Introduction
Cisplatin (cis-diamminedichloroplatinum II,
CDDP) is one of the most effective chemotherapeutic agents used in
the treatment of advanced cervical cancer. However, chemoresistance
is the major reason of treatment failure. Therefore, in order to
improve the clinical outcome, more effective and tolerable
combination treatment strategies are required (1).
DNA damage induced by cisplatin triggers cell cycle
arrest and this then leads to apoptosis. Nevertheless, it has been
demonstrated that various survival signals, including
mitogen-activated protein kinase (MAPK) and phosphoinositide
3-kinase (PI3K)/protein kinase B (PKB/AKT) signaling, are activated
by cisplatin treatment and may thus be responsible for the
chemoresistance (2). There are 3
major subfamilies of the MAPK family: the extracellular
signal-regulated kinases (ERKs), the c-Jun, N-terminal kinases
(JNKs) and the p38 kinases, regulating a variety of physiological
processes such as cell growth, metabolism, differentiation and cell
death; however, MAPK signal dysfunction could results in
tumorigenesis and drug resistance. PKB/AKT, a serine/threonine
kinase, functions as an oncogene and has also been implicated in
resistance to chemotherapy drugs. It has been reported that the
inhibition of AKT enhances the therapeutic activity of paclitaxel
against cervical carcinomas (3,4).
3,4,20,40-Tetrahydroxychalcone (butein), as a
polyphenolic compound, is used as a food additive and a traditional
herbal medicine to alleviate pain, and in the treatment of
parasitic and thrombotic diseases (5). Previous studies, including ours have
demonstrated that butein exerts anticancer activity, and suppresses
the proliferation of a number of human cancers, including breast
carcinoma, colon caner, hepatocellular carcinoma and bladder cancer
(6–10). The anticancer activity of butein
has been reported to involve the regulation of AKT/MAPK signaling.
Butein has been shown to inhibit the activation of ERK, JNK and p38
in human hepatocellular carcinoma and breast cancer cells (9,11).
Moreover, butein has been shown to inhibit AKT phosphorylation,
resulting in the suppression of in breast cancer and prostate
cancer cell growth (7,12). Based on the above-mentioned facts,
we hypothesized that butein may sensitize cervical cancer cells to
cisplatin by suppressing the activation of the MAPK and PI3K/AKT
signaling pathways.
Materials and methods
Drugs and antibodies
Butein and cisplatin (CDDP) were obtained from Sigma
(St. Louis, MO, USA), and were dissolved in dimethyl sulfoxide
(DMSO) and stored at −20°C until use. The ERK inhibitor, U0126, p38
inhibitor, SB203580, and the AKT inhibitor, LY294002, were obtained
from Sigma and used at final concentrations of 10, 20 and 20
µM, respectively. The following antibodies were used in
western blot analysis: anti-ERK1/2 (#9102; 1:1,000), anti-p-ERK1/2
(Thr202/Tyr204) (#9101; 1:1,000), anti-p38 (#9212; 1:1,000),
anti-p-p38 (Thr180/Tyr182) (#9211; 1:1,000), anti-Akt (#9272;
1:1,000) and anti-p-Akt (Ser413) (#9271; 1:1,000) (all purchased
from Cell Signaling Technology, Danvers, MA, USA). β-actin
(sc-4778; 1:5,000), forkhead box O3a (FoxO3 or FoxO3a; sc-9812;
1:1,000), Bax (sc-7480; 1:1,000), Bcl-2 (sc-7382; 1:1,000), p27
(sc-1641; 1:1,000) and cyclin D1 (sc-718; 1:1,000) antibodies were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Horseradish peroxidase (HRP)-conjugated goat anti-mouse/anti-rabbit
immunoglobulin (IgG; ab6721; 1:1,000) was obtained from Abcam (Hong
Kong, China).
Cell culture
HeLa human cervical carcinoma cells were obtained
from the Shanghai Cell Bank of Chinese Academy of Sciences
(Shanghai, China). The cells were cultured in RPMI-1640 medium
(Gibco-BRL, Grand Island, NY, USA) supplemented with 10% bovine
calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin, and
maintained at 37°C in a humidified atmosphere of 5%
CO2.
Cell viability assay
The viability of the HeLa cells following treatment
with butein and cisplatin, alone or in combination, was determined
using the methylthiazol tetrazolium (MTT) assay. The Cells were
digested and diluted to 1×105/ml. Subsequently, 200
µl of the single cell suspension were seeded into 96-well
culture plates. Following overnight incubation, the cells were
washed with phosphate-buffered saline (PBS) and divided into groups
[control (untreated), butein-treated, cisplatin-treated and butein
+ cisplatin-treated cells]; each group had 6-wells in a single
line. The groups were placed in starvation medium with 0.2% DMSO or
the drugs and incubated for a further 48 h. The drug-containing
medium was then replaced with fresh medium. MTT solution (500
mg/ml) was added to the medium and this was maintained at 37°C for
4 h. The cells were cultured at 37°C for 4 h, 150 µl DMSO
was added, and the 570 nm wavelength absorption values were
measured using an EnSpire Multimode plate reader (Perkin-Elmer,
Waltham, MA, USA). All experiments were performed in triplicate,
and repeated 3 times. Cell viability and growth inhibition were
calculated as follows: cell viability rate = A570 value of the drug
treated group/A570 value of the control untreated group ×100;
growth inhibition rate = 1 - cell viability rate. The interaction
between the 2 drugs was judged according to a method described in
the study by Jin (13). Briefly,
a q-value was calculated according to the formula: q = Ea + b/(Ea +
Eb − Ea × Eb). The 2 drugs were considered to have additive effects
if 0.85<q<1.15, or synergistic effects if q>1.15, and are
antagonistic if q<0.85.
Apoptosis assay
Apoptosis was assessed using the Annexin V-FITC
apoptosis detection kit according to the manufacturer's
instructions (Sigma-Aldrich, Oakville, ON, Canada). Approximately
106 cells were seeded onto sterile flat-bottom
25-cm2 culture flasks. The cells were treated with
cisplatin and butein according to the experimental design.
Following incubation for 48 h, the cells were collected, washed in
PBS and resuspended in 500 ml of 1X Annexin V binding buffer and
then incubated at room temperature with teh Annexin V-FITC and PI
stain in the absence of light. Following a 10-min incubation, the
cells were immediately analyzed by flow cytometry. Annexin V
staining was detected as green fluorescence and PI as red
fluorescence. The percentage of cells undergoing apoptosis was
determined by 3 independent experiments.
Cell cycle analysis
Cells were seeded at a density of 2×105
cells/well in 6-well plates. Following overnight incubation, the
cells were then exposed to butein and/or cisplatin for 48 h.
Following incubation, the cells were then fixed for 1 h in ice-cold
70% ethanol and incubated for 30 min at 37°C with 0.5 U of RNase A
(Sigma-Aldrich). DNA was then stained for 10 min with 50
µg/ml of PI and the cells analyzed using a flow cytometer
(FACSCalibur; BD Biosciences, San Jose, CA, USA).
Western blot analysis
The cells were lysed with RIPA buffer (150 mM NaCl,
1.0% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris,
pH 8.0) containing protease inhibitor cocktail (Roche Applied
Science, Mannheim, Germany). Following centrifugation for 10 min at
10000 × g at 4°C, the supernatant was collected for western blot
analysis. The protein concentration were determined using the
Bradford protein assay kit (Bio-Rad Laboratories, Hercules, CA,
USA). Equal amounts of protein were loaded onto a 10% SDS-PAGE gel
and then transferred onto nitrocellulose membranes (Pall Life
Sciences-Pall Corp., Port Washington, NY, USA) using a wet
transmembrane device (Amersham Pharmacia Biotech, Piscataway, NJ,
USA). The membranes were blocked with 5% non-fat milk at room
temperature for 1 h, probed overnight with primary antibodies
followed by incubation with the appropriate HRP-conjugated
secondary antibody for 2 h at room temperature. ECL reagent (Santa
Cruz Biotechnology) was used to develop the blots. All values were
normalized to those of β-actin.
Immunofluorescence staining
The cfells were grown on glass coverslips and
exposed to various concentrations of the drugs for the indicated
periods of time. The cells were fixed with methanol for 10 min at
−20°C and permeabilized with 0.5% Triton X-100 in PBS for 5 min at
room temperature, and were then blocked with goat serum for 30 min
at room temperature, incubated with primary antibody to FoxO3a
(1:100; Santa Cruz Biotechnology;) diluted in PBS overnight, and
then incubated with fluorescent secondary antibody for 30 min at
room temperature. The nucleus was counterstained with DAPI (0.5
µg/ml) for 10 min in dark. The coverslips with cells were
examined and photographed under a fluorescence microscope (Axio
Observer Inverted Microscope; Carl Zeiss, Oberkochen, Germany).
Transfection with siRNA
The HeLa cells were transfected with siRNA specific
to FoxO3a (5′-ACUCCGGGUCCAGCU CCAC-3′) (synthesized by Shanghai
GenePharma Co., Ltd., Shanghai, China) using Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer's
instructions. A control non-specific siRNA
(5′-UUCUCCGAACGUGUCACGUTT-3′) was used in parallel experiments as a
negative control.
Animal studies
Nude mice were obtained from the Animal Institute of
Xi'an Jiaotong University, China (XJTU). In total, 12 female 6- or
7-week old nude mice were raised in autoclave cages and supplied
with unlimited water and 5% fatty food. Room temperature and
humidity were maintained at 26–28°C and 40–60%, respectively. All
the animal-related procedures were approved by the Ethics Committee
of the First Affiliated Hospital, and were adherent to the
institutional guidelines and ethical standards. Suspensions of
1×106 HeLa cells were injected subcutaneously into the
flanks of nude mice. When the tumor volume was ≥0.1 cm3,
the mice were treated intraperitoneally with butein (2 mg/kg/2
days, n=4) or butein (2 mg/kg every 2 days, n=4) + cisplatin (2
mg/kg every 2 days, n=4) for 3 weeks. Body weight and clinical
symptoms of the mice were determined every other day. Tumor volume
was calculated according to the formula: V = 0.5236 × (L ×
W2), where V represents the tumor volume, L represents
the length and W represents the width. The animals were euthanized
on day 22 following the therapeutic injection.
Histological examination
Tumor samples were fixed in 4% paraformaldehyde for
24 h and embedded in paraffin blocks. The sections were dewaxed in
xylene, hydrated through an upgraded ethanol series and stained
with H&E, and for immunohistochemical analysis, the sections
were then heated in 0.01 M citrate buffer (pH 6.0) in a steamer for
1.5 min to retrieve the antigen binding sites. The detection of
antigens was carried out by incubation with primary antibody
(FoxO3a, 1:200) for 2 h at room temperature, followed by incubation
with HRP-labeled secondary antibody (MaxVision HRP-Polymer
anti-Mouse/Rabbit IHC kit, 1:200) at room temperature for 30 min
and color development with DAB. The negative control specimens were
incubated in PBS without the primary antibody under the same
conditions. Digital images were acquired on an Olympus BH-2
microscope (Olympus, Tokyo, Japan) installed with a DeltaPix camera
and software (Delta Pix, Maalov, Denmark).
Statistical analysis
Data are expressed as the means ± SD. All
statistical analyses were performed using the SPSS 18.0 statistical
software package. Statistical differences were determined by the
Student's t-test. Differences were considered statistically
significant at P<0.05 and highly significant at P<0.01 for
all comparisons.
Results
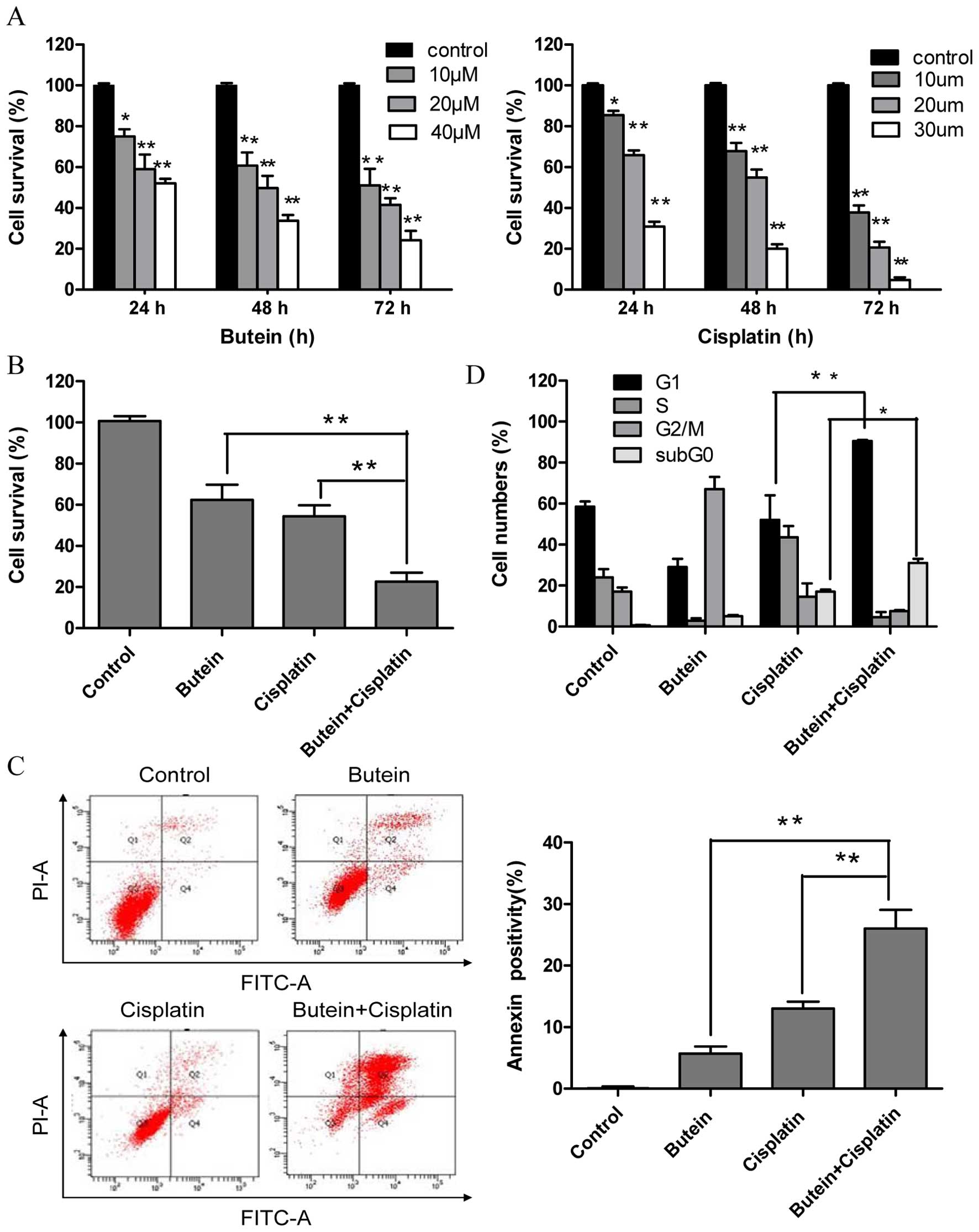
Butein synergistically enhances the cell
growth inhibitory and apoptosis-inducing effects of cisplatin
To investigate the effectiveness of combined
treatment with butein and cisplatin, the HeLa cells were treated
with various concentrations of butein (10, 20 and 40 µm)
and/or cisplatin (10, 20 and 30 µm) for different periods of
time, and cytotoxicity was evaluated by MTT assay and the
interaction index. Treatment with both butein and cisplatin alone
inhibited cell growth in a dose- and time-dependent manner
(Fig. 1A). Combined treatment
with 20 µm butein and 20 µm cisplatin for 48 h
induced a marked synergistic cytotoxic effect (q-values were 1.17;
Fig. 1B). The apoptosis of the
HeLa cells induced by butein in combination with cisplatin was
further investigated by Annexin V/PI staining. As shown in Fig. 1C, treatment with butein or
cisplatin alone slightly increased the percentage of apoptotic
cells compared to the untreated controls, while the combinatino of
both drugs significantly enhanced apoptosis. These results were in
concordance with those of MTT assay, illustrating the synergistic
effects of butein and cisplatin.
Co-treatment with butein and cisplatin
increases G1 phase arrest
A previous study demonstrated that butein induced
G2/M arrest (14). Thus, in this
study, we investigated whether the synergistic effects of butein
and cisplatin are due to G2/M cell cycle arrest, as induced by
butein. In contrast to our expectations, co-treatment with butein
and cisplatin induced G1 phase arrest, which differed from the
effects on the cell cycle induced by treatment with butein or
cisplatin alone (Fig. 1D).
Furthermore, the sub-G0 DNA content was taken as a measure of the
apoptotic cell population. We observed an enhancement in apoptosis
in the cells treated with both butein and cisplatin compared to
those treated with cisplatin alone, which is in agreement with our
above-mentioned findings.
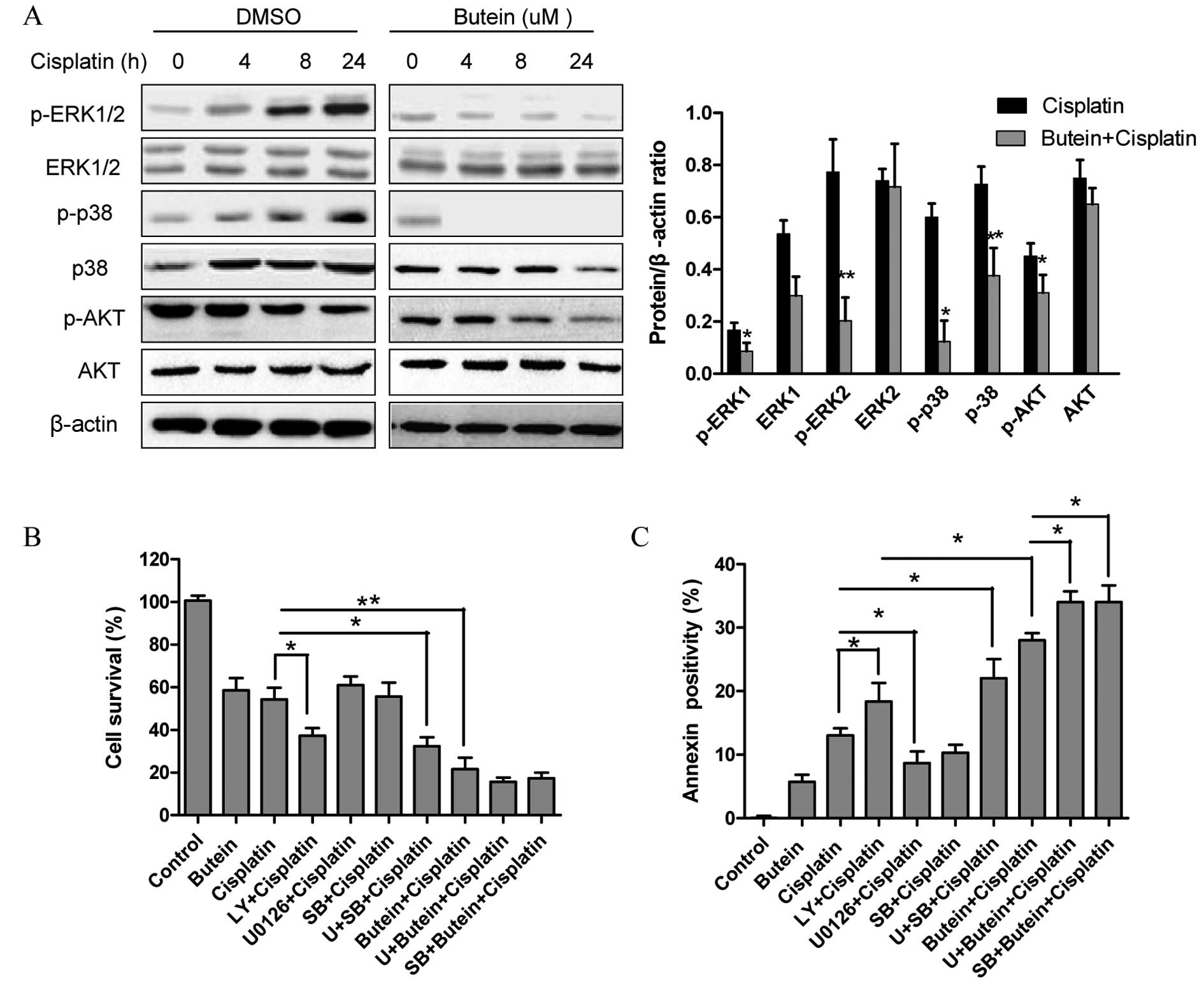
The AKT, ERK and p38 MAPK pathways are
involved in the synergistic effects of butein and cisplatin
The role of the MAPK and AKT pathways in resistance
to cisplatin has been previously reported (4). Since butein has been shown to exert
its anticancer effects by inhibiting the AKT and MAPK signaling
pathways (11,12), in this study, we investigated
whether these signaling pathways are associated with the enhanced
growth inhibitory effects on HeLa cells following combined
treatment with butein and cisplatin. As shown in Fig. 2A, butein significantly inhibited
the phosphorylation of ERK and p38 induced by cisplatin, but had
obvious effect on JNK expression (data not shown). Moreover,
treatment with either cisplatin or butein alone or in combination
inhibited AKT activation; however, p-AKT was inhibited to a greater
extent following treatment with both drugs than with cisplatin
alone.
Next, ERK, p38 and AKT signaling inhibitors were
used to examine the functional specificity of these signaling
pathways in the synergistic effects of butein and cisplatin
(Fig. 2B). The HeLa cells were
pre-treated with U0126 (10 µm)/SB203580 (20
µm)/LY294002 (20 µm) for 8 h and then treated with
cisplatin and/or butein for 48 h. A significant enhancement in the
cytotoxic effects of cisplatin was observed following treatment
with the AKT inhibitor (LY294002), even though these cytotoxic
effects (with AKT inhibitor plus cisplatin) were not as prominent
as those observed following treatment with both cisplatin and
butein, demonstrating that the inhibition of AKT, to a certain
extent, plays a role in the synergistic effects of these drugs.
Notably, a slight increase in the cytotoxicity induced by cisplatin
was observed when the cells were co-treated with the ERK (U0126) or
p38 MAPK (SB203580) inhibitor as well as cisplatin; however,
enhanced cytotoxic effects were observed following treatment with
the ERK or p38 MAPK inhibitor in combination with butein and
cisplatin. These findings were further supported by the results of
apoptosis assay (Fig. 2C).
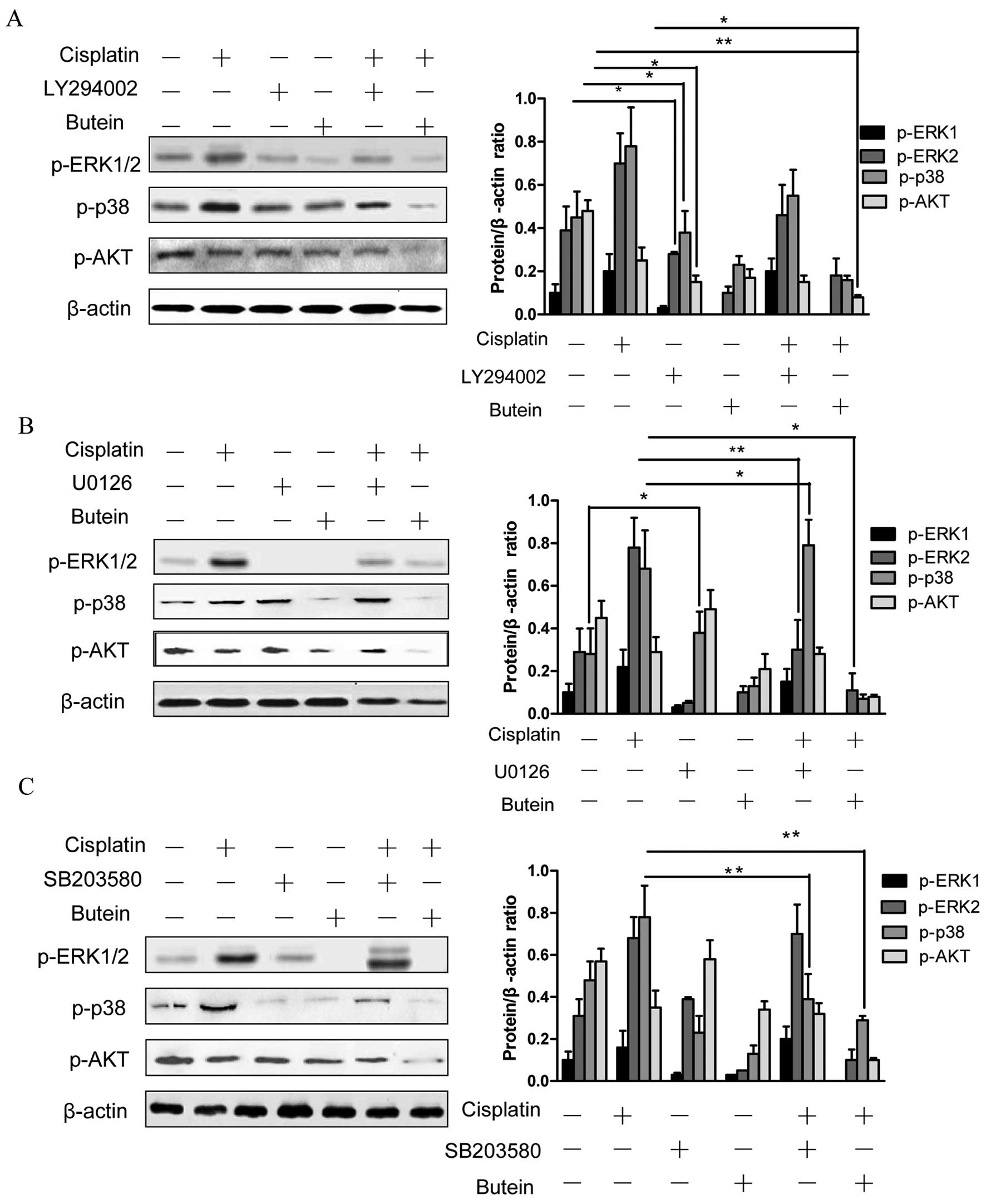
Furthermore, we observed that the inhibition of AKT (with the
inhibitor LY294002) decreased the phosphorylation of ERK and p38
(Fig. 3A). In addition, the
inhibition of ERK by U0126 increased the phosphorylation of p38,
and the inhibition of p38 by SB203580 activated ERK1/2 (with
cisplatin treatment); however, the inhibition of both ERK and p38
had no obvious effect on p-AKT (Fig.
3B and C).
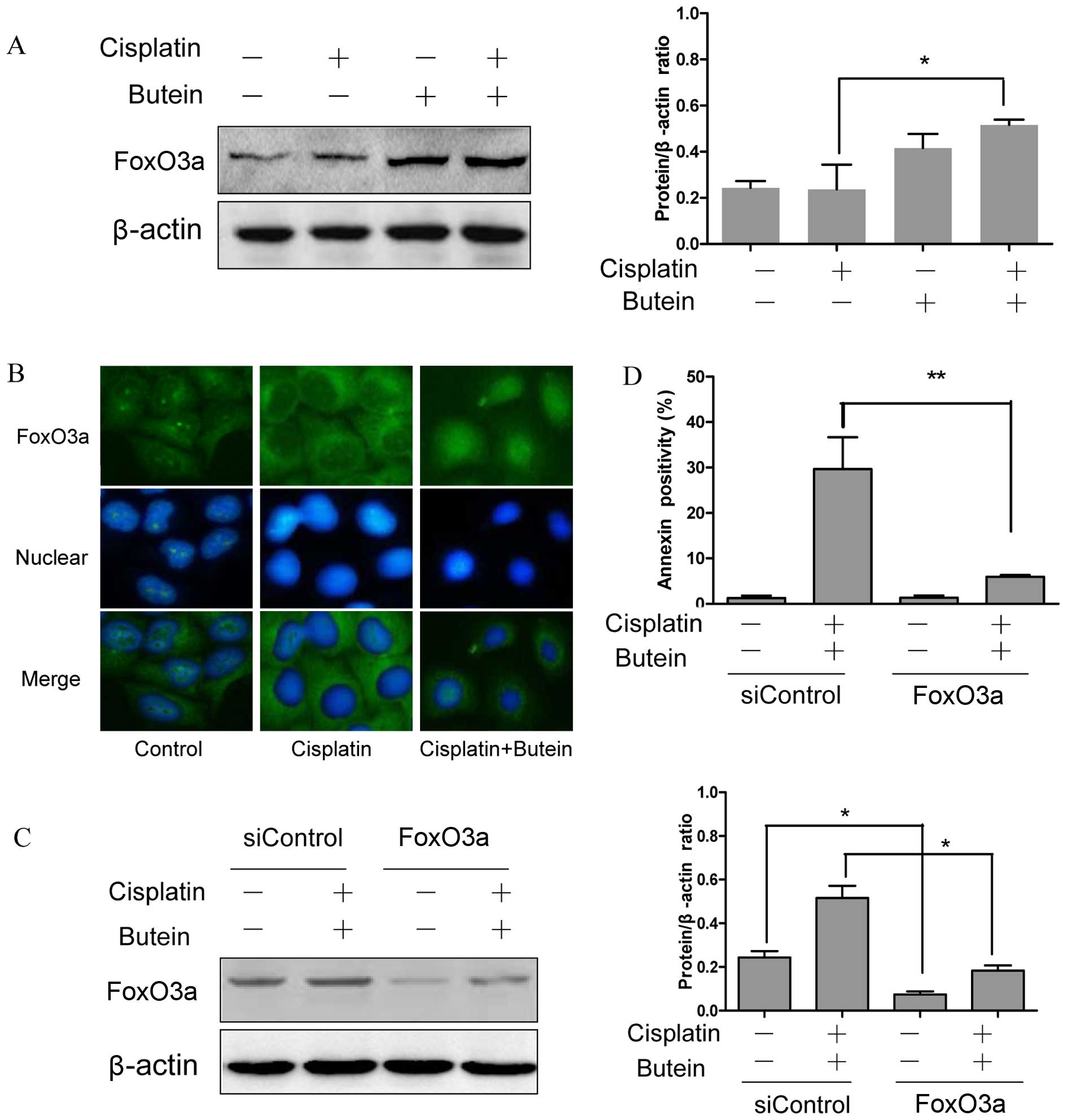
FoxO3a and its downstream molecules play
a role in the synergistic effects of butein and cisplatin
FoxO3a, as a tumor suppressor protein, is involved
in the resistance to cisplatin (15); FoxO3a can be phosphorylated and in
turn degraded by AKT and ERK (16,17). Moreover, a recent study revealed
that the inhibition of p38α in combination with chemotherapeutic
agents promoted the activation of FoxO3a (18). Based on the above observations, we
hypothesized that FoxO3a may be assoicated with the synergistic
effects of butein and cisplatin. To examine this hypothesis, the
subcellular localization and expression of FoxO3a were determined
by immunofluorescence staining and western blot analysis. The
results revealed that treatment of the cells with both agents led
to an increased expression level of FoxO3a and to its translocation
from the cytoplasm to the nucleus compared to the controls and the
cells treated with cisplatin alone (Fig. 4A and B).
In order to further confirm the role of FoxO3a in
the synergistic effects of cisplatin and butein, the HeLa cells
were incubated for 48 h with siRNA targeting FoxO3a and then
treated with butein and cisplatin for 48 h. The RNAi-mediated
downregulation of FoxO3a significantly decreased the apoptosis
induced by combined treatment with butein and cisplatin in the HeLa
cells (Fig. 4C and D). These data
provide evidence that FoxO3a plays a role in the synergistic
apoptotic effects induced by butein and cisplatin.
Previous studies have demonstrated that FoxO3a
functions as a tumor suppressor by regulating the expression of
genes involved in apoptosis, cell cycle arrest, oxidative stress,
resistance and autophagy (15–17). Thus, we also examined the
expression of several molecules involved in cell cycle arrest and
apoptosis, which are known as downstream targets of FoxO3a. As
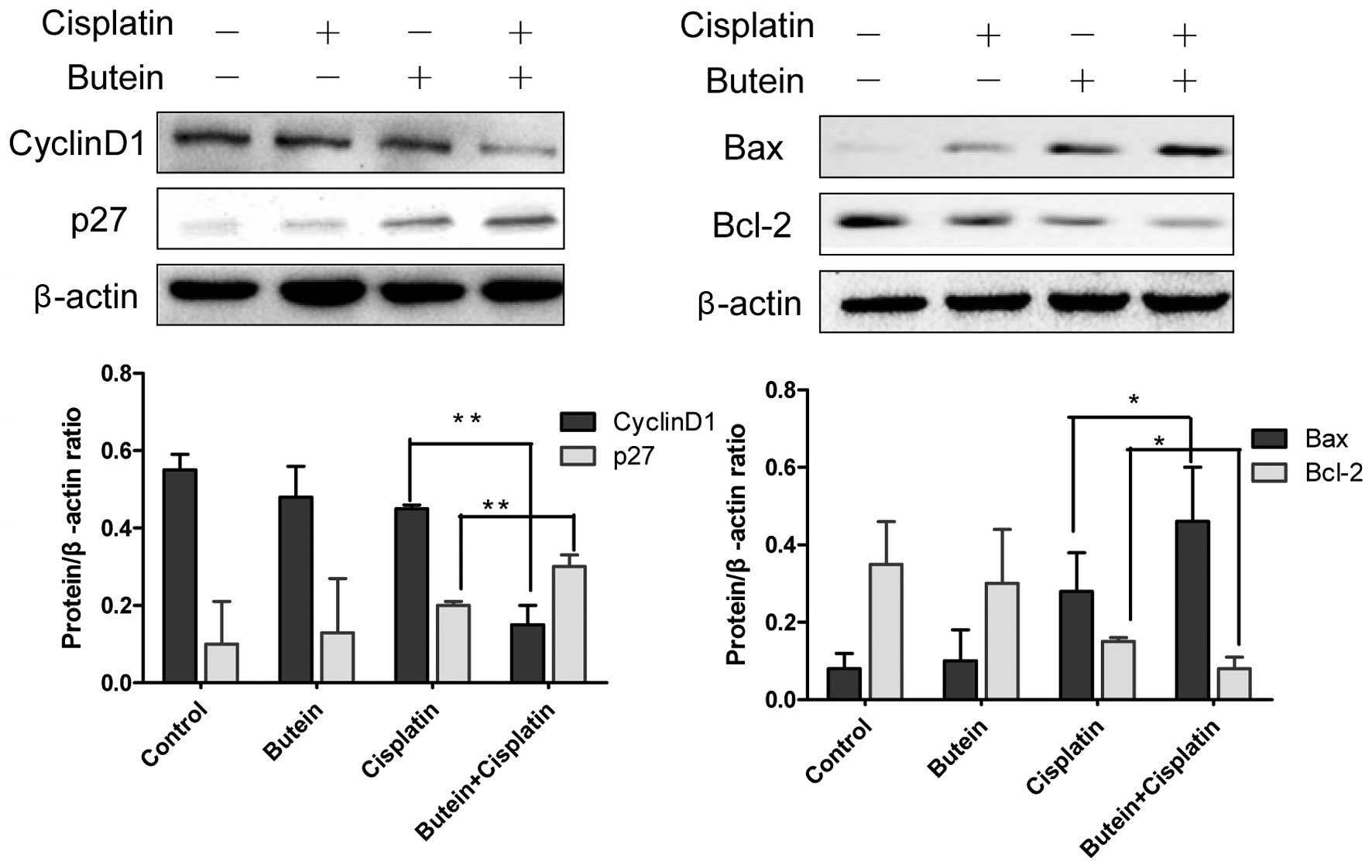
shown in Fig. 5, butein, in
combination with cisplatin, significantly enhanced the expression
of p27 and decreased that of cyclin D1 compared to treatment with
cisplatin alone, but had no effect on the expression of p21 (data
not shown). Bax and Bcl-2 are important members of Bcl-2 family
proteins and regulate mitochondrial involvement in apoptosis
(19,20). Co-treatment with butein and
cisplatin for 48 h resulted in increased expression levels of Bax,
but reduced protein levels of Bcl-2 compared to treatment with
cisplatin alone (Fig. 5).
Butein in combination with cisplatin
suppresses tumor growth and increases FoxO3a expression in
vivo
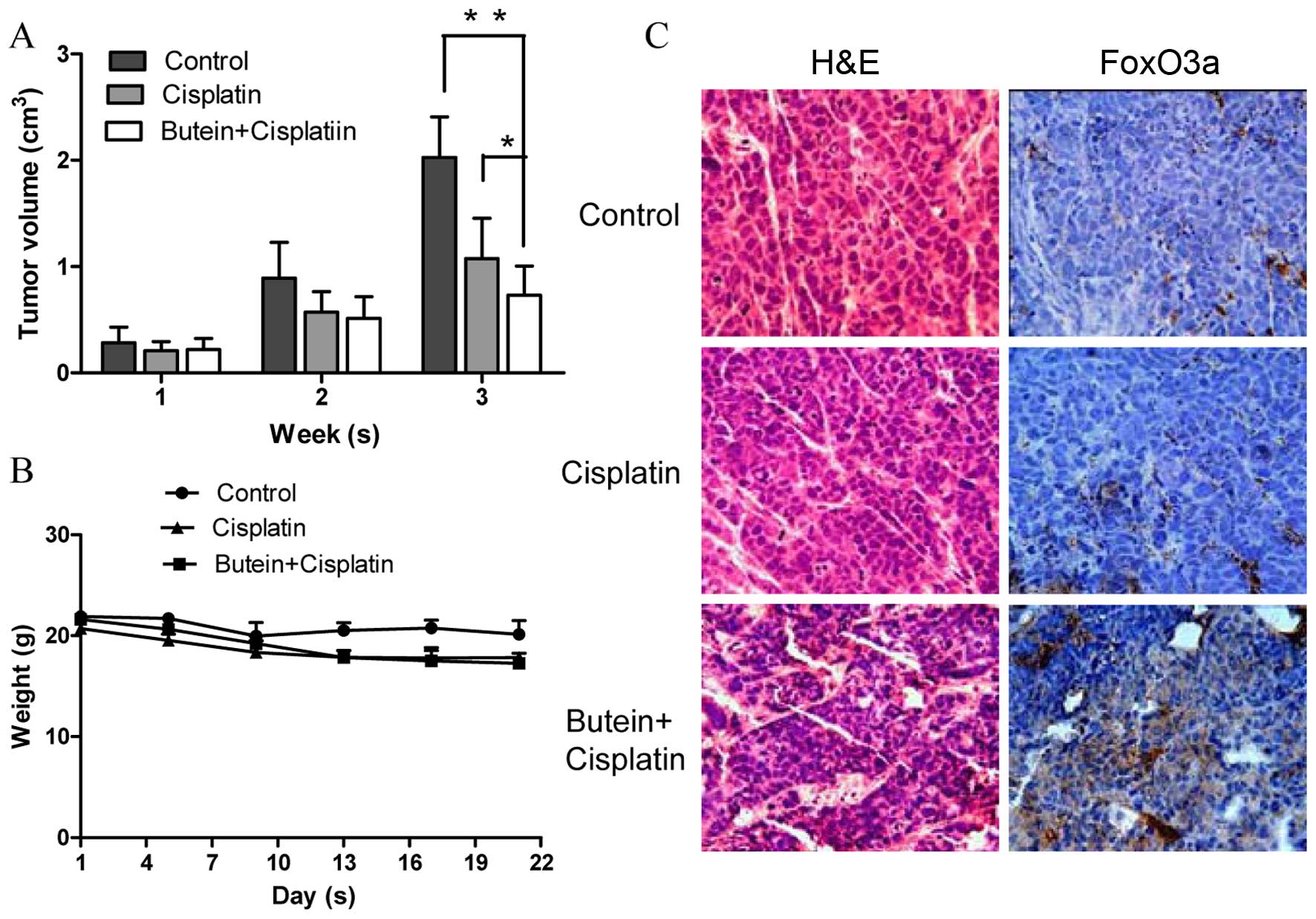
To determine the synergistic antitumor potential of
butein and cisplatin in vivo, a nude mouse tumor xenograft
growth model was created. The animals were administered cisplatin
and/or butein by intraperitoneal injection every 2 days when the
average tumor volume was ≥0.1 cm3. The primary tumor
sizes were monitored each week. At necropsy on day 22 following
treatment, the observed inhibitory effects of cisplatin on tumor
volume were significant, as compared with the controls. However, we
found that combined treatment (butein + cisplatin) had the most
prominent effect on tumor volume (Fig. 6A), while there was no obvious
difference observed in body weight between the mice in the control
group, and those treated with cisplatin alone or both drugs
(Fig. 6B). We further evaluated
the effects of butein on the FoxO3a expression level in the tumor
tissues by immunohistochemical staining and found that the
expression of FoxO3a was substantially increased in the mice
treated with both agents compared with the controls and the mice
treated with cisplatin alone (Fig.
6C).
Discussion
Drug resistance remains a major challenge in cancer
therapy and has attracted increasing attention. In the present
study, to the best of our knowledge, we investigated for the first
time the synergistic effects of butein and cisplatin on cervical
cancer cell growth inhibition and apoptosis in vitro and
in vivo, and further explored the possible mechanisms
responsible for their synergistic effects.
The anticancer effects of butein have been well
documented in various types of cancer (6–10).
Our results also indicated that butein inhibited HeLa cell
proliferation in a dose- and time-dependent manner. However, to the
best of our knowledge, no previous studies on the possible
synergistic anticancer effects of butein and cisplatin have been
published to date. In the present study, we found that butein
enhanced the growth inhibitory and apoptotic effects induced by
cisplatin. The underlying molecular mechanisms were also explored.
Although the signaling pathways activated by DNA damage are
different from the types of DNA damage, the activation of these
pathways has similar results, including cell cycle arrest and
ultimately, either cell survival or cell death. In this study,
butein induced G2/M arrest, which is in accordance with the
findings of a previous study (14). We speculated that the synergistic
effects of butein and cisplatin may be due to G2/M cell cycle
arrest. In contrast to our expectations, co-treatment with butein
and cisplatin increased G1 phase arrest, suggesting that enhanced
G1 phase arrest plays a role in the synergistic apoptotic effects
induced by combined treatment with butein and cisplatin.
It has been demonstrated that chemoresistance is due
to survival signaling pathways activated during chemotherapy
(2,21). In the present study, we found that
butein markedly reduced the phosphorylation and activation of ERK,
p38 and AKT in the presence of cisplatin, but had no obvious effect
on JNK (data not shown). Therefore, it is possible that butein
blocks the signaling circuit involving ERK/p38 MAPK and AKT, acting
as a chemosensitizer. The ERK pathway is widely accepted as an
important survival regulator, and the chemical inhibition of the
ERK pathway has been shown to sensitize cells to cisplatin
(22,23). However, studies have produced
different data and have shown that ERK activation is required in
cisplatin-induced apoptosis (24,25); the inhibition of ERK activation
has also been shown to markedly attenuate cisplatin-induced cell
death (26,27). The biological effects of p38
activation are also highly conflicting. The inhibition of p38 by
SB203580 has been shown to significantly block Met-induced
apoptosis in A549/CDDP cells (28), while other researchers have found
that the inhibition of p38 sensitizes breast and gastric cancer
cells to cisplatin-induced apoptosis (29,30) and enhanced p38 activation has been
associated with a poor overall survival in patients with breast
cancer (31) and hepatocellular
carcinoma (32). It has been
reported that p38 enhances cancer cell growth after the acquisition
of the malignant phenotype, and acts as a tumor suppressor, mainly
at the onset of cellular transformation (33,34). Thus, the role of ERK and p38
appears to be dependent upon the cellular context and stimuli. The
constitutive activation of AKT has also been implicated in
chemoresistance (35), while the
inactivation of AKT signaling by chemicals sensitizes human cancer
cells to cisplatin (36).
To further identify the functional specificities of
these pathways in the synergistic effects of butein and cisplatin,
in this study, we used AKT and ERK/p38 MAPK inhibitors. The results
revealed that the AKT inhibitor enhanced the apoptotic effects of
cisplatin, although the apoptotic effects induced by the AKT
inhibitor in combination with cisplatin were not as prominent as
those induced by combined treatment with butein and cisplatin.
These findings suggest that butein exerts its sensitizing effects
on cisplatin, to a certain extent, through the regulation of AKT,
and that other signaling pathways are involved in the synergistic
effects. We then found the dual inhibition of ERK/p38 MAPK and ERK
or p38 in combination with butein enhanced the apoptosis induced by
cisplatin, indicating that the inhibition of ERK and p38 by butein
also plays a role in the synergistic effects of butein and
cisplatin. Of note, we found that the inhibition of ERK promoted
p38 activation, and the slight activation of ERK was also observed
upon the inhibition of p38 in combination with cisplatin treatment,
although no significant difference was observed. In fact, ERK
inhibition triggers p38 activation in HeLa cells (37), and p38 inhibition has also been
reported to upregulate the activation of the MEK-ERK1/2 survival
pathway (38). A recent study by
Chiacchiera et al (38)
demonstrated that the combined inhibition of p38α and MEK
specifically induced apoptosis through caspase-3 in colorectal
cancer cells. These data indicate that there is a crosstalk between
the ERK and p38 pathways, which is crucial for the therapeutic
response. In addition, we observed that the AKT inhibitor decreased
the phosphorylation of ERK and p38, while the inhibition of ERK and
p38 had no effects on AKT activation, indicating that AKT may be
the upstream signal of ERK and p38.
FoxO3a has been investigated as a crucial protein
that is involved in the regulation of cell survival and
proliferation, contributing to tumor suppression (16). The AKT and ERK-mediated
phosphorylation of FoxO3a stimulate its ubiquitination, resulting
in proteasomal degradation (17,40). AKT directly phosphorylates FoxO3a
at S253, which is a crucial residue regulating the
nuclear/cytoplasmic shuttling of FoxO3a. FoxO3a localization in the
cytoplasm is a key step leading to FoxO3a deactivation and
degradation, and correlates with poor survival in patients with
breast cancer (39). Studies have
found that FoxO3a may be a key molecule of the p38 pathway and may
be involved in drug resistance (41,42). Since butein inhibited the AKT, ERK
and p38 MAPK pathways, which are all involved in the regulation of
FoxO3a, we subsequently examined whether FoxO3a is a key molecule
involved in the synergistic effects of butein and cisplatin. We
found that combined treatment with butein and cisplatin increased
the nuclear translocation and expression of FoxO3a compared to
treatment with cisplatin alone, and the downregulation of FoxO3a by
RNAi significantly inhibited the synergistic effects of butein and
cisplatin in HeLa cells, suggesting that butein exerts its
chemosensitizing effects, in part through FoxO3a activation. Our
in vivo findings revealed that butein and cisplatin exerted
similar inhibitory effects on tumor growth, by increasing the
FoxO3a protein level.
Activated FoxO3a is able to bind to promoters and
induces the transcription of target genes, which include p21, p27
and cyclin D1 for cell cycle arrest, and Bim, Bcl-2 and Bax for
cell apoptosis (43–45). Alterations in cell cycle
progression in various tumors are often due to mutations or the
overexpression of genes. As an inhibitor of cyclin E-Cdk2, p27
plays a pivotal role in controlling cell G1-S phase transition
during development and tumorigenesis. In addition, cyclin D1
mediates the G1/S transition by binding to Cdk4 and also by
sequestering a Cdk inhibitor of p21 and p27 (46). The present study demonstrated that
the upregulation of p27 and the downregulation of cyclin D1
expression was induced by combined treatment with butein and
cisplatin compared to treatmetn with cisplatin alone, which
coincided with G1 phase arrest. This suggests that cyclin D1 and
p27, two important regulators of the cell cycle, are intracellular
targets of the butein-mediated anti-proliferative effects on HeLa
cells through FoxO3a activation. The Bcl-2 family of proteins are
important regulators of apoptosis (47,48). In this study, the apoptosis
induced by butein in HeLa cells was associated with the
downregulation of anti-apoptotic Bcl-2 expression and an increased
Bax expression.
Overall, the findings of this study reveal a new
function of butein that enhances the sensitization of cervical
cancer cells to cisplatin in vitro and in vivo, which
may be related to the AKT and ERK/p38 MAPK pathways, at least to a
certain extent, through the regulation of FoxO3a. These data shed
some light on the synergistic antitumor effects of butein and
cisplatin and verify the potential clinical use of butein.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81202055); the
Key Sci-tech Project of Shaanxi Province (no. 2012SF2-03) and the
Sci-tech Project of Xi'an City (no. SF1204(56).
References
|
1
|
Basu A and Krishnamurthy S: Cellular
responses to cisplatin-induced DNA damage. J Nuleic Acids 2010:
Article ID 201367. 2010.
|
|
2
|
Brozovic A and Osmak M: Activation of
mitogen-activated protein kinases by cisplatin and their role in
cisplatin-resistance. Cancer Lett. 251:1–16. 2007. View Article : Google Scholar
|
|
3
|
Wang Z, Hou J, Lu L, Qi Z, Sun J, Gao W,
Meng J, Wang Y, Sun H, Gu H, et al: Small ribosomal protein subunit
S7 suppresses ovarian tumorigenesis through regulation of the
PI3K/AKT and MAPK pathways. PLoS One. 8:e791172013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bava SV, Puliappadamba VT, Deepti A, Nair
A, Karunagaran D and Anto RJ: Sensitization of taxol-induced
apoptosis by curcumin involves down-regulation of nuclear
factor-kappaB and the serine/threonine kinase Akt and is
independent of tubulin polymerization. J Biol Chem. 280:6301–6308.
2005. View Article : Google Scholar
|
|
5
|
Kang DG, Lee AS, Mun YJ, Woo WH, Kim YC,
Sohn EJ, Moon MK and Lee HS: Butein ameliorates renal concentrating
ability in cisplatin-induced acute renal failure in rats. Biol
Pharm Bull. 27:366–370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Chan FL, Chen S and Leung LK: The
plant polyphenol butein inhibits testosterone-induced proliferation
in breast cancer cells expressing aromatase. Life Sci. 77:39–51.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cho SG, Woo SM and Ko SG: Butein
suppresses breast cancer growth by reducing a production of
intracellular reactive oxygen species. J Exp Clin Cancer Res.
33(51)2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yit CC and Das NP: Cytotoxic effect of
butein on human colon adenocarcinoma cell proliferation. Cancer
Lett. 82:65–72. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma CY, Ji WT, Chueh FS, Yang JS, Chen PY,
Yu CC and Chung JG: Butein inhibits the migration and invasion of
SK-HEP-1 human hepatocarcinoma cells through suppressing the ERK,
JNK, p38, and uPA signaling multiple pathways. J Agric Food Chem.
59:9032–9038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang L, Chen W and Li X: A novel
anticancer effect of butein: Inhibition of invasion through the
ERK1/2 and NF-kappa B signaling pathways in bladder cancer cells.
FEBS Lett. 582:1821–1828. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lau GT, Huang H, Lin SM and Leung LK:
Butein downregulates phorbol 12-myristate 13-acetate-induced COX-2
transcriptional activity in cancerous and non-cancerous breast
cells. Eur J Pharmacol. 648:24–30. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khan N, Adhami VM, Afaq F and Mukhtar H:
Butein induces apoptosis and inhibits prostate tumor growth in
vitro and in vivo. Antioxid Redox Signal. 16:1195–1204. 2012.
View Article : Google Scholar :
|
|
13
|
Jin ZJ: About the evaluation of drug
combination. Acta Pharmacol Sin. 25:146–147. 2004.PubMed/NCBI
|
|
14
|
Moon DO, Kim MO, Choi YH, Hyun JW, Chang
WY and Kim GY: Butein induces G(2)/M phase arrest and apoptosis in
human hepatoma cancer cells through ROS generation. Cancer Lett.
288:204–213. 2010. View Article : Google Scholar
|
|
15
|
Shiota M, Yokomizo A, Kashiwagi E, Tada Y,
Inokuchi J, Tatsugami K, Kuroiwa K, Uchiumi T, Seki N and Naito S:
Foxo3a expression and acetylation regulate cancer cell growth and
sensitivity to cisplatin. Cancer Sci. 101:1177–1185. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khatri S, Yepiskoposyan H, Gallo CA,
Tandon P and Plas DR: FOXO3a regulates glycolysis via
transcriptional control of tumor suppressor TSC1. J Biol Chem.
285:15960–15965. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang W, Dolloff NG and El-Deiry WS: ERK
and MDM2 prey on FOXO3a. Nat Cell Biol. 10:125–126. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Germani A, Matrone A, Grossi V, Peserico
A, Sanese P, Liuzzi M, Palermo R, Murzilli S, Campese AF,
Ingravallo G, et al: Targeted therapy against chemoresistant
colorectal cancers: Inhibition of p38α modulates the effect of
cisplatin in vitro and in vivo through the tumor suppressor FoxO3A.
Cancer Lett. 344:110–118. 2014. View Article : Google Scholar
|
|
19
|
Quast SA, Berger A, Plötz M and Eberle J:
Sensitization of melanoma cells for TRAIL-induced apoptosis by
activation of mitochondrialpathways via Bax. Eur J Cell Biol.
93:42–48. 2014. View Article : Google Scholar
|
|
20
|
Kim MJ, Yun HS, Hong EH, Lee SJ, Baek JH,
Lee CW, Yim JH, Kim JS, Park JK, Um HD and Hwang SG: Depletion of
end-binding protein 1 (EB1) promotes apoptosis of human
non-small-cell lung cancer cells via reactive oxygen species and
Bax-mediated mitochondrial dysfunction. Cancer Lett. 339:15–24.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsu HH, Cheng LH, Ho TJ, Kuo WW, Lin YM,
Chen MC, Lee NH, Tsai FJ, Tsai KH and Huang CY: Apicidin-resistant
HA22T hepato-cellular carcinoma cells massively promote
pro-survival capability via IGF-IR/PI3K/Akt signaling pathway
activation. Tumour Biol. 35:303–313. 2014. View Article : Google Scholar
|
|
22
|
Persons DL, Yazlovitskaya EM, Cui W and
Pelling JC: Cisplatin-induced activation of mitogen-activated
protein kinases in ovarian carcinoma cells: Inhibition of
extracellular signal-regulated kinase activity increases
sensitivity to cisplatin. Clin Cancer Res. 5:1007–1014.
1999.PubMed/NCBI
|
|
23
|
Wang J, Zhou JY and Wu GS: Bim protein
degradation contributes to cisplatin resistance. J Biol Chem.
286:22384–22392. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Govind S, Sajankila SP, Mi L, Roy
R and Chung FL: Phenethyl isothiocyanate sensitizes human cervical
cancer cells to apoptosis induced by cisplatin. Mol Nutr Food Res.
55:1572–1581. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sheridan C, Brumatti G, Elgendy M, Brunet
M and Martin SJ: An ERK-dependent pathway to Noxa expression
regulates apoptosis by platinum-based chemotherapeutic drugs.
Oncogene. 29:6428–6441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guégan JP, Ezan F, Théret N, Langouët S
and Baffet G: MAPK signaling in cisplatin-induced death:
Predominant role of ERK1 over ERK2 in human hepatocellular
carcinoma cells. Carcinogenesis. 34:38–47. 2013. View Article : Google Scholar
|
|
28
|
Wang Y, Lin B, Wu J, Zhang H and Wu B:
Metformin inhibits the proliferation of A549/CDDP cells by
activating p38 mitogen-activated protein kinase. Oncol Lett.
8:1269–1274. 2014.PubMed/NCBI
|
|
29
|
Pereira L, Igea A, Canovas B, Dolado I and
Nebreda AR: Inhibition of p38 MAPK sensitizes tumour cells to
cisplatin-induced apoptosis mediated by reactive oxygen species and
JNK. EMBO Mol Med. 5:1759–1774. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Feng R, Zhai WL, Yang HY, Jin H and Zhang
QX: Induction of ER stress protects gastric cancer cells against
apoptosis induced by cisplatin and doxorubicin through activation
of p38 MAPK. Biochem Biophys Res Commun. 406:299–304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Esteva FJ, Sahin AA, Smith TL, Yang Y,
Pusztai L, Nahta R, Buchholz TA, Buzdar AU, Hortobagyi GN and Bacus
SS: Prognostic significance of phosphorylated P38 mitogen-activated
protein kinase and HER-2 expression in lymph node-positive breast
carcinoma. Cancer. 100:499–506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang SN, Lee KT, Tsai CJ, Chen YJ and Yeh
YT: Phosphorylated p38 and JNK MAPK proteins in hepatocellular
carcinoma. Eur J Clin Invest. 42:1295–1301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dolado I, Swat A, Ajenjo N, De Vita G,
Cuadrado A and Nebreda AR: p38alpha MAP kinase as a sensor of
reactive oxygen species in tumorigenesis. Cancer Cell. 11:191–205.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Voisset E, Oeztuerk-Winder F, Ruiz EJ and
Ventura JJ: p38α negatively regulates survival and malignant
selection of transformed bronchioalveolar stem cells. PLoS One.
8:e789112013. View Article : Google Scholar
|
|
35
|
Piaggi S, Raggi C, Corti A, Pitzalis E,
Mascherpa MC, Saviozzi M, Pompella A and Casini AF: Glutathione
transferase omega 1–1 (GSTO1–1) plays an anti-apoptotic role in
cell resistance to cisplatin toxicity. Carcinogenesis. 31:804–811.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ishitsuka A, Fujine E, Mizutani Y, Tawada
C, Kanoh H, Banno Y and Seishima M: FTY720 and cisplatin
synergistically induce the death of cisplatin-resistant melanoma
cells through the down-regulation of the PI3K pathway and the
decrease in epidermal growth factor receptor expression. Int J Mol
Med. 34:1169–1174. 2014.PubMed/NCBI
|
|
37
|
Berra E, Diaz-Meco MT and Moscat J: The
activation of p38 and apoptosis by the inhibition of Erk is
antagonized by the phosphoinositide 3-kinase/Akt pathway. J Biol
Chem. 273:10792–10797. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chiacchiera F, Grossi V, Cappellari M,
Peserico A, Simonatto M, Germani A, Russo S, Moyer MP, Resta N,
Murzilli S, et al: Blocking p38/ERK crosstalk affects colorectal
cancer growth by inducing apoptosis in vitro and in preclinical
mouse models. Cancer Lett. 324:98–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang
F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, et al: IkappaB kinase
promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell.
117:225–237. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang JY, Zong CS, Xia W, Yamaguchi H, Ding
Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC, et al: ERK promotes
tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation.
Nat Cell Biol. 10:138–148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chiacchiera F, Matrone A, Ferrari E,
Ingravallo G, Lo Sasso G, Murzilli S, Petruzzelli M, Salvatore L,
Moschetta A and Simone C: p38alpha blockade inhibits colorectal
cancer growth in vivo by inducing a switch from HIF1alpha- to
FoxO-dependent transcription. Cell Death Differ. 16:1203–1214.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chiacchiera F and Simone C: Inhibition of
p38alpha unveils an AMPK-FoxO3A axis linking autophagy to
cancer-specific metabolism. Autophagy. 5:1030–1033. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ho KK, Myatt SS and Lam EW: Many forks in
the path: Cycling with FoxO. Oncogene. 27:2300–2311. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schmidt M, Fernandez de Mattos S, van der
Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM and Medema RH:
Cell cycle inhibition by FoxO forkhead transcription factors
involves downregulation of cyclin D. Mol Cell Biol. 22:7842–7852.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee MT, Ho SM, Tarapore P, Chung I and
Leung YK: Estrogen receptor β isoform 5 confers sensitivity of
breast cancer cell lines to chemotherapeutic agent-induced
apoptosis through interaction with Bcl2L12. Neoplasia.
15:1262–1271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kiyoshima T1, Yoshida H, Wada H, Nagata K,
Fujiwara H, Kihara M, Hasegawa K, Someya H and Sakai H:
Chemoresistance to concanamycin A1 in human oral squamous cell
carcinoma is attenuated by an HDAC inhibitor partly via suppression
of Bcl-2 expression. PLoS One. 8:e809982013. View Article : Google Scholar : PubMed/NCBI
|