1. Introduction
Alzheimer's disease (AD), also known as the most
common form of dementia, is a progressive, neurodegenerative
disorder in adults, afflicting 35.6 million individuals worldwide
(1). Although a variety of
therapies have been developed over the years to treat patients with
AD, none of these treatments are able to provide a cure, but merely
alleviate the AD-associated symptoms. After recognizing the
contributing role of synaptic loss in memory deficits, researchers
have attributed the poor therapeutic effects in patients with AD to
the pathological features of this disease. These features include
extensive extracellular deposits of β-amyloid (Aβ) plaques,
intracellular neurofibrillary tangles (NFTs), as well as subsequent
neuronal and synaptic loss, which often begin several years prior
to memory loss and the damage is alread irreversible at the time of
diagnosis.
In order to explore more effective treatments, a
more complete understanding of the pathophysiology of AD is
required. As the pathological changes associated with AD are
initiated several years prior to the onset of clinical signs
(2,3), it is impossible to probe the human
neuropathology at this stage. However, on the positive side, animal
models are valuable tools which may be used to simulate the
pathological changes associated with human AD and then to decipher
the pathogenesis, particularly in the pre-symptomatic stage. In
particular, the advent of transgenic animal models, in combination
with non-transgenic animals, has allowed researchers to conduct
in-depth studies of AD (4,5).
Animal models cam reproduce the overt changes occurring in patients
with AD; however, complex in vivo conditions may limit the
accessibility to the tissue of interest and prevent real-time and
spatial measurements of biological changes (6). Consequently, several in vitro
experimental models of AD (7,8),
which provide detailed regional and cell-level information, have
been developed to enhance the usefulness of in vivo animal
models. All these models are of value for deciphering the
fundamental mechanisms underlying AD pathology and also, for the
testing of novel therapies targeted against this disease.
However, the currently available experimental animal
models are not without drawbacks, and none of these models
replicate all the features of AD. In addition, it is necessary to
take into account the extent to which the results obtained from the
experimental models may be applicable to humans, as well as
consideration of how to use these models to the best of their
potential. In this review, we summarize the experimental models of
AD that are currently in use, focusing on the similarities and
differences to human disease presented by each of these models, for
the purpose of generating more useful information on how to use
these models when testing novel therapeutics. Furthermore, we hope
to provide the basis for developing more appropriate models for AD
research.
2. Pathogenesis of AD
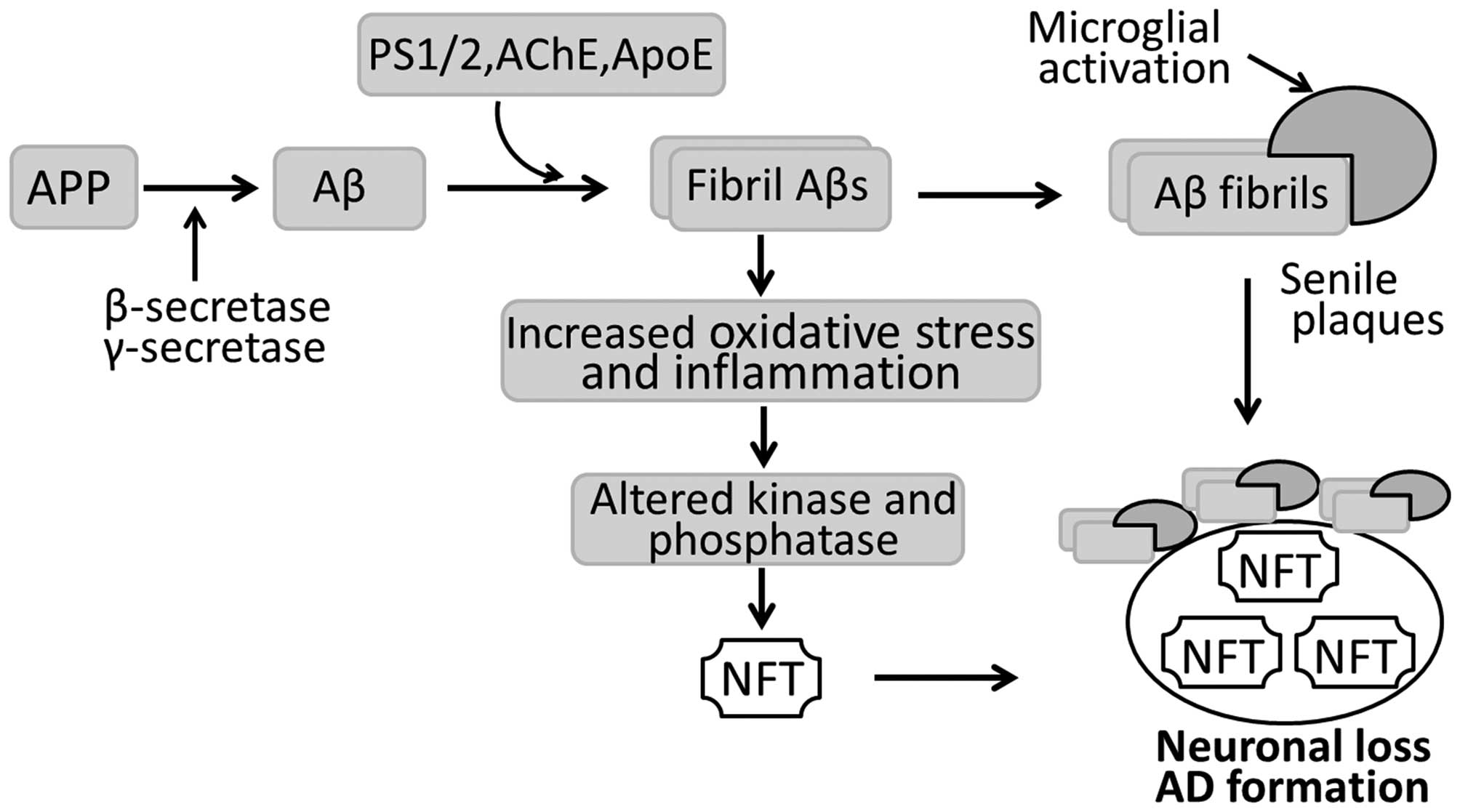
In pathological terms, the AD-affected brain is
characterized by widespread senile plaques of extracellular Aβ
peptides, intracellular NFTs, as well as neuronal and synaptic loss
(Fig. 1). Senile plaque is
composed of a central core formed by Congo red-positive Aβ proteins
and degenerating nerve endings surrounding the Aβ core. NFTs are
neuronal inclusions of the microtubule-associated protein (MAP) tau
and consist of aggregated, hyperphosphorylated tau. In AD
pathology, senile plaques and NFTs appear in the hippocampus, the
entorhinal and polymodal association cortices, and the basal
forebrain. These brain regions are also severely affected by
neuronal and synaptic loss. In addition to the neuropathological
changes, there are also a number of pathophysiological disturbances
occurring in the AD-affected brain, including inflammatory
reactions, such as angiogenesis and gliogenesis (9).
Nowadays, it is recognized that the loss of synapses
mostly correlates with the cognitive deficits and dementia
associated with AD (10,11). With respect to the causes of
synaptic loss, there are several hypotheses although a general
consensus exists in favor of the 'amyloid cascade hypothesis': Aβ,
particularly highly fibrillogenic Aβ developed following the
hydrolysis of amyloid precursor protein (APP), initiates a
neurotoxic cascade, consequently causing neurodegeneration and
ultimately, AD (Fig. 1). APP, an
ubiquitously expressed transmembrane protein, is involved in the
regulation of neuronal proliferation, migration and differentiation
under normal conditions. In AD, overexpressed APP is hydrolyzed by
β-secretase and then γ-secretase into Aβ peptides. Aβ peptides are
toxic to neurons and induce neuronal and synaptic damage when
assembled into amyloid fibrils (Fig.
1). It has also been reported that acetylcholinesterase (AChE),
an enzyme responsible for the hydrolysis of the neurotransmitter
acetylcholine (ACh), may promote Aβ assembly (12) and form stable complexes with Aβ
fibrils (13). The Aβ/AChE
complex has proven to be more toxic to cultured neurons than Aβ
fibrils alone (14). This may
explain why cholinergic neurons are the major cell types destroyed
in patients with AD. In addition, presenilin (PS)1 and PS2, as well
as apolipoprotein E (ApoE) bind to soluble Aβ in vitro, and
promote the formation of Aβ fibrils in an isoform-specific manner.
Aβ fibrils can also induce tau hyperphosphorylation by activating
several protein kinases, including glycogen synthase kinase-3β
(GSK-3β), MAP kinase and microtubule affinity-regulating kinase
(15). Hyperphosphorylated tau,
combined with unassembled microtubules, accumulates to form NFTs in
neurons and dendrites, leading to neuronal degeneration and
ultimately, AD (Fig. 1).
The tendency of familial aggregation and early-onset
of AD in patients has urged researchers to investigate the genetic
etiology of this disease. Currently, it has been recognized that
some forms of AD are caused by the inheritance of mutant genes,
including genes encoding APP (16), PS1 (17) and PS2 (18), tau (19) ApoE (20) and β- and γ-secretase (21). It has also been reported that
individuals harboring α-macroglobulin (22) and endothelial nitric oxide
synthase-3 mutant genes (23) are
more susceptible to AD than others.
3. Transgenic animal models of AD
Since the discovery of AD-associated genes, a number
of transgenic animal models have been generated by introducing
mutant genes into the existing genetic makeup or modifying genes of
interest using gene targeting technology so as to model some
features of human AD (5)
(Table I). Mice are the most
commonly used species for transgenic modeling due to their easy
manipulation and accessibility. Transgenic models have also been
created in rats, although thye are not so widely available
(24). Different transgenic
animal models are discussed in more detail below.
 | Table ISummary of transgenic animal models
of AD mentioned in this review.a |
Table I
Summary of transgenic animal models
of AD mentioned in this review.a
| Models | Transgene | Pathological
changes | Neurological
deficits | Refs. |
|---|
| APP transgenic
mice |
| PDAPP | hAPP695(Ind), PDGF
promoter | Aβs; plaques at 6–9
months; synaptic loss; astrocytosis; no NFTs | Cognitive deficits
at 6 months | (27) |
| APP23 | hAPP751(Swe), Thy-1
promoter | Plaques at 6
months; vascular Aβ deposits; synaptic loss; inflammation; no
NFTs | Cognitive deficits
at 3 months) | (29) |
| Tg2576 | hAPP695(Swe) HamPrP
promoter | Extensive plaques
in cortical and limbic structures at 8 months; synaptic loss; no
NFTs | Cognitive
impairment at 9 months | (28) |
| PS2APP | hAPP695(Swe),
PSEN2(N141I), Thy-1.2 promoter | Severe plaques at 5
months; synaptic loss, inflammation; glial activation; no NFTs | Behavioral and
cognitive decline at 8 months | (30) |
| Tau transgenic
mice |
| PrP-Tau | hTau(P301L),
MurinePrP promoter | Age- and
gene-dependent development of NFTs in brain and spinal cord 6
months | Behavioral and
motor deficits at 6 months | (34,39) |
| Thy-Tau22 | hTau(P301S),
Thy-1.2 promoter | NFTs; mild
astrogliosis; synaptic loss | Decline in learning
at 3 months, memory at 6 months | (10) |
| GFAP-Tau | hTau(P301L), GFAP
promoter | Astrocytic tau
pathology; mild BBB disruption; focal neuron damage | Motor deficits | (36) |
| APP and tau double
transgenic mice |
| APP/tau | hAPP695(Swe),
hTau(P301L); PrP promoter | Aβ deposits; NFTs;
nerve loss at 8–15 months | Motor
disturbance | (38) |
| Triple transgenic
mice |
| 3xTg-AD | hAPP695(Swe) Thy1
promoter, hTau(P301L) Thy1 promoter, PSEN1(M146V)mPS1 promoter | Aβ deposits at 6
months; NFTs at 10–12 months; neuronal and synaptic loss;
microglial activation; inflammation | Age-related
behavior deficits | (40) |
| Five transgenic
mice |
| 5xTg-AD | hAPP695(Swe, Lon,
Flo), PSEN1(M146L, L28V); Thy1 promoter | Excessive Aβ42; Aβ
plaque at 2 months; gliosis; synaptic loss; no NFTs | Memory and
cognitive loss | (43) |
| Transgenic rats for
AD |
|
APPSwe | hAPP751(Swe), PDGF
promoter | Increased Aβ40 and
Aβ42 peptide at 17–18 months; plaques | Learning and memory
decline | (46) |
| PS1APP | hAPP695(Swe),
PSEN1(M146V), synapsin I promoter | Aβ deposits at 7
months; astrocytic and microglial activation; increased p-tau
reactivity; no NFTs | Behavioral
deficits | (57) |
| Thy-Tau | hTau truncated
(151–391), Thy1 promoter | Increased p-tau,
tangles in cortex; no neuron loss | Decreased
lifespan | (47) |
APP transgenic mice
The first, widely used transgenic mouse models of AD
were developed to express the transgene of human APP (Table I). In earlier studies (25,26), transgenic mice harboring APP
mutations merely exhibited an accumulation of intracellular Aβ and
occasional behavior deficits, but no senile plaques, suggesting
that cognitive impairment occur independently of plaque formation,
but correlate with Aβ fibril formation. The lack of senile plaques
in these models was supposed be associated with the low expression
levels of the APP gene. In subsequent studies, a mutant human APP
gene controlled by platelet-derived growth factor-β promoter
(PDAPP) was used as a transgene in mice, which produced extensive
extracellular Aβ deposits, synaptic loss and astrocytosis, with
plaque formation at 6–9 months of age (27). Consistent with these observations,
other transgenic models, including Tg2576 and APP23 (both carrying
a Swedish double mutation, APPSwe), demonstrated APP
overexpression in addition to neuronal loss in the CA1 region, and
learning deficits as was observed in the PDAPP transgenic mice
(28). Moreover, Tg2576 mice
developed Aβ plaques at 8 months of age, with a 5-fold increase in
the concentrations of Aβ40 and a 14-fold increase in those of Aβ42
(28), whereas plaque formation
occurred in APP23 mice at 6 months of age, together with a notable
decrease in the CA1 pyramidal neurons which correlated inversely
with the plaque loads in this region (29). When a human mutant presenilin gene
was introduced, TgAPPSwe mice displayed abundant plaques
in the neocortex, olfactory bulb, thalamus and hypothalamus at an
earlier age (30). These mice
also exhibited progressive plaque accumulation, astrocytic and
microglial activation, as well as behavioral deficits related to
Aβ42 loads in the hippocampus.
These findings provide support for the 'amyloid
cascade hypothesis' as an explanation for the pathogenesis of AD,
and the experimental models provide valuable tools for the testing
of Aβ-modifying drugs. However, APP transgenic mice seldom present
with NFTs and neuronal loss, which are the hallmarks of AD. This
may be associated with the fact that multiple genes, rather than
APP alone, are involved in the occurrence of AD and information
learned from studies of familial AD (fAD) may not be applicable to
sporadic AD (sAD) (31).
Tau transgenic mice
In order to elucidate the role of tau in the
pathology of AD, transgenic mouse models have been developed to
express human tau protein using different promoters. As there are 6
tau isoforms in humans, numerous transgenic models have been
created. In a previous study, when a human gene encoding a 4-repeat
form of tau (P301L) under the PrP promoter was expressed in mice,
the tau levels were significantly increased in the neurons of the
somatosensory cortex and the motor neurons of the spinal cord
(32). The majority of these
mice, however, exhibited motor deficits or hind limb paralysis and
exhibited no NFT-like changes (33), which was inconsistent with the
changes observed in humans. Thus, this model doses not induce a
phenotype representing human AD (34). In another study, extensive NFTs
were also observed in a new mouse model (Thy-Tau22) that expresses
4-repeat human tau mutated at two sites of G272V and P301S under a
Thy1.2 promoter, and synaptic dysfunction and synapse loss were
also evident (10). Behavioral
analysis of the diseased mice revealed progressive anxiety, delayed
learning, as well as spatial memory without motor deficits. In this
mouse model, neuropathologies occurred at 3 months of age, and NFTs
were formed mainly in the hippocampus (10). Of particular note, neuronal loss
in the P301L tau mice was lower than that in the P301S tau mice,
echoing the fact that patients with early onset of fAD carry the
P301S mutation (35).
Glial tau inclusions are commonly observed in
patients with fAD. This can be modeled in mice using P301L and
wide-type human tau which are driven by 2′,3′-cyclic nucleotide
3′-phosphodiesterase and glial fibrillary acidic protein (GFAP)
promoters, respectively (36,37) (Table
I). These two mouse models exhibit neuronal dysfunction and
axonal degeneration, suggesting that tau pathology affects
neurons.
Taken together, tau transgenic models have proven
that mutations in the tau gene can accelerate tau aggregation and
cause nerve-cell dysfunction or loss in vivo, thereby
imitating an important aspect of human fAD. These models provide
valuable tools for AD research with respect to the role and
mechanism of tau hyperphosphorylation and aggregation in regulating
AD pathology.
APP and tau double transgenic mice
As mentioned above, Aβ deposits and NFTs are two
features of AD pathology; however, the interaction between Aβ
deposits and NFTs remains to be elucidated (31). APP and tau double transgenic mice
are candidate models containing both pathological features of AD,
which can be used to investigate the interaction between Aβ and tau
(Table I). By crossing APP Tg2576
transgenic mice with VLW lines expressing human mutant tau,
transgenic mice carrying both APP and tau genes were obtained
(32). These double transgenic
mice demonstrated a 7-fold increase in NFT formation accompanied by
enhanced Aβ deposits in the olfactory bulb and amygdala compared
with that observed in the tau transgenic mice alone; however, no
significant difference in the tau level was observed between the
single and double transgenic mice (32,38). These results suggest that the Aβ
deposits promote NFT formation in AD in double transgenic mice and
then enhance the neuropathological correlates of AD brain. This is
consistent with the results of other studies demonstrating that
Thy1.2 promoter-driven P301L mice injected with Aβ42 exhibited a
5-fold increase in NFT formation at 18 days after the injection of
Aβ into the amygdala (39). Taken
together, these findings challenge the idea that tauopathy is
merely a downstream effect of Aβ deposition, and confirm that Aβ
deposits interact with tau protein in vivo, and indicate
that the interaction between Aβ and tau should be further studied
as a potential target for AD therapies.
Triple transgenic mice
Despite plaque and NFT formation, the APP and tau
double transgenic mice exhibited the same level of Aβ deposits
compared to that in the single APP transgenic mice, indicating that
these mouse models do not accurately reflect AD. Triple transgenic
mice (3xTg-AD), expressing APPSwe, PS1M146V and tauP301L
transgenes, have been shown to develop extracellular Aβ deposits
from 6 months of age and a tau-related pathology at 10-12 months of
age, which began in the hippocampus and then expanded to the
neocortex, closely mimicking the development pattern of Aβ and
tauopathy in the human AD brain (40) (Table
I). At the same time, amyloidosis and tauopathy were
accompanied by neuronal and synaptic loss, and age-related behavior
deficits were also present prior to the development of plaques and
tangle pathology (40). In this
model, microglial activation and the upregulation of inflammatory
markers were observed at 3 months of age, in parallel with the
increase in Aβ deposits (41). It
appears that this model simulates the key pathological features of
human AD and has the potential to be used to validate therapies
targeting the mechanisms underlying AD pathology. This model has
been widely used in the study of AD pathophysiology and the testing
of potential therapies.
5xFAD transgenic mice
Although triple transgenic mice present a
neuropathology similar to that observed in AD, most transgenic
models develop plaques slowly (31). To accelerate plaque formation and
the effects of very high cerebral Aβ levels, investigators
generated new transgenic mice that co-expressed five fAD mutant
genes (5xTg-AD) and Aβ plaque formation was increased at 2 months
of age (42,43). These 5xTg-AD mice produced
excessive amounts of Aβ42, exceeding that of Aβ40, suggesting that
fAD mutations, when combined together, have an additive effect,
resulting in increased Aβ42 production and the promotion of plaque
formation at an early age (43).
This model exhibits an extensive accumulation of intraneuronal Aβ42
within the neuronal soma and neurites at 1.5 months before plaque
formation, indicating that intracellular Aβ deposits may be
involved in plaque formation, neurodegeneration and neuronal loss
(31). Furthermore, 5xTg-AD mice
exhibited marked gliosis, reduced synaptic markers, and notable
memory and cognitive loss. However, abnormal tau
hyperphosphorylation was not observed in the 5xTg-AD mice (42,43). These findings suggest that more
complex mechanisms than the 'amyloid cascade hypothesis' alone are
involved in the pathogenesis of AD and further analysis of
pathological tau epitopes is required (31). The rapid development of amyloid
pathology in the 5xTg-AD mice renders them candidate models for
studying AD-like amyloi-dosis. However, the condition that this
model represents rarely occurs since no cases of AD have been
reported to carry all five familial mutations (31); this should be taken into account
when using the data obtained from this model.
Transgenic rats
Transgenic rats offer distinctive advantages over
mice in many aspects, including rich, well-characterized behaviors,
as well as being closer genetically, physiologically and
morphologically to humans than mice. Thus, great efforts have been
made to develop models of AD in transgenic rats, as well as in
transgenic mice (Table I). The
earliest transgenic rat models of AD were developed by using
wide-type APP transgene, which exhibited an accumulation of
intracellular Aβ without extracellular senile plaques (44) until 17–18 months of age (45,46). When a human PS1 transgene was
introduced, the resulting rats exhibited progressive plaque
accumulation, astrocytic and microglial activation, tau
hyperphosphorylation, as well as behavioral deficits related to
Aβ42 loads in the hippocampus, with the onset of Aβ deposition at 7
months (45). The first
transgenic rat models developing progressive NFTs expressed a human
3-repeat tau protein (47), which
formed NFTs in neocortices at 9 months of age. Unfortunately, this
model exhibited no neuronal damage in the hippocampus and
neocortex, despite largest accumulations of tangles in these
regions.
As a whole, the currently available transgenic rat
models recapitulate some, but not all aspects of AD, and thus, the
extrapolation of obtained conclusions to humans requires a measure
of caution. Furthermore, rat embryo cells are difficult to acquire
due to technical reasons, and the survival of embryos after
injection is low, thus rendering rat transgenesis more demanding
and transgenic rats less accessible for AD research (4).
4. Non-transgenic animal models of AD
Natural AD models of AD
Memory loss is often the first, and common feature
of old age. Thus, aged animals may potentially be used as natural
models of memory deficits and dementia. Various animals ranging
from rodents to non-human primates have been shown to spontaneously
develop AD neuropathology followed by cognitive impairments similar
to those observed in patients with AD (21,48). These animals models are valuable
tools that may be used to reveal the natural pathophysiology of AD,
since they are non-invasive and without any neurochemical
manipulations (Table II).
| Table IISummary of non-transgenic animal
models of AD.a |
Table II
Summary of non-transgenic animal
models of AD.a
| Models | Pathophysiological
changes | Advantages | Limitations | When to use for
mimicking AD | Refs. |
|---|
| Natural AD
model |
| Rodents | Aβs; memory and
learning deficits | Small size; short
life-span; accessible; low cost; assessable behaviors | Different Aβ
sequence and profile; Aβs initiate in neocortex | For APP processing;
impact of Aβs; pathophysiological changes after therapies | (51) |
| SAMP8 mice OXYS
rats | Refer to the
above | Accelerated
senescence; assessable behaviors; age-related cognitive
deficits | Refer to the
above | For genetic and
proteomic alterations under age-related memory and learning
deficits | (114) |
| Dog | Aβ deposits;
cognitive deficits | Similar Aβ
sequences and distributions to humans; correlates between Aβs and
cognitive deficits | No NFTs; need
predict which dog will develop AD | For new diagnostic
tools; preclinical testing of drugs | (63,64) |
| Non-human
primates | Aβ deposits; NFTs,
brain atrophy; cognitive deficits | Comparable brain
anatomy and genetic proximity, similar AD-pathology to humans | Long developmental
period; low output; high costs; ethical constraints | For diagnostic
markers; testing safety and effectiveness of new drugs | (75,76) |
| High-fat diet
induced model |
| Rodents | Aβ deposits;
cognitive deficits | Easy manipulation;
abnormalities in genes and proteins | Costs time and
vigor; devoid other metabolic abnormalities; no NFTs | For the role and
mechanism of different dietary components in AD | (77,79) |
| Intervention
model |
| Chemically-induced
model | Specific
neurotransmitter pathway damage; cognitive deficits | Rapid; easy to
obtain; specific; similar cognitive deficits to humans | No
neurodegenerative process | For the complex
mechanisms of dementia; testing symptomatic treatments | (89) |
| Lesion model | Memory and learning
deficits | Refer to the
above | No plaques and
NFTs; only some models show neurodegeneration | Refer to the
above | (92,93) |
The rodent is the most frequently used animal in
experimental research due to the low cost, availability and ease of
manipulation, as well as well-characterized behaviors (Table II). Prior studies conducted on
rats have advanced our understanding of APP processing and revealed
that neprilysin and insulin-degrading enzyme are the primary
proteases implicated in Aβ degradation (49,50). Rats have also been used to
investigate the spatial and temporal expression of different APP
isoforms (51), the impact of Aβ
aggregation on cholinergic and glutamine neurons (52), the influence of age or diet on the
effects of intracerebral injections of Aβ peptides, correlations
between cognitive deficits and AD pathology, as well as the
cognitive improvements after new therapies. However, the primary
structure and complete profile of Aβ peptide generated from APP
processing in rats is different from that in humans (53), limiting their use in investigating
the long-term effects of APP manipulation. Guinea pigs, another
type of rodent, are similar to humans in terms of the Aβ sequence
and patterns of APP processing, making them suitable models for the
study of the physiological functions of APP and Aβ processing.
Nonetheless, limitations still exist as to the use of this animal
as there are no typical senile plaques and NFTs in the diseased
brain and a lack of good behavioral assessments for this species
(54).
Another example of a rodent model includes the
senescence-accelerated mouse (SAM) model which simulates several of
the silent features of AD (55).
Early in 1981, SAM models were developed through phenotypic
selection from a common genetic pool of AKP/J strain of mice,
including nine senescence-accelerated mouse-prone (SAMP) substrains
that underwent accelerated aging and three senescence-accelerated
mouse-resistant substrains that underwent normal aging processes
(56). In subsequent studies, the
SAMP8 strain was found to be a reliable model for dementia,
developing deficits in memory and learning at an early age
(57). OXYS rats have reportedly
been used as senescence-accelerated models of AD-like pathology as
their cognitive deficits are associated with the overexpression of
APP, the accumulation of Aβ, as well as hyperphosphorylated tau in
the brain (58). It has been
demonstrated that senescence-accelerated rodents present with
alterations in various genes and proteins that are involved in
neuroprotection, signal transduction, oxygen species production
(59) and immune reactions
(60). Thus, these animals may be
used to explore the underlying mechanisms behind age-dependent
learning and memory deficits at the gene and the protein level
(21).
The dog has been proposed as a particularly suitable
model for studying human AD due to the close phylogenetic proximity
of canines to humans (61). Dogs
have a conserved Aβ sequence identical to humans and may
spontaneously develop age-associated cognitive dysfunction
resembling aspects of AD, and measures of cognitive function have
been well characterized. It has been reported that dogs develop
diffuse Aβ deposits and age-associated plaques, and the extent of
Aβ deposits correlates well with cognitive decline in the absence
of NFTs (62,63). In previous studies,
histopathological analysis of a canine model of AD revealed that
that the foremost and consistently impaired regions were within the
prefrontal cortex, including the hippocampus and gyrus proteus
(63,64), and the anatomical spreading of Aβ
accumulation, beginning from the deep brain and progressing to the
neocortex (64), in contrast to
that observed in human AD. Due to the correlation between
age-associated Aβ deposits and cognitive decline, dogs are the most
widely used animals for the pre-clinical validation of a variety of
drugs (66), and almost all
therapies currently used for patients with AD, including
cholinesterase inhibitors, have been tested on dogs (67). However, NFTs and dense neuritic
plaques do not appear consistently in all aging dogs, and Aβ
accumulation is merely observed in a few dogs rather than appearing
as a natural consequence of normal aging (68). In addition, some dogs develop
heavy Aβ burdens in their brains, but present with no apparent
cognitive decline (69).
Furthermore, the development of neuropathology takes a long period
of time in dogs due to their relatively longer lifespan. This
requires the exploitation of new diagnostic tools to predict which
dog will develop AD-like symptoms. As such, dogs offer a useful
model in the search for indicators for predicting AD.
Primates share many diseases of aging with humans
and thereby are among the few animal species that develop
age-related neuropathologies similar to human AD (Table II). Of the primates, the mouse
lemur serves as a reliable model that presents with Aβ plaques,
NFTs and other AD-related pathologies (70), accounting for a more complete
disease model. Moreover, this animal has a relatively short
lifespan of 8-10 years, and approximately 20% of mouse lemurs aged
5 years or older have been found to exhibit notable cerebral
atrophy, extensive Aβ accumulation, tauopathy and cholinergic
dysfunction (71). Further
studies on mouse lemurs have revealed that brain atrophy is
associated with Aβ accumulation in parallel with cognitive
impairment, and the age-related cytoskeletal abnormalities caused
by NFTs are similar to those observed in humans (72). However, it should be noted that
the Aβ distribution in mouse lemurs differs from that in humans, in
that Aβ accumulation begings in the hippocampus of humans, but in
the neocortices of mouse lemurs (73). In contrast to mouse lemurs,
monkeys have a closer genetic proximity to humans and suffer
significant AD-like lesions in the brain regions suspected to
affect cognitive function when elderly (over 19 years of age)
(74). These advantages make
primates ideal models to identify diagnostic markers and to develop
safe and effective treatments for human AD (75,76). However, their long developmental
period, low reproductive output, high costs and ethical constraints
limit their wider application in studies of AD.
Models of high-fat diet (HFD)-induced
AD
In these models, animals are fed a HFD, a process
that results in hypercholesteremia (77) (Table II). Despite the presence of
hypercholesteremia, these animals are not true models of AD as they
are often devoid of other attendant metabolic abnormalities
associated with AD (78).
Dementia is always induced in HFD-fed rats over a 3-month period,
with increased Aβ deposits present at the same time (79). It has been shown that a HFD
increases the brain cholesterol concentration by producing a higher
serum cholesterol level, and there is a positive feed-back
association between cerebral and serum cholesterol levels (80). As cholesterol plays an important
role in the clearance and deposition of Aβ peptide, increased
cholesterol levels in the brain promote the generation and
deposition of Aβ peptides (81),
and Aβ deposits can cause cognitive decline by inducing
inflammatory responses, as well as oxidative and nitrosative stress
(82). During this process,
insulin sensitivity and glucose intolerance are also decreased in
HFD-fed animals, which increases the risk of susceptibility to AD
(83). This model is often used
to investigate the importance of different dietary components in AD
pathophysiology. However, the experimental procedure involved with
the use of this model is fat too time-consuming (84).
Intervention models of AD
The intervention model was established to explore
the role of one specific neurotransmitter pathway in AD
pathophysiology, and in particular to test the cholinergic
hypothesis of AD (4). To
establish such models, animals are subjected to intracranial
injections of chemical substances or the induction of lesions into
specific brain regions to reproduce AD (Table II). The established models
represent some characteristics of AD, such as deficits in
cholinergic function and learning impairments (4).
The chemically-induced models of AD were established
initially in rats (85) in order
to investigate the effects of injected Aβ on the surrounding
neurons (52). Other substances
used to induce an AD-like pathology include the relatively specific
scopolamine for cholinergic neurons (86), L-methionine for activating NMDA
receptors (87), okadaic acid for
tau phosphorylation (88), sodium
azide for mitochondrial dysfunction (89), as well as heavy metal for
increasing reactive oxygen species (90). These substances induce dementia in
animals by blocking neuronal or neurotransmitter pathways, which
provide a distinctive means to study the effects of damage to a
specific neurotransmitting pathway on cognitive function and also
to test crucial drugs targeted at these pathways.
Lesion models were established based on previous
findings showing that injury to specific brain regions, such as the
medial temporal lobe produced memory loss (91). When a bilateral transection was
performed in the hippocampal fimbria-fornix, memory and learning
deficits were observed in the injured rats (92). In light of these findings,
researchers imposed radiofrequency lesions to the lateral internal
medullary lamina region of the thalamus in rats, which led to a
significant cognitive decline in the diseased animals (93), and in addition, neurodegeneration
similar to human AD was found in the brain regions of these rats
distant to the site of injury (93). These lesion models offer novel
insight into the multiple mechanisms of memory and learning loss,
and are of particular use to develop symptomatic treatments for
dementia.
As noted above, the intervention models replicate
many of the clinical symptoms associated with AD, suggesting
complex mechanisms underlie memory and learning loss. Due to a lack
of typical senile plaques and NFTs, most of these models do not,
however, represent accurate modeling systems for AD. Although
cerebral injections of Aβ produced some features of AD, such as
cholinergic dysfunction and behavioral deficits, the progressive
neurodegeneration characteristic of AD was not observed. Thus,
therapies designed to halt the pathological progression of AD
should not be tested on these models.
5. In vitro models of AD
In contrast to in vivo animal models, in
vitro models of AD provide a prompt and direct manner with
which to reveal the pathological changes occurring in AD and also
to study the responses of nerve cells to treatments at molecular
and cellular levels. With the technological advances in cell
biology, in vitro AD models have been extensively
developed.
Tissue models
Brain tissue, cultured in vitro, provides a
good platform for studying the impact of AD-associated substances,
such as Aβ on nerve cells (Table
III). In an earlier study, Yankner et al (7) treated cultured hippocampal neurons
with various synthetic Aβ fragments, and found that Aβ peptides
were neurotrophic to undifferentiated neurons, but neurotoxic to
mature neurons, wherein the proportion of Aβ25-35 mediated the
trophic and toxic effects of Aβs. They also showed that the effects
of Aβ can be mimicked by tachykinin antagonists and be completely
reversed by specific tachykinin agonists. Similar findings were
obtained in the study by Itokazu et al (8). In other studies, in vitro
cultured neurons were employed as a model to reproduce the
pathophysiological changes involved in AD, including inflammatory
reactions, enzyme kinetics, cellular ion homeostasis, altered
signal pathways, as well as epigenetic regulations (94,95). Due to the ease of manipulation and
the controllable environments, these models are also used to test
therapeutics targeted at AD pathophysiology (96) and to reveal the underlying
molecular mechanisms (97,98).
| Table IIISummary of identified aspects of AD
by different in vitro models and the associated
limitations. |
Table III
Summary of identified aspects of AD
by different in vitro models and the associated
limitations.
| Models | Identified aspects
of AD | Advantages | Limitations | Refs. |
|---|
| Tissue models |
| Cultured
tissues | Biochemical
changes; p-Tau; amyloidosis; neuron degeneration | Show the AD
mechanism at molecular and cellular level; controllable
environment | No neurological
changes; no plaques; no NFTs | (7) |
| Brain slices | Refer to the
above | Show pathology
biochemically and histologically; natural physiological
environment | Refer to the
above | (99) |
| Cell models |
| iPSC | Biochemical
changes; p-Tau; amyloidosis; neuron degeneration | Similar genetic
background to humans | No pathological and
neurological changes | (104) |
| Neuroblastoma
cell | Specific molecular
pathway like APP processing | Rapid and
direct | Refer to the
above | (106) |
| Molecular
simulation model |
| Antibubble
biomachinery | Inflammatory
reactivation; neuronal death | Dissect molecular
functions of endogenous regulatory pathways in AD | No pathological and
neurological changes | (110) |
Moreover, investigators have developed metabolically
competent brain slices in order to model the regulation of AD
pathophysiology (Table III). In
a previous study, Gong et al (99) treated the metabolically active rat
brain slices with okadaic acid to inhibit protein phosphatase 2A,
and found that the hyperphosphorylation of tau at many sites was
observed in AD, as well as the accumulation of hyperphosphorylated
tau in pyramidal neurons located in the hippocampus and cortex. The
phosphorylation of MAP protein was also displayed with this model
(99). Thus, this model not only
recapitulates the pathological alterations associated with AD
biochemically, but also reveals their regional distribution
histologically (99,100). In another study, Li et al
(101) demonstrated that
memantine inhibited and reversed the Alzheimer type abnormal tau
hyper-phosphorylation and associated neurodegeneration using the
metabolically active rat brain slices. Taken together, the findings
of these studies demonstrate that brain slices may be used to model
AD at a biochemical level and test potential therapies.
Furthermore, neurons in the brain slices, unlike cultured cells,
reside in the physiological environment of the brain, consisting of
natural extracellular matrix, neuronal connectivity, and
neuronal-glial interactions, which points to a more accurate model
of AD (99).
Cell models
Progress in stem cell techniques has broadened our
horizon for in vitro disease modeling (Table III). For AD, disease-specific
induced pluripotent stem cell (iPSC) lines have been generated from
patients with sAD or fAD (102),
providing an effective approach for disease modeling and drug
discovery. In the study by Yagi et al (103), iPSCs were acquired from patients
with fAD with mutations in PS1 and PS2, which demonstrated
increased levels of Aβ42 in all iPSC lines, supporting the role of
Aβ as an initiating factor in AD pathogenesis. Using the same
method, Israel et al (104) generated iPSCs not only from
patients with fAD, but also sporadic cases. In their study, the
iPSC-derived neurons exhibited significantly higher levels of Aβ40,
phospho-tau, GSK-3β, and endosomes (104). Phenotypic analysis revealed that
neurons with the genome of sAD cases exhibited the phenotypes
observed in fAD samples, suggesting that genetics plays an
important, albeit complex, role in sAD. In a previous study, when
docosahexaenoic acid (DHA) was used to treat AD iPSC-derived
neurons (105), intracellular Aβ
loads were notably reduced in sAD and fAD AD neurons, while the
stress response of neurons was alleviated in only one patient with
sAD, suggesting that heterogeneity exists between patients with sAD
and fAD, and indicating that DHA may be effective in some patients
with AD. These results indicate that iPSCs provide a unique
platform for observing AD-associated phenotypes and therapeutic
screening. As iPSC lines from patients with sAD carry different
genetic variants, the generation of sufficient stem cells is
required to fully guide drug development and enhance our
understanding of AD.
Human neuroblastoma cell lines are also used as a
model to simulate AD pathophysiology (Table III). Macias et al
(106) reported the modeling of
the APP-processing pathway in human neuroblastoma cells, which
expressed critical components of the amyloidogenic cascade as
observed in AD. In a previous study, when carnosic acid (CA) was
used to treat human neuroblastoma cells exposed to Aβ42, it was
found that CA attenuated cell apoptosis by suppressing the
activated caspase cascades due to intracellular Aβ aggregation
(107). Likewise, the dual
blockade of A1 and A2A adenosine receptors has also been
demonstrated to prevent Aβ toxicity in neuroblastoma cells exposed
to aluminum chloride (108).
These results indicate that human neuroblastoma cell lines can be
modified to model certain aspects of AD pathophysiology so as to
explore the underlying mechanisms and to develop potential
treatment drugs.
Molecular simulation models
Recently, researchers have attempted to simulate the
molecular events in the pathophysi-ology of AD in vitro, so
as to establish efficient model systems for speeding up the drug
development process (Table
III). Denis (109) created
an NADPH oxidase-nitric oxide system as an antibubble biomachinery,
to imitate the inflammatory cascade seen in AD. This biomachinery
induced neuronal death by triggering a guanylyl-cyclase-mediated
inflammatory cascade. Researchers have also reported the
establishment of molecular dynamics stimulations of model systems
that are comprised of an Aβ40 peptide in water in interaction with
β-sheet breakers mimicking the 17-21 region of Aβ40 sequence
(110), showing that β-sheet
breakers inhibit Aβ40 aggregation by stabilizing its native
structure. These studies have provided us with streamlined
approaches to dissect the molecular functions of endogenous
regulatory pathways involved in AD, permitting the direct
mechanistic studies of the modulators of these pathways. These
approaches are also used to screen possible therapeutic compounds
efficiently and evidently.
6. Current status and future development of
experimental models of AD
Current status of experimental models of
AD and strategies for model selection
Experimental models of AD have greatly enhanced our
understanding of the disease process of AD and promoted the
development of novel therapies. While transgenic animal models
enable researchers to explore the genetic etiology of AD, and have
led to the establishment of more complete models of AD which may be
used for genetic manipulation and the identification of novel
therapies, non-transgenic animal models recapitulate the natural
process of AD including its occurrence, evolution and prognosis,
which has shown a high application value in earlier studies. By
contrast, in vitro models isolate specific molecular
pathways from others in AD, permitting therapeutic screening in a
rapid and direct way. Among them, transgenic models are the most
popularly used in AD research.
However, there are obvious limitations with current
experimental models of AD. To date, none of the existing models
reproduce all aspects of human AD. Although transgenic animals for
modeling AD produce Aβ plaques and NFTs with a spatial and temporal
distribution similar to humans, most of these models mimic fAD
closely but not sAD conditions, and the latter accounts for >90%
of AD cases (111). There are
certain pathophysiological differences between fAD and sAD, in
which one is driven primarily by Aβ overproduction and the other is
not. This may explain why so many treatments have shown significant
benefits by suppressing the β-secretase in animal models carrying
hAPPSwe genes, yet fail to clinically treat sAD. Human
iPSCs isolated from patients with AD may serve as a valuable tool
to bridge the gap between animal studies and clinical testing.
However, sAD is characterized by its multifactorial nature and
various clinical phenotypes, which may differ further during the
long period of pathology development; this may lead to
inconsistencies in different iPSC lines even from the same patient.
In addition, there are marked differences in the genetic
background, biochemical composition and metabolism among different
animals that make it difficult to compare the resulting data across
species. Due to the complexity of AD conditions, further studies
are warranted to define the pathological signatures of different
forms of AD in humans and in model animals.
The selection of one particular model for AD
research should be based on the research objectives and the
advantages of each model. In general, transgenic animals are of the
first choice for modeling AD pathophysiology and testing new
therapeutics, although new treatments should be tested in natural
models, such as primates prior to clinical trials. In vitro
models provide a fast and efficient means of exploring the
molecular mechanisms behind AD and to preliminarily screen new
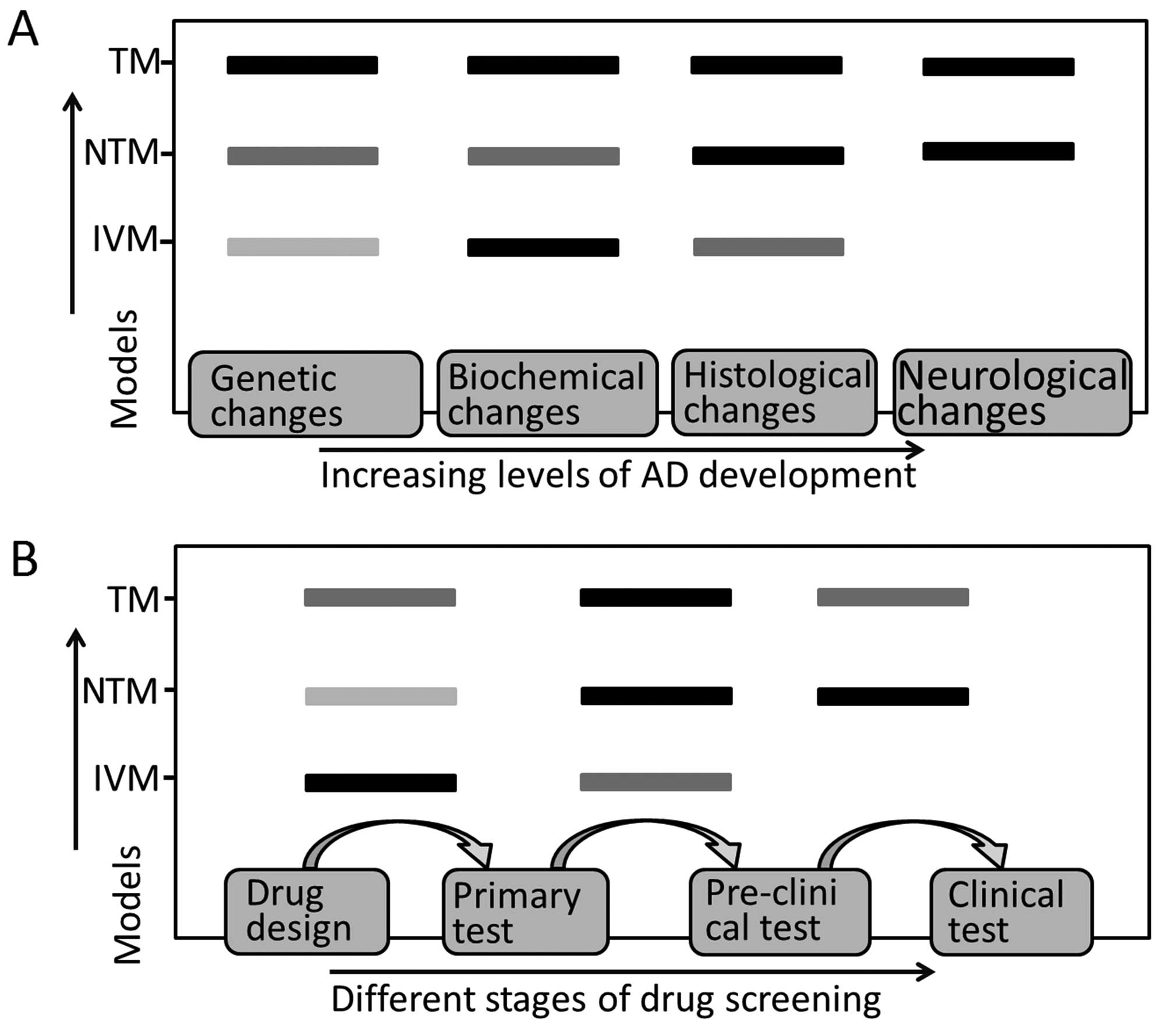
therapeutics. Each type of AD model mimics specific stages of AD
pathophysiological changes (Fig.
2A) and shows different efficiency in therapeutic screening
(Fig. 2B), which should be taken
into account in AD model selection.
The future of experimental models of
AD
The advent of interference RNAs and antibody arrays
may make it possible to acquire 'perfect' models in the years to
come by using these substances through epigenetic mechanisms. It is
expected that there will be a wider application of genetic
technology to models of AD (111). Furthermore, the development of
optogenetics provides an appealing method with which to evaluate
the physiological changes of model cells at a cellular level with a
level of high accuracy (94). The
use of this technique in AD will allow the observation of the
'behavioral responses' of cell models or tissue models; however,
there are limited studies on this aspect to date (94) and further investigations into this
matter are thus required. In addition, computational technology
combined with the molecular simulation model may be used to mimi
molecular structures, such as Aβ peptide, and screen novel
therapeutics efficiently (110,112). It is likely that in the coming
years, these techniques will be widely used in the research of
AD.
Recently, attention has been focused on combined
approaches that rely on the limited existing models in order to
induce a pronounced pathology. The models of one type may not
accurately reproduce the anatomical distribution of lesions
observed in humans, although biochemically, they are quite similar
to human conditions. In terms of neurological impairments,
neuroanatomy and endocrine systems, animals models are superior to
in vitro models. By contrast, in vitro models exhibit
significant advantages over animal models in dissecting signaling
pathways, performing modifier screenings and analyzing families of
mutations. Using a combination of multiple models has been shown to
accelerate the pathogenic disclosure and drug development process
in AD (113). This may be one
direction for modeling AD in future studies.
7. Conclusion
Over the past decades, great progress has been made
in the understanding of the athophysiology of AD and the underlying
genetic and biochemical alterations. In parallel, a variety of
experimental models of AD, including animal models and in
vitro models, have been developed. Animal models cast light on
the neurodegeneration conditions and facilitate the exploration of
pathological, physiological, and behavioral changes occurring in
AD. In vitro models allow accurate and reproducible control
of extracellular environments, providing additional information for
animal models. All these models are particularly suitable for
studying the pathophysiology of AD and evaluating potential
therapies. Each model offers particular benefits, as well as
specific limitations. Selecting an experimental model is dependent
on both the research goals and the underlying objectives of the
study.
As a whole, the experimental models of AD discussed
in this review have contributed to our understanding of AD.
Nevertheless, none of these models are able to reproduce all
aspects of disease progression in the vast majority of AD subtypes.
Thus, current models require additional modifications so as to
fully replicate the complex conditions of human AD. Undoubtedly,
experimental models will continue to play a vital role in future AD
research.
Acknowledgments
This study was supported by the National High
Technology Research and Development Program ('973' program, no.
2014CB541603; '863' program, no. 2013AA020106) and the National
Natural Science Foundation (nos. 81301061 and 81200916) of
China.
References
|
1
|
Li XY, Bao XJ and Wang RZ: Potential of
neural stem cell-based therapies for Alzheimer's disease. J
Neurosci Res. 93:1313–1324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bateman RJ, Xiong C, Benzinger TL, Fagan
AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al
Dominantly Inherited Alzheimer Network: Clinical and biomarker
changes in dominantly inherited Alzheimer's disease. N Engl J Med.
367:795–804. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fleisher AS, Chen K, Quiroz YT, Jakimovich
LJ, Gomez MG, Langois CM, Langbaum JB, Ayutyanont N, Roontiva A,
Thiyyagura P, et al: Florbetapir PET analysis of amyloid-β
deposition in the presenilin 1 E280A autosomal dominant Alzheimer's
disease kindred: a cross-sectional study. Lancet Neurol.
11:1057–1065. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Do Carmo S and Cuello AC: Modeling
Alzheimer's disease in transgenic rats. Mol Neurodegener. 8:372013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elder GA, Gama Sosa MA and De Gasperi R:
Transgenic mouse models of Alzheimer's disease. Mt Sinai J Med.
77:69–81. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mattsson MO and Simkó M: Is there a
relation between extremely low frequency magnetic field exposure,
inflammation and neurodegenerative diseases? A review of in vivo
and in vitro experimental evidence. Toxicology. 301:1–12. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yankner BA, Duffy LK and Kirschner DA:
Neurotrophic and neurotoxic effects of amyloid beta protein:
reversal by tachykinin neuropeptides. Science. 250:279–282. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Itokazu Y, Kato-Negishi M, Nakatani Y,
Ariga T and Yu RK: Effects of amyloid β-peptides and gangliosides
on mouse neural stem cells. Neurochem Res. 38:2019–2027. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Blasko I, Stampfer-Kountchev M, Robatscher
P, Veerhuis R, Eikelenboom P and Grubeck-Loebenstein B: How chronic
inflammation can affect the brain and support the development of
Alzheimer's disease in old age: the role of microglia and
astrocytes. Aging Cell. 3:169–176. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schindowski K, Bretteville A, Leroy K,
Bégard S, Brion JP, Hamdane M and Buée L: Alzheimer's disease-like
tau neuropathology leads to memory deficits and loss of functional
synapses in a novel mutated tau transgenic mouse without any motor
deficits. Am J Pathol. 169:599–616. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Terry RD, Masliah E, Salmon DP, Butters N,
DeTeresa R, Hill R, Hansen LA and Katzman R: Physical basis of
cognitive alterations in Alzheimer's disease: synapse loss is the
major correlate of cognitive impairment. Ann Neurol. 30:572–580.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Inestrosa NC, Alvarez A, Pérez CA, Moreno
RD, Vicente M, Linker C, Casanueva OI, Soto C and Garrido J:
Acetylcholinesterase accelerates assembly of amyloid-beta-peptides
into Alzheimer's fibrils: possible role of the peripheral site of
the enzyme. Neuron. 16:881–891. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alvarez A, Opazo C, Alarcón R, Garrido J
and Inestrosa NC: Acetylcholinesterase promotes the aggregation of
amyloid-beta-peptide fragments by forming a complex with the
growing fibrils. J Mol Biol. 272:348–361. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alvarez A, Alarcón R, Opazo C, Campos EO,
Muñoz FJ, Calderón FH, Dajas F, Gentry MK, Doctor BP, De Mello FG
and Inestrosa NC: Stable complexes involving acetylcholinesterase
and amyloid-beta peptide change the biochemical properties of the
enzyme and increase the neurotoxicity of Alzheimer's fibrils. J
Neurosci. 18:3213–3223. 1998.PubMed/NCBI
|
|
15
|
Yamada K and Nabeshima T: Animal models of
Alzheimer's disease and evaluation of anti-dementia drugs.
Pharmacol Ther. 88:93–113. 2000. View Article : Google Scholar
|
|
16
|
Chartier-Harlin MC, Crawford F, Houlden H,
Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J
and Mullan M: Early-onset Alzheimer's disease caused by mutations
at codon 717 of the beta-amyloid precursor protein gene. Nature.
353:844–846. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rogaev EI, Sherrington R, Rogaeva EA,
Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et
al: Familial Alzheimer's disease in kindreds with missense
mutations in a gene on chromosome 1 related to the Alzheimer's
disease type 3 gene. Nature. 376:775–778. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Levy-Lahad E, Wijsman EM, Nemens E,
Anderson L, Goddard KA, Weber JL, Bird TD and Schellenberg GD: A
familial Alzheimer's disease locus on chromosome 1. Science.
269:970–973. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goedert M and Spillantini MG: Tau
mutations in frontotemporal dementia FTDP-17 and their relevance
for Alzheimer's disease. Biochim Biophys Acta. 1502:110–121. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Levy-Lahad E, Lahad A, Wijsman EM, Bird TD
and Schellenberg GD: Apolipoprotein E genotypes and age of onset in
early-onset familial Alzheimer's disease. Ann Neurol. 38:678–680.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Neha, Sodhi RK, Jaggi AS and Singh N:
Animal models of dementia and cognitive dysfunction. Life Sci.
109:73–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liao A, Nitsch RM, Greenberg SM, Finckh U,
Blacker D, Albert M, Rebeck GW, Gomez-Isla T, Clatworthy A, Binetti
G, et al: Genetic association of an alpha2-macroglobulin
(Val1000lle) polymorphism and Alzheimer's disease. Hum Mol Genet.
7:1953–1956. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dahiyat M, Cumming A, Harrington C,
Wischik C, Xuereb J, Corrigan F, Breen G, Shaw D and St Clair D:
Association between Alzheimer's disease and the NOS3 gene. Ann
Neurol. 46:664–667. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lithner CU, Hedberg MM and Nordberg A:
Transgenic mice as a model for Alzheimer's disease. Curr Alzheimer
Res. 8:818–831. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Quon D, Wang Y, Catalano R, Scardina JM,
Murakami K and Cordell B: Formation of beta-amyloid protein
deposits in brains of transgenic mice. Nature. 352:239–241. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Higgins LS, Rodems JM, Catalano R, Quon D
and Cordell B: Early Alzheimer disease-like histopathology
increases in frequency with age in mice transgenic for beta-APP751.
Proc Natl Acad Sci USA. 92:4402–4406. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Games D, Adams D, Alessandrini R, Barbour
R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T,
Gillespie F, et al: Alzheimer-type neuropathology in transgenic
mice overex-pressing V717F beta-amyloid precursor protein. Nature.
373:523–527. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hsiao K, Chapman P, Nilsen S, Eckman C,
Harigaya Y, Younkin S, Yang F and Cole G: Correlative memory
deficits, Abeta elevation, and amyloid plaques in transgenic mice.
Science. 274:99–102. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Calhoun ME, Wiederhold KH, Abramowski D,
Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B
and Jucker M: Neuron loss in APP transgenic mice. Nature.
395:755–756. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Richards JG, Higgins GA, Ouagazzal AM,
Ozmen L, Kew JN, Bohrmann B, Malherbe P, Brockhaus M, Loetscher H,
Czech C, et al: PS2APP transgenic mice, coexpressing hPS2mut and
hAPPswe, show age-related cognitive deficits associated with
discrete brain amyloid deposition and inflammation. J Neurosci.
23:8989–9003. 2003.PubMed/NCBI
|
|
31
|
Braidy N, Muñoz P, Palacios AG,
Castellano-Gonzalez G, Inestrosa NC, Chung RS, Sachdev P and
Guillemin GJ: Recent rodent models for Alzheimer's disease:
clinical implications and basic research. J Neural Transm.
119:173–195. 2012. View Article : Google Scholar
|
|
32
|
Lewis J, Dickson DW, Lin WL, Chisholm L,
Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, et al:
Enhanced neuro-fibrillary degeneration in transgenic mice
expressing mutant tau and APP. Science. 293:1487–1491. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Götz J, Probst A, Spillantini MG, Schäfer
T, Jakes R, Bürki K and Goedert M: Somatodendritic localization and
hyperphos-phorylation of tau protein in transgenic mice expressing
the longest human brain tau isoform. EMBO J. 14:1304–1313.
1995.
|
|
34
|
James ND, Davis DR, Sindon J, Hanger DP,
Brion JP, Miller CC, Rosenberg MP, Anderton BH and Propst F:
Neurodegenerative changes including altered tau phosphorylation and
neurofilament immunoreactivity in mice transgenic for the
serine/threonine kinase Mos. Neurobiol Aging. 17:235–241. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Allen B, Ingram E, Takao M, Smith MJ,
Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, et al:
Abundant tau filaments and nonapoptotic neurodegeneration in
transgenic mice expressing human P301S tau protein. J Neurosci.
22:9340–9351. 2002.PubMed/NCBI
|
|
36
|
Forman MS, Lal D, Zhang B, Dabir DV,
Swanson E, Lee VM and Trojanowski JQ: Transgenic mouse model of tau
pathology in astrocytes leading to nervous system degeneration. J
Neurosci. 25:3539–3550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Higuchi M, Zhang B, Forman MS, Yoshiyama
Y, Trojanowski JQ and Lee VM: Axonal degeneration induced by
targeted expression of mutant human tau in oligodendrocytes of
transgenic mice that model glial tauopathies. J Neurosci.
25:9434–9443. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ribé EM, Pérez M, Puig B, Gich I, Lim F,
Cuadrado M, Sesma T, Catena S, Sánchez B, Nieto M, et al:
Accelerated amyloid deposition, neurofibrillary degeneration and
neuronal loss in double mutant APP/tau transgenic mice. Neurobiol
Dis. 20:814–822. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Götz J, Chen F, van Dorpe J and Nitsch RM:
Formation of neurofibrillary tangles in P301l tau transgenic mice
induced by Abeta 42 fibrils. Science. 293:1491–1495. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Oddo S, Caccamo A, Shepherd JD, Murphy MP,
Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y and LaFerla
FM: Triple-transgenic model of Alzheimer's disease with plaques and
tangles: Intracellular Abeta and synaptic dysfunction. Neuron.
39:409–421. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Janelsins MC, Mastrangelo MA, Oddo S,
LaFerla FM, Federoff HJ and Bowers WJ: Early correlation of
microglial activation with enhanced tumor necrosis factor-alpha and
monocyte chemoattractant protein-1 expression specifically within
the entorhinal cortex of triple transgenic Alzheimer's disease
mice. J Neuroinflammation. 2:232005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kimura R and Ohno M: Impairments in remote
memory stabi-lization precede hippocampal synaptic and cognitive
failures in 5XFAD Alzheimer mouse model. Neurobiol Dis. 33:229–235.
2009. View Article : Google Scholar :
|
|
43
|
Oakley H, Cole SL, Logan S, Maus E, Shao
P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik
L, et al: Intraneuronal beta-amyloid aggregates, neurodegeneration,
and neuron loss in transgenic mice with five familial Alzheimer's
disease mutations: Potential factors in amyloid plaque formation. J
Neurosci. 26:10129–10140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Clarke J, Thornell A, Corbett D, Soininen
H, Hiltunen M and Jolkkonen J: Overexpression of APP provides
neuroprotection in the absence of functional benefit following
middle cerebral artery occlusion in rats. Eur J Neurosci.
26:1845–1852. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Flood DG, Lin YG, Lang DM, Trusko SP,
Hirsch JD, Savage MJ, Scott RW and Howland DS: A transgenic rat
model of Alzheimer's disease with extracellular Abeta deposition.
Neurobiol Aging. 30:1078–1090. 2009. View Article : Google Scholar
|
|
46
|
Ruiz-Opazo N, Kosik KS, Lopez LV,
Bagamasbad P, Ponce LR and Herrera VL: Attenuated
hippocampus-dependent learning and memory decline in transgenic
TgAPPswe Fischer-344 rats. Mol Med. 10:36–44. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Filipcik P, Zilka N, Bugos O, Kucerak J,
Koson P, Novak P and Novak M: First transgenic rat model developing
progressive cortical neurofibrillary tangles. Neurobiol Aging.
33:1448–1456. 2012. View Article : Google Scholar
|
|
48
|
Van Dam D and De Deyn PP: Animal models in
the drug discovery pipeline for Alzheimer's disease. Br J
Pharmacol. 164:1285–1300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Iwata N, Tsubuki S, Takaki Y, Watanabe K,
Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E,
Sekine-Aizawa Y and Saido TC: Identification of the major
Abeta1-42-degrading catabolic pathway in brain parenchyma:
suppression leads to biochemical and pathological deposition. Nat
Med. 6:143–150. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vekrellis K, Ye Z, Qiu WQ, Walsh D,
Hartley D, Chesneau V, Rosner MR and Selkoe DJ: Neurons regulate
extracellular levels of amyloid beta-protein via proteolysis by
insulin-degrading enzyme. J Neurosci. 20:1657–1665. 2000.PubMed/NCBI
|
|
51
|
Solà C, García-Ladona FJ, Sarasa M, Mengod
G, Probst A, Palacios G and Palacios JM: Beta APP gene expression
is increased in the rat brain after motor neuron axotomy. Eur J
Neurosci. 5:795–808. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gonzalo-Ruiz A, González I and
Sanz-Anquela JM: Effects of beta-amyloid protein on serotoninergic,
noradrenergic, and cholinergic markers in neurons of the
pontomesencephalic tegmentum in the rat. J Chem Neuroanat.
26:153–169. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Du P, Wood KM, Rosner MH, Cunningham D,
Tate B and Geoghegan KF: Dominance of amyloid precursor protein
sequence over host cell secretases in determining beta-amyloid
profiles studies of interspecies variation and drug action by
internally standardized immunoprecipitation/mass spectrometry. J
Pharmacol Exp Ther. 320:1144–1152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Beck M, Bigl V and Rossner S: Guinea pigs
as a nontransgenic model for APP processing in vitro and in vivo.
Neurochem Res. 28:637–644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen Y, Wei G, Nie H, Lin Y, Tian H, Liu
Y, Yu X, Cheng S, Yan R, Wang Q, et al: β-Asarone prevents
autophagy and synaptic loss by reducing ROCK expression in
asenescence-accelerated prone 8 mice. Brain Res. 1552:41–54. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Takeda T, Hosokawa M, Takeshita S, Irino
M, Higuchi K, Matsushita T, Tomita Y, Yasuhira K, Hamamoto H and
Shimizu K: A new murine model of accelerated senescence. Mech
Ageing Dev. 17:183–194. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Flood JF and Morley JE: Learning and
memory in the SAMP8 mouse. Neurosci Biobehav Rev. 22:1–20. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Stefanova NA, Kozhevnikova OS, Vitovtov
AO, Maksimova KY, Logvinov SV, Rudnitskaya EA, Korbolina EE,
Muraleva NA and Kolosova NG: Senescence-accelerated OXYS rats: a
model of age-related cognitive decline with relevance to
abnormalities in Alzheimer disease. Cell Cycle. 13:898–909. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Stefanova NA, Muraleva NA, Skulachev VP
and Kolosova NG: Alzheimer's disease-like pathology in
senescence-accelerated OXYS rats can be partially retarded with
mitochondria-targeted antioxidant SkQ1. J Alzheimers Dis.
38:681–694. 2014.
|
|
60
|
Poon HF, Calabrese V, Scapagnini G and
Butterfield DA: Free radicals: key to brain aging and heme
oxygenase as a cellular response to oxidative stress. J Gerontol A
Biol Sci Med Sci. 59:478–493. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bosch MN, Pugliese M, Gimeno-Bayón J,
Rodríguez MJ and Mahy N: Dogs with cognitive dysfunction syndrome:
A natural model of Alzheimer's disease. Curr Alzheimer Res.
9:298–314. 2012. View Article : Google Scholar
|
|
62
|
Head E, Callahan H, Muggenburg BA, Cotman
CW and Milgram NW: Visual-discrimination learning ability and
beta-amyloid accumulation in the dog. Neurobiol Aging. 19:415–425.
1998. View Article : Google Scholar
|
|
63
|
Head E, McCleary R, Hahn FF, Milgram NW
and Cotman CW: Region-specific age at onset of beta-amyloid in
dogs. Neurobiol Aging. 21:89–96. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hou Y, White RG, Bobik M, Marks JS and
Russell MJ: Distribution of beta-amyloid in the canine brain.
Neuroreport. 8:1009–1012. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Satou T, Cummings BJ, Head E, Nielson KA,
Hahn FF, Milgram NW, Velazquez P, Cribbs DH, Tenner AJ and Cotman
CW: The progression of beta-amyloid deposition in the frontal
cortex of the aged canine. Brain Res. 774:35–43. 1997. View Article : Google Scholar
|
|
66
|
Cuyckens F, Balcaen LI, De Wolf K, De
Samber B, Van Looveren C, Hurkmans R and Vanhaecke F: Use of the
bromine isotope ratio in HPLC-ICP-MS and HPLC-ESI-MS analysis of a
new drug in development. Anal Bioanal Chem. 390:1717–1729. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Head E: Combining an antioxidant-fortified
diet with behavioral enrichment leads to cognitive improvement and
reduced brain pathology in aging canines: strategies for healthy
aging. Ann NY Acad Sci. 1114:398–406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Papaioannou N, Tooten PC, van Ederen AM,
Bohl JR, Rofina J, Tsangaris T and Gruys E: Immunohistochemical
investigation of the brain of aged dogs. I. Detection of
neurofibrillary tangles and of 4-hydroxynonenal protein, an
oxidative damage product, in senile plaques. Amyloid. 8:11–21.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pugliese M, Mascort J, Mahy N and Ferrer
I: Diffuse beta-amyloid plaques and hyperphosphorylated tau are
unrelated processes in aged dogs with behavioral deficits. Acta
Neuropathol. 112:175–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Languille S, Blanc S, Blin O, Canale CI,
Dal-Pan A, Devau G, Dhenain M, Dorieux O, Epelbaum J, Gomez D, et
al: The grey mouse lemur: a non-human primate model for ageing
studies. Ageing Res Rev. 11:150–162. 2012. View Article : Google Scholar
|
|
71
|
Bons N, Rieger F, Prudhomme D, Fisher A
and Krause KH: Microcebus murinus: a useful primate model for human
cerebral aging and Alzheimer's disease? Genes Brain Behav.
5:120–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kraska A, Dorieux O, Picq JL, Petit F,
Bourrin E, Chenu E, Volk A, Perret M, Hantraye P, Mestre-Frances N,
et al: Age-associated cerebral atrophy in mouse lemur primates.
Neurobiol Aging. 32:894–906. 2011. View Article : Google Scholar
|
|
73
|
Giannakopoulos P, Silhol S, Jallageas V,
Mallet J, Bons N, Bouras C and Delaère P: Quantitative analysis of
tau protein-immunoreactive accumulations and beta amyloid protein
deposits in the cerebral cortex of the mouse lemur, Microcebus
murinus. Acta Neuropathol. 94:131–139. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Laurijssens B, Aujard F and Rahman A:
Animal models of Alzheimer's disease and drug development. Drug
Discov Today Technol. 10:e319–e327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Bélanger N, Grégoire L, Bédard PJ and Di
Paolo T: DHEA improves symptomatic treatment of moderately and
severely impaired MPTP monkeys. Neurobiol Aging. 27:1684–1693.
2006. View Article : Google Scholar
|
|
76
|
Yue F, Lu C, Ai Y, Chan P and Zhang Z:
Age-associated changes of cerebrospinal fluid amyloid-β and tau in
cynomolgus monkeys. Neurobiol Aging. 35:1656–1659. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Molteni R, Barnard RJ, Ying Z, Roberts CK
and Gómez-Pinilla F: A high-fat, refined sugar diet reduces
hippocampal brain-derived neurotrophic factor, neuronal plasticity,
and learning. Neuroscience. 112:803–814. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Demetrius LA and Driver J: Alzheimer's as
a metabolic disease. Biogerontology. 14:641–649. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Herculano B, Tamura M, Ohba A, Shimatani
M, Kutsuna N and Hisatsune T: β-alanyl-L-histidine rescues
cognitive deficits caused by feeding a high fat diet in a
transgenic mouse model of Alzheimer's disease. J Alzheimers Dis.
33:983–997. 2013.
|
|
80
|
Haley RW and Dietschy JM: Is there a
connection between the concentration of cholesterol circulating in
plasma and the rate of neuritic plaque formation in Alzheimer
disease? Arch Neurol. 57:1410–1412. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Gibson Wood W, Eckert GP, Igbavboa U and
Müller WE: Amyloid beta-protein interactions with membranes and
cholesterol: Causes or casualties of Alzheimer's disease. Biochim
Biophys Acta. 1610:281–290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wu YY, Wang X, Tan L, Liu D, Liu XH, Wang
Q, Wang JZ and Zhu LQ: Lithium attenuates scopolamine-induced
memory deficits with inhibition of GSK-3β and preservation of
post-synaptic components. J Alzheimers Dis. 37:515–527. 2013.
|
|
83
|
Vandal M, White PJ, Tremblay C, St-Amour
I, Chevrier G, Emond V, Lefrançois D, Virgili J, Planel E, Giguere
Y, et al: Insulin reverses the high-fat diet-induced increase in
brain Aβ and improves memory in an animal model of Alzheimer
disease. Diabetes. 63:4291–4301. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Pinton S, Brüning CA, Sartori Oliveira CE,
Prigol M and Nogueira CW: Therapeutic effect of organoselenium
dietary supplementation in a sporadic dementia of Alzheimer's type
model in rats. J Nutr Biochem. 24:311–317. 2013. View Article : Google Scholar
|
|
85
|
Nakamura S, Murayama N, Noshita T, Annoura
H and Ohno T: Progressive brain dysfunction following
intracerebroventricular infusion of beta(1-42)-amyloid peptide.
Brain Res. 912:128–136. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Winslow JT and Camacho F: Cholinergic
modulation of a decrement in social investigation following
repeated contacts between mice. Psychopharmacology (Berl).
121:164–172. 1995. View Article : Google Scholar
|
|
87
|
Sain H, Sharma B, Jaggi AS and Singh N:
Pharmacological investigations on potential of peroxisome
proliferator-activated receptor-gamma agonists in
hyperhomocysteinemia-induced vascular dementia in rats.
Neuroscience. 192:322–333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kamat PK, Rai S and Nath C: Okadaic acid
induced neuro-toxicity: An emerging tool to study Alzheimer's
disease pathology. Neurotoxicology. 37:163–172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhang J, Li P and Wang Y, Liu J, Zhang Z,
Cheng W and Wang Y: Ameliorative effects of a combination of
baicalin, jasminoidin and cholic acid on ibotenic acid-induced
dementia model in rats. PLoS One. 8:e566582013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Bonda DJ, Lee HG, Blair JA, Zhu X, Perry G
and Smith MA: Role of metal dyshomeostasis in Alzheimer's disease.
Metallomics. 3:267–270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Squire LR and Zola-Morgan S: Memory: Brain
systems and behavior. Trends Neurosci. 11:170–175. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Liu J, Zhang Z, Li JT, Zhu YH, Zhou HL,
Liu S and Wang TH: Effects of NT-4 gene modified fibroblasts
transplanted into AD rats. Neurosci Lett. 466:1–5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Savage LM, Sweet AJ, Castillo R and
Langlais PJ: The effects of lesions to thalamic lateral internal
medullary lamina and posterior nuclei on learning, memory and
habituation in the rat. Behav Brain Res. 82:133–147. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Avaliani N, Sørensen AT, Ledri M, Bengzon
J, Koch P, Brüstle O, Deisseroth K, Andersson M and Kokaia M:
Optogenetics reveal delayed afferent synaptogenesis on grafted
human-induced pluripotent stem cell-derived neural progenitors.
Stem Cells. 32:3088–3098. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Green KN, Smith IF and Laferla FM: Role of
calcium in the pathogenesis of Alzheimer's disease and transgenic
models. Subcell Biochem. 45:507–521. 2007. View Article : Google Scholar
|
|
96
|
Nepovimova E, Uliassi E, Korabecny J,
Peña-Altamira LE, Samez S, Pesaresi A, Garcia GE, Bartolini M,
Andrisano V, Bergamini C, et al: Multitarget drug design strategy:
quinone-tacrine hybrids designed to block amyloid-β aggregation and
to exert anticholinesterase and antioxidant effects. J Med Chem.
57:8576–8589. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Akhter R, Sanphui P and Biswas SC: The
essential role of p53-up-regulated modulator of apoptosis (Puma)
and its regulation by FoxO3a transcription factor in
β-amyloid-induced neuron death. J Biol Chem. 289:10812–10822. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Matlack KE, Tardiff DF, Narayan P,
Hamamichi S, Caldwell KA, Caldwell GA and Lindquist S: Clioquinol
promotes the degradation of metal-dependent amyloid-β (Aβ)
oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc
Natl Acad Sci USA. 111:4013–4018. 2014. View Article : Google Scholar
|
|
99
|
Gong CX, Lidsky T, Wegiel J, Grundke-Iqbal
I and Iqbal K: Metabolically active rat brain slices as a model to
study the regulation of protein phosphorylation in mammalian brain.
Brain Res Brain Res Protoc. 6:134–140. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Roder S, Danober L, Pozza MF, Lingenhoehl
K, Wiederhold KH and Olpe HR: Electrophysiological studies on the
hippocampus and prefrontal cortex assessing the effects of
amyloidosis in amyloid precursor protein 23 transgenic mice.
Neuroscience. 120:705–720. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Li L, Sengupta A, Haque N, Grundke-Iqbal I
and Iqbal K: Memantine inhibits and reverses the Alzheimer type
abnormal hyperphosphorylation of tau and associated
neurodegeneration. FEBS Lett. 566:261–269. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Jang J, Yoo JE, Lee JA, Lee DR, Kim JY,
Huh YJ, Kim DS, Park CY, Hwang DY, Kim HS, et al: Disease-specific
induced pluripotent stem cells: A platform for human disease
modeling and drug discovery. Exp Mol Med. 44:202–213. 2012.
View Article : Google Scholar :
|
|
103
|
Yagi T, Ito D, Okada Y, Akamatsu W, Nihei
Y, Yoshizaki T, Yamanaka S, Okano H and Suzuki N: Modeling familial
Alzheimer's disease with induced pluripotent stem cells. Hum Mol
Genet. 20:4530–4539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu
Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al:
Probing sporadic and familial Alzheimer's disease using induced
pluripotent stem cells. Nature. 482:216–220. 2012.PubMed/NCBI
|
|
105
|
Kondo T, Asai M, Tsukita K, Kutoku Y,
Ohsawa Y, Sunada Y, Imamura K, Egawa N, Yahata N, Okita K, et al:
Modeling Alzheimer's disease with iPSCs reveals stress phenotypes
associated with intracellular Aβ and differential drug
responsiveness. Cell Stem Cell. 12:487–496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Macias MP, Gonzales AM, Siniard AL, Walker
AW, Corneveaux JJ, Huentelman MJ, Sabbagh MN and Decourt B: A
cellular model of amyloid precursor protein processing and
amyloid-β peptide production. J Neurosci Methods. 223:114–122.
2014. View Article : Google Scholar :
|
|
107
|
Meng P, Yoshida H, Tanji K, Matsumiya T,
Xing F, Hayakari R, Wang L, Tsuruga K, Tanaka H, Mimura J, et al:
Carnosic acid attenuates apoptosis induced by amyloid-β 1-42 or
1-43 in SH-SY5Y human neuroblastoma cells. Neurosci Res. 94:1–9.
2015. View Article : Google Scholar
|
|
108
|
Giunta S, Andriolo V and Castorina A: Dual
blockade of the A1 and A2A adenosine receptor prevents amyloid beta
toxicity in neuroblastoma cells exposed to aluminum chloride. Int J
Biochem Cell Biol. 54:122–136. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Denis PA: Alzheimer's disease: A gas
model. The NADPH oxidase-Nitric Oxide system as an antibubble
biomachinery. Med Hypotheses. 81:976–987. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Minicozzi V, Chiaraluce R, Consalvi V,
Giordano C, Narcisi C, Punzi P, Rossi GC and Morante S:
Computational and experimental studies on β-sheet breakers
targeting Aβ1-40 fibrils. J Biol Chem. 289:11242–11252. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Götz J and Ittner LM: Animal models of
Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci.
9:532–544. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Viet MH, Chen CY, Hu CK, Chen YR and Li
MS: Discovery of dihydrochalcone as potential lead for Alzheimer's
disease: In silico and in vitro study. PLoS One. 8:e791512013.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Lo AC, Iscru E, Blum D, Tesseur I,
Callaerts-Vegh Z, Buée L, De Strooper B, Balschun D and D'Hooge R:
Amyloid and tau neuro-pathology differentially affect prefrontal
synaptic plasticity and cognitive performance in mouse models of
Alzheimer's disease. J Alzheimers Dis. 37:109–125. 2013.
|
|
114
|
Okuma Y and Nomura Y:
Senescence-accelerated mouse (SAM) as an animal model of senile
dementia: pharmacological, neurochemical and molecular biological
approach. Jpn J Pharmacol. 78:399–404. 1998. View Article : Google Scholar
|