Introduction
Doxorubicin (DOX) is an anthracycline antibiotic
that is effective in treating a wide spectrum of cancer types,
including leukemia, lymophoma, soft tissue sarcoma and solid tumors
(1). However, the toxic
side-effects of DOX, particularly those involving the heart,
require dose limitations and thus, this reduces the effectiveness
of DOX administration (2).
DOX-induced cardiac abnormalities have been reported in a wide
range of patients (3,4). The mechanisms underlying DOX-induced
cardiotoxicity are thought to involve complex multifactorial
processes, including oxidative stress (5,6),
mitochondrial biogenesis (7) and
autophagy (8). However, the role
of endoplasmic reticulum (ER) stress has received little attention,
and therefore has not been completely elucidated, despite
indications that ER stress plays a key role in the development of
DOX-induced cardiotoxicity and in cell death pathways (18–21).
The ER is one of the critical organelles within
cells responsible for maintaining metabolism, lipoprotein secretion
and calcium homeostasis. Accordingly, the disruption of ER
homeostasis or the induction of ER stress has profound effects on
cell survival. Perturbations in cellular physiological and/or
various pathological processes, such as increased protein
synthesis, alterations in the redox status or disturbances in
calcium storage, trigger ER stress. These stresses are sensed by
cells through three ER-resident transmembrane proteins, namely
inositol-requiring enzyme 1 (IRE1), double-stranded RNA-activated
protein kinase-like ER kinase (PERK) and activating transcription
factor 6 (ATF6) (9–11). These proteins, which are
collectively referred to as an unfolded protein response (UPR),
trigger downstream signaling pathways to restore ER homeostasis.
Initially, the UPR facilitates adaptation to acute cellular
perturbations and re-establishes ER homeostasis, and thus has
cell-protecting activites (12).
Gluocose-regulated protein 78 (GRP78), also known as binding
immunoglobulin protein (BiP), is a key mediator of the UPR. The
accumulation of unfolded proteins within the ER leads to the
dissociation of GRP78 from these three transmembrane proteins,
thereby inducing their activation (13,14). If these adaptive responses of UPR
are insufficient in attenuating ER stress, the UPR switches to a
pro-apoptotic signal (15). The
resultant activation of pro-apoptotic proteins, such as C/EBP
homologous protein (CHOP), also known as GADD153 (CHOP/GADD153),
caspase-12 and Bax ultimately leads to cell death (9). Under physiological conditions, GRP78
and CHOP are expressed at low levels, whereas they are strongly
expressed in response to ER stress (10,11,16). Therefore, they serve as critical
indicators of ER stress (17).
During the past decade, much attention has been
directed towards examining the roles of ER stress in
anthracycline-induced cardiac injury (18–20). DOX inactivates GRP78 leading to an
increase in misfolded proteins, ER stress, the activation of UPR
sensors and increased CHOP expression (21). Lu et al (21) reported that the expression of
non-functional GRP78 isoforms and CHOP in the heart were increased
with DOX treatment. In a previous study, in rat H9c2
cardiomyocytes, DOX induced a decrease in cell viability and
markedly enhanced the expression of caspase-12, another marker of
ER stress (18). Thus, the
DOX-induced inactivation of GRP78 and the enhanced expression of
CHOP in heart tissue may represent a mechanistic pathway for the
inhibitory effects of DOX on UPR and protein synthesis, thereby
serving as a basis for DOX-induced cardiotoxicity.
Resveratrol (RV; 3,4′,5-trihydroxystilbene), a
stilbenoid found in grapes and red wine, is a potent antioxidant
and has been studied for its benefical effects on cardiovascular
diseases (22–25). Mounting evidence indicates that RV
plays both physiological and pathophysiological roles in regulating
cardiovascular function. Recently, it was shown that RV plays a
protective role in attenuating DOX-induced cardiac injury in mice,
by decreasing left ventricular dysfunction and remodeling (26). RV has also been shown to improve
cardiac function, reduce mortality following myocardial infarction
(MI) and to increase the expression of AMP-activated protein kinase
(AMPK) in a rat model of MI (25). In addition, it has been reported
that RV exerts anti-cardiotoxic effects through the inhibition of
cardiac apoptosis and mitochondrial stabilization via the Sirt1
pathway in DOX-treated rat ventricular myocytes (7).
Sirt1, a NAD-dependent class III histone
deacetylase, is an important regulator of cell survival and life
span (27). Sirt1 catalyzes the
deacetylation of numerous proteins and generates nicotinamide (NIC)
as a by-product, which then functions as a negative regulator of
Sirt1 activity (28,29). Sirt1 has been shown to increase
cell resistance and survival from stress through a number of
pathways (29–31). The cardiac-specific overexpression
of Sirt1 protects the heart from ischemia/reperfusion injury by
negatively regulating pro-apoptotic molecules, such as caspase-3,
an ER stress downstream activator (32). Sirt1 has been shown to be
activated by oxidative stress and RV treatment (31,33–36). The recent findings of Liu et
al provide evidence linking Sirt1 expression and ER-related
protein activation, thereby strongly indicating that the
anti-apoptotic effects of RV against ethanol-induced ER stress
involve a Sirt1-dependent process (37). Although it is known that RV
activates Sirt1, only recently was this effect demonstrated in H9c2
cells subjected to cardiotoxicity (38).
Whether exogenous RV protects cardiomyocytes against
DOX-induced ER stress through a Sirt1-dependent mechanism is not
yet known. To examine this possibility, in this study, we
investigated the effects of RV on Sirt1 activity as a means of
modulating ER stress responses in vitro and the resultant
impact on cardiomyocyte apoptosis. Our data demonstrated that the
treatment of H9c2 cells with RV attenuated DOX-induced
cardiomyocyte apoptosis, alleviated cardiotoxicity, upregulated
Sirt1 expression and ultimately suppressed the ER stress-induced
overexpression of GRP78 and CHOP.
Materials and methods
Materials
RV, DOX and dimethyl sulfoxide (DMSO) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). NIC was purchased from
Aladdin Industrial Corp. (Shanghai, China). GRP78 (#3183), CHOP
(#2895) and Sirt1 (#9475) antibodies were provided by Cell
Signaling Technology, Inc. (Lake Placid, NY, USA), and the β-actin
antibody was obtained from Proteintech (Chicago, IL, USA). The Cell
Counting kit-8 (CCK-8) was purchased from Dojindo Laboratories Co.,
Ltd. (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM,
F12) and fetal bovine serum (FBS) were purchased from HyClone
Laboratories, Inc. (Logan, UT, USA). Penicillin and streptomycin
were purchased from Beijing Solarbio Science & Technology Co.,
Ltd. (Beijing, China). Trypsin was purchased from Gibco-BRL
(Calsbad, CA, USA). RIPA buffer was purchased from the Beyotime
Institute of Biotechnology (Jiangsu, China). TRIzol reagent was
purchased from Tiangen Biotech (Beijing) Co., Ltd. (Beijing,
China).
Cell culture
The rat heart tissue-derived H9c2 embryonic cardiac
myoblast cell line (H9c2 cells) was purchased from the Peking Union
Medical College, Experimental Cell Resource Center (IBMS, Beijing,
China). The cells were cultured in DMEM-high glucose medium
supplemented with 10% FBS and 1% penicillin/streptomycin
antibiotics. The cells were incubated at 37°C in an atmosphere of
5% CO2 and 95% O2 with saturated humidity.
When the H9c2 cells reached 70% confluency, they were cultured in
DMEM with or without various concentrations of DOX (0, 1, 5 or 10
µM) for 24 h. In order to investigate the potential
involvement of the ER stress signaling pathway in response to DOX,
the H9c2 cells were pre-treated with various doses of RV (0, 10,
25, 50 or 75 µM) for 24 h, followed by treatment with DOX (5
µM). This preparation was then combined with the
pharmacological Sirt1 inhibitor, NIC (20 mM), for 24 h prior to
treatment with DOX. The cells and supernatants were harvested and
stored at −80°C until use.
Cell viability assay
The viability of the H9c2 cells was determined using
the CCK-8 assay according to the manufacturer's instructions. In
brief, 100 µl of H9c2 cell suspensions (5,000 cells/well)
were dispensed in a 96-well plate. The plates were pre-incubated
for 24 h in a humidified incubator at 37°C with 5% CO2.
Following treatment with drug-containing media for various periods
of time, 10 µl of CCK-8 solution were added to each well
followed by a further 3 h of incubation at 37°C. The absorbance was
measured at 450 nm using a microplate reader (BioTek Instruments,
Inc., Winooski, VT, USA). The mean optical density (OD) of 5–7
wells in the indicated groups was used to calculate the percentage
of viable cells according to the following formula: percentage of
viable cells = OD of treatment group/OD of control group ×100.
Western blot analysis
Following treatment with assay media for 24 h, the
cell samples were harvested and lysed with ice-cold RIPA buffer.
The total protein concentrations were determined with the use of
the BCA Protein Assay kit (Beyotime Institute of Biotechnology).
Equal amounts of protein were separated by electrophoresis on 12%
SDS-polyacrylamide gels and then transferred onto polyvinylidene
fluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA). The
membranes were blocked in blocking buffer [Tiangen Biotech
(Beijing) Co., Ltd.] overnight at 4°C and then incubated with
primary antibodies to GRP78 (1:1,000) and CHOP (1:1,000) at room
temperature for 4 h. Following 3 washes with Tris-buffered saline
plus Tween-20 (TBST), the membranes were incubated with horseradish
peroxidase (HRP)-conjugated anti-rabbit IgG (ZB-2301) and
HRP-conjugated anti-mouse IgG (ZB-2305) secondary antibodies
(1:50,000) (ZsBio, Beijing, China) for 1 h at room temperature.
Signals were detected using an ECL kit plus reagents (Hangzhou Fude
Biological Technology Co., Ltd., Hangzhou, China).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
according to the manufacturer's instructions. Total RNA (1
µg) from each sample was used for cDNA synthesis using the
HiFi-MMLV cDNA kit (Beijing CoWin Biotech Co., Ltd., Beijing,
China). qPCR was performed using the Applied Biosystems 7500
Real-Time PCR System with the Ultra SYBR Mixture (Beijing CoWin
Biotech Co., Ltd.). For normalization, the housekeeping gene,
GAPDH, was used as a reference. The primer sequences used were as
follows: GRP78 forward, 5′-GAATCCCTCCTGCTCCCCGT-3′ and reverse,
5′-TTGG TCATTGGTGATGGTGATTTTG-3′; CHOP forward,
5′-CTTCACTACTCTTGACCCTG-3′ and reverse,
5′-TGAGCCATAGAACTCTGACTGGAATC-3′; and GAPDH forward,
5′-TGGAGTCTACTGGCGTCTT-3′ and reverse, 5′-TGTCATATTT
CTCGTGGTTCA-3′. The comparative critical thresh old (CT) method,
also referred to as the 2−ΔΔCT method, was used to
quantify gene expression. Changes in the expression of target genes
(GRP78 and CHOP) were measured relative to the mean CT values of
the GAPDH gene.
Statistical analysis
All data are expressed as the means ± standard
deviation (SD). Differences among groups were analyzed by analysis
of variance (ANOVA). A value of p<0.05 was considered to
indicate a statistically significant difference.
Results
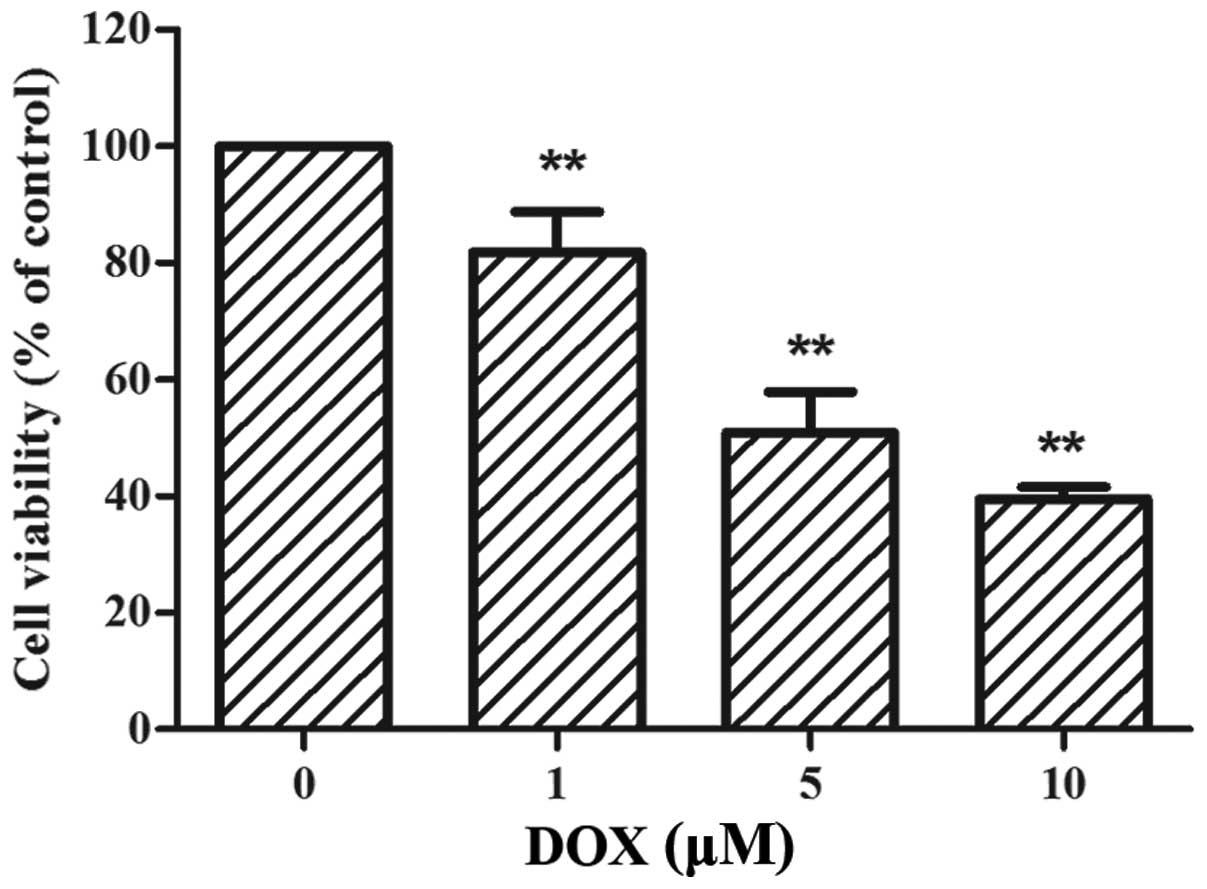
DOX reduces the viability of rat H9c2
cells
The H9c2 cells were treated with increasing
concentrations of DOX (0–10 µM) for 24 h, followed by CCK-8
assay. As shown in Fig. 1, the
viability of the H9c2 cells was significantly reduced by DOX
treatment in a dose-dependent manner. Following treatment with 5
µM DOX, cell viability was decreased by approximately 50%
compared to the control (50.90±6.98%, p<0.01 vs. control).
Therefore, the dose of 5 µM served as an effective
injury-inducing factor for the following experiments.
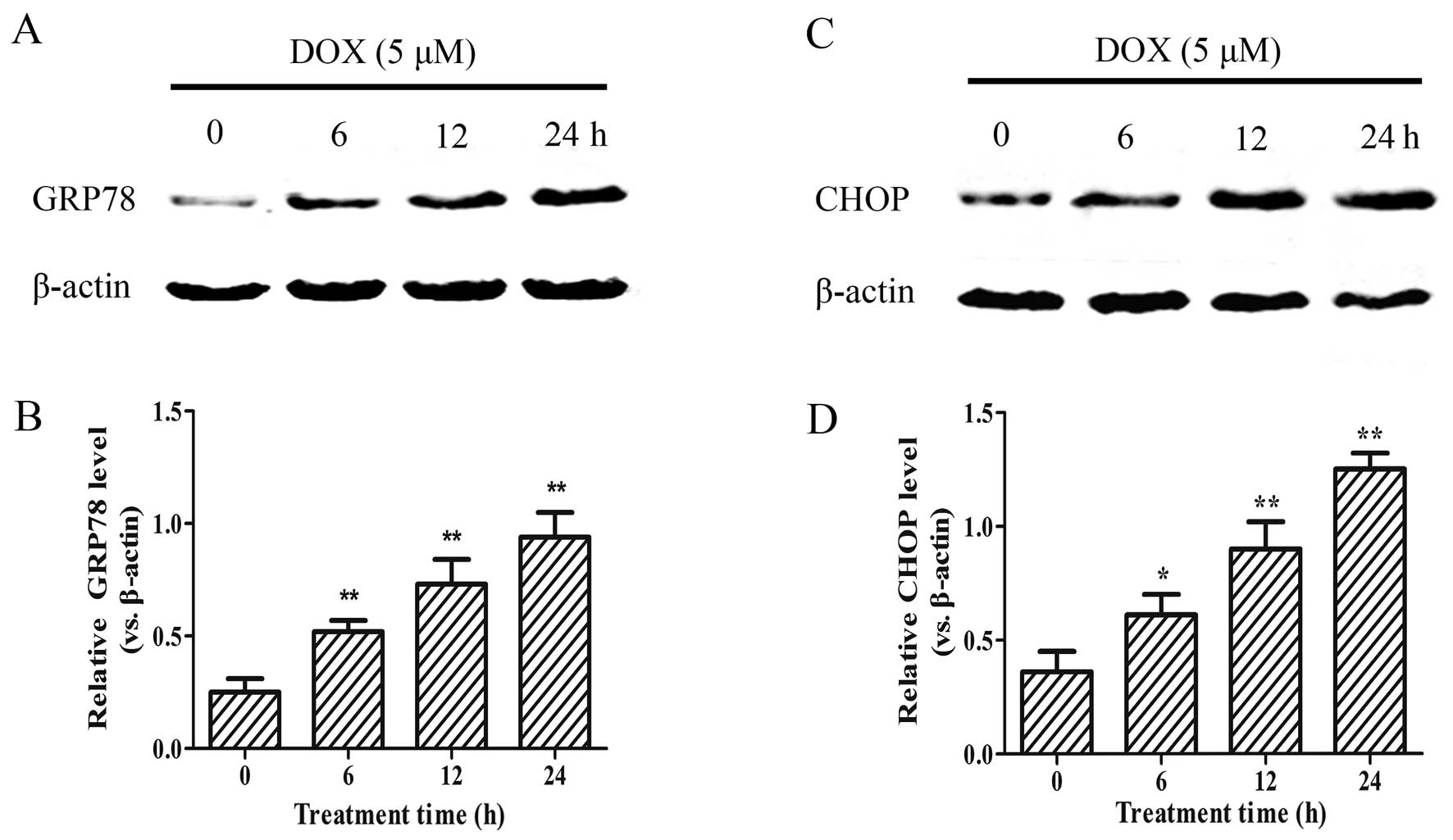
DOX enhances the expression of ER
stress-related proteins
As DOX reduced the viability of the H9c2 cells in a
concentration-dependent manner, we examined whether this effect is
associated with an increased expression of the ER stress-related
apoptotic proteins, GRP78 and CHOP. As shown in Fig. 2, following treatment with 5
µM DOX for 0–24 h, the expression of the GRP78 and CHOP
apoptotic proteins significantly increased in the H9c2 cells in a
time-dependent manner (p<0.01). These results suggest that
DOX-induced myocardial injury enhances the ER stress response in
H9c2 cells.
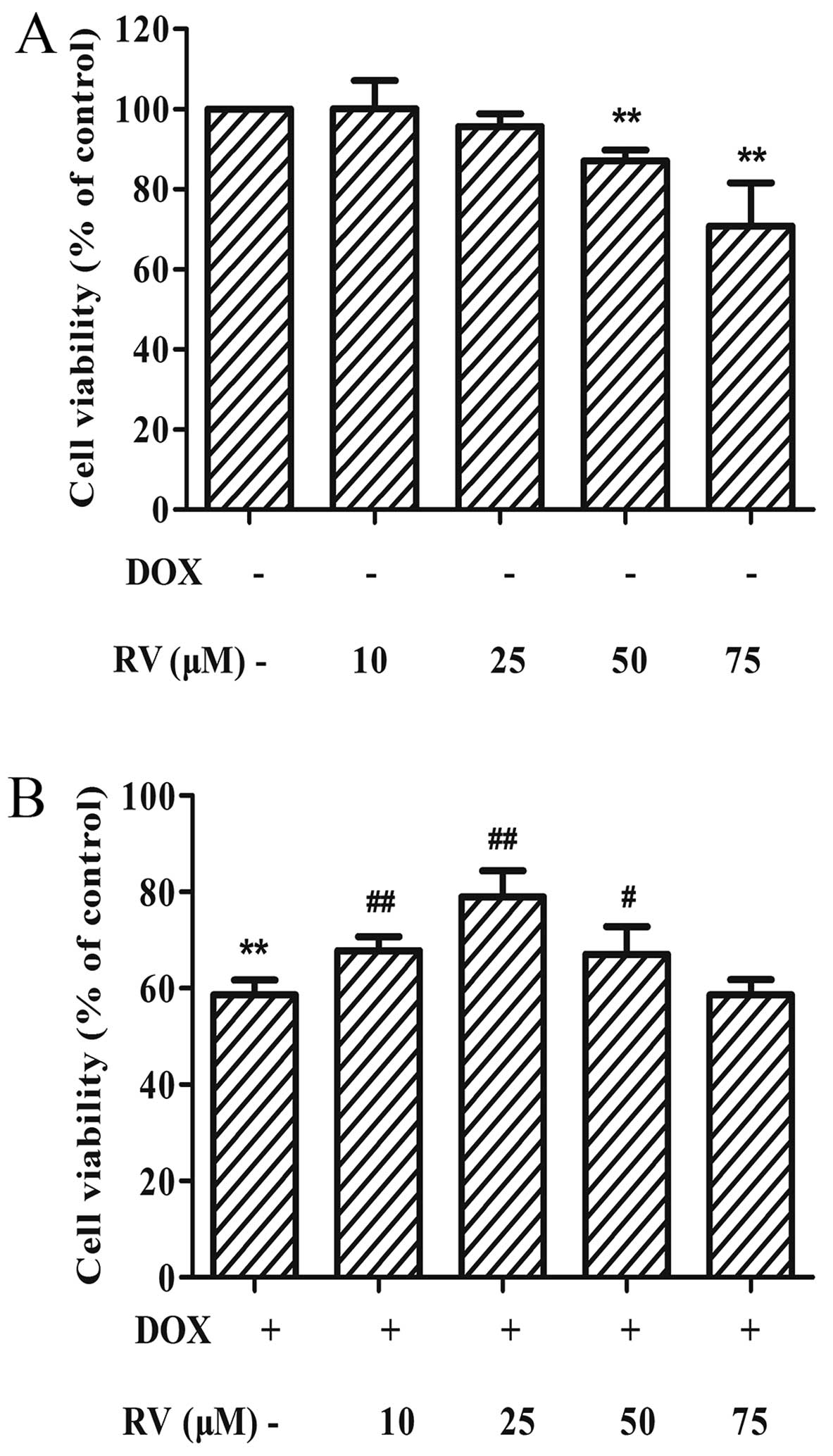
RV attenuates DOX-induced cardiomyocyte
apoptosis
The results of the two previous sets of experiments
described above demonstrate that DOX induces the epxression of ER
stress-related apoptotic proteins and, subsequently, cell death.
Thus, to determine whether RV alters the effects of DOX in
cardiomyocytes, we performed CCK-8 assay. When used alone, RV at
the dose of 0–25 µM had no significant effects on cell
viability; at the doses of 50 and 75 µM, RV decreased cell
viabilty (p<0.01 vs. controls). Thus, RV at 25 µM was
used in the subsequent experiments. When used with DOX, RV
attenuated the DOX-induced decrease in cell viability. The maximal
attenuation of the detrimental effects of DOX on cell viability was
obtained with a dose of 25 µM RV (78.92±5.48%, p<0.01)
compared to treatment with DOX alone (58.64±3.10%, p<0.01;
Fig. 3B). RV alone, at doses of
10 or 25 µM, did not alter the viability of H9c2 cells
(Fig. 3A).
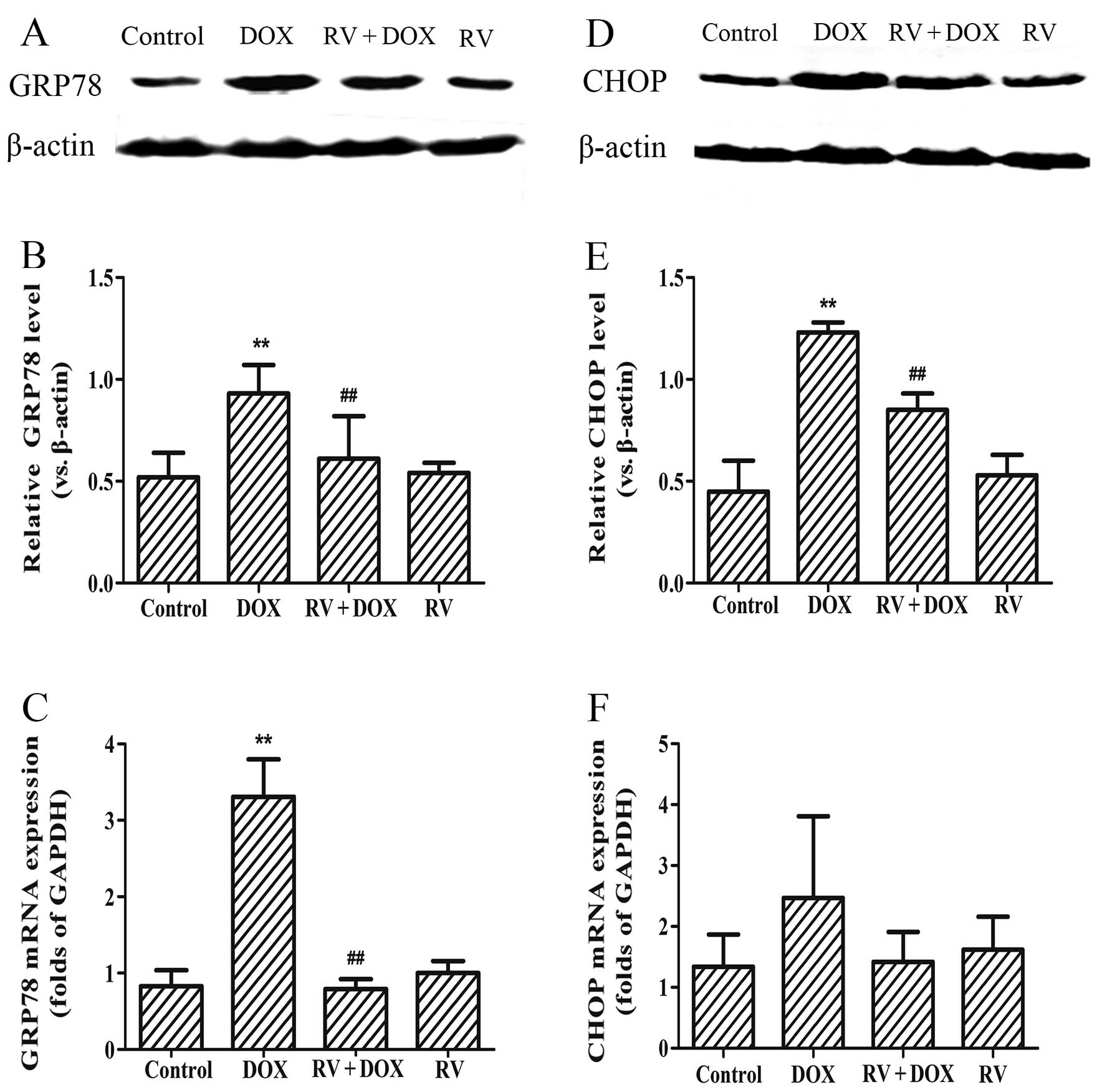
RV inhibits the DOX-induced protein
expression of ER stress markers
The results presented above suggest that RV prevents
DOX-induced cell death. Therefore, we hypothesized that RV may
decrease the DOX-induced ER stress responses and cell apoptosis. To
examine this hypothesis, the protein and mRNA expression levels of
GRP78 and CHOP were measured by western blot analasis and RT-qPCR,
respectively. As shown in Fig. 4,
the protein expression levels of GRP78 (0.93±0.14, p<0.01 vs.
controls) and CHOP (1.23±0.06, p<0.01 vs. controls), together
with mRNA levels of GRP78 (3.31±0.49, p<0.01 vs. controls), but
not those of CHOP (2.47±1.34, p=0.127 vs. controls), were increased
significantly in the DOX-treated group. By contrast, treatment with
RV (25 µM) at 24 h prior to the exposure of the H9c2 cells
to DOX (5 µM) significantly downregulated the GRP78
(Fig. 4A and B) and CHOP
(Fig. 4D and E) protein
expression levels (p<0.01 for both vs. the group treated with
DOX only). The mRNA levels of GRP78 were also decreased in the RV +
DOX group (Fig. 4C; p<0.01 vs.
the group treated with DOX only); however, the mRNA levels of CHOP
were only slightly decreased in the RV + DOX group and failed to
achieve statistical significance (Fig. 4F; p=0.13 vs. the group treated
with DOX only). Treatment with RV alone did not affect the basal
expression of GRP78 and CHOP in the H9c2 cells. These results
suggest that the cytoprotective effects of RV are associated with
the inhibition of ER stress in H9c2 cells.
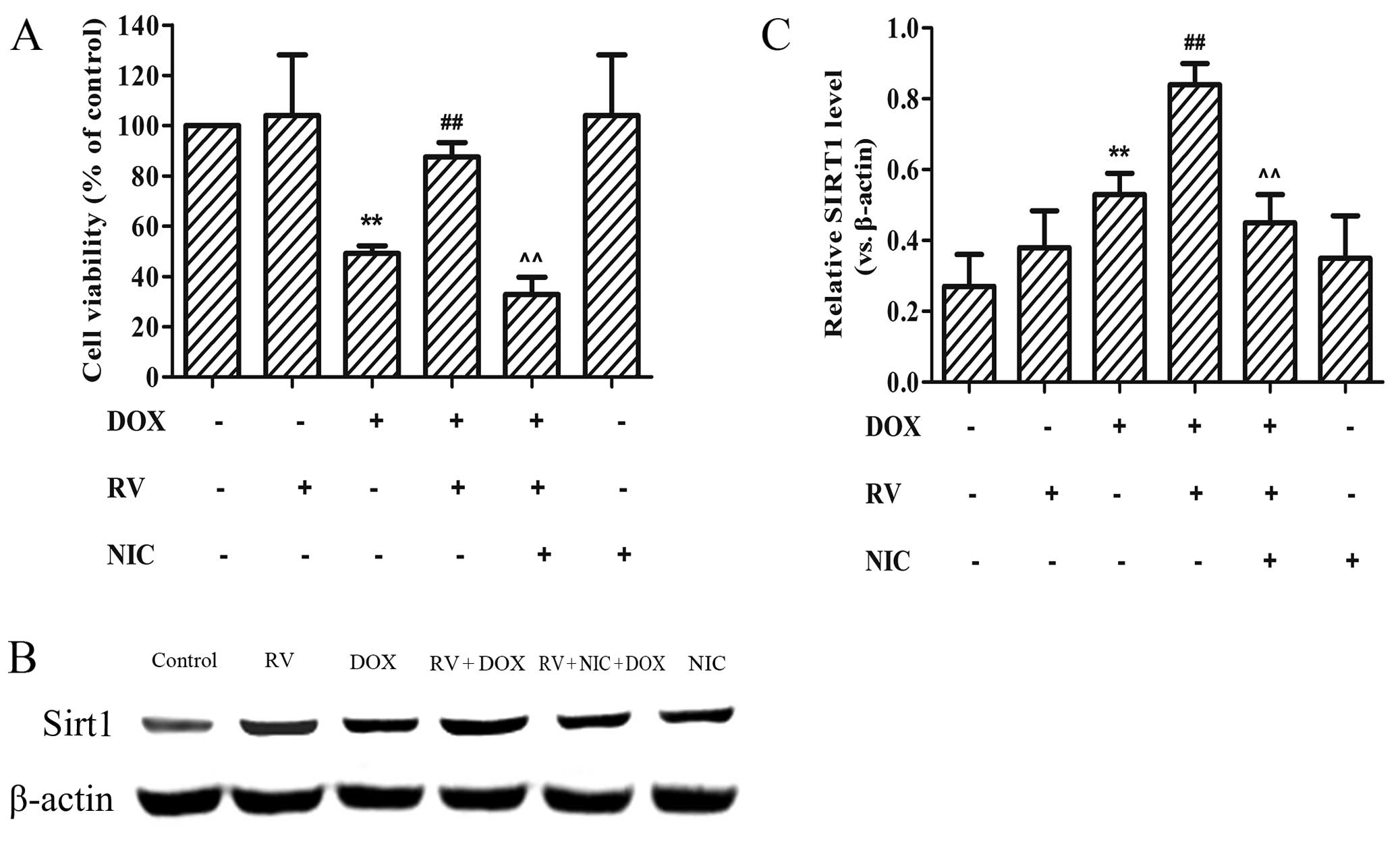
RV induces Sirt1 protein overexpression
and prevents DOX-induced cell death
The findings of our previous experiments indicated
that RV decreased ER stress and protected the cardiomyocytes from
DOX-induced cell death. To investigate the potential role of the
Sirt1 pathway in the protective effects of RV, we determined
whether treatment with NIC, a known Sirt1 inhibitor, affects cell
viability (which was increased by RV) in the DOX-treated cells.
Cell viability in the DOX, RV + DOX and RV + NIC + DOX groups was
49.23±3.02%, 87.58±5.65%, 32.89±6.88% of the controls,
respectively. Thus, based upon the cell viability rates determined
using CCK-8 assay, NIC abolished the protective effects of RV
against the DOX-induced decrease in cell viability of the H9c2
cells (Fig. 5A).
To verify that RV activates Sirt1 in H9c2 cells, we
measured the expression levels of Sirt1 by western blot anlaysis
(Fig. 5B). Moderate levels of
Sirt1 were detected in the control group, and the Sirt1 protein
levels were increased following treatment with RV or DOX alone
(Fig. 5B and C). Notably, a
significant increase in the Sirt1 protein levels was observed in
the RV + DOX group (0.84±0.06, p<0.01), while the addition of
NIC to this group significantly decreased the protein expression of
Sirt1 (0.45±0.08, p<0.01; Fig. 5B
and C).
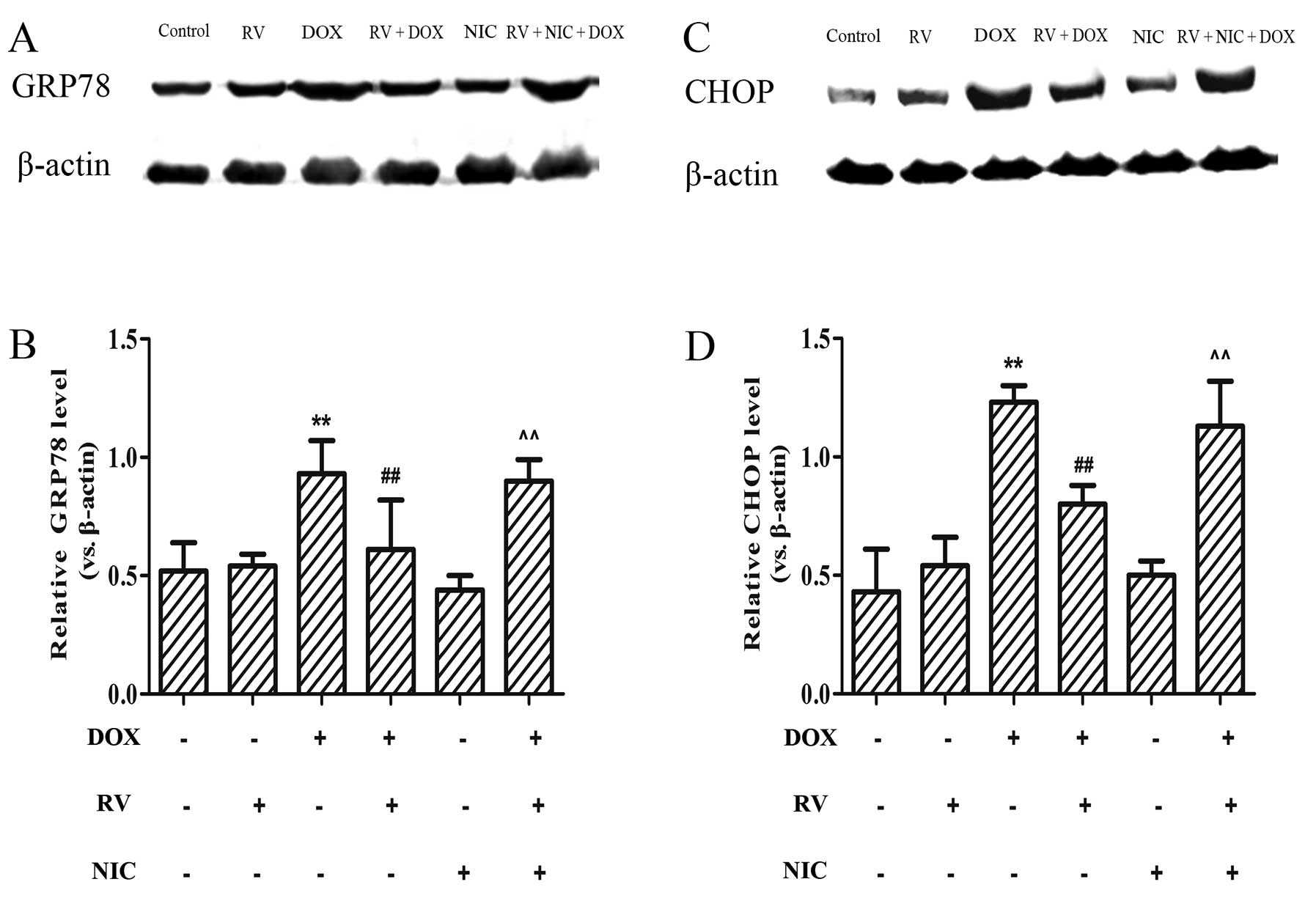
RV inhibits the DOX-induced increase in
the expression of ER stress-related apoptotic proteins through the
activation of the Sirt1 pathway
To determine whether RV exerts its cytoprotective
effects against DOX-induced ER stress through the activation of
Sirt1, the expression levels of downstream targets of ER
stress-related protein were measured by western blot analysis. As
shown in Fig. 6A and C, moderate
levels of GRP78 and CHOP were detected in the RV and NIC groups.
However, these protein levels increased following treatment with
DOX and significantly decreased by pre-treatment with RV (p<0.01
vs. the DOX group). These levels significantly increased when the
cells were treated with NIC, as well as RV + DOX (p<0.01 vs. the
RV + DOX group; Fig. 6). These
findings indicate that the protective effects of RV against
DOX-induced cardiotoxicity involve the alleviation of ER
stress-induced injury and homeostasis, at least in part, through
the activation of the Sirt1 pathway.
Discussion
RV, a well-known antioxidant and anti-inflammatory
compound, is found abundantly in grapes and red wine. It exerts a
number of pharmacological effects within the cardiovascular system
and is known to reduce mortality in a variety of heart-related
diseases (39–43). In this study, to establish a means
for assessing the potential protective effects of RV against
DOX-induced cardiotoxicity, we developed an in vitro cell
model of DOX-induced myocardial injury. Our findings demonstrated
that DOX significantly decreased the viability of H9c2 cells and
induced the overexpression of ER stress-related proteins in a
time-dependent manner. These results are consistent with those of a
recent study indicating that both GRP78 and CHOP expression was
enhanced in DOX-treated H9c2 cells (44). Moreover, the exogenous
administration of RV prior to the exposure of H9c2 cells to DOX (5
µM) was effective in protecting the H9c2 cells against
DOX-induced myocardial injury. Therefore, our results provide clear
support for the hypothesis that RV attenuates DOX-induced
cardiotoxicity.
The findings of previous studies have provided
evidence demonstrating that the activation of an apoptotic pathway
represents an important mechanism in DOX-induced cardiotoxicity
(45,46). The specific role of apoptosis in
the DOX-treated myocardium remains undetermined. Therefore, the
elucidation of the mechanisms involved is an important area of
investigation. According to previous research, ER stress may serve
as a central mode of apoptosis in DOX-induced cardiotoxicity
(44,47). It has recently been demonstrated
that the treatment of cardiomyoblasts with DOX significantly
increased the ER load, indicating a substantial elevation in ER
stress (47). Furthermore, DOX
not only translocates to the nucleus, but also shows a modest
affinity for ER binding (47).
Another study linked ER stress with DOX-induced cardiac insults, as
shown by an elevated expression of GRP78 and CHOP accompanied by
heart dysfunction and the decreased activity of antioxidant enzymes
in the hearts of DOX-treated mice (19). RV has been shown to reduce
cardiomyocyte apoptosis resulting from various forms of cardiac
injury, such as oxidative stress (48) and ischemic reperfusion injury
(49–52), and. Another study showed that the
DOX-induced apoptotic index decreased from 11.8 to 7% in response
to RV treatment (47). In the
present study, we assessed the viability of H9c2 cells using a
CCK-8 assay. Our findings are consistent with those of previous
studies showing that RV protects cardiomyocyte from DOX-induced
apoptosis (7,26,47). There are data indicating that RV
is associated with the induction of an anti-apoptotic signal that
results in cardioprotection (53).
One of the primary aims of the present study was to
examine the hypothesis that DOX-induced cell death occurs through a
mechanism that possibly begins with ER stress and results in the
activation of GRP78 and CHOP. RV may then alleviate ER stress and
decrease cardiomyocyte apoptosis through the involvement of the
Sirt1-dependent pathway. Sirt1 has been shown to be involved in
various cellular functions, ranging from gene silencing, the
control of the cell cycle and apoptosis to energy homeostasis
(54). Moreover, the results of
previous studies have indicated that Sirt1 increases cell viability
and oxidative stress resistance through a variety of pathways
(7,30,31), and that the moderate
overexpression of Sirt1 protects the heart from oxidative stress
(32,33,55–57). It has been well established that
Sirt1 downregulates ER stress-related genes believed to be involved
in determining life spans in organisms (58,59), indicating that Sirt1 contributes
to the maintenance of ER homeostasis and consequently enhances cell
viability. According to a recent study by Liu et al, RV
alleviated ethanol-induced ER stress through the activation of
Sirt1 in hepatocytes (37). Of
particular significance to the present study, Sirt1 has been shown
to be activated by RV treatment (31,35). Given this background information,
in this study, we examined the effects of RV on the expression of
ER stress-related apoptotic proteins, GRP78 and CHOP, as induced by
DOX, as well as its resultant effects on DOX-induced
cardiotoxicity. Our data indicated that RV significantly attenuated
the ER stress response and enhanced Sirt1 expression in DOX-treated
H9c2 cells. Furthermore, pre-treatment of the RV + DOX-treated
cells with NIC reversed these effects, suggesting that this
anti-apoptotic effect of RV protects against DOX-induced ER stress
through the Sirt1 pathway.
In addition to its known function as an anticancer
agent, RV may also function as a cardioprotective compound as
revealed by its capacity to decrease DOX-induced cardiotoxicity
(60,61). Therefore, an important implication
resulting from these findings is the potential of RV not only to
protect cells against DOX-induced cardiotoxicity by preventing ER
stress and cell death, but also the potential of RV to be used in
conjunction with decreasing therapeutic doses of DOX. The role of
RV as an effective cardioprotective agent against DOX-induced
cardiotoxicity is supported by previous findings demonstrating that
RV has antitumor properties and, when combined with DOX, enhances
its effectiveness as a therapeutic cancer agent (61). With findings indicating increased
survival rates of cancer patients treated with combinations of RV
with DOX (61), and the
recognition of DOX-induced cardiotoxicity, the need for a
cardioprotective agent to be administered in conjunction with DOX
is evident. Furthermore, a modest administration of RV to patients
receiving DOX-based therapy may provide a significant benefit by
reducing the risk for chemotherapy-induced left ventricular
dysfunction (24).
In conclusion, the results of this study demonstrate
that DOX impairs the survival of H9c2 cells at least partly by
triggering the ER stress response, whereas RV ameliorates these
effects of DOX and preserves cell viability. We established that
one of the protective effects of RV against DOX-induced
cardiotoxicity involves the attenuation of ER stress injury partly
through the Sirt1 pathway. Our findings have important implications
as they suggest that DOX-induced cardiac complications may be
diminished with the adjuvant administration of RV. Further
research, including whole animal studies, is required before any
definitive conclusion regarding the protective effects of RV
against DOX-induced ER stress can be drawn.
Acknowledgments
The ED-IT Editorial Service (edit.service@yahoo.com) was
used for the English revision of this manuscript.
References
|
1
|
Muggia FM and Green MD: New anthracycline
antitumor antibiotics. Crit Rev Oncol Hematol. 11:43–64. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weiss RB: The anthracyclines: Will we ever
find a better doxorubicin? Semin Oncol. 19:670–686. 1992.PubMed/NCBI
|
|
3
|
Lipshultz SE, Colan SD, Gelber RD,
Perez-Atayde AR, Sallan SE and Sanders SP: Late cardiac effects of
doxorubicin therapy for acute lymphoblastic leukemia in childhood.
New Engl J Med. 324:808–815. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shan K, Lincoff AM and Young JB:
Anthracycline-induced cardiotoxicity. Ann Intern Med. 125:47–58.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Doroshow JH: Effect of anthracycline
antibiotics on oxygen radical formation in rat heart. Cancer Res.
43:460–472. 1983.PubMed/NCBI
|
|
6
|
Olson RD and Mushlin PS: Doxorubicin
cardiotoxicity: Analysis of prevailing hypotheses. FASEB J.
4:3076–3086. 1990.PubMed/NCBI
|
|
7
|
Danz ED, Skramsted J, Henry N, Bennett JA
and Keller RS: Resveratrol prevents doxorubicin cardiotoxicity
through mitochondrial stabilization and the Sirt1 pathway. Free
Radic Biol Med. 46:1589–1597. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dirks-Naylor AJ: The role of autophagy in
doxorubicin-induced cardiotoxicity. Life Sci. 93:913–916. 2013.
View Article : Google Scholar
|
|
9
|
Rasheva VI and Domingos PM: Cellular
responses to endoplasmic reticulum stress and apoptosis. Apoptosis.
14:996–1007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
11
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schröder M: The unfolded protein response.
Mol Biotechnol. 34:279–290. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zu K, Bihani T, Lin A, Park YM, Mori K and
Ip C: Enhanced selenium effect on growth arrest by Bip/GRP78
knockdown in p53-null human prostate cancer cells. Oncogene.
25:546–554. 2006.
|
|
15
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Mao W, Iwai C, Fukuoka S, Vulapalli
R, Huang H, Wang T, Sharma VK, Sheu SS, Fu M and Liang CS: Adoptive
passive transfer of rabbit beta1-adrenoceptor peptide immune
cardiomyopathy into the Rag2-/- mouse: Participation of the ER
stress. J Mol Cell Cardiol. 44:304–314. 2008. View Article : Google Scholar
|
|
17
|
Mandl J and Bánhegyi G: Endoplasmic
reticulum stress - common pathomechanism of different diseases? Orv
Hetil. 148:1779–1785. 2007.In Hungarian. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chua CC, Liu X, Gao J, Hamdy RC and Chua
BH: Multiple actions of pifithrin-alpha on doxorubicin-induced
apoptosis in rat myoblastic H9c2 cells. Am J Physiol Heart Circ
Phys. 290:H2606–H2613. 2006. View Article : Google Scholar
|
|
19
|
Lai HC, Yeh YC, Ting CT, Lee WL, Lee HW,
Wang LC, Wang KY, Lai HC, Wu A and Liu TJ: Doxycycline suppresses
doxorubicin-induced oxidative stress and cellular apoptosis in
mouse hearts. Eur J Pharmacol. 644:176–187. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reeve JL, Szegezdi E, Logue SE, Ní
Chonghaile T, O'Brien T, Ritter T and Samali A: Distinct mechanisms
of cardiomyocyte apoptosis induced by doxorubicin and hypoxia
converge on mitochondria and are inhibited by Bcl-xL. J Cell Mol
Med. 11:509–520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu M, Merali S, Gordon R, Jiang J, Li Y,
Mandeli J, Duan X, Fallon J and Holland JF: Prevention of
Doxorubicin cardiopathic changes by a benzyl styryl sulfone in
mice. Genes Cancer. 2:985–992. 2011. View Article : Google Scholar
|
|
22
|
Das DK, Sato M, Ray PS, Maulik G, Engelman
RM, Bertelli AA and Bertelli A: Cardioprotection of red wine: Role
of polyphenolic antioxidants. Drugs Exp Clin Res. 25:115–120.
1999.PubMed/NCBI
|
|
23
|
Orallo F, Alvarez E, Camiña M, Leiro JM,
Gómez E and Fernández P: The possible implication of
trans-Resveratrol in the cardioprotective effects of long-term
moderate wine consumption. Mol Pharmacol. 61:294–302. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sabe AA, Sadek AA, Elmadhun NY, Dalal RS,
Robich MP, Bianchi C and Sellke FW: Investigating the effects of
resveratrol on chronically ischemic myocardium in a Swine model of
metabolic syndrome: A proteomics analysis. J Med Food. 18:60–66.
2015. View Article : Google Scholar
|
|
25
|
Gu XS, Wang ZB, Ye Z, Lei JP, Li L, Su DF
and Zheng X: Resveratrol, an activator of SIRT1, upregulates AMPK
and improves cardiac function in heart failure. Genet Mol Res.
13:323–335. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dolinsky VW, Rogan KJ, Sung MM, Zordoky
BN, Haykowsky MJ, Young ME, Jones LW and Dyck JR: Both aerobic
exercise and resveratrol supplementation attenuate
doxorubicin-induced cardiac injury in mice. Am J Physiol Endocrinol
Metab. 305:E243–E253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brachmann CB, Sherman JM, Devine SE,
Cameron EE, Pillus L and Boeke JD: The SIR2 gene family, conserved
from bacteria to humans, functions in silencing, cell cycle
progression, and chromosome stability. Genes Dev. 9:2888–2902.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bitterman KJ, Anderson RM, Cohen HY,
Latorre-Esteves M and Sinclair DA: Inhibition of silencing and
accelerated aging by nicotinamide, a putative negative regulator of
yeast sir2 and human SIRT1. J Biol Chem. 277:45099–45107. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Blander G and Guarente L: The Sir2 family
of protein deacetylases. Ann Rev Biochem. 73:417–435. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brunet A, Sweeney LB, Sturgill JF, Chua
KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et
al: Stress-dependent regulation of FOXO transcription factors by
the SIRT1 deacetylase. Science. 303:2011–2015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen CJ, Yu W, Fu YC, Wang X, Li JL and
Wang W: Resveratrol protects cardiomyocytes from hypoxia-induced
apoptosis through the SIRT1-FoxO1 pathway. Biochem Biophys Res
Commun. 378:389–393. 2009. View Article : Google Scholar
|
|
32
|
Hsu CP, Zhai P, Yamamoto T, Maejima Y,
Matsushima S, Hariharan N, Shao D, Takagi H, Oka S and Sadoshima J:
Silent information regulator 1 protects the heart from
ischemia/reperfusion. Circulation. 122:2170–2182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alcendor RR, Gao S, Zhai P, Zablocki D,
Holle E, Yu X, Tian B, Wagner T, Vatner SF and Sadoshima J: Sirt1
regulates aging and resistance to oxidative stress in the heart.
Circ Res. 100:1512–1521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Howitz KT, Bitterman KJ, Cohen HY, Lamming
DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL,
et al: Small molecule activators of sirtuins extend Saccharomyces
cerevisiae lifespan. Nature. 425:191–196. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lagouge M, Argmann C, Gerhart-Hines Z,
Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P,
Elliott P, et al: Resveratrol improves mitochondrial function and
protects against metabolic disease by activating SIRT1 and
PGC-1alpha. Cell. 127:1109–1122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wood JG, Rogina B, Lavu S, Howitz K,
Helfand SL, Tatar M and Sinclair D: Sirtuin activators mimic
caloric restriction and delay ageing in metazoans. Nature.
430:686–689. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu LQ, Fan ZQ, Tang YF and Ke ZJ: The
resveratrol attenuates ethanol-induced hepatocyte apoptosis via
inhibiting ER-related caspase-12 activation and PDE activity in
vitro. Alcohol Clin Exp Res. 38:683–693. 2014. View Article : Google Scholar
|
|
38
|
Li YG, Zhu W, Tao JP, Xin P, Liu MY, Li JB
and Wei M: Resveratrol protects cardiomyocytes from oxidative
stress through SIRT1 and mitochondrial biogenesis signaling
pathways. Biochem Biophys Res Commun. 438:270–276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen B, Xue J, Meng X, Slutzky JL, Calvert
AE and Chicoine LG: Resveratrol prevents hypoxia-induced arginase
II expression and proliferation of human pulmonary artery smooth
muscle cells via Akt-dependent signaling. Am J Physiol Lung Cell
Mol Physiol. 307:L317–L325. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Arafa MH, Mohammad NS, Atteia HH and
Abd-Elaziz HR: Protective effect of resveratrol against
doxorubicin-induced cardiac toxicity and fibrosis in male
experimental rats. J Physiol Biochem. 70:701–711. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang JP, Huang SS, Deng JY, Chang CC, Day
YJ and Hung LM: Insulin and resveratrol act synergistically,
preventing cardiac dysfunction in diabetes, but the advantage of
resveratrol in diabetics with acute heart attack is antagonized by
insulin. Free Radic Biol Med. 49:1710–1721. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang DL, Zhang HG, Xu YL, Gao YH, Yang XJ,
Hao XQ and Li XH: Resveratrol inhibits right ventricular
hypertrophy induced by monocrotaline in rats. Clin Exp Pharmacol
Physiol. 37:150–155. 2010. View Article : Google Scholar
|
|
43
|
Chen YR, Yi FF, Li XY, Wang CY, Chen L,
Yang XC, Su PX and Cai J: Resveratrol attenuates ventricular
arrhythmias and improves the long-term survival in rats with
myocardial infarction. Cardiovasc Drugs Ther. 22:479–485. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang XY, Yang CT, Zheng DD, Mo LQ, Lan AP,
Yang ZL, Hu F, Chen PX, Liao XX and Feng JQ: Hydrogen sulfide
protects H9c2 cells against doxorubicin-induced cardiotoxicity
through inhibition of endoplasmic reticulum stress. Mol Cell
Biochem. 363:419–426. 2012. View Article : Google Scholar
|
|
45
|
Zhang C, Feng Y, Qu S, Wei X, Zhu H, Luo
Q, Liu M, Chen G and Xiao X: Resveratrol attenuates
doxorubicin-induced cardiomyocyte apoptosis in mice through
SIRT1-mediated deacetylation of p53. Cardiovasc Res. 90:538–545.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ueno M, Kakinuma Y, Yuhki K, Murakoshi N,
Iemitsu M, Miyauchi T and Yamaguchi I: Doxorubicin induces
apoptosis by activation of caspase-3 in cultured cardiomyocytes in
vitro and rat cardiac ventricles in vivo. J Pharmacol Sci.
101:151–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sishi BJ, Loos B, van Rooyen J and
Engelbrecht AM: Doxorubicin induces protein ubiquitination and
inhibits proteasome activity during cardiotoxicity. Toxicology.
309:23–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lv XC and Zhou HY: Resveratrol protects
H9c2 embryonic rat heart derived cells from oxidative stress by
inducing autophagy: Role of p38 mitogen-activated protein kinase.
Can J Physiol Pharmacol. 90:655–662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lekli I, Szabo G, Juhasz B, Das S, Das M,
Varga E, Szendrei L, Gesztelyi R, Varadi J, Bak I, et al:
Protective mechanisms of resveratrol against
ischemia-reperfusion-induced damage in hearts obtained from Zucker
obese rats: The role of GLUT-4 and endothelin. Am J Physiol Heart
Circ Physiol. 294:H859–H866. 2008. View Article : Google Scholar
|
|
50
|
Das S, Falchi M, Bertelli A, Maulik N and
Das DK: Attenuation of ischemia/reperfusion injury in rats by the
anti-inflammatory action of resveratrol. Arzneimittelforschung.
56:700–706. 2006.
|
|
51
|
Goh SS, Woodman OL, Pepe S, Cao AH, Qin C
and Ritchie RH: The red wine antioxidant resveratrol prevents
cardiomyocyte injury following ischemia-reperfusion via multiple
sites and mechanisms. Antioxid Redox Signal. 9:101–113. 2007.
View Article : Google Scholar
|
|
52
|
Hung LM, Su MJ, Chu WK, Chiao CW, Chan WF
and Chen JK: The protective effect of resveratrols on
ischaemia-reperfusion injuries of rat hearts is correlated with
antioxidant efficacy. Br J Pharmacol. 135:1627–1633. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
El-Mowafy AM and White RE: Resveratrol
inhibits MAPK activity and nuclear translocation in coronary artery
smooth muscle: Reversal of endothelin-1 stimulatory effects. FEBS
Lett. 451:63–67. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yamamoto H, Schoonjans K and Auwerx J:
Sirtuin functions in health and disease. Mol Endocrinol.
21:1745–1755. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Becatti M, Taddei N, Cecchi C, Nassi N,
Nassi PA and Fiorillo C: SIRT1 modulates MAPK pathways in
ischemic-reperfused cardiomyocytes. Cell Mol Life Sci.
69:2245–2260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ungvari Z, Labinskyy N, Mukhopadhyay P,
Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P and Csiszar A:
Resveratrol attenuates mitochondrial oxidative stress in coronary
arterial endothelial cells. Am J Physiol Heart Circ Physiol.
297:H1876–H1881. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hsu CP, Odewale I, Alcendor RR and
Sadoshima J: Sirt1 protects the heart from aging and stress. Biol
Chem. 389:221–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Viswanathan M, Kim SK, Berdichevsky A and
Guarente L: A role for SIR-2.1 regulation of ER stress response
genes in determining C. elegans life span. Dev Cell. 9:605–615.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li Y, Xu S, Giles A, Nakamura K, Lee JW,
Hou X, Donmez G, Li J, Luo Z, Walsh K, et al: Hepatic
overexpression of SIRT1 in mice attenuates endoplasmic reticulum
stress and insulin resistance in the liver. FASEB J. 25:1664–1679.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jang M, Cai L, Udeani GO, Slowing KV,
Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta
RG, et al: Cancer chemopreventive activity of resveratrol, a
natural product derived from grapes. Science. 275:218–220. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rezk YA, Balulad SS, Keller RS and Bennett
JA: Use of resveratrol to improve the effectiveness of cisplatin
and doxorubicin: Study in human gynecologic cancer cell lines and
in rodent heart. Am J Obstet Gynecol. 194:e23–e26. 2006. View Article : Google Scholar : PubMed/NCBI
|