Introduction
Ischemia-reperfusion injury increases oxidative
stress and inflammation as well as provoking oxidative damage to
the hippocampal CA1 region of the brain (1). Oxidative stress leads to
mitochondrial dysfunction, calcium accumulation, and the increased
production of reactive oxygen species (ROS) resulting in
post-ischemic reperfusion neuronal death (2,3).
The pathophysiology of ischemia-reperfusion injury includes the
excessive production of ROS that contributes to tissue damage, both
directly through lipid peroxidation, protein oxidation, and DNA
damage as well as indirectly through the activation of apoptotic
pathways (4,5).
Oxidative stress is known to induce apoptosis, which
has been implicated in various types of cell death including
ischemic damage to the heart and brain (6,7).
Apoptosis may be initiated through the two major pathways of
apoptosis, the extrinsic and the intrinsic pathways (8). The intrinsic apoptotic pathway is
initiated by stress signals that lead to the translocation of Bax
to mitochondria (9), the release
of cytochrome c (10) and
the associated activation of caspase-3 (11). The extrinsic apoptotic pathway
activates death receptors, resulting in death-inducing signaling
complex (DISC) formation that leads to the activation of caspase-8,
followed by the activation of caspase-3 (11). p53 is a transcription factor that
has been found to induce growth arrest through the transactivation
of p21cip1/waf1 and also, to initiate apoptosis in
response to DNA damage (12).
Caspase-2 is known to mediate intrinsic apoptotic pathway signaling
and may be activated by oxidative stress (7,13).
The NOL3 [nucleolar protein 3 (apoptosis repressor
with CARD domain)] protein is highly expressed in the heart,
skeletal muscle and the brain, and plays a role in the inhibition
of physiological apoptotic pathways (7). NOL3 protein contains two functional
domains: the CARD and the proline/glutamic acid (P/E) domains. The
CARD domain is similar to the pyrin domain, a small helical death
domain that is involved in protein-protein interactions. The CARD
domain of the NOL3 protein is capable of downregulating the
activity of caspase-2 and -8 by CARD-CARD interactions (14,15).
Protein transduction domains (PTDs) including Tat
peptide deliver proteins into cells and tissues by crossing
membranes or the blood brain barrier (BBB). Many studies, including
those conducted by our group have demonstrated that the Tat fusion
proteins efficiently transduce into cells/tissues where they
protect against various types of oxidative stress-induced cell
death including neuronal cell death (16–25).
Under conditions of oxidative stress, the regulation
of ROS, caspases and pro-apoptotic proteins may be crucial
therapies for ischemic brain injury. To address this hypothesis, we
have demonstrated the protective effect of cell permeable Tat-fused
NOL3 protein (Tat-NOL3) on oxidative stress-induced
hippo-(Tat-NOL3) on oxidative stress-induced hippocampal HT22
neuronal death and ischemic brain injury.
Materials and methods
Cell lines and reagents
The mouse hippocampal neuronal cell line, HT22 (from
ATCC, Manassas, VA, USA), was cultured in Dulbecco's modifed
Eagle's medium (DMEM) (Lonza/BioWhittaker, Walkersville, MD, USA)
containing 10% fetal bovine serum (FBS) and antibiotics (100
µg/ml streptomycin, 100 U/ml penicillin and 100 µg/ml
gentamicin sulfate) and cultured in an incubation chamber
(37°C, 95% air and 5% CO2).
FBS and the antibiotics were purchased from
Gibco-BRL (Grand Island, NY, USA). We purchased a
Ni2+-nitrilotriacetic acid Sepharose superflow column
from Qiagen, Inc. (Valencia, CA, USA) and Tat peptide from Peptron,
Inc (Daejeon, Korea). The primary, secondary, and β-actin
antibodies were obtained from Cell Signaling Technology, Inc.
(Beverly, MA, USA) and Santa Cruz Biotechnology, Inc. (Santa Cruz,
CA, USA). All other chemicals and reagents were of the highest
quality grade available.
Animals
Mongolian gerbils (Meriones unguiculatus;
body weight, 65–75 g, 6 months of age) were obtained from the
Experimental Animal Center at Hallym University (Chuncheon, Korea).
The animals were housed at a constant temperature (23°C) and
relative humidity (60%) under a fixed 12-h light:12-h dark cycle
with unlimited access to food and water. All experimental
procedures involving the animals and their care conformed to the
Guide for the Care and Use of Laboratory Animals of the National
Veterinary Research and Quarantine Service of Korea, and were
approved by the Hallym Medical Center Institutional Animal Care and
Use Committee (Chuncheon, Korea).
Expression and purification of Tat-NOL3
protein
The construction of the Tat expression vector was
performed as described in our previous study (26). Human NOL3 cDNA was obtained
through PCR amplification with the following specific primers:
sense primer, 5′-CTCGAGGGCAACGCGCAG-3′; and antisense primer,
5′-GGATCCTCAGGAATCTTCGGA CTC-3′. The PCR product was subcloned in
the Tat expression vector with an N-terminal 6His-tag. To obtain
the control NOL3 (con-NOL3) plasmid, Tat leader cDNA was deleted
from the Tat-NOL3 plasmid. The recombinant Tat-NOL3 plasmid was
transformed into Escherichia coli BL21(DE3; Novagen,
Madison, WI, USA) cells and then incubated with 0.1 mM isopropyl
β-D-thiogalactoside (IPTG; Duchefa Biochemie, Haarlem, The
Netherlands) at 20°C for 20 h. The cell homogenates were
purified by chromatography using an
Ni2+-nitrilotriacetic acid Sepharose affinity column and
a PD-10 column (Amersham, Braunschweig, Germany). Quantification of
the purified protein was measured by performing a Bradford assay
(Bio-Rad Laboratories, Hercules, CA, USA) using bovine serum
albumin (BSA) as the standard (27).
Transduction of Tat-NOL3 protein
To analyze the transduction of Tat-NOL3 protein into
cells, the HT22 cells were exposed to various concentrations of
purified Tat-NOL3 protein (1–14 µM, for 1 h) or for various
time periods (10–60 min, at 14 µM). The measurement of the
quantity of transduced Tat-NOL3 protein was performed by western
blot analysis (28,29). We performed 15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to analyze
the protein. After the proteins were transferred from the gel to a
nitrocellulose membrane, the membrane was blocked with phosphate
buffered saline (PBS) with Tween 20 (PBS-T) containing 5% non-fat
dry milk or BSA. The membrane was probed with a rabbit
anti-histidine polyclonal antibody (1:5,000; sc-804, Santa Cruz
Biotechnology, Inc.). In addition, to detect apoptosis
related-signals in the whole cell lysates, the membrane was
immunoblotted using specific antibodies to p21 (2947), p-p53
(9284), p53 (9282), PARP (9532), cleaved PARP (9544), Bax (2772),
Bcl-2 (2876), caspase-3 (9662), cleaved caspase-3 (9661), caspase-2
(2224) and caspase-8 (4927) (1:1,000; Cell Signaling Technology,
Inc.). In all cases, a horseradish peroxidase-conjugated secondary
antibody was used (1:10,000; Cell Signaling Technology, Inc.). The
proteins were detected with enhanced chemiluminescence reagents
(Amersham, Franklin Lakes, NJ, USA).
To determine the intracellular stability of Tat-NOL3
protein, the cells were treated with Tat-NOL3 for 1 h and were then
incubated further for 1–72 h. Subsequently, the cells were
harvested and transduced Tat-NOL3 protein was detected using a
rabbit anti-histidine polyclonal antibody.
Immunostaining analysis
The cells were cultured on coverslips placed in
wells of a tissue culture plate and treated with Tat-NOL3 protein
(14 µM). The samples were incubated at 37°C for 1 h
and washed with PBS twice after which they were then fixed with 4%
paraformaldehyde for 5 min at room temperature. Subsequently, the
cells were incubated with 3% BSA, 0.1% Triton X-100 and PBS
(PBS-BT) for 30 min at room temperature, and washed twice with
PBS-BT. The primary antibody (His-probe; Santa Cruz Biotechnology,
Inc.) was diluted 1:2,000 and the cells were exposed for 2 h at
room temperature. The cells were incubated in the dark for 1 h with
Alexa Fluor 488-conjugated secondary antibody (Invitrogen,
Carlsbad, CA, USA) diluted 1:15,000. To stain the nuclei, 1
µg/ml DAPI (Roche Applied Science, Basel, Switzerland) was
added for 2 min. Analysis was performed under a confocal
fluorescence microscope (FA-300; Olympus, Tokyo, Japan) (23,30).
Cell viability assay
Cell viability was evaluated as described previously
(31,32). Briefly, the HT22 cells were
exposed to 1 µM hydrogen peroxide
(H2O2) for 16 h following treatment with
Tat-NOL3 or con-NOL3 protein. Cell viability was determined using a
WST-1 assay kit according to the manufacturer's instructions (Daeil
Lab Service Co., Seoul, Korea). Absorbance was measured at 450 nm
with an enzyme-linked immunosorbent assay (ELISA) microplate reader
(Multiskan MCC/340; Thermo Labsystems Oy., Helsinki, Finland). Cell
viability is presented as a percentage of the control.
Measurement of ROS levels
The ROS level was evaluated as previously described
(29). Briefly, the HT22 cells
were treated with 0.5 mM H2O2 for 20 min
following treatment with Tat-NOL3 or con-NOL3 protein. To measure
the intracellular ROS levels, the cells were treated with
2′-,7′-dichlorofluorescein diacetate (DCF-DA) which is converted
into 2′-,7′-dichlorofluorescein (DCF) by
H2O2. Cell fluorescence image intensity was
measured at 485-nm excitation and 538-nm emission using a
Fluoroskan ELISA plate reader (Thermo Labsystems Oy.). Control cell
images were obtained using differential interference contrast
(DIC), which is used in images of cell morphology.
Analysis of DNA fragmentation by terminal
deoxynucleotidyl transferase (TdT)-mediated biotinylated dUTP
nick-end labeling (TUNEL) assay
Tat-NOL3 protein (14 µM) or con-NOL3 protein
was transduced into the HT22 cells for 1 h prior to exposure to 0.5
mM H2O2 for 16 h. DNA fragmentation was
detected by TUNEL staining using a Cell Death Detection kit (Roche
Applied Science). Images were captured using a fluorescence
microscope (Nikon Eclipse 80i; Nikon, Tokyo, Japan) (23).
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazoly-carbocyanine
iodine (JC-1) mitochondrial membrane potential (ΔΨm)
assay
To examine ΔΨm, 14 µM Tat-NOL3
protein or 14 µM con-NOL3 protein was transduced into the
HT22 cells for 1 h and subsequently, they were exposed to 0.5 mM
H2O2 for 2 h. ΔΨm was assessed as
described previously (33,34).
Briefly, 100 µl of JC-1 staining solution per ml of culture
medium was added and the cells were incubated in a CO2
incubator at 37°C for 15 min. JC-1 staining was performed
according to the manufacturer's instructions using a JC-1 assay kit
(Cayman Chemical Company, Inc., Ann Arbor, MI, USA). The cells were
then analyzed with a fluorescence microscope using 507 nm and 530
nm as excitation and emission wavelengths, respectively. To analyze
ΔΨm, the ratio of the reading at 590 nm to the reading
at 530 nm was considered as the relative ΔΨm value.
Images of each sample were captured using a fluorescence microscope
(Nikon Eclipse 80i).
Establishment of an animal model of
forebrain ischemia
A model of forebrain ischemia was established as
described in previous studies (35,36). Briefly, the animals were
anesthetized, common carotid arteries were isolated, freed of nerve
fibers, and occluded with non-traumatic aneurysm clips. Complete
interruption of blood flow was confirmed by observing the retinal
artery using an ophthalmoscope (HEINE K180®, Heine
Optotechnik, Herrsching, Germany). After 5 min of occlusion, the
aneurysm clips were removed.
To explore the protective effects of Tat-NOL3
protein against ischemic damage, the animals were divided into 5
groups (n=10/group); sham-operated group, vehicle (saline)-treated
group, Tat peptide-treated group, con-NOL3-treated group, and
Tat-NOL3-treated group (each 2 mg/kg) with ischemic surgery. Tat
peptide, con-NOL3 protein, and Tat-NOL3 proteins were administered
intraperitoneally 30 min after ischemia-reperfusion.
Immunohistochemical analysis
To examine the effects of Tat-NOL3 on ischemic
damage, brain tissue samples were obtained at 7 days after
ischemia-reperfusion. Immunohistochemistry was performed as
described previously (33,35,36).
The animals were anesthetized with 30 mg/kg Zoletil 50 (Virbac
Laboratories, Carros, France) at 7 days after ischemia-reperfusion
and perfused transcardially with 0.1 M PBS (pH 7.4) followed by 4%
paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The brains
were removed and post-fixed in the same fixative for 6 h. The brain
tissue samples were cryoprotected by infiltration with 30% sucrose
overnight. The frozen tissues were then serially sectioned on a
cryostat (Leica, Wetzlar, Germany) into 30 µm coronal
sections to investigate the morphological and neuronal changes in
the hippocampus, and subsequently, the sections were collected into
6-well plates containing PBS.
For the histological analysis, the sectioned brains
were incubated with mouse anti-neuronal nuclei (NeuN; 1:1,000;
Chemicon International, Inc., Temecula, CA, USA), rabbit
anti-ionized calcium-binding adapter molecule 1 (Iba-1; 1:500;
Wako, Osaka, Japan), and rabbit anti-glial fibrillary acidic
protein (GFAP; 1:1,000; Chemicon International, Inc.) for 48 h at
20°C. They were then exposed to biotinylated rabbit
anti-goat IgG (1:200; Vector Laboratories, Inc., Burlingame, CA,
USA) or goat anti-mouse IgG and streptavidin-peroxidase complex and
visualized with 3,3′-diaminobenzidine (Sigma-Aldrich, St. Louis,
MO, USA). Cresyl violet (CV; Junsei Chemical Co. Ltd., Saitama,
Japan) and Fluoro-Jade B (FJB; Millipore Co., Temecula, CA, USA)
staining were performed as previously described (23,33). Additionally, we performed
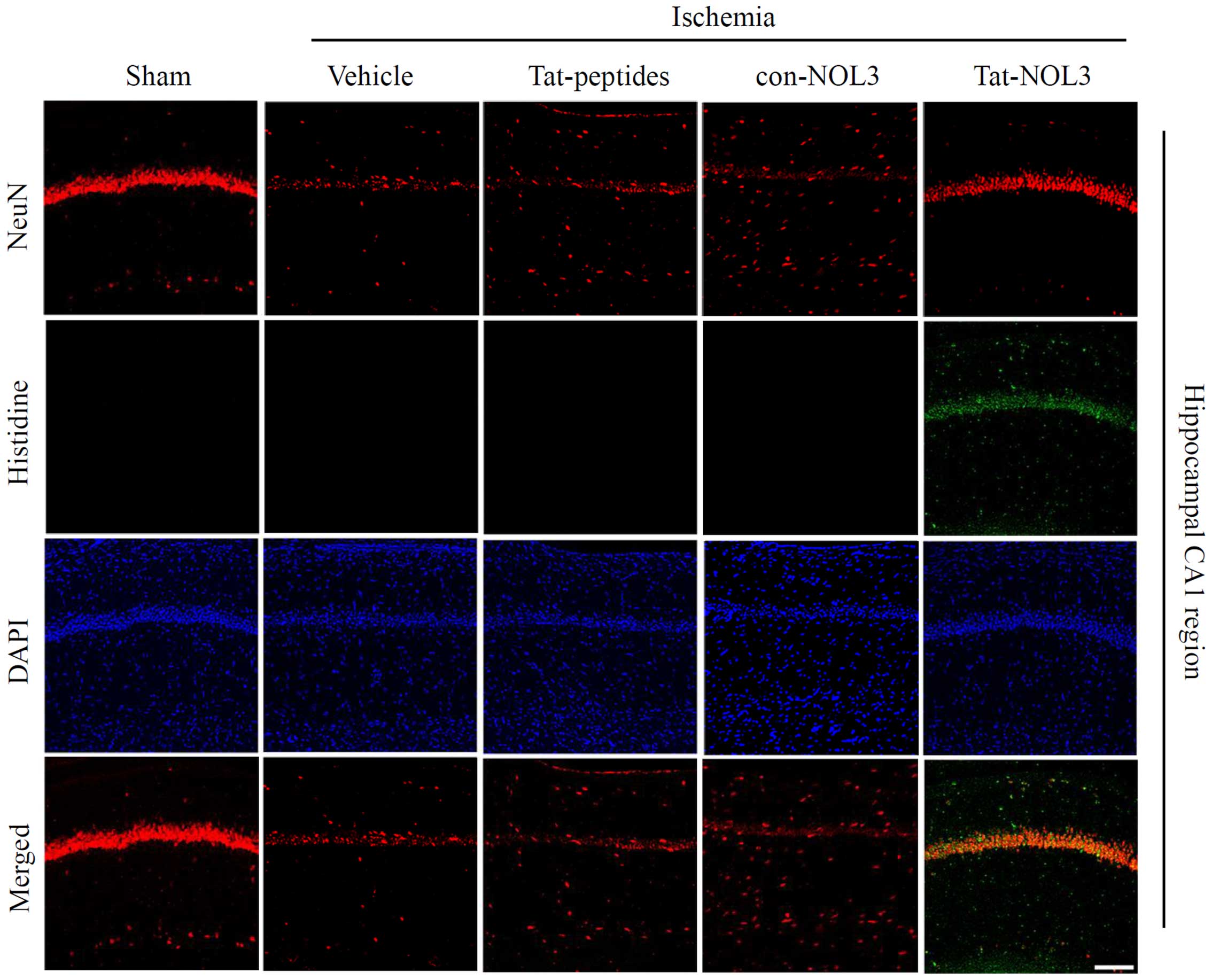
immunostaining with a rabbit anti-histidine antibody and DAPI to
detect the transduction of Tat-NOL3 protein in the brain
hippocampus.
Statistical analysis
The data presented represent the means ± SD from
three independent experiments. One way ANOVA and the Student's
t-test were used for comparisons between the groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
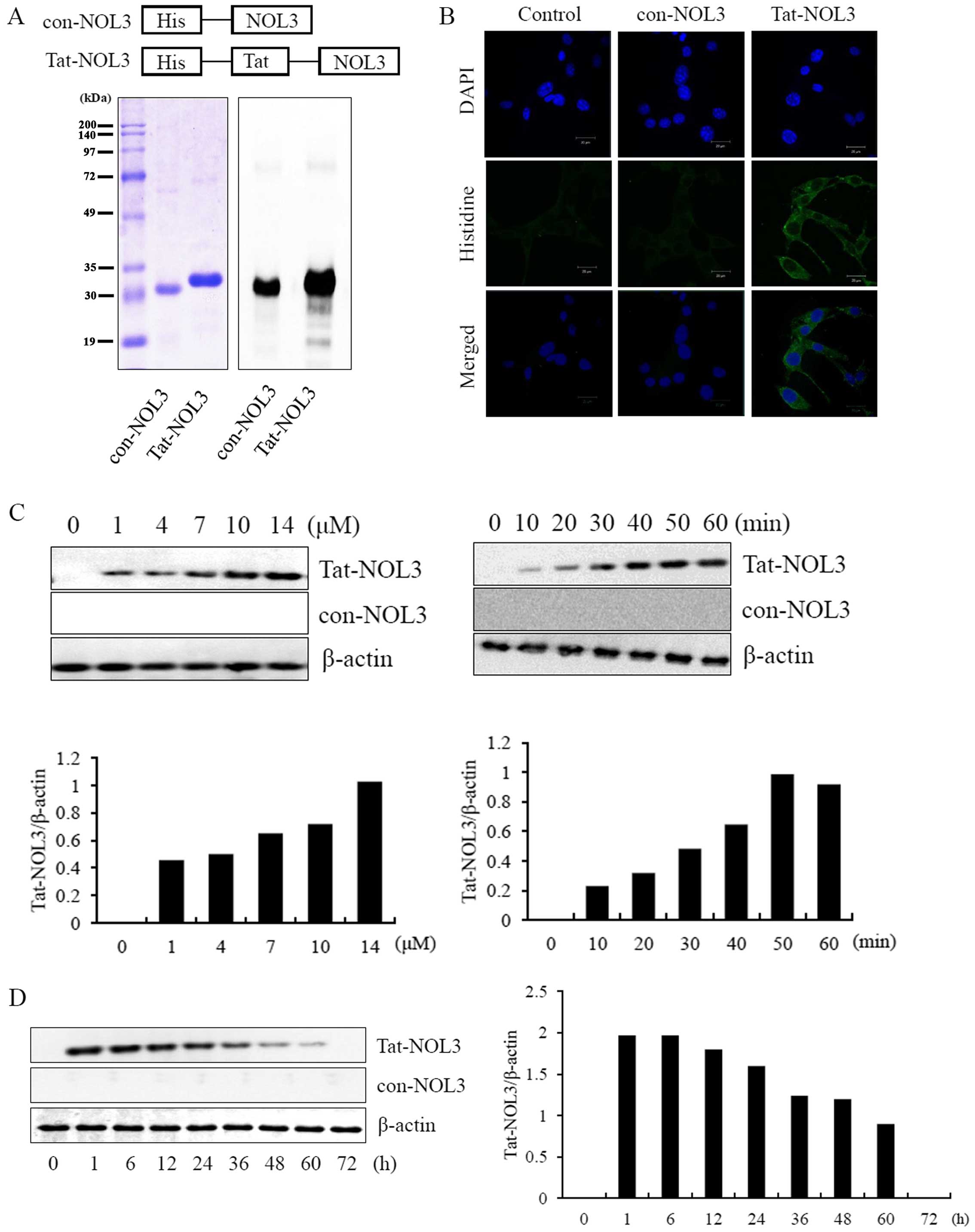
Purification and transduction of Tat-NOL3
protein into HT22 cells
Transducible Tat-NOL3 proteins were generated by
cloning cDNA of NOL3 into the 6His tags of a Tat-expression vector.
To obtain con-NOL3 proteins, the Tat domain was deleted from the
Tat-NOL3 construct. Recombinant Tat-NOL3 and con-NOL3 proteins were
overexpressed in E. coli BL21(DE3) cells by IPTG induction.
The Tat-NOL3 and con-NOL3 proteins were purified using an
Ni2+-nitrilotriacetic acid Sepharose affinity column and
each Tat-NOL3 and con-NOL3 protein was evaluated by SDS-PAGE and
western blot analysis using an anti-rabbit polyhistidine antibody
(Fig. 1A). To examine the
localization of Tat-NOL3 protein in the cells, transduced Tat-NOL3
proteins were stained with nuclear and cytosolic markers, DAPI and
Alexa Fluor 488-conjugated secondary antibody, respectively. As
shown in Fig. 1B, con-NOL3
proteins were undetectable in the HT22 cells. Differing from the
con-NOL3 proteins, the transduced Tat-NOL3 proteins were mainly
detected in the cytoplasm by fluorescence microscopy. We also
evaluated the permeability of Tat-NOL3 proteins and con-NOL3
proteins in the hippocampal neuronal HT22 cells. Tat-NOL3 protein
transduction into the HT22 cells gradually increased in a
dose-(1–14 µM) and time-dependent (10–60 min) manner
(Fig. 1C). In addition,
intracellular stability of transduced Tat-NOL3 protein existed in
the cells for a maximum of 60 h (Fig.
1D).
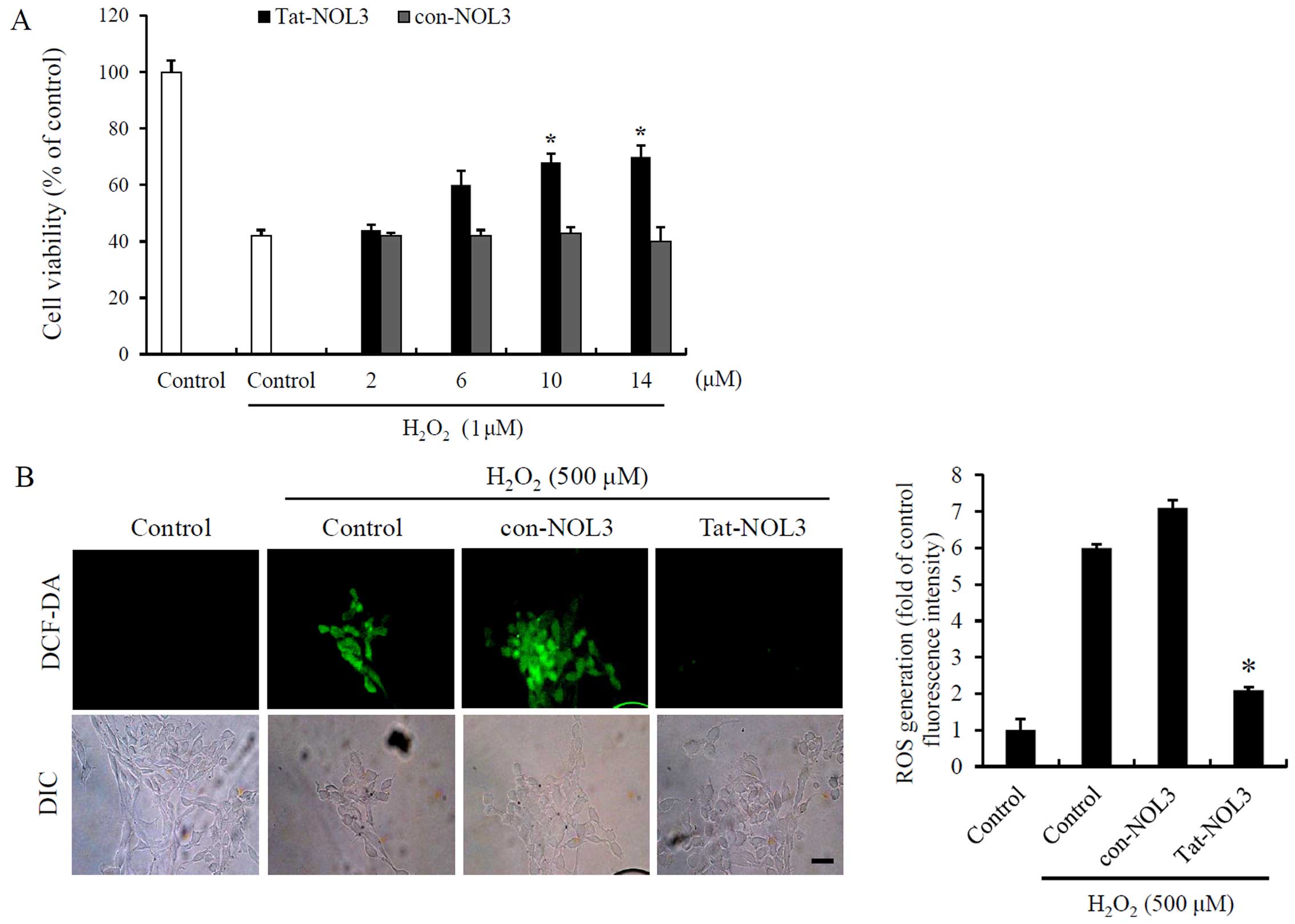
Tat-NOL3 protein increases cell viability
and inhibits ROS-induced DNA fragmentation and mitochondrial
dysfunction in HT22 cells
Ischemia-reperfusion is associated with ROS
generation in tissues (37). To
determine the effects of Tat-NOL3 protein on
H2O2-exposed HT22 cells, we treated the HT22
cells with Tat-NOL3 or con-NOL3 prior to H2O2
exposure. We then measured the ability of Tat-NOL3 protein to
inhibit H2O2-induced cell death and ROS
generation. The cell viability of the
H2O2-treated cells was 42%. However, cell
viability increased in a dose-dependent manner up to 70% in the
cells treated with Tat-NOL3 protein whereas con-NOL3 protein did
not exert a protective effect against
H2O2-induced cell death (Fig. 2A). In addition, we measured the
degree of ROS generation using fluorescence microscopy. In the
H2O2-and con-NOL3 protein-treated HT22 cells,
fluorescence signals were strongly detected. However, the
fluorescence signals were markedly decreased in the Tat-NOL3
protein treated cells (Fig. 2B).
These results indicate that transduced Tat-NOL3 prevents cell death
caused by H2O2-induced ROS production.
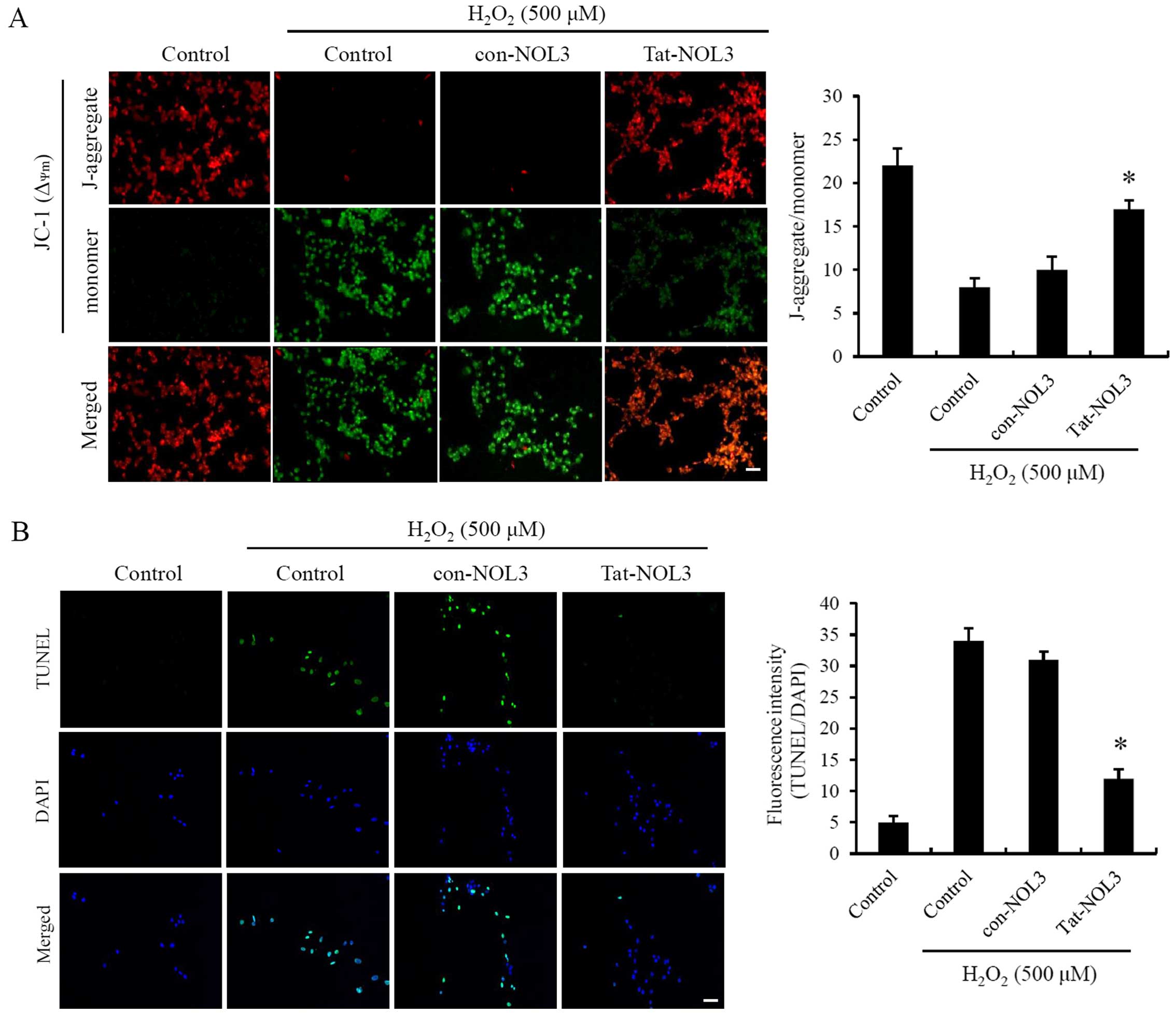
Mitochondria are a major cellular source of ROS
production. Ischemia-reperfusion is known to induce changes in
ΔΨm resulting in mitochondrial dysfunction (3,9).
To examine whether Tat-NOL3 protein exerts an effect on
ΔΨm, the HT22 cells were exposed to
H2O2. As shown in Fig. 3A, ΔΨm changes in the
cells treated with con-NOL3 protein were strongly induced following
exposure to H2O2 whereas transduced Tat-NOL3
protein protected against ΔΨm changes in the
H2O2-exposed HT22 cells and ΔΨm
changes were similar to those observed in the normal control group.
Thus, it seems reasonable to conclude that transduced Tat-NOL3
protein plays a role in protecting cells against mitochondrial
dysfunction.
ROS production contributes to apoptotic cell death
and DNA fragmentation after ischemia-reperfusion injury (38,39). Tat-NOL3 protein suppressed
ROS-induced mitochondrial dysfunction in the HT22 cells. Consistent
with this, we determined the effect of Tat-NOL3 protein on
H2O2-induced DNA fragmentation in the HT22
cells by performing a TUNEL assay. TUNEL-positive cells were
detected in the H2O2-treated control group
and the con-NOL3 protein-treated group. However, TUNEL-positive
cells were only slightly detected in the Tat-NOL3 protein-treated
group (Fig. 3B). These results
indicate that Tat-NOL3 inhibits ROS-induced DNA fragmentation in
HT22 cells.
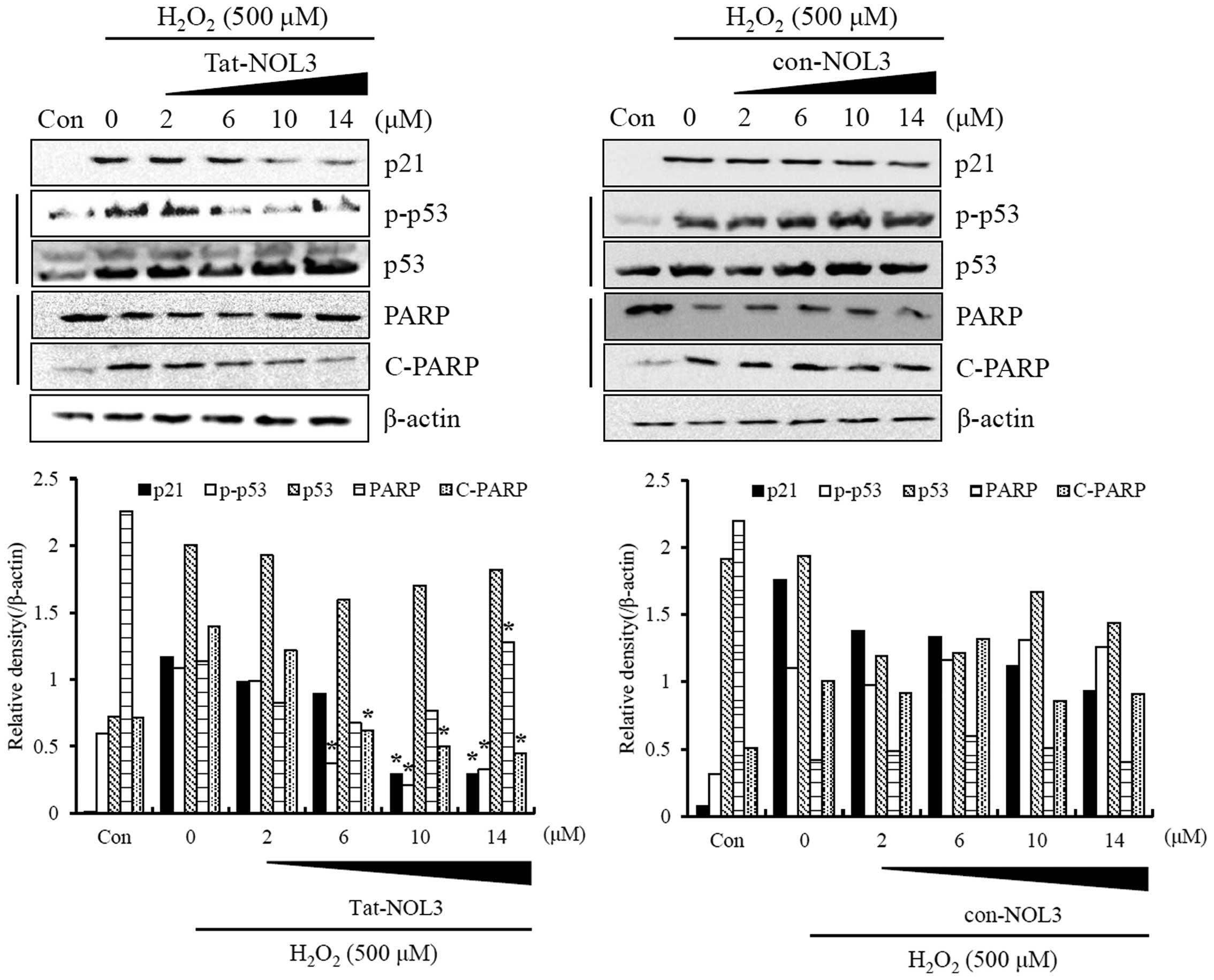
The activation of p53 by elevated ROS levels elicits
cell cycle arrest or apoptosis (40). Oxidative DNA damage, which may
result in DNA fragmentation causing cells to undergo cycle arrest
followed by apoptotic cell death (38). It has been demonstrated that
excessive ROS levels induce increased levels of
p21cip1/waf1 (p21) and decreased levels of cleaved-PARP
in a p53 dose-dependent manner (41). Thus, we examined the effect of
Tat-NOL3 protein on p53 function in
H2O2-stimulated HT22 cells. Tat-NOL3 protein
inhibited the phosphorylation of p53 in the presence of
H2O2, whereas con-NOL3 protein demonstrated
no effect on H2O2-stimulated p53
phosphorylation. However, Tat-NOL3 protein did not change p53
expression levels in the H2O2-exposed HT22
cells (Fig. 4). In addition, we
examined the correlation between Tat-NOL3 protein and oxidative DNA
damage in H2O2-exposed HT22 cells. With
gradually increasing doses of Tat-NOL3 protein, the levels of p21
and cleaved-PARP markedly decreased whereas PARP levels increased.
However, con-NOL3 protein did not demonstrate the same effects on
p21 and PARP expression levels (Fig.
4). These findings suggest that Tat-NOL3 inhibited p53-induced
oxidative DNA damage through the regulation of p21 and PARP
expression levels in the HT22 cells.
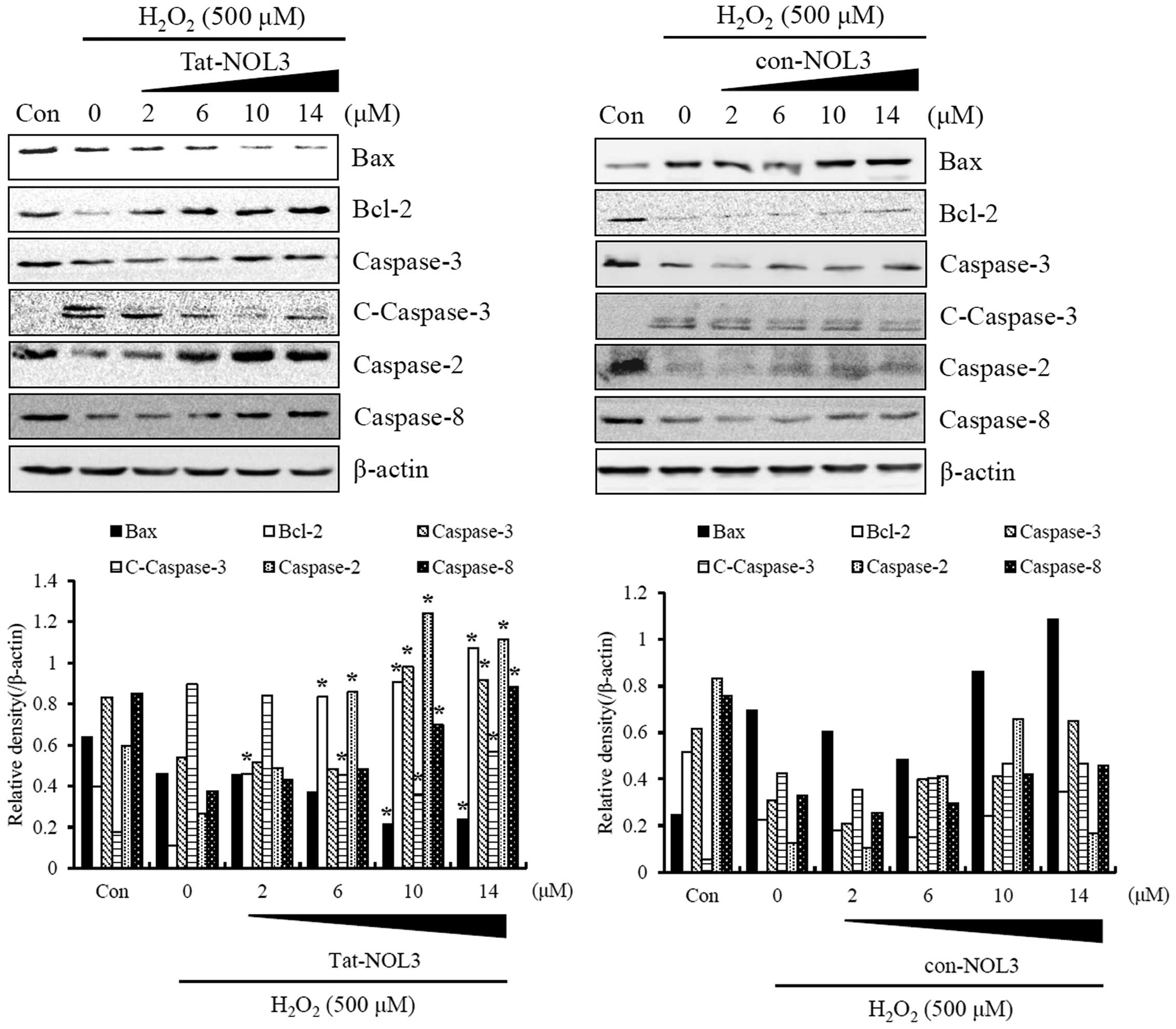
Tat-NOL3 protein inhibits ROS-induced
apoptosis in HT22 cells
ROS are associated with the caspase-dependent
apoptotic pathways and H2O2 is known to be a
potent pro-apoptotic stimulus (42). To examine the effect of Tat-NOL3
protein on ROS-induced apoptosis in HT22 cells, the cells were
transiently exposed to 0.5 mM H2O2 for 20 min
and the expression of Bax, Bcl-2, caspase-2 and -8, cleaved
caspase-3, and caspase-3 were detected using western blot analysis.
The expression levels of Bax and cleaved-caspase-3 were increased
whereas the expression levels of Bcl-2, caspase-2 and -3 were
decreased by H2O2. Notably, transduced
Tat-NOL3 protein markedly decreased the expression levels of Bax
and cleaved caspase-3, whereas it increased the expression levels
of Bcl-2, caspase-3 and -8. However, con-NOL3 protein did not
affect the expression of apoptosis-related proteins in the
H2O2-exposed HT22 cells (Fig. 5). These results indicate that
H2O2 acts as pro-apoptotic stimulus, whereas
transduced Tat-NOL3 protein selectively suppressed both ROS-induced
intrinsic and extrinsic apoptotic pathways through the
downregulation of pro-apoptotic proteins and the upregulation of
anti-apoptotic proteins.
Transduced Tat-NOL3 protects against
neuronal cell death in a model of ischemic injury
Ischemia-reperfusion results in a decrease of
neurons in the CA1 region of the hippo-campus (43). NeuN is used as a marker for
neurons (44). Thus, we examined
whether transduced Tat-NOL3 protein protects against ischemic
injury-induced neuronal cell death in the hippocampal CA1 regions.
Cell viability was determined by immunohistochemistry using a His
antibody and NeuN stains. As shown in Fig. 6, transduced Tat-NOL3 protein
significantly inhibited neuronal cell death in the CA1 regions
compared with the vehicle-, Tat peptide-, and con-NOL3-treated
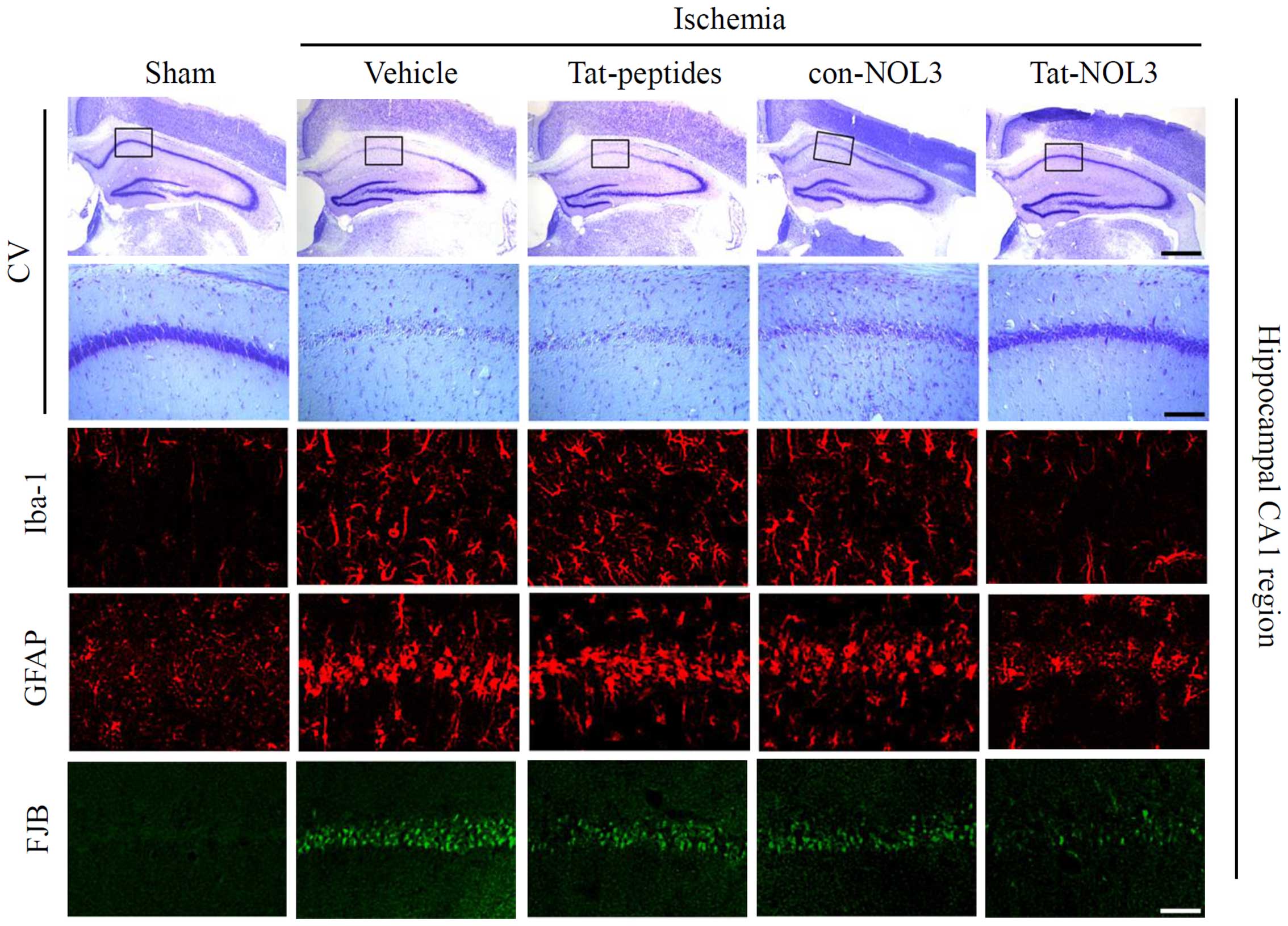
groups. Consistent with these results, CV staining also revealed
that transduced Tat-NOL3 protein markedly increased neuronal cell
viability in the hippocampal CA1 region. However, Tat peptide- and
con-NOL3 protein-treated groups did not show the same effect on
neuronal cell viability and showed similar levels to the vehicle
control group (Fig. 7).
Iba-1 is expressed in microglia and may be
associated with the activation of microglia in the ischemic brain
(45). Neuronal injury was
stained and detected using FJB. GFAP is a specific marker for
astrocytes, which are one of the predominant types of glial cells
(46). Thus, we also performed
Iba-1, GFAP, and FJB staining in the hippocampal CA1 regions of the
animal brains (Fig. 7). In the
vehicle-, con-NOL3 protein-, and Tat peptide-treated groups, Iba-1,
GFAP, and FJB fluorescence was strongly detected. However, Iba-1,
GFAP, and FJB fluorescence were significantly decreased by
transduced Tat-NOL3 protein. These results indicate that ischemic
injury induced microglial activation (Iba-1), astrocyte
accumulation (GFAP), and neuronal injury (FJB) in the CA1 region of
the hippocampus. However, transduced Tat-NOL3 protein protected
against neuronal cell death resulting from ischemic injury in the
hippocampal CA1 region by reducing the activation of astrocytes and
microglia.
Discussion
Ischemia-reperfusion injury causes neuronal death in
the hippocampal CA1 region through oxidative stress-induced
apoptosis. Oxidative stress is associated with mitochondrial
dysfunction and increased ROS production resulting from the
ischemic cascade initiated due to oxygen reperfusion following
ischemia (47).
Ischemia-reperfusion triggers two apoptotic pathways. The intrinsic
mitochondrial pathway causes the activation of Bcl-2 and the
translocation of Bax into mitochondria which results in the release
of cytochrome c and the activation of caspase-3 (48). The extrinsic apoptotic pathway is
mediated by the activation of cell surface death receptors which
activate caspase-8 and subsequently activate caspase-3 (49). It is important to note that p53 is
involved in both the intrinsic and the extrinsic apoptotic pathways
(50).
Excessive ROS are generated from mitochondria and
are also stimuli of the intrinsic mitochondrial pathway (3), whereas the effect of intracellular
ROS on the extrinsic apoptotic pathway remains unclear. Notably,
recent evidence suggests that ROS activate sphingomyelinase which
is responsible for the generation of ceramide that is involved in
the induction of Fas receptor and Fas ligand. Moreover, ROS
sensitize cancer cells to TRAIL-induced apoptosis (5,20).
This implies that ROS may affect not only the intrinsic
mitochondrial pathway but also the extrinsic apoptotic pathway. In
this study, we examined whether transduced Tat-NOL3 protein plays a
role in the intrinsic and extrinsic apoptotic pathways under
conditions of oxidative stress.
To transduce NOL3 protein into hippocampal neuronal
HT22 cells as well as gerbil brains in a model of forebrain
ischemia, we prepared Tat-NOL3. Tat-NOL3 protein transduced into
the HT22 cells in a time- and dose-dependent manner. To examine the
effects of Tat-NOL3 protein on stress-induced HT22 cell death, we
exposed the cells to H2O2. The
H2O2-exposed HT22 cells exhibited
intracellular ROS accumulation and disruption of ΔΨm
leading to intrinsic mitochondrial apoptosis. However, the
transduction of Tat-NOL3 protein into the HT22 cells exposed to
H2O2, resulted in reduced levels of
intracellular ROS and recovery of ΔΨm to normal levels,
suggesting that Tat-NOL3 protein inhibits mitochondrial
dysfunction. Although the exact mechanisms responsible for the
protective effects of Tat-NOL3 in
H2O2-induced neuronal cell death warrant
further study, these results indicate that Tat-NOL3 may control
complex cascades of H2O2 production that are
mediated by the inhibition of intrinsic mitochondrial apoptosis.
Consistent with this observation, the transduction of Tat-NOL3
protein to the cytosol of the H2O2-exposed
HT22 cells resulted in decreased DNA fragmentation and the
inactivation of p53. Additionally, the expression levels of
oxidative DNA damage-related proteins, p21cip1/waf1 and
cleaved PARP, were regulated by Tat-NOL3 protein. These results
indicate that Tat-NOL3 inhibited the
H2O2-mediated loss of p21 and cleavage of
PARP.
Next, we examined the interplay between Tat-NOL3
protein and the intrinsic and extrinsic apoptotic pathways through
interactions with caspases and apoptosis-related proteins.
Transduced Tat-NOL3 protein protected against the oxidative-stress
induced activation of the intrinsic apoptotic pathway through the
inhibition of mitochondrial damage and the regulation of pro- or
anti-apoptotic protein expression including Bax and Bcl-2 protein.
Tat-NOL3 protein increased anti-apoptotic Bcl-2 protein expression
levels whereas pro-apoptotic protein Bax expression levels were
reduced by the inhibition of excessive ROS generation. Furthermore,
Tat-NOL3 protein inhibited the extrinsic apoptotic pathway by
activating the caspase cascade including caspase-3.
Apoptotic cell death is initiated through the
extrinsic and intrinsic pathways. Previous research has shown that
NOL3 inhibits the extrinsic pathway by activating caspase-8 while
activating the intrinsic pathway through the regulation of Bax,
p53, and mitochondrial responses to stress stimuli (51). Although further study is merited
in order to understand the precise mechanism, other studies have
suggested that NOL3 protects against cell death by regulating the
extrinsic and intrinsic apoptosis pathways through a
multifunctional mechanism (52).
In agreement with the other studies, the results of this study
demonstrate that transduced Tat-NOL3 protein protected against
oxidative stress-induced cell death by inhibiting the intrinsic and
extrinsic apoptotic pathways and suggest that transduced Tat-NOL3
protein inhibited H2O2-induced neuronal cell
death through the regulation of apoptotic signal pathways.
The ischemia-reperfusion model leads to the delayed
degeneration of pyramidal neurons in the CA1 region of the
hippocampus (53). In previous
studies, we demonstrated that transduced PTD fusion proteins
protect against delayed pyramidal neuronal degeneration in the
hippocampal CA1 region in an animal model of ischemia (23,33). Thus, the protective effects of
Tat-NOL3 protein against ischemic damage were determined by
immunohistochemistry. To confirm that transduced Tat-NOL3 protein
protects against neuronal damage in an animal model of ischemia,
Tat-NOL3 proteins were intraperitoneally administered 30 min after
the induction of ischemia. Seven days after ischemia, the
protective effects of Tat-NOL3 proteins were confirmed by CV and
NeuN immunohistochemistry. Neuronal cell viability was
significantly increased in the hippocampal CA1 regions of the
Tat-NOL3 protein-treated group. These results indicate that
Tat-NOL3 protein transduced into the CA1 region of the animal brain
and protected against neuronal cell death. We also demonstrated
that CA1 pyramidal neurons were protected against ischemic damage
in the Tat-NOL3 protein-treated groups. Activated astrocytes and
microglia were significantly decreased in the Tat-NOL3
protein-treated groups compared with the vehicle-treated groups.
These results indicate that transduced Tat-NOL3 protein protected
against ischemic damage by reducing the activation of astrocytes
and microglia. Thus, we suggest that transduced Tat-NOL3 protein
may be a useful agent for preventing ischemic damage.
In conclusion, we demonstrated that Tat-NOL3
protein transduced into HT22 cells and protected against neuronal
cell death induced by oxidative stress through the regulation of
apoptotic signaling pathways. In addition, Tat-NOL3 protein
markedly protected against neuronal cell death in the CA1 region of
the hippocampus by reducing the activation of astrocytes and
microglia. Thus, we suggest that Tat-NOL3 protein may be a
therapeutic agent against ischemia and oxidative stress-induced
neuronal cell death.
Acknowledgments
The present study was supported by a Priority
Research Centers Program grant (no. 2009-0093812) through the
National Research Foundation of Korea funded by the Ministry of
Science, ICT and Future Planning, and in part by the BioGreen21
Program (no. PJ01121401) of the Rural Development Administration,
Republic of Korea, and by a Hallym University Research Fund (no.
HRF-G-2015-2).
References
|
1
|
Smith JA, Park S, Krause JS and Banik NL:
Oxidative stress, DNA damage, and the telomeric complex as
therapeutic targets in acute neurodegeneration. Neurochem Int.
62:764–775. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Simonian NA and Coyle JT: Oxidative stress
in neurodegenerative diseases. Annu Rev Pharmacol Toxicol.
36:83–106. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Niizuma K, Endo H and Chan PH: Oxidative
stress and mitochondrial dysfunction as determinants of ischemic
neuronal death and survival. J Neurochem. 109(Suppl 1): 133–138.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schaller B and Graf R: Cerebral ischemia
and reperfusion: the pathophysiologic concept as a basis for
clinical therapy. J Cereb Blood Flow Metab. 24:351–371. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
D'Autréaux B and Toledano MB: ROS as
signalling molecules: mechanisms that generate specificity in ROS
homeostasis. Nat Rev Mol Cell Biol. 8:813–824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mronga T, Stahnke T, Goldbaum O and
Richter-Landsberg C: Mitochondrial pathway is involved in
hydrogen-peroxide-induced apoptotic cell death of oligodendrocytes.
Glia. 46:446–455. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang YQ and Herman B: ARC protects rat
cardiomyocytes against oxidative stress through inhibition of
caspase-2 mediated mitochondrial pathway. J Cell Biochem.
99:575–588. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang JX, Li Q and Li PF: Apoptosis
repressor with caspase recruitment domain contributes to
chemotherapy resistance by abolishing mitochondrial fission
mediated by dynamin-related protein-1. Cancer Res. 69:492–500.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lejay A, Meyer A, Schlagowski AI, Charles
AL, Singh F, Bouitbir J, Pottecher J, Chakfé N, Zoll J and Geny B:
Mitochondria: mitochondrial participation in ischemia-reperfusion
injury in skeletal muscle. Int J Biochem Cell Biol. 50:101–105.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ow YP, Green DR, Hao Z and Mak TW:
Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol.
9:532–542. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li J and Yuan J: Caspases in apoptosis and
beyond. Oncogene. 27:6194–6206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Norbury CJ and Zhivotovsky B: DNA
damage-induced apoptosis. Oncogene. 23:2797–2808. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhivotovsky B and Orrenius S: Caspase-2
function in response to DNA damage. Biochem Biophys Res Commun.
331:859–867. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Neuss M, Monticone R, Lundberg MS, Chesley
AT, Fleck E and Crow MT: The apoptotic regulatory protein ARC
(apoptosis repressor with caspase recruitment domain) prevents
oxidant stress-mediated cell death by preserving mitochondrial
function. J Biol Chem. 276:33915–33922. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Razorenova OV, Castellini L, Colavitti R,
Edgington LE, Nicolau M, Huang X, Bedogni B, Mills EM, Bogyo M and
Giaccia AJ: The apoptosis repressor with a CARD domain (ARC) gene
is a direct hypoxia-inducible factor 1 target gene and promotes
survival and proliferation of VHL-deficient renal cancer cells. Mol
Cell Biol. 34:739–751. 2014. View Article : Google Scholar :
|
|
16
|
Embury J, Klein D, Pileggi A, Ribeiro M,
Jayaraman S, Molano RD, Fraker C, Kenyon N, Ricordi C, Inverardi L
and Pastori RL: Proteins linked to a protein transduction domain
efficiently transduce pancreatic islets. Diabetes. 50:1706–1713.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wadia JS and Dowdy SF: Protein
transduction technology. Curr Opin Biotechnol. 13:52–56. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kubo E, Fatma N, Akagi Y, Beier DR, Singh
SP and Singh DP: TAT-mediated PRDX6 protein transduction protects
against eye lens epithelial cell death and delays lens opacity. Am
J Physiol Cell Physiol. 294:C842–C855. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dietz GP: Cell-penetrating peptide
technology to deliver chaperones and associated factors in diseases
and basic research. Curr Pharm Biotechnol. 11:167–174. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim DW, Lee SH, Jeong MS, Sohn EJ, Kim MJ,
Jeong HJ, An JJ, Jang SH, Won MH and Hwang IK: Transduced Tat-SAG
fusion protein protects against oxidative stress and brain ischemic
insult. Free Radic Biol Med. 48:969–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
van den Berg A and Dowdy SF: Protein
transduction domain delivery of therapeutic macromolecules. Curr
Opin Biotechnol. 22:888–893. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim DW, Lee SH, Ku SK, Lee JE, Cha HJ,
Youn JK, Kwon HY, Park JH, Park EY, Cho SW, et al: The effects of
PEP-1-FK506BP on dry eye disease in a rat model. BMB Rep.
48:153–158. 2015. View Article : Google Scholar :
|
|
23
|
Shin MJ, Kim DW, Lee YP, Ahn EH, Jo HS,
Kim DS, Kwon OS, Kang TC, Cho YJ, Park J, et al: Tat-glyoxalase
protein inhibits against ischemic neuronal cell damage and
ameliorates ischemic injury. Free Radic Biol Med. 67:195–210. 2014.
View Article : Google Scholar
|
|
24
|
Eom SA, Kim DW, Shin MJ, Ahn EH, Chung SY,
Sohn EJ, Jo HS, Jeon SJ, Kim DS, Kwon HY, et al: Protective effects
of PEP-1-Catalase on stress-induced cellular toxicity and
MPTP-induced Parkinson's disease. BMB Rep. 48:395–400. 2015.
View Article : Google Scholar :
|
|
25
|
Kim HR, Kim DW, Jo HS, Cho SB, Park JH,
Lee CH, Choi YJ, Yeo EJ, Park SY, Kim ST, et al: Tat-biliverdin
reductase A inhibits inflammatory response by regulation of MAPK
and NF-κB pathways in Raw 264.7 cells and edema mouse model. Mol
Immunol. 63:355–366. 2015. View Article : Google Scholar
|
|
26
|
Kwon HY, Eum WS, Jang HW, Kang JH, Ryu J,
Ryong Lee B, Jin LH, Park J and Choi SY: Transduction of
Cu,Zn-superoxide dismutase mediated by an HIV-1 Tat protein basic
domain into mammalian cells. FEBS Lett. 485:163–167. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ju SM, Youn GS, Cho YS, Choi SY and Park
J: Celastrol ameliorates cytokine toxicity and pro-inflammatory
immune responses by suppressing NF-κB activation in RINm5F beta
cells. BMB Rep. 48:172–177. 2015. View Article : Google Scholar :
|
|
29
|
Shehzad A, Lee J and Lee YS: Autocrine
prostaglandin E2 signaling promotes promonocytic
leukemia cell survival via COX-2 expression and MAPK pathway. BMB
Rep. 48:109–114. 2015. View Article : Google Scholar :
|
|
30
|
Park G and Oh MS: Acceleration of heat
shock-induced collagen breakdown in human dermal fibroblasts with
knockdown of NF-E2-related factor 2. BMB Rep. 48:467–472. 2015.
View Article : Google Scholar :
|
|
31
|
Im CN and Seo JS: Overexpression of tumor
necrosis factor receptor-associated protein 1 (TRAP1), leads to
mitochondrial aberrations in mouse fibroblast NIH/3T3 cells. BMB
Rep. 47:280–285. 2014. View Article : Google Scholar :
|
|
32
|
Lee YJ, Lee YJ and Lee SH: Resveratrol and
clofarabine induces a preferential apoptosis-activating effect on
malignant mesothelioma cells by Mcl-1 down-regulation and caspase-3
activation. BMB Rep. 48:166–171. 2015. View Article : Google Scholar :
|
|
33
|
Kim YN, Jung HY, Eum WS, Kim DW, Shin MJ,
Ahn EH, Kim SJ, Lee CH, Yong JI, Ryu EJ, et al: Neuroprotective
effects of PEP-1-carbonyl reductase 1 against
oxidative-stress-induced ischemic neuronal cell damage. Free Radic
Biol Med. 69:181–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee SJ and Park JW: Enhancement of UVB
radiation-mediated apoptosis by knockdown of cytosolic
NADP+-dependent isocitrate dehydrogenase in HaCaT cells.
BMB Rep. 47:209–214. 2014. View Article : Google Scholar :
|
|
35
|
Hwang IK, Yoo KY, Kim DW, Lee CH, Choi JH,
Kwon YG, Kim YM, Choi SY and Won MH: Changes in the expression of
mitochondrial peroxiredoxin and thioredoxin in neurons and glia and
their protective effects in experimental cerebral ischemic damage.
Free Radic Biol Med. 48:1242–1251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jeong HJ, Yoo DY, Kim DW, Yeo HJ, Cho SB,
Hyeon J, Park JH, Park J, Eum WS, Hwang HS, et al: Neuroprotective
effect of PEP-1-peroxiredoxin2 on CA1 regions in the hippocampus
against ischemic insult. Biochim Biophys Acta. 1840:2321–2330.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Choi DW: Ischemia-induced neuronal
apoptosis. Curr Opin Neurobiol. 6:667–672. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
MacManus JP, Buchan AM, Hill IE, Rasquinha
I and Preston E: Global ischemia can cause DNA fragmentation
indicative of apoptosis in rat brain. Neurosci Lett. 164:89–92.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Zhang Z and Yan H: Simvastatin
inhibits ischemia/reperfusion injury-induced apoptosis of retinal
cells via downregulation of the tumor necrosis factor-α/nuclear
factor-κB pathway. Int J Mol Med. 36:399–405. 2015.PubMed/NCBI
|
|
40
|
Fussenegger M and Bailey JE: Molecular
regulation of cell-cycle progression and apoptosis in mammalian
cells: implications for biotechnology. Biotechnol Prog. 14:807–833.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Martinez LA, Yang J, Vazquez ES,
Rodriguez-Vargas MC, Olive M, Hsieh JT, Logothetis CJ and Navone
NM: p21 modulates threshold of apoptosis induced by DNA-damage and
growth factor withdrawal in prostate cancer cells. Carcinogenesis.
23:1289–1296. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gough DR and Cotter TG: Hydrogen peroxide:
a Jekyll and Hyde signalling molecule. Cell Death Dis. 2:e213–e218.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hara K, Yasuhara T, Matsukawa N, Maki M,
Masuda T, Yu G, Xu L, Tambrallo L, Rodriguez NA, Stern DM, et al:
Hippocampal CA1 cell loss in a non-human primate model of transient
global ischemia: A pilot study. Brain Res Bull. 74:164–171. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schmued LC and Hopkins KJ: Fluoro-Jade B:
a high affinity fluorescent marker for the localization of neuronal
degeneration. Brain Res. 874:123–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Broughton BRS, Reutens DC and Sobey CG:
Apoptotic mechanisms after cerebral ischemia. Stroke. 40:e331–e339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rossi DJ, Brady JD and Mohr C: Astrocyte
metabolism and signaling during brain ischemia. Nat Neurosci.
10:1377–1386. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chong ZZ, Li F and Maiese K: Oxidative
stress in the brain: novel cellular targets that govern survival
during neurodegenerative disease. Prog Neurobiol. 75:207–246. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Swanton E, Savory P, Cosulich S, Clarke P
and Woodman P: Bcl-2 regulates a caspase-3/caspase-2 apoptotic
cascade in cytosolic extracts. Oncogene. 18:1781–1787. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li YZ, Lu DY, Tan WQ, Wang JX and Li PF:
p53 initiates apoptosis by transcriptionally targeting the
antiapoptotic protein ARC. Mol Cell Biol. 28:564–574. 2008.
View Article : Google Scholar :
|
|
51
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wu L, Nam YJ, Kung G, Crow MT and Kitsis
RN: Induction of the apoptosis inhibitor ARC by Ras in human
cancers. J Biol Chem. 285:19235–19245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Konaka K, Ueda H, Li JY, Matsumoto M,
Sakoda S and Yanagihara T: N-acetylaspartate to total creatine
ratio in the hippocampal CA1 sector after transient cerebral
ischemia in gerbils: influence of neuronal elements, reactive
gliosis, and tissue atrophy. J Cereb Blood Flow Metab. 23:700–708.
2003. View Article : Google Scholar : PubMed/NCBI
|