
RNA editing site of the P protein results in the frameshift variant
protein V (13). The V protein of
NDV has been shown to inhibit the IFN response in birds in two
ways: i) through the inhibition of IFN signaling by targeting STAT1
for degradation (14); and ii)
through interaction with melanoma differentiation-associated gene 5
(MDA5), leading to the inhibition of interferon regulatory factor 3
(IRF-3) activation and IFN-β induction (15).
Of note, it has been demonstrated that the V protein
of NDV is a determinant of host range restriction. Recombinant NDV
(rNDV) mutants, which are defective in the expression of V protein,
grow poorly in embryonated chicken eggs and chicken embryo
fibroblasts compared to wild-type (WT) rNDV. Furthermore, the NDV V
protein has been shown to play an important role in preventing
apoptosis in a species-specific manner. It has been suggested that
the host range of NDV is limited by the specificity of its V
protein for bird proteins to efficiently prevent innate host
defenses, such as the IFN response and apoptosis (16–18).
In recent years, NDV has drawn a lot of research
interest, not only due to the fact that it is an important pathogen
affecting poultry, but also that in man, it exerts fascinating
oncolytic and immune stimulatory effects. It also has potential for
use as a novel vaccine vector for the treatment of diseases in
humans and animals (10).
NDV in mouse and man
The NDV-induced type I IFN
response
Upon the infection of mouse or human cells with the
NDV bird virus, an uninhibited type I IFN response (19) is initiated, as the viral V protein
cannot interact with the proteins from mammalian cells. It is of
particular significance that the activation of a rapid and strong
type I IFN response by NDV in normal mouse or human cells prevents
viral replication, cytotoxic effects and disease pathology in
normal tissues. Thus, NDV is not considered a pathogen in mouse or
man and shows a high safety profile as regards its clinical
application.
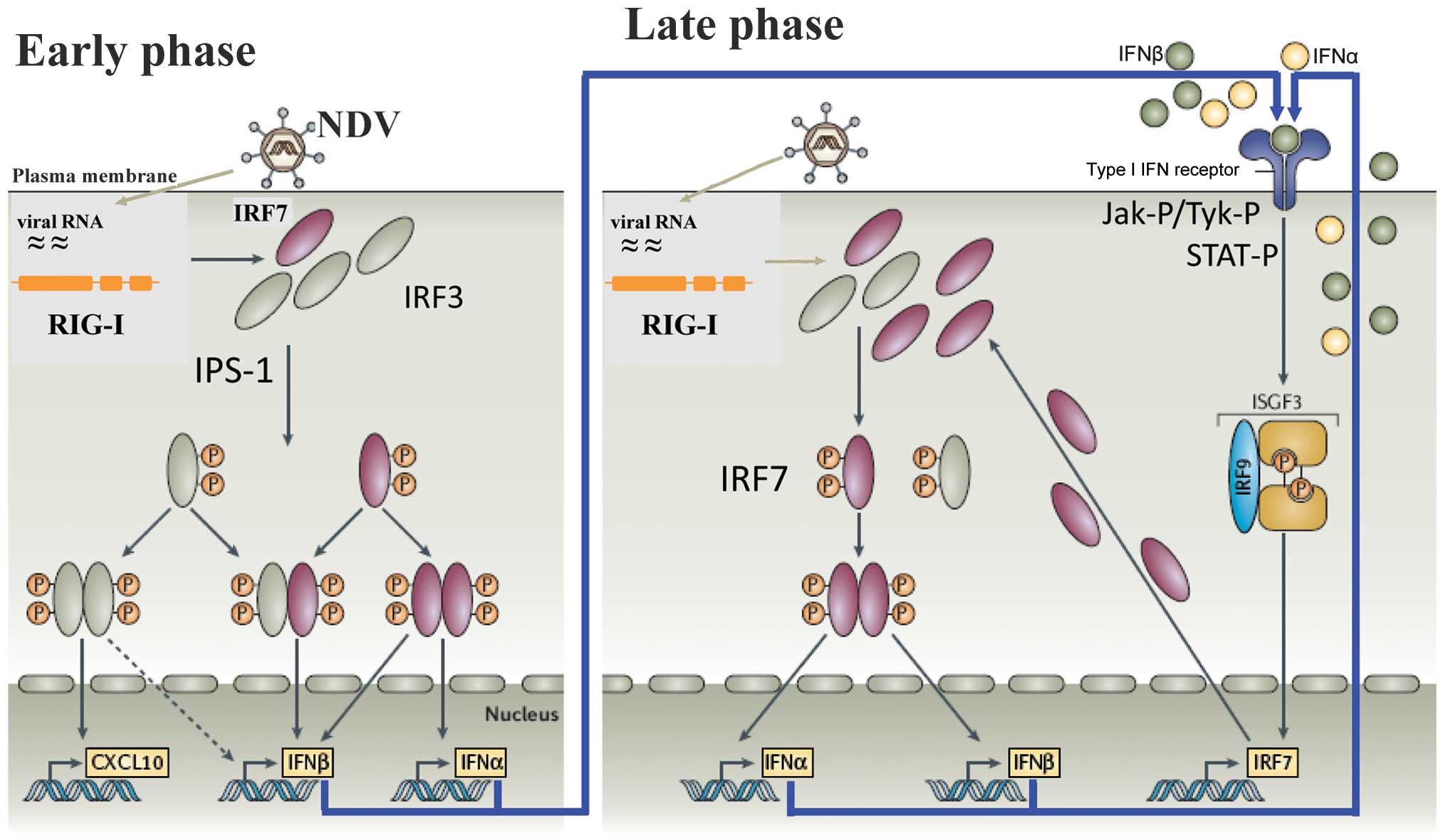
A diagram of the cellular response in mouse and man
is illustrated in Fig. 1. It
shows an early- and a late-phase response. During the early phase,
the antiviral response of normal (non-tumor) cells is initiated
through the recognition of viral RNA by two types of pathogen
recognition receptors: i) endosomal Toll-like receptors (TLRs),
particularly TLR3 and ii) cytoplasmic RIG-I-like receptors (RLRs).
RIG-I has been shown to be the cytoplasmic viral RNA receptor for
NDV (20,21). Of note, RIG-I binds specifically
to RNA containing 5′-phosphate, such as viral RNA, while mammalian
RNA is either capped or contains base modifications (22). Once activated, RIG-I binds to the
adaptor protein IFN-β promoter stimulator-1 (IPS-1) which, after a
further signaling cascade, activates, IRF-3 during the early phase.
This transcription factor (TF) is then phosphorylated, translocates
to the nucleus and induces the IFN response (23). IRF-3 plays an important role in
the IFN response of mouse macrophages to NDV infection (24).
RIG-I triggering does not only involve the
upregulation of RNA copies. RIG has the structural combination of
an N-terminal caspase recruitment domain (CARD) not and a
C-terminal RNA helicase domain with which it interacts with the
viral non-capped RNA (25).
Following the recognition of small viral RNAs, RIG-I elicits
signaling cascades, which eventually lead to the activation of the
nuclear factor (NF)-κB and IRF-3 TFs. NF-κB regulates the
production of most cytokines and chemokines (26), while IRF-3 is central to the
development of an antiviral state through the induction of
antiviral genes (27). The rapid
and robust expression of type I IFN genes is a prerequisite for the
induction of numerous antiviral proteins that modulate protein
synthesis, growth arrest and apoptosis (28).
During the late phase of the IFN response, the type
I IFN molecules secreted during the early phase interact with the
cell surface, express IFNAR and initiate an amplification loop of
the IFN response, which involves STAT proteins and IRF-7 (29). IFNAR (2) is expressed by virtually all cells in
the body and consists of IFNAR1 and IFNAR2 chains. The cytoplasmic
domains of IFNAR1 and IFNAR2 are physically associated with the
JAKs, Tyk2 and JAK1, respectively. Ligands binding IFNAR represent
a large family of four-helix bundle cytokines, numbering close to
20 in humans and mice. Upon ligand binding, Tyk2 and JAK1 become
activated by transphosphorylation (6). These, in turn, phosphorylate
receptor tyrosine(s), directing the recruitment of inactive STAT1
and STAT2. At the receptor, STAT1 and STAT2 are activated by
phosphorylation (29,30). The STAT proteins then
heterodimerize and with the IFN regulatory factor IRF-9, to form a
complex known as ISGF3 (Fig. 1).
This translocates to the nucleus to bind to the IFN-stimulated
response element (ISRE). This DNA-binding complex directs the
expression of IFN-stimulated genes (ISGs) that create the antiviral
state in the target cells and block viral replication.
Importance of the IFN receptor-mediated
feedback amplification loop
IFNAR plays a crucial role in the IFN feedback
amplification loop and its consequences. This is illustrated in
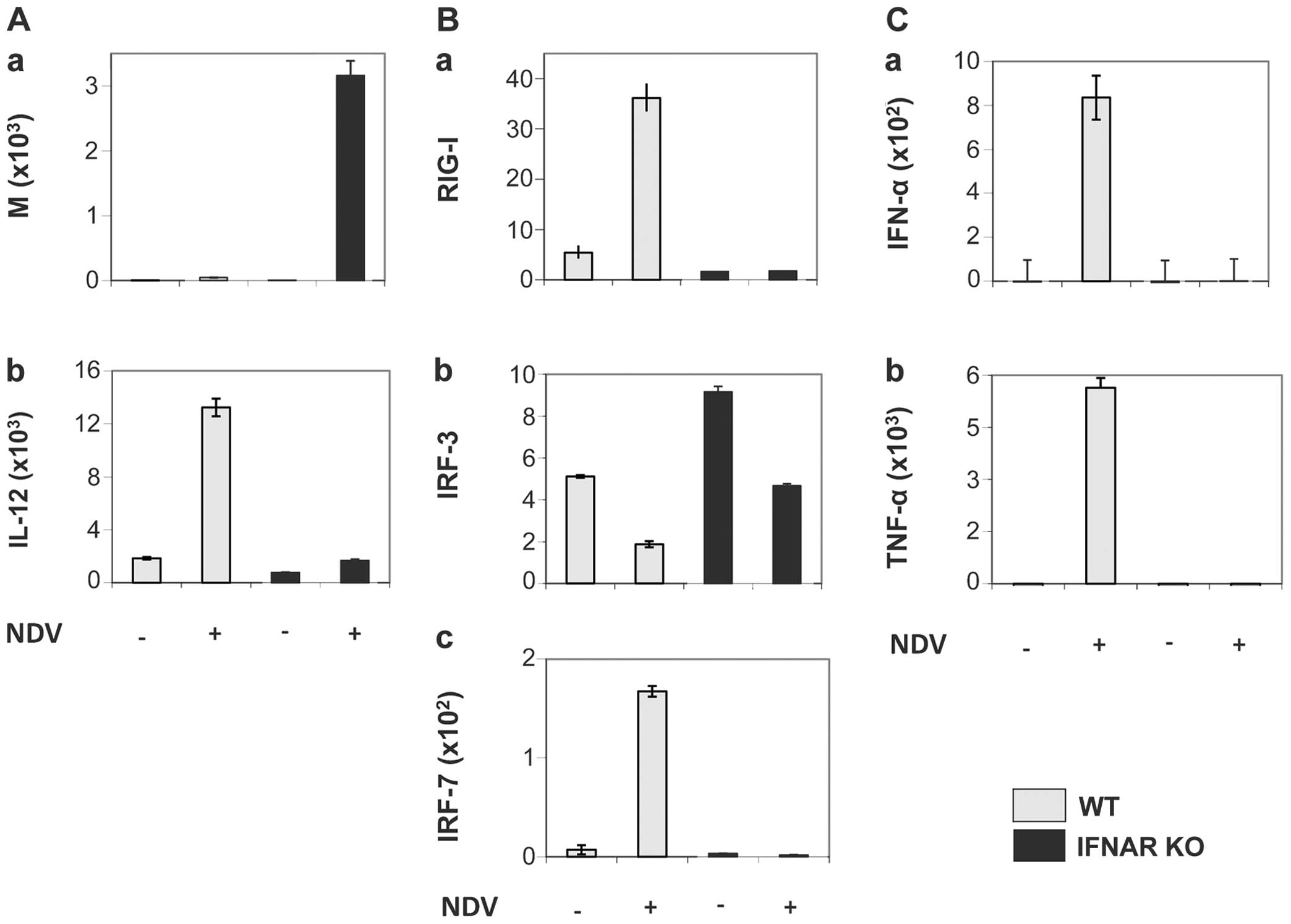
Fig. 2, which shows the results
of the NDV infection of murine DCs from either C57BL/6 WT mice or
IFNAR gene knockout (KO) mice. Fig.
2A (panel a) shows viral replication 10.5 h following infection
by NDV (strain Ulster) assessed by RT-PCR of the viral M gene. Over
3,000 M gene copies were obtained from the DCs of KO mice in
comparison to <100 copies from cells obtained from WT mice. Cell
culture supernatants were tested for the content of the cytokines,
Il-12, IFN-α and tumor necrosis factor (TNF)-α. The NDV infection
of DCs from WT mice induced a strong expression of genes coding for
IL-12p70 (Fig. 2A, panel b),
IFN-α and TNF-α (Fig. 2C, panels
a and b), while this was not the case with DCs from KO mice.
In DCs from WT mice, the produced IFN-α caused a
feedback loop stimulation through IFNAR. This resulted in the
suppression of viral replication and the expression of
pro-inflammatory cytokines, such as TNF-α and IL-12. In the cells
from KO mice, neither RIG-I nor IRF-7 were upregulated upon NDV
infection, in contrast to the cells from WT mice (Fig. 2B, panels a and c). Further results
with DCs from mice deficient in either IRF-3 or IRF-7, or from
IRF-3/-7 double KO mice revealed that RIG-I triggering by NDV
replication in mouse DCs induced IL-12 production independently of
the IRF-3 and IRF-7 pathway (31).
DCs function to maintain tissue-specific tolerance
or they can be immunogenic and initiate antigen-specific T cell
immunity. It depends on the microenvironment in vivo and on
inflammatory stimuli whether immature DCs with functional
plasticity differentiate into tolerogenic or immunogenic DCs with
stable phenotypes. Murine DCs, upon infection with NDV,
differentiate into the immunogenic phenotype DC1 characterized by
the secretion of pro-inflammatory cytokines, in particular IL-12
and IFN-α/β (31). The priming or
programming for DC1 involves two receptor-initiated signaling
cascades, the first one initiated through cytoplasmic RIG-I, and
the second through membrane-expressed IFNAR (21,32).
NDV, a prototype avian virus that may be
used to study an uninhibited cellular response to viral infection
in human DCs
We investigated the effects of NDV on human DCs by
analyzing the release of cytokines important for Th1 or Th2
polarization. Human monocyte-derived DCs were found to become
polarized towards DC1 by in vitro stimulation with NDV
(33).
Pathogenic viruses subvert normal immune functions
in DCs through the expression of immune antagonists (18,34). Understanding how these antagonists
interact with the host immune response requires knowledge of the
underlying genetic regulatory network that operates during an
uninhibited antiviral response. Such a network was identified by
studying human DCs and their response to infection by NDV (19), knowing that this virus is able to
stimulate innate immunity and DC maturation through the activation
of RIG-I signaling and lacks the ability to evade the human IFN
response.
A new approach was developed, integrating
genome-wide expression kinetics and time-dependent promoter
analysis. It was found that the genetic program underlying the
antiviral cell-state transition during the first 18 h
post-infection can be explained by a single convergent regulatory
network. Gene expression changes were driven by a step-wise
multi-factor cascading control mechanism, where the specific TFs
controlling expression changed over time. This study of systems
biology involved, among others, microarray experiments, microarray
analysis, transcription factor binding site analysis,
time-dependent promoter analysis, electromobility shift assay and
regulatory network construction (19). Through all these new tools, this
analysis revealed a robust antiviral transcriptional network that
may be induced in human DCs by infection with NDV. Table I lists the 24 critical TFs and
their time of expression following infection with NDV. The timing
of this program appeared as highly conserved.
 | Table INDV infection of human DCs: Kinetics
of upregulated transcription factors. |
Table I
NDV infection of human DCs: Kinetics
of upregulated transcription factors.
| Transcription | Hours | Transcription | Hours |
|---|
| factor | p.i. | factor | p.i |
| IRF-1 | 2.5 | IRF-7 | 3 |
| STAT1 | 4 | STAT2 | 4 |
| ATF3 | 5 | IRF-2 | 6 |
| CREM | 6 | MAX | 7–8 |
| STAT3 | 7–8 | RUNX3 | 8.5 |
| RELA | 8.5 | FOXC1 | 8.5 |
| IRF-8 | 9–10 | ALX1 | 9–10 |
| TGIF1 | 10–11 | IRF | 11–12 |
| STAT4 | 11–12 | NFB | 12 |
| NF-κB2 | 12 | EGR4 | 12.5 |
| FOX03 | 13.5 | ZEB1 | 14 |
| REL | 14.5 | STAT5A | 18 |
The described network of TFs spans virtually the
entire time-period analyzed. Of the 24 TFs, 18 appear in the known
general pathogen response signature (35) or in the core DC response signature
(36). The TFs are predicted to
regulate 779 of the 1,351 upregulated genes. The network contains
both feed-forward links, which propagate the transcriptional signal
through time, as well as feedback links, where TFs may influence
the activity of targets that have previously been upregulated. It
was concluded that the proposed network is effective in capturing
the underlying biology and produces a pattern that is consistent
with a step-wise transcriptional signal propagation.
Tumor-selective replication, safety and
the activation of immune cells
Integral to the life cycle of all RNA viruses is the
formation of double-stranded RNA (dsRNA), which activates a
spectrum of cellular defence mechanisms involving IFN-α/β. Tumors
from mouse or man provide a relatively permissive substrate for the
propagation of RNA viruses, such as NDV as mutations in tumor cells
often cripple the IFN system to allow uninhibited proliferation and
to provide resistance to apoptosis (37). The first step of infection with
NDV takes place in all cell types of mouse or man, whereas the
second step (which corresponds to viral replication) occurs only in
tumor cells since it is stopped rapidly in normal cells. The
replication of NDV involves the use of the full-length viral
antigenome as a template. This step is prevented in non-tumorigenic
mouse or human cells (37).
An inverse correlation was found between the
expression of four antiviral genes and the susceptibility of cells
to infection with NDV: i) RIG-I, ii) IRF-3, iii) IFN-β and iv)
IRF-7 (20). In addition, the
membrane receptor IFNAR was demonstrated to be of great importance
(21). The basic or induced
levels of the four aforementioned genes were higher in the normal
cells than in the tumor cells. In addition, signaling through IFNAR
was often found to not be fully functional in tumor cells. Taken
together, these observations explain the high safety profile of NDV
upon human application (10,11).
The activation of immune cells is a further factor
for the safety of this virus. In vitro infection with NDV
has been shown to cause the activation of human NK cells (38), human monocytes (39) and had a co-stimulatory effect on
CD4 (40) and CD8 (41) mouse and human T cells. The
activated cells exerted cytotoxic effects through NKp46 (38), TNF-related apoptosis-inducing
ligand (TRAIL) (39) and released
nitric oxide (NO) (42) and
pro-inflammatory cytokines (31).
3. Ebola virus
EBOV in man
The Filoviridae family of viruses includes
the genera EBOV and Marburg virus (43). EBOV was first discovered in 1976.
With a diameter of 80 nm and a length of up to 14,000 nm, EBOV
belongs to the largest known RNA viruses. Similar to NDV, the
genome consists of a negative ssRNA. It codes for eight proteins,
two of which are the viral proteins, VP24 and VP35, which are
discussed below.
EBOV infects primates (gorillas, chimpanzees and
humans). The primary target cells are macrophages and DCs (44). The zoonotic transmission of EBOV
to humans causes severe and often times lethal hemorrhagic fever.
The disease characteristics are systemic inflammatory response
syndrome (SIRS), disseminated intravascular coagulation (DIC),
systemic hemorrhage and multiple organ failure (45). EBOV shuts down the host’s innate
and adaptive immune systems. It then replicates uncontrollably and
causes a cytokine storm in the host (46).
Filoviral infections in primates are associated with
ineffective innate antiviral responses as a result of virally
encoded immune antagonists. These render the host incapable of
mounting effective innate or adaptive immune responses. During the
first 3 weeks after infection, the release of endogenous pyrogenes
(IL-1β, IL-6 and TNF-α) is prevented. Several filoviral encoded
components target type I IFN responses. Many of these innate immune
suppression mechanisms that are important for viral replication and
pathogenesis are species-specific (47).
EBOV: a virus-inhibited IFN response
Two of the eight viral proteins of EBOV are involved
in immunosuppression (48,49).
They prevent type I IFN signaling in multiple ways, which is also
the topic of the present review. While VP35 antagonizes the early
phase of the IFN response, VP24 is an antagonist of the late phase.
The EBOV VP35 protein binds directly to dsRNA and inhibits several
antiviral signaling pathways. It acts as a component of the viral
RNA polymerase complex, a viral assembly factor and an inhibitor of
host IFN production. Mutation of selected basic residues within the
C-terminal half of VP35 abrogates its dsRNA-binding activity,
impairs VP35-mediated IFN antagonism, and attenuates EBOV growth
in vitro and in vivo. The structure of the C-terminal
VP35 IFN inhibitory domain (IID), solved to a resolution of 1.4 Å,
revealed a unique fold centered on Arg-312 (48). In an analogous study, Marburg
virus VP35 proteins were demonstrated to be capable of fully
coating the backbone and capping the ends of dsRNA for IFN
antagonism (50). Furthermore, it
was shown that conserved basic residues in the IID recognize the
dsRNA backbone, whereas the dsRNA blunt ends are ̔end-capped̓ by a
pocket of hydrophobic residues that mimic the RLR recognition of
blunt-end dsRNA, the initiation step of the early-phase response
(Fig. 1, early phase) (51).
VP35 also blocks virus-induced IRF-3
phosphorylation, subsequent IRF-3 dimerization and nuclear
translocation. VP35 thus inhibits the early induction of antiviral
genes, including the IFN-β gene (Fig.
1, early phase). VP35 is also capable of preventing the
IRF-3-dependent activation of the IFN-α4 promoter in response to
viral infection (52). It is also
able to inhibit the antiviral response induced by IFN-α (Fig. 1, late phase). The phosphorylation
of the dsRNA-dependent protein kinase (PKR) and of the elongation
initiation factor eIF-2α was also suppressed in cells expressing
VP35 (53). A single amino acid
change in the VP35 protein was demonstrated to be capable of
reversing the inhibition of host innate immune responses. Thus,
infection with a mutated recombinant virus (recEbo-VP35/R312A)
resulted in a strong innate immune response, including the
increased expression of MDA5, RIG-1, regulated on activation,
normal T cell expressed and secreted (RANTES), monocyte
chemoattractant protein-1 (MCP-1), ISG15, ISG54, ISG56, ISG60,
STAT1, IFN-β, 2,5-oligoad-enylate synthetase (OAS) and myxovirus
(influenza virus) resistance 1 (MX1) (54).
During antiviral defence, IFNAR signaling triggers
the nuclear transport of tyrosine-phosphorylated STAT1 (PY-STAT1),
which occurs through a subset of karyopherin α (KPNA) nuclear
transporters. EBOV VP24 (eVP24) binds directly to STAT1 and
inhibits its nuclear translocation, a step of the late-phase
feedback loop (Fig. 1). Recently,
it has been demonstrated that eVP24 targets a unique nuclear
localization signal (NLS) binding site on KPNA to selectively
compete with nuclear import of PY-STAT1 (49). It leaves the transport of other
cargo that may be required for viral replication unaffected. New
crystal structures of VP24 derived from pathogenic and
non-pathogenic EBOV revealed a pyramidal fold with sites required
for virulence and for STAT1 binding (55). Such studies offer templates for
drug design, and provide the three-dimensional framework necessary
for the biological dissection of the many functions of VP24 in the
virus life cycle.
4. Conclusions
The comparison of the two viruses, NDV and EBOV, in
this review demonstrates that RNA viruses with a similar genome may
exert completedly different effects in man. Depending on whether
they are derived from birds or primates, they can exert either
beneficial or detrimental effects. Signals delivered through RIG-I
and IFNAR cause immune activation, which is the case with NDV, or
they are antagonized causing immune evasion, which is the case with
EBOV.
The infection of human DCs with NDV induces a robust
uninhibited antiviral response which prevents viral replication,
and differentiates and polarizes the cells towards DC1-activating
Th1 cells. When NVD infects mouse or human cells it activates a
multitude of genes, cells and cellular activities. In vivo,
such viral ‘priming’ of cells may lead to immune system stimulation
or immune system conditioning, which mostly relies on the effects
of the induced type I IFN response.
Signaling through RIG-I and IFNAR may have
far-reaching consequences for the antiviral immune response. The
activation of the RIG-I pathway is able to reduce the antigen
requirement by 10- to 100-fold in inducing optimal
influenza-specific cellular and humoral responses, including
protective immunity. These effects include an enhanced germinal
center reaction, T follicular helper cell responses, antibody
affinity maturation and plasma cell responses in draining lymph
nodes, the spleen and bone marrow. These effects are dependent on
type I IFN and IPS-1 signaling, but are independent of the MyD88-
and TLR3-mediated pathways (56).
Since type I IFN receptors are expressed by virtually every type of
cell in the body, the ligand-induced IFN signaling cascades have
far-reaching consequences. For instance, type I IFN-mediated
crosstalk between plasmacytoid DCs on one side and macrophages and
conventional DCs on the other, secure the control of fatal
cytopathic mouse hepatitis virus (57).
Primate-derived EBOV succeeds in antagonizing the
human IFN response by two proteins, VP35 and VP24, which
specifically target signals through RIG-I and IFNAR, respectively,
thereby demonstrating the importance of these coordinated signaling
systems. The bird-derived virus, NDV, has only one protein, the
frameshift variant V protein, with which to antagonize the IFN
response. Perhaps this difference can be accounted for by the
difference in the respective time periods available for adaptation
during evolution. EBOV may be more potent than NDV in antagonizing
the IFN response as it has two, instead of only one, inhibitory
proteins. However, it is likely that it is not the quantity but the
quality of inhibitory proteins that matters (58).
5. Prospects
Due to its high safety profile in human application
(10,11), NDV may be employed not only as an
oncolytic agent in cancer patients, but also as an agent for immune
stimulation and for prophylacting conditioning of the host immune
system against the risk of viral infection. This may be
particularly relevant for individuals whow come into contact with
patients infected with EBOV. Such immune-conditioning pre-treatment
would ̔prime̓ the cells and establish a state of increased viral
resistance. In this way, one virus, the avian NDV, may exert
beneficial effects against the other virus, EBOV. Beneficial
effects can be expected in particular during the early
non-symptomatic phase of infection. It has been suggested that
applying a non-specific antiviral approach during the incubation
period of viral infection is an essential protective approach which
renders the host immune sytem into an alert state, thus attenuating
viral replication (46). The use
of an IFN-inducing agent, such as NDV, may be thus more effective
and may cause less severe side-effects than the use of IFN-β, which
has also been suggested (59).
Other immunological means for counteracting EBOV
infection may be based on neutralizing antibodies, particularly
when isolated from memory B cells (60) of patients who have survived the
infection. In addition, libraries of memory T cells may be
developed from such patients to screen the T cell immune
repertoires for protective activity, function and specificity
(61). This is supported by a
recent immune analytical study (62).
In connection with the EBOV epidemic in West Africa
in 2014, a vaccine was developed by the Canadian National
Microbiology Laboratory on the basis of recombinant vesicular
stomatitis virus (rVSV) expressing the filovirus glycoprotein of
the lethal Zaire ebolavirus (ZEBOV). Antibodies were reported as
necessary for rVSV/ZEBOV-GP-mediated protection against lethal EBOV
challenge in non-human primates (63). VSV, similar to NDV and EBOV, is a
negative ssRNA virus (64).
Similar to rabies, it belongs to the family of
Rhabdoviridae. It causes stomatitis vesicularis, an
infectious disease affecting hooved mammals (cattle, horse and
pig). In humans, it can cause flu-like symptoms, swelling of the
lymph nodes and neurological side-effects.
It is conceivable to design a recombinant vaccine
vector against EBOV based on NDV. This would ensure an uninhibited
IFN response with all its positive consequences as discussed in the
present review. Whether a mammalian virus such as VSV can exert an
uninhibited and similarly strong IFN response as that of NDV in
other mammals, including man, remains to be investigated.
Acknowledgments
The author acknowledges financial support from the
German Cancer Research Center, Heidelberg, at which the study on
murine KO cells was performed.
References
|
1
|
Pestka S, Krause CD and Walter MR:
Interferons, interferon-like cytokines, and their receptors.
Immunol Rev. 202:8–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levy DE and García-Sastre A: The virus
battles: IFN induction of the antiviral state and mechanisms of
viral evasion. Cytokine Growth Factor Rev. 12:143–156. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
González-Navajas JM, Lee J, David M and
Raz E: Immunomodulatory functions of type I interferons. Nat Rev
Immunol. 12:125–135. 2012.PubMed/NCBI
|
|
4
|
Kadowaki N, Antonenko S, Lau JY and Liu
YJ: Natural interferon alpha/beta-producing cells link innate and
adaptive immunity. J Exp Med. 192:219–226. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taniguchi T, Mantei N, Schwarzstein M, et
al: Human leukocyte and fibroblast interferons are structurally
related. Nature. 285:547–549. 1980. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shuai K and Liu B: Regulation of JAK-STAT
signalling in the immune system. Nat Rev Immunol. 3:900–911. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barber GN: Host defense, viruses and
apoptosis. Cell Death Differ. 8:113–126. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jarvis ED, Mirarab S, Aberer AJ, Li B,
Houde P, Li C, Ho SY, Faircloth BC, Nabholz B, Howard JT, et al:
Whole-genome analyses resolve early branches in the tree of life of
modern birds. Science. 346:1320–1331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alexander DJ: Newcastle Disease. Kluwer
Academic Publishers; Boston, MA: 1988, View Article : Google Scholar
|
|
10
|
Schirrmacher V and Fournier P: Newcastle
disease virus: A promising vector for viral therapy, immune
therapy, and gene therapy of cancer (Review). Methods Mol Biol.
542:565–605. 2009. View Article : Google Scholar
|
|
11
|
Fournier P and Schirrmacher V: Oncolytic
Newcastle Disease Virus as cutting edge between tumor and host.
Biology (Basel). 2:936–975. 2013.
|
|
12
|
Hanson RP and Brandly CA: Identification
of vaccine strains of Newcastle disease virus. Science.
122:156–157. 1955.PubMed/NCBI
|
|
13
|
Mebatsion T, de Vaan LT, de Haas N,
Römer-Oberdörfer A and Braber M: Identification of a mutation in
editing of defective Newcastle disease virus recombinants that
modulates P-gene mRNA editing and restores virus replication and
pathogenicity in chicken embryos. J Virol. 77:9259–9265. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang Z, Krishnamurthy S, Panda A and
Samal SK: Newcastle disease virus V protein is associated with
viral pathogenesis and functions as an alpha interferon antagonist.
J Virol. 77:8676–8685. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park MS, Shaw ML, Muñoz-Jordan J, Cros JF,
Nakaya T, Bouvier N, Palese P, García-Sastre A and Basler CF:
Newcastle disease virus (NDV)-based assay demonstrates
interferon-antagonist activity for the NDV V protein and the Nipah
virus V, W, and C proteins. J Virol. 77:1501–1511. 2003. View Article : Google Scholar :
|
|
16
|
Park MS, García-Sastre A, Cros JF, Basler
CF and Palese P: Newcastle disease virus V protein is a determinant
of host range restriction. J Virol. 77:9522–9532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Horvath CM: Weapons of STAT destruction.
Interferon evasion by paramyxovirus V protein. Eur J Biochem.
271:4621–4628. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hengel H, Koszinowski UH and Conzelmann
KK: Viruses know it all: new insights into IFN networks. Trends
Immunol. 26:396–401. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zaslavsky E, Hershberg U, Seto J, Pham AM,
Marquez S, Duke JL, Wetmur JG, Tenoever BR, Sealfon SC and
Kleinstein SH: Antiviral response dictated by choreographed cascade
of transcription factors. J Immunol. 184:2908–2917. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wilden H, Fournier P, Zawatzky R and
Schirrmacher V: Expression of RIG-I, IRF3, IFN-beta and IRF7
determines resistance or susceptibility of cells to infection by
Newcastle Disease Virus. Int J Oncol. 34:971–982. 2009.PubMed/NCBI
|
|
21
|
Fournier P, Wilden H and Schirrmacher V:
Importance of retinoic acid-inducible gene I and of receptor for
type I interferon for cellular resistance to infection by Newcastle
disease virus. Int J Oncol. 40:287–298. 2012.
|
|
22
|
Hornung V, Ellegast J, Kim S, Brzózka K,
Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, et al:
5′-Triphosphate RNA is the ligand for RIG-I. Science. 314:994–997.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taniguchi T and Takaoka A: The
interferon-alpha/beta system in antiviral responses: A multimodal
machinery of gene regulation by the IRF family of transcription
factors. Curr Opin Immunol. 14:111–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wilden H, Schirrmacher V and Fournier P:
Important role of interferon regulatory factor (IRF)-3 in the
interferon response of mouse macrophages upon infection by
Newcastle disease virus. Int J Oncol. 39:493–504. 2011.PubMed/NCBI
|
|
25
|
Goubau D, Schlee M, Deddouche S,
Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen
AG, Fujimura T, Rehwinkel J, et al: Antiviral immunity via
RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature.
514:372–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang X, Li S, Luo Y, Chen Y, Cheng S,
Zhang G, Hu C, Chen H and Guo A: Mycobacterium bovis and BCG induce
different patterns of cytokine and chemokine production in
dendritic cells and differentiation patterns in CD4+ T
cells. Microbiology. 159:366–379. 2013. View Article : Google Scholar
|
|
27
|
Grandvaux N, Servant MJ, tenOever B, Sen
GC, Balachandran S, Barber GN, Lin R and Hiscott J: Transcriptional
profiling of interferon regulatory factor 3 target genes: direct
involvement in the regulation of interferon-stimulated genes. J
Virol. 76:5532–5539. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sen GC and Peters GA: Viral
stress-inducible genes. Adv Virus Res. 70:233–263. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tailor P, Tamura T and Ozato K: IRF family
proteins and type I interferon induction in dendritic cells. Cell
Res. 16:134–140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ivashkiv LB and Donlin LT: Regulation of
type I interferon responses. Nat Rev Immunol. 14:36–49. 2014.
View Article : Google Scholar :
|
|
31
|
Parks GD and Alexander-Miller MA:
Paramyxovirus activation and inhibition of innate immune responses.
J Mol Biol. 425:4872–4892. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fournier P, Arnold A, Wilden H and
Schirrmacher V: Newcastle disease virus induces pro-inflammatory
conditions and type I interferon for counter-acting Treg activity.
Int J Oncol. 40:840–850. 2012.
|
|
33
|
Fournier P, Arnold A and Schirrmacher V:
Polarization of human monocyte-derived dendritic cells to DC1 by in
vitro stimulation with Newcastle Disease Virus. J BUON. 14(Suppl):
S111–S122. 2009.
|
|
34
|
Weber F, Kochs G and Haller O: Inverse
interference: how viruses fight the interferon system. Viral
Immunol. 17:498–515. 2004. View Article : Google Scholar
|
|
35
|
Jenner RG and Young RA: Insights into host
responses against pathogens from transcriptional profiling. Nat Rev
Microbiol. 3:281–294. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang Q, Liu D, Majewski P, Schulte LC,
Korn JM, Young RA, Lander ES and Hacohen N: The plasticity of
dendritic cell responses to pathogens and their components.
Science. 294:870–875. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fiola C, Peeters B, Fournier P, Arnold A,
Bucur M and Schirrmacher V: Tumor selective replication of
Newcastle disease virus: Association with defects of tumor cells in
antiviral defence. Int J Cancer. 119:328–338. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jarahian M, Watzl C, Fournier P, Arnold A,
Djandji D, Zahedi S, Cerwenka A, Paschen A, Schirrmacher V and
Momburg F: Activation of natural killer cells by newcastle disease
virus hemagglutinin-neuraminidase. J Virol. 83:8108–8121. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Washburn B, Weigand MA, Grosse-Wilde A,
Janke M, Stahl H, Rieser E, Sprick MR, Schirrmacher V and Walczak
H: TNF-related apoptosis-inducing ligand mediates tumoricidal
activity of human monocytes stimulated by Newcastle disease virus.
J Immunol. 170:1814–1821. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Termeer CC, Schirrmacher V, Bröcker EB and
Becker JC: Newcastle disease virus infection induces
B7-1/B7-2-independent T-cell costimulatory activity in human
melanoma cells. Cancer Gene Ther. 7:316–323. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ertel C, Millar NS, Emmerson PT,
Schirrmacher V and von Hoegen P: Viral hemagglutinin augments
peptide-specific cytotoxic T cell responses. Eur J Immunol.
23:2592–2596. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Umansky V, Shatrov VA, Lehmann V and
Schirrmacher V: Induction of NO synthesis in macrophages by
Newcastle disease virus is associated with activation of nuclear
factor-kappa B. Int Immunol. 8:491–498. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Klenk HD: Marburg and Ebola Viruses.
Current Topics in Microbiol and Immunol 235. Springer; 1999
|
|
44
|
Bray M and Geisbert TW: Ebola virus: The
role of macrophages and dendritic cells in the pathogenesis of
Ebola hemorrhagic fever. Int J Biochem Cell Biol. 37:1560–1566.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
WHO Ebola Response Team: Ebola virus
disease in West Africa. New Engl J Med. 371:1481–1495. 2014.
View Article : Google Scholar
|
|
46
|
Zhang L, Wang H and Zhang YQ: Against
Ebola: Type I interferon guard risk and mesenchymal stromal cell
combat sepsis. J Zhejiang Univ Sci B. 16:1–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ramanan P, Shabman RS, Brown CS,
Amarasinghe GK, Basler CF and Leung DW: Filoviral immune evasion
mechanisms. Viruses. 3:1634–1649. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Leung DW, Ginder ND, Fulton DB, Nix J,
Basler CF, Honzatko RB and Amarasinghe GK: Structure of the Ebola
VP35 interferon inhibitory domain. Proc Natl Acad Sci USA.
106:411–416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xu W, Edwards MR, Borek DM, Feagins AR,
Mittal A, Alinger JB, Berry KN, Yen B, Hamilton J, Brett TJ, et al:
Ebola virus VP24 targets a unique NLS binding site on karyopherin
alpha 5 to selectively compete with nuclear import of
phosphorylated STAT1. Cell Host Microbe. 16:187–200. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bale S, Julien JP, Bornholdt ZA, Kimberlin
CR, Halfmann P, Zandonatti MA, Kunert J, Kroon GJ, Kawaoka Y,
MacRae IJ, et al: Marburg virus VP35 can both fully coat the
backbone and cap the ends of dsRNA for interferon antagonism. PLoS
Pathog. 8:e10029162012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Leung DW, Prins KC, Borek DM, Farahbakhsh
M, Tufariello JM, Ramanan P, Nix JC, Helgeson LA, Otwinowski Z,
Honzatko RB, et al: Structural basis for dsRNA recognition and
interferon antagonism by Ebola VP35. Nat Struct Mol Biol.
17:165–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Basler CF, Mikulasova A, Martinez-Sobrido
L, Paragas J, Mühlberger E, Bray M, Klenk HD, Palese P and
García-Sastre A: The Ebola virus VP35 protein inhibits activation
of interferon regulatory factor 3. J Virol. 77:7945–7956. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Feng Z, Cerveny M, Yan Z and He B: The
VP35 protein of Ebola virus inhibits the antiviral effect mediated
by double-stranded RNA-dependent protein kinase PKR. J Virol.
81:182–192. 2007. View Article : Google Scholar :
|
|
54
|
Hartman AL, Ling L, Nichol ST and Hibberd
ML: Whole-genome expression profiling reveals that inhibition of
host innate immune response pathways by Ebola virus can be reversed
by a single amino acid change in the VP35 protein. J Virol.
82:5348–5358. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang AP, Bornholdt ZA, Liu T, Abelson DM,
Lee DE, Li S, Woods VL Jr and Saphire EO: The ebola virus
interferon antagonist VP24 directly binds STAT1 and has a novel,
pyramidal fold. PLoS Pathog. 8:e10025502012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kulkarni RR, Rasheed MA, Bhaumik SK,
Ranjan P, Cao W, Davis C, Marisetti K, Thomas S, Gangappa S,
Sambhara S, et al: Activation of the RIG-I pathway during influenza
vaccination enhances the germinal center reaction, promotes T
follicular helper cell induction, and provides a dose-sparing
effect and protective immunity. J Virol. 88:13990–14001. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cervantes-Barragán L, Kalinke U, Züst R,
König M, Reizis B, López-Macías C, Thiel V and Ludewig B: Type I
IFN-mediated protection of macrophages and dendritic cells secures
control of murine coronavirus infection. J Immunol. 182:1099–1106.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ayllon J and García-Sastre A: The NS1
protein: A multitasking virulence factor. Curr Top Microbiol
Immunol. 386:73–107. 2015.
|
|
59
|
Reder AT, Oger JF, Kappos L, O’Connor P
and Rametta M: Short-term and long-term safety and tolerability of
interferon β-1b in multiple sclerosis. Mult Scler Relat Disord.
3:294–302. 2014. View Article : Google Scholar
|
|
60
|
Corti D and Lanzavecchia A: Broadly
neutralizing antiviral antibodies. Annu Rev Immunol. 31:705–742.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Corti D, Sallusto F and Lanzavecchia A:
High throughput cellular screens to interrogate the human T and B
cell repertoires. Curr Opin Immunol. 23:430–435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sobarzo A, Eskira Y, Herbert AS, Kuehne
AI, Stonier SW, Ochayon DE, Fedida-Metula S, Balinandi S, Kislev Y,
Tali N, et al: Immune memory to Sudan virus: Comparison between two
separate disease outbreaks. Viruses. 7:37–51. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Marzi A, Engelmann F, Feldmann F,
Haberthur K, Shupert WL, Brining D, Scott DP, Geisbert TW, Kawaoka
Y, Katze MG, et al: Antibodies are necessary for
rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus
challenge in nonhuman primates. Proc Natl Acad Sci USA.
110:1893–1898. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Barber GN: VSV-tumor selective replication
and protein translation. Oncogene. 24:7710–7719. 2005. View Article : Google Scholar : PubMed/NCBI
|