Introduction
The common pathological pathway of heart failure is
myocardial remodeling, including the hypertrophy of cardiomyocytes
and hyperplasia of fibroblasts that subsequently causes the
accumulation of extracellular matrix components, such as collagen
and leads to myocardial fibrosis (1). Severe fibrosis results in increased
myocardial stiffness, causing ventricular dysfunction and,
ultimately, heart failure (2). No
effective treatment for cardiac fibrosis has yet been established
(3).
Isoproterenol, a selective β-adrenoceptor agonist,
has been used to induce a mouse model of cardiac fibrosis (4). Adrenergic stimulation induces
cardiac fibrosis partly by stimulating fibroblast proliferation
(5). Nitric oxide (NO) produced
by endothelial NO synthase (eNOS) inhibits adrenergic stimulation
(6). Human eNOS gene delivery has
been shown to protect against cardiac remodeling following
myocardial infarction (7).
However, the effect of eNOS activation on isoproterenol-induced
myocardial fibrosis is not fully understood.
Intracellular calcium and calmodulin-dependent
protein kinase play an important role in the activation of eNOS
(8,9). Changes in the concentration of free
cytosolic Ca2+ are of fundamental importance in eNOS
activation (10). The transient
receptor potential (TRP) superfamily consists of a large number of
cation channels that are modulators of intracellular
Ca2+ signaling (11).
TRP vanilloid subtype 1 (TRPV1), a non-selective cation channel
with significant permeability to Ca2+, was initially
identified as the receptor of capsaicin, with high expression in
sensory neurons and heart cells, including cardiac fibroblasts
(12). Although TRPV1 has been
shown to be involved in the regulation of several types of
cardiovascular diseases, including hypertension, atherosclerosis
and cardiac arrhythmias (13),
the role of TRPV1 in isoproterenol-induced cardiac fibrosis has not
been yet elucidated.
Collectively, we hypothesized that TRPV1 may
regulate cardiac fibrosis by regulating Ca2+ entry and
the eNOS/NO signaling pathway. The present study was designed to
determine whether the transgenic overexpression of TRPV1 affects
isoproterenol-induced cardiac fibrosis in mice.
Materials and methods
Transgenic mice
The handling of animals and all experimental
procedures were approved by the Institutional Animal Care and Use
Committee of the Chengdu Military General Hospital. Transgenic
founder mice were generated on a C57BL/6J background according to
our previous study (14). The
cDNA of TRPV1 driven by the elongation factor 1α (EF1α) promoter
was cloned into the pDONR221 vector (Cyagen Biosciences Inc.,
Guangzhou, China). The resulting plasmid, pDown-TRPV1, was
confirmed by restriction enzyme analysis and DNA sequencing. The
linearized pRP.EX3d-TRPV1 was purified from agarose gel using a
QIAquick Gel Extraction kit (Qiagen, Chatsworth, CA, USA), adjusted
to a final concen tration of 1 mg/ml in Tris-EDTA buffer and used
as a DNA solution for microinjection. The fertilized one-cell eggs
from hormonally superovulated female C57BL/6J mice (from Cyagen
Biosciences Inc.; n=20) were micro-injected with the DNA solution
under a microscope. The injected fertilized eggs were transplanted
into the oviducts of pseudo-pregnant mice (foster mothers; C57BL/6J
mice), which gave birth to offspring. The offspring mice carrying
the TRPV1 cDNA detected by PCR analysis were considered the
transgenic founder mice.
Animal care
Male C57BL/6J wild-type mice were purchased from
Dashuo Biotech Inc., Chengdu, China. The transgenic mice were mated
with the C57BL/6J wild-type mice, and their offspring were
genotyped to obtain the transgenic and wild-type mice for the
experiments. Male C57BL/6J wild-type and TRPV1 transgenic mice 6–8
weeks of age were housed under a 12/12-h day/night cycle, with
access to food and water ad libitum. The mice were randomly
assigned to the following groups: i) the control group (n=16): mice
received a daily injection of 0.9% saline subcutaneously; ii) the
isoproterenol group (n=16): mice received 3 mg/kg isoproterenol
subcutaneously once daily, as previously described (15); and iii) the isoproterenol plus
Nω-nitro-L-arginine methyl ester (L-NAME) (Sigma-Aldrich,
St. Louis, MO, USA) group (n=16): mice received L-NAME (1 mg/ml) in
their drinking water, as previously described (16). At 2 weeks after the treatment, the
mice were sacrificed by cervical dislocation under deep anesthesia
with pentobarbital (60 mg/kg) and the hearts of 8 mice from each
group were harvested and weighed to calculate the heart/body weight
ratio. The atrial tissue and right ventricle were then removed from
the hearts to obtain the left ventricle for calculating the left
ventricular/body weight ratio and for further measurements.
Additionally, the hearts from the other 8 mice were used for
histological examination.
Measurement of blood pressure (BP)
Indirect systolic and diastolic BP and heart rate
measurements were performed in conscious, restrained mice by
tail-cuff plethysmography (BP-2010A blood pressure system; Softron
Biotechnology, Beijing, China).
Hemodynamics
The mice were anesthetized with urethane (1.2 g/kg,
intraperitoneally). A microconductance pressure catheter (ARIA
SPR-853; Millar Instruments, Inc., Houston, TX, USA) was introduced
through the right carotid artery into the left ventricle as
previously described (15). Data
were collected on Chart via PowerLab (AD Instruments Pty, Ltd.,
Castle Hill, Australia). The left ventricular end-systolic pressure
(LVESP), left ventricular end-diastolic pressure (LVEDP) and left
ventricular dP/dtmax/min were calculated.
Histological analysis
The hearts were excised and retrograde-perfused with
phosphate-buffered saline and embedded in paraffin. The
paraffin-embedded hearts were cut into 5-µm-thick sections
which were stained with Masson's trichrome. The blue-stained area
presents fibrosis (15).
Culture of cardiac fibroblasts
Primary adult mouse cardiac fibroblasts were
isolated by proteolytic dissociation of the ventricular tissue of
the transgenic mice and the wild-type littermates and cultured
using standard protocols as previously described (15). Cardiac fibroblasts were harvested
and cultured in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% FBS, 2 mM L-glutamine, 100 µg/ml
penicillin G, 100 µg/ml streptomycin and 100 U/ml
penicillin. Cardiac fibroblasts during early passages were used.
One day before the experiments, the cells were serum-starved. TRPV1
expression in the cultured cardiac fibroblast was detected by
immunohistochemical staining with primary anti-TRPV1 antibody
(1:200 dilution) (ab111973; Abcam, Cambridge, UK) and stained using
a DAB detection kit. The cardiac fibroblasts were incubated with
the vehicle (saline), 10 µM isoproterenol or isoproterenol
plus 100 µM L-NAME for 24 h.
Measurement of cytosolic calcium
The cytosolic free calcium concentration was
measured in the cultured fibroblasts as previously described
(17). Briefly, the cells were
loaded with 2 µM of fura2/AM (Sigma-Aldrich) at room
temperature for 60 min and washed to remove the extraneous dye.
Fluorescence was recorded at 510 nm emission with excitation
wavelengths of 340 and 380 nm in baseline and following stimulation
with capsaicin (100 nM) and the fluorescence excitation ratio was
then calculated.
Western blot analysis
Protein lysates were obtained by homogenizing
myocardial tissues or cardiac fibroblasts with lysis buffer
according to our previous study (18). The protein concentration was
determined with Bio-Rad protein assay reagent (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Equal amounts of protein
extracts were separated by SDS-polyacrylamide gels (8–12%). The
samples were then transferred onto PVDF membranes (Millipore Corp.,
Billerica, MA, USA). The membranes were blocked with TBS-T
containing 5% skim powdered milk for 1 h and then incubated with
anti-TRPV1 (1:500 dilution; ab74855) (Abcam), anti-collagen I
(1:250 dilution; sc-8784), anti-collagen III (1:300 dilution;
sc-8781), anti-fibronectin (1:300 dilution; sc-9068) (all from
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
anti-phospho-Akt (1:800 dilution; 9271), anti-Akt (1:700 dilution;
9272), anti-phospho-eNOS (1:1,000 dilution; 9571), anti-eNOS
(1:1,000 dilution; 9572) (all from Cell Signaling Technology, Inc.,
Danvers, MA, USA), and anti-GAPDH (1:200; BA2913) (Boster, Wuhan,
China) antibodies overnight at 4°C. The membranes were incubated
with a horseradish peroxidase-conjugated secondary antibody
(1:1,000 dilution) (Santa Cruz Biotechnology, Inc.). The membranes
were then analyzed using an ECL chemiluminescence kit (Amersham
Biosciences, Uppsala, Sweden). The results of western blot analysis
were quantified using NIH Image software version 1.61. Values are
expressed as relative value units.
Real-time polymerase chain reaction
(RT-PCR)
RT-PCR was performed using the One Step SYBR
PrimeScript RT-PCR kit II (RR086A) (Takara Bio, Inc., Otsu, Japan).
The relative amount of mRNA was calculated by 2−ΔΔCT and
was normalized to the housekeeping gene, GAPDH. Each sample was run
and analyzed in triplicate. The sequences of the primers used for
PCR were as follows: mouse collagen I forward, 5′-gcg agt gct gtg
ctt tct g-3′ and reverse, 5′-tcc ctc gac tcc tac atc ttc-3′; mouse
collagen III forward, 5′-ccc aac cca gag atc cca tt-3′ and reverse,
5′-gaa gca cag gag cag gtg tag a-3′; mouse fibronectin forward,
5′-tct ggg aaa tgg aaa agg gga atg g-3′ and reverse, 5′-cac tga agc
agg ttt cct cgg tgt-3′; and mouse GAPDH forward, 5′-gac atc aag aag
gtg gtg aag c-3′ and reverse, 5′-gaa ggt gga aga gtg gga
gtt-3′.
Measurements of cardiac NO and cyclic
guanosine monophosphate (cGMP) levels
The frozen left ventricles excised from the mice and
the cardiac fibroblasts were homogenized in lysis buffer for the
measurements of NO and cGMP levels. The NO levels were determined
using the Total Nitrate/Nitrite Fluorometric assay kit, and the
cGMP levels were quantified using the acetylation protocol for a
competitive cGMP enzyme immunoassay (both from Cayman Chemical Co.,
Ann Arbor, MI, USA).
Cell proliferation assay
Cell proliferation was measured using a CCK-8 cell
proliferation kit (Dojindo Laboratories, Kumamoto, Japan) as
previously described (19). The
cells were seeded onto a 96-well plate with 100 µl complete
medium and cultured at 37°C. CCK-8 solution (10 µl) was
added to each well. The plates were incubated at 37°C for 2 h, and
the absorbance at 450 nm was then measured using a microplate
reader (Multiskan MK3-Thermo labsystems; Thermo Fisher Scientific,
Waltham, MA, USA).
Statistical analysis
Data are presented as the means ± SEM. Comparisons
between groups were determined by one-way ANOVA with the Student's
t-test post hoc test (SPSS Inc., Chicago, IL, USA). Results were
considered significant when the P-value was <0.05.
Results
Establishment of transgenic mice
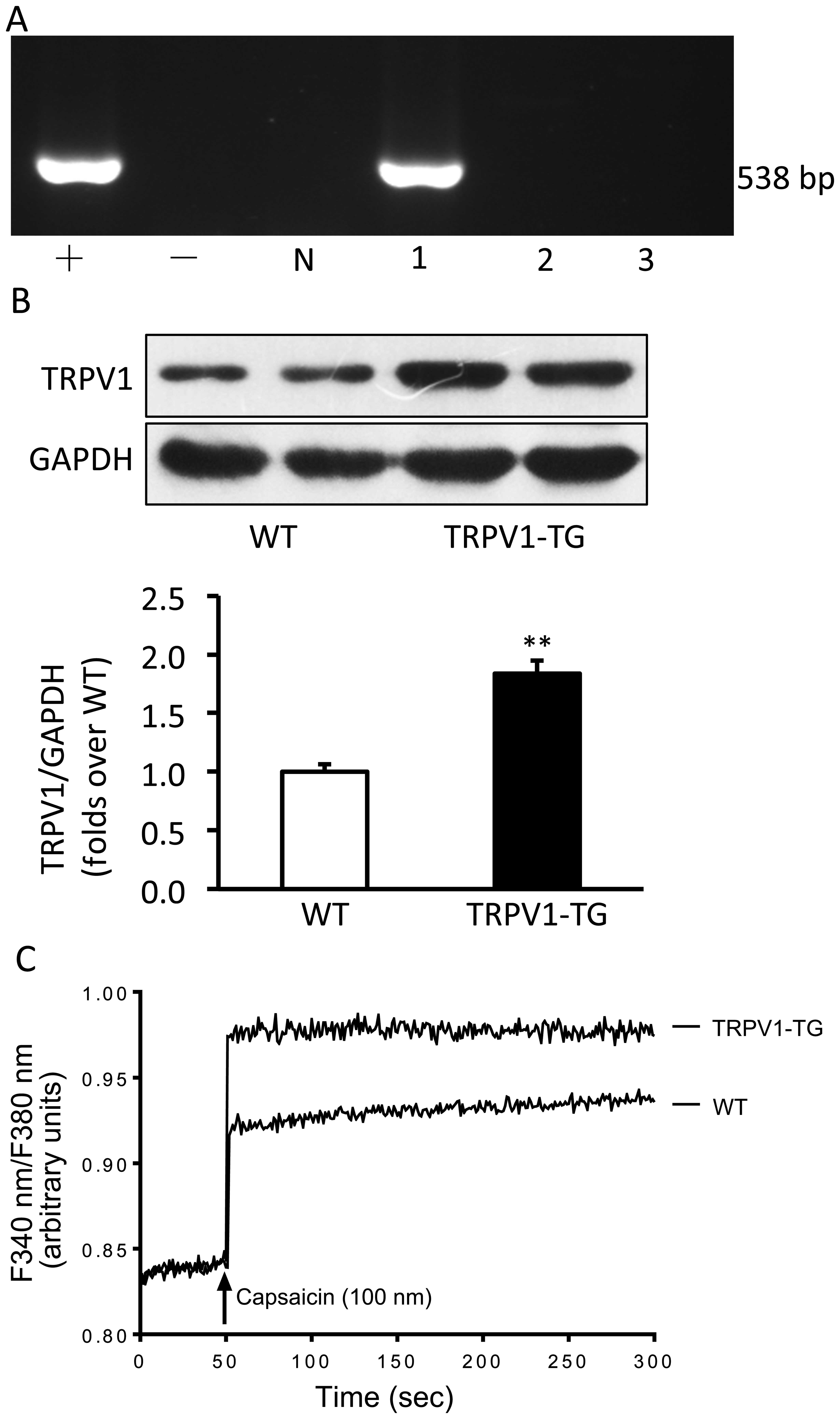
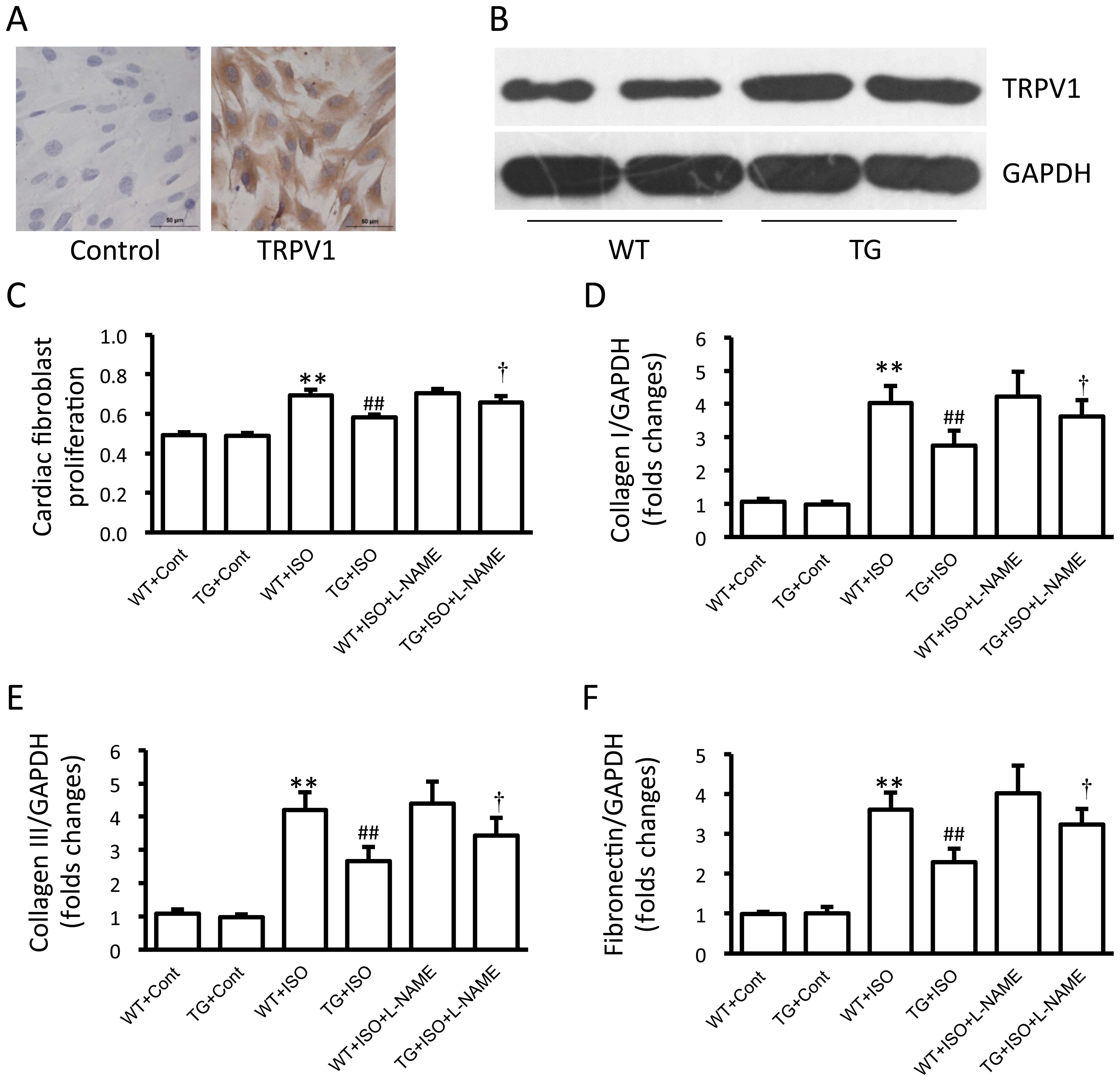
Human TRPV1 cDNA was micro-injected into the male
pronuclei of fertilized oocytes of C57BL/6J mice. The injected eggs
were implanted into the oviducts of pseudo-pregnant foster mothers,
which gave birth to offspring. The offspring mice carrying the
TRPV1 cDNA detected by PCR analysis were considered the transgenic
founder mice (Fig. 1A). Western
blot analysis confirmed that TRPV1 protein was significantly
overexpressed in the hearts of the transgenic mice compared with
those of the wild-type mice (p<0.01; Fig. 1B). Additionally, calcium entry
induced by capsaicin was also enhanced in the cardiac fibroblasts
from the TRPV1 transgenic mice compared with those from the
wild-type mice (Fig. 1C).
Effect of TRPV1 overexpression on left
ventricular function
At 2 weeks, there were no significant differences
observed in heart weight and hemodynamics between the wild-type
mice and the transgenic mice (Table
I). The wild-type mice exhibited a significant increase in
heart/body weight ratio and left ventricle/body weight ratio at 2
weeks following the administration of isoproterenol (both
p<0.01; Table I). However,
this increase was significantly attenuated in the TRPV1 transgenic
mice when compared to the wild-type mice (both p<0.01; Table I). As expected, the
isoproterenol-treated wild-type mice exhibited a significant
increase in LVEDP (p<0.01) and a marked decrease in
dP/dtmin (p<0.01); these effects were blunted in the
TRPV1 transgenic mice (Table I).
However, isoproterenol failed to affect LVESP and
dP/dtmax in both types of mice (Table I).
 | Table IHeart weight and hemodynamics of the
mice at 2 weeks. |
Table I
Heart weight and hemodynamics of the
mice at 2 weeks.
| Parameter | WT-Cont | TG-Cont | WT-ISO | TG-ISO | WT-ISO-L-NAME | TG-ISO-L-NAME |
|---|
| HW/BW (mg/g) | 4.96±0.08 | 4.98±0.04 | 6.59±0.23a | 5.67±0.21b | 6.65±0.19 | 6.29±0.20c |
| LVW/BW (mg/g) | 3.47±0.07 | 3.44±0.06 | 4.93±0.23a | 4.12±0.18b | 4.97±0.18 | 4.69±0.19c |
| HR (bpm) | 449±12 | 451±14 | 552±13a | 546±14 | 560±16 | 557±17 |
| SBP (mmHg) | 105±4 | 104±5 | 111±4 | 108±5 | 115±6 | 115±7 |
| DBP (mmHg) | 71±2 | 69±3 | 74±2 | 72±4 | 75±4 | 75±4 |
| MAP (mmHg) | 82±3 | 81±3 | 86±3 | 84±4 | 89±4 | 88±5 |
| LVESP (mmHg) | 107.6±4.4 | 106.5±5.0 | 112.2±4.8 | 116.8±7.3 | 117.1±7.9 | 119.6±7.4 |
| LVEDP (mmHg) | 2.8±0.4 | 2.7±0.3 | 9.4±0.9a | 5.6±0.6b | 9.9±1.2 | 8.1±1.2c |
| dP/dtmax
(mmHg/sec) | 11889±685 | 12174±745 | 12834±1030 | 12335±751 | 12740±950 | 12188±635 |
| dP/dtmin
(mmHg/sec) | −11301±645 | −11551±731 | −7416±359a | −9547±480b | −7259±335 | −7933±398c |
Effect of TRPV1 overexpression on cardiac
fibrosis
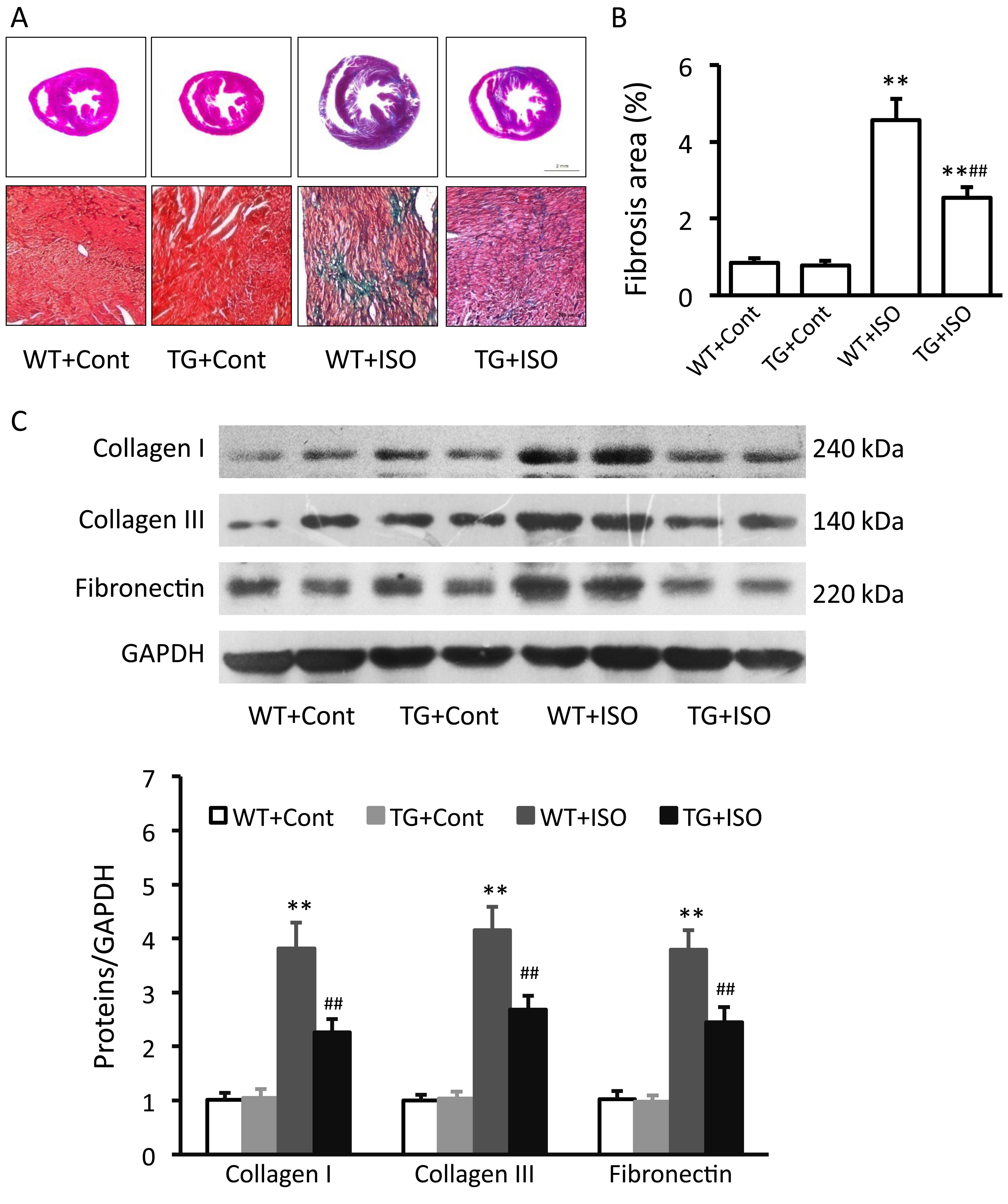
Masson trichrome staining revealed severe
interstitial fibrosis within the myocardium of the
isoproterenol-treated wild-type mice, while isoproterenol-induced
myocardial fibrosis was obviously attenuated in the TRPV1
transgenic mice (Fig. 2A and B).
Isoproterenol induced a significant increase in the protein
expression levels of collagen I and III, as well as in the
expression of fibronectin in the heart tissues from the wild-type
mice (all p<0.01; Fig. 2C).
However, this increase was significantly prevented in the TRPV1
transgenic mice (all p<0.01; Fig.
2C).
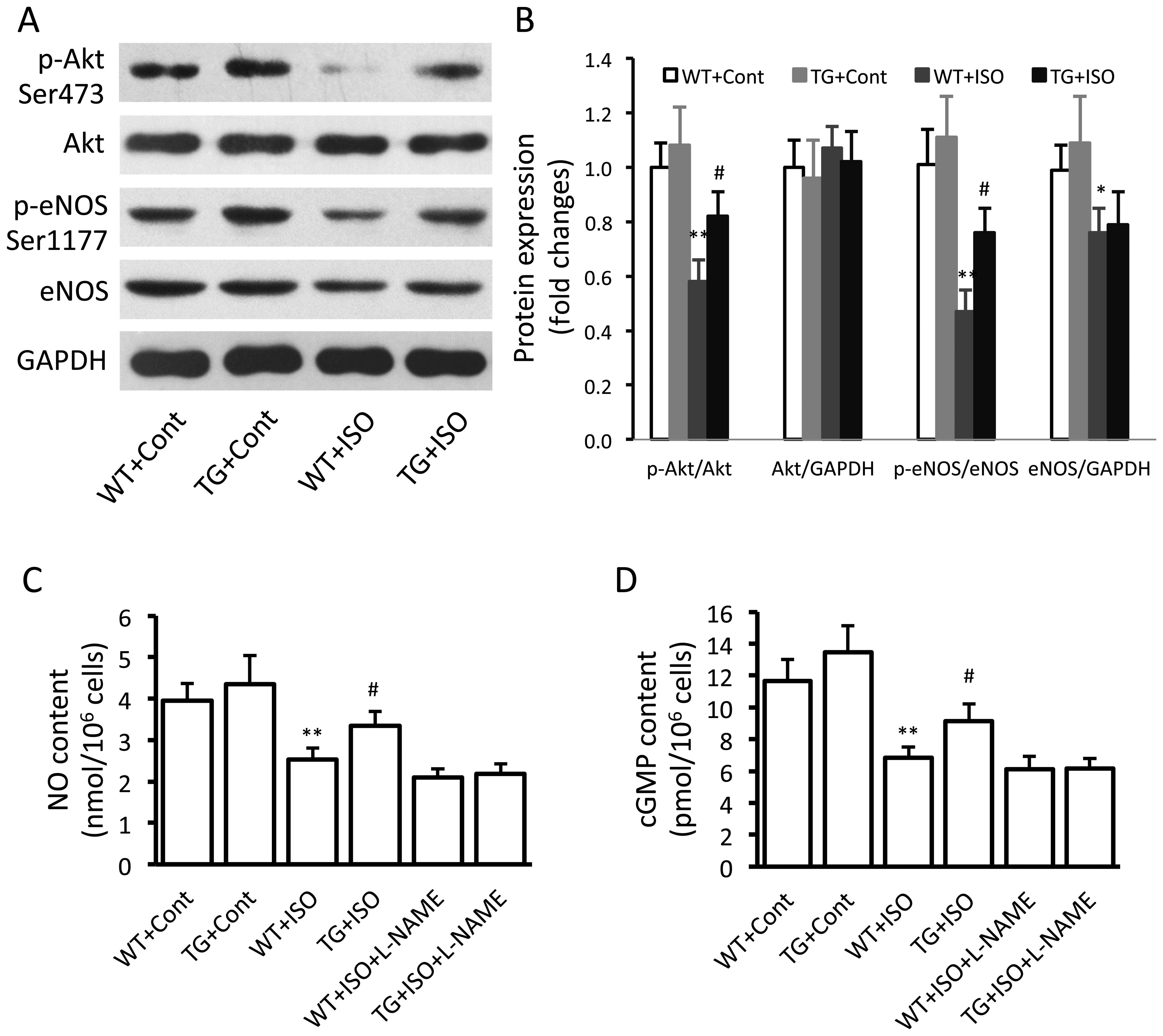
Effect of TRPV1 overexpression on the
eNOS signaling pathway
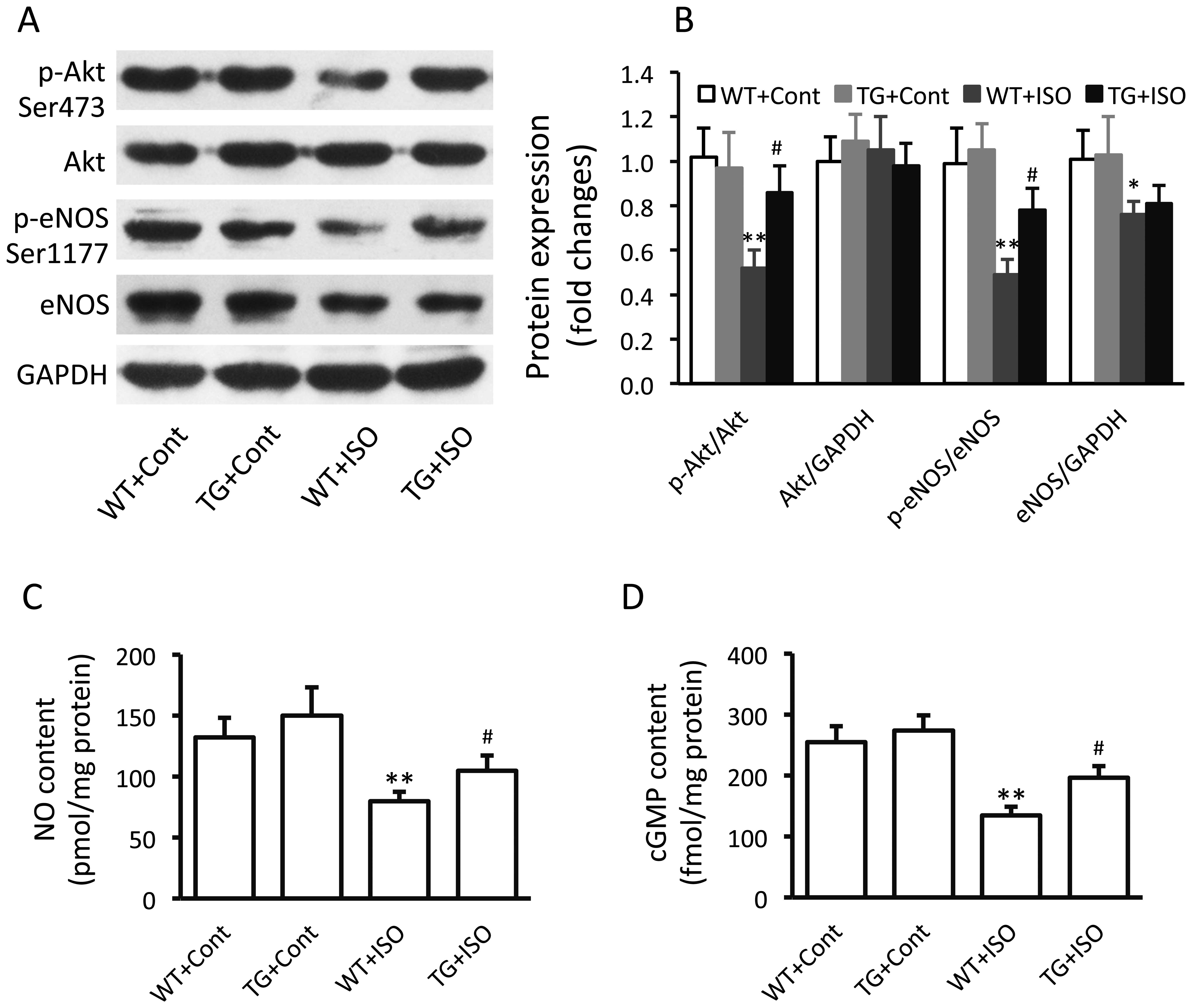
In the isoproterenol-treated wild-type mice,
significantly decreased expression levels of p-Akt (p<0.01),
p-eNOS (p<0.01) and total eNOS (p<0.05) were noted in the
ventricular tissues (Fig. 3A and
B). However, the levels of p-Akt and p-eNOS were significantly
increased in the TRPV1 transgenic mice compared with the wild-type
mice following the administration of isoproterenol (Fig. 3B). Similar results were obtained
in the measurements of the myocardial NO and cGMP levels (Fig. 3C and D).
Influence of eNOS on the beneficial
effects of TRPV1 overexpression
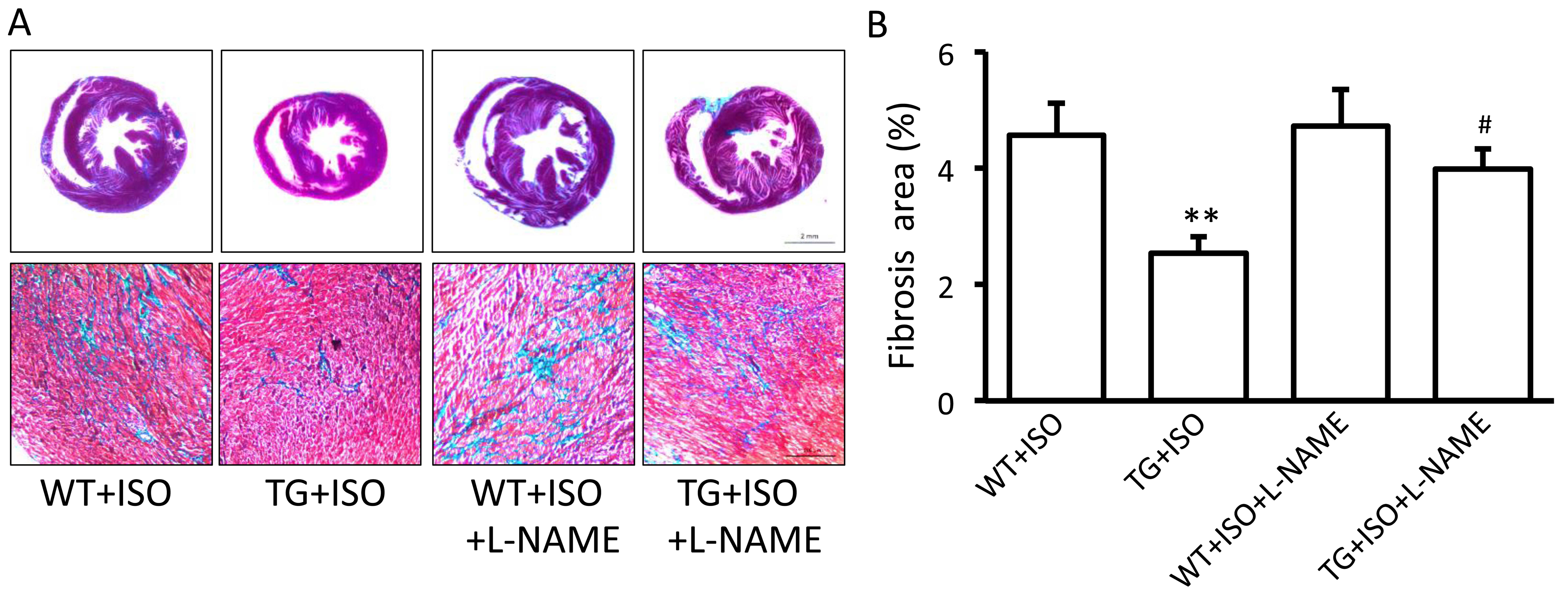
In order to confirm the role of eNOS in the
beneficial effects induced by TRPV1 overexpression, the eNOS
inhibitor, L-NAME, was administered to the mice receiving
isoproterenol. We found that the administration of L-NAME for 2
weeks significantly attenuated the TRPV1 overexpression-induced
decrease in LVEDP (Table I) and
the increase in dP/dtmin (Table I). Moreover, L-NAME also
attenuated the TRPV1 overexpression-induced suppression of fibrosis
(Fig. 4A and B).
Analysis of cardiac fibroblasts
The expression of TRPV1 in the cultured cardiac
fibroblasts was confirmed by immunohistochemical staining (Fig. 5A). Moreover, TRPV1 expression was
markedly increased in the cardiac fibroblasts from the transgenic
mice compared with those from the wild-type mice (Fig. 5B). Treatment with isoproterenol
induced a significant increase in cell proliferation and in the
expression of collagen I and III, and fibronectin in the cardiac
fibroblasts from the wild-type mice, and these effects were
markedly attenuated in the cardiac fibroblasts from the transgenic
mice (Fig. 5C–F). Additionally,
treatment with L-NAME attenuated the TRPV1 overexpression-induced
suppression of fibroblast proliferation and the expression of
collagen I and III and fibronectin (Fig. 5C–F). Isoproterenol induced a
significant downregulation in the levels of p-Akt and p-eNOS and a
decrease in the levels of NO and cGMP in the cardiac fibroblasts
from wild-type mice, and these effects were partially reversed in
the cells from the transgenic mice (Fig. 6A–D). Similarly, L-NAME abolished
the TRPV1 overexpression-caused increase in the NO and cGMP levels
(Fig. 6C and D).
Discussion
There are three novel findings in the present study:
firstly, the transgenic overexpression of TRPV1 attenuated the
development of cardiac fibrosis induced by isoproterenol. Secondly,
the overexpression of TRPV1 attenuated the isoproterenol-induced
impairment of the eNOS/NO/cGMP pathway in myocardial tissue and
cardiac fibroblasts. Thirdly, the beneficial effects of TRPV1 on
fibrosis and collagen deposition were blunted by the administration
of the eNOS inhibitor, L-NAME. These results suggest that TRPV1
overexpression may be able to ameliorate isoproterenol-induce
myocardial fibrosis partly through the activation of the eNOS/NO
pathway in cardiac fibroblasts.
TRPV1 has been implicated in several types of
cardiovascular diseases and metabolic disorders, including
hypertension (20),
atherosclerosis (21), vascular
dysfunction (22), diabetes
(23) and obesity (24). The beneficial effects of TRPV1 on
ischemia reperfusion injury and on myocardial remodeling and
dysfunction have been previously reported by our group and others
(19,25–29). Moreover, TRPV1 has also been shown
involved in low ambient temperature-induced cardiac hypertrophy and
contractile dysfunction and high-salt diet-induced cardiac
hypertrophy and fibrosis (30,31). However, there are also a few
studies that have demonstrated that TRPV1 may have harmful effects
on cardiac hypertrophy and fibrosis using TRPV1-deficient mice
(32,33). Therefore, the role of TRPV1 in
cardiac hypertrophy and fibrosis has yet to be determined. The
present study demonstrated that the overexpression of TRPV1
attenuates isoproterenol-induced cardiac fibrosis. TRPV1 is
expressed in several types of cells in the cardiovascular system,
including cardiomyocytes, cardiac fibroblasts and endothelial cells
(17,30,34). We confirmed the expression of
TRPV1 in cardiac fibroblasts and found that the overexpression of
TRPV1 exerted anti-fibrotic effects in cell culture experiments.
However, we cannot confirm the anti-fibrotic effects induced by
TRPV1 overexpression in other types of cells or other organs, since
we used global transgenic mice. Moreover, we also cannot rule out
the effects of other calcium channels which may be regulated by
human TRPV1 overexpression.
The eNOS/NO system provides an ubiquitous protection
of the cardiovascular system. It has been reported that
isoproterenol reduces the expression and phosphorylation of eNOS
(35). The activation of the
eNOS/NO pathway plays a beneficial role in either angiotensin II-
or isoproterenol-induced cardiac disorders (36–39). Previous studies have demonstrated
that Ca2+ signal is essential for the phosphorylation
and activation of eNOS. TRP channels, as Ca2+-permeable
channels, play a beneficial role in cardiovascular diseases.
Moreover, the TRPV1 channel has been included in the activation of
eNOS in several previous studies (40–43). The present study demonstrated that
TRPV1 overexpression increased the phosphorylation of eNOS in the
myocardium from isoproterenol-treated mice. We also showed that
L-NAME suppressed the beneficial effects of TRPV1 overexpression on
fibrosis. A previous study demonstrated that L-NAME enhanced
cardiac fibrosis (44). However,
L-NAME failed to further increase the fibrosis in
isoproterenol-treated mice, suggesting that both L-NAME and
isoproterenol may cause fibrosis through the inhibition of eNOS
activity.
Collectively, the present study demonstrates that
the overexpression of TRPV1 ameliorates isoproterenol-induced
cardiac fibrosis and that fibroblasts may play a role in this
anti-fibrotic effect.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81170081 awarded
to D.Y.), the Military Medical Science Youth Development Project
(no. 13QNP058 awarded to Q.W.), the Sichuan Scientific and
Technical Innovation Development Project (no. 2015016 awarded to
Q.W.), the Chengdu Military Health Academic Leader Foundation
(awarded to D.Y.), and the Foundation for Talents of Scientific
Research from Chengdu Military General Hospital (awarded to
D.Y.).
References
|
1
|
Cohn JN, Ferrari R and Sharpe N: Cardiac
remodeling - concepts and clinical implications: a consensus paper
from an international forum on cardiac remodeling. Behalf of an
International Forum on Cardiac Remodeling. J Am Coll Cardiol.
35:569–582. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Biernacka A and Frangogiannis NG: aging
and cardiac fibrosis. Aging Dis. 2:158–173. 2011.PubMed/NCBI
|
|
3
|
Leask A: Potential therapeutic targets for
cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF,
partners in fibroblast activation. Circ Res. 106:1675–1680. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Osadchii OE: Cardiac hypertrophy induced
by sustained beta-adrenoreceptor activation: pathophysiological
aspects. Heart Fail Rev. 12:66–86. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nichtova Z, Novotova M, Kralova E and
Stankovicova T: Morphological and functional characteristics of
models of experimental myocardial injury induced by isoproterenol.
Gen Physiol Biophys. 31:141–151. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ozaki M, Kawashima S, Yamashita T, Hirase
T, Ohashi Y, Inoue N, Hirata K and Yokoyama M: Overexpression of
endothelial nitric oxide synthase attenuates cardiac hypertrophy
induced by chronic isoproterenol infusion. Circ J. 66:851–856.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Smith RS Jr, Agata J, Xia CF, Chao L and
Chao J: Human endothelial nitric oxide synthase gene delivery
protects against cardiac remodeling and reduces oxidative stress
after myocardial infarction. Life Sci. 76:2457–2471. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park JY, Shin HK, Choi YW, Lee YJ, Bae SS,
Han J and Kim CD: Gomisin A induces Ca2+-dependent
activation of eNOS in human coronary artery endothelial cells. J
Ethnopharmacol. 125:291–296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Park JH, Lee S, Cho DH, Park YM, Kang DH
and Jo I: Far-infrared radiation acutely increases nitric oxide
production by increasing Ca(2+) mobilization and
Ca(2+)/calmodulin-dependent protein kinase II-mediated
phosphorylation of endothelial nitric oxide synthase at serine
1179. Biochem Biophys Res Commun. 436:601–606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gail MH, Boone CW and Thompson CS: A
calcium requirement for fibroblast motility and prolifertion. Exp
Cell Res. 79:386–390. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gees M, Colsoul B and Nilius B: The role
of transient receptor potential cation channels in Ca2+
signaling. Cold Spring Harb Perspect Biol. 2:a0039622010.
View Article : Google Scholar
|
|
12
|
Thilo F, Liu Y, Schulz N, Gergs U, Neumann
J, Loddenkemper C, Gollasch M and Tepel M: Increased transient
receptor potential vanilloid type 1 (TRPV1) channel expression in
hypertrophic heart. Biochem Biophys Res Commun. 401:98–103. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marshall NJ, Liang L, Bodkin J,
Dessapt-Baradez C, Nandi M, Collot-Teixeira S, Smillie SJ, Lalgi K,
Fernandes ES, Gnudi L and Brain SD: A role for TRPV1 in influencing
the onset of cardiovascular disease in obesity. Hypertension.
61:246–252. 2013. View Article : Google Scholar
|
|
14
|
Ma S, Li D, Yang D, Tan Y, Tang B, Jin F,
Jiang S, Li X and Yang Y: Establishment of a conditional transgenic
mouse model expressing human uncoupling protein 2 in vascular
smooth muscle cells. Exp Ther Med. 4:545–547. 2012.PubMed/NCBI
|
|
15
|
Ma S, Yang D, Wang K, Tang B, Li D and
Yang Y: Cryptotanshinone attenuates isoprenaline-induced cardiac
fibrosis in mice associated with upregulation and activation of
matrix metal-loproteinase-2. Mol Med Rep. 6:145–150.
2012.PubMed/NCBI
|
|
16
|
Mukai Y, Rikitake Y, Shiojima I, Wolfrum
S, Satoh M, Takeshita K, Hiroi Y, Salomone S, Kim HH, Benjamin LE,
et al: Decreased vascular lesion formation in mice with inducible
endothelial-specific expression of protein kinase Akt. J Clin
Invest. 116:334–343. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang D, Luo Z, Ma S, Wong WT, Ma L, Zhong
J, He H, Zhao Z, Cao T, Yan Z, et al: Activation of TRPV1 by
dietary capsaicin improves endothelium-dependent vasorelaxation and
prevents hypertension. Cell Metab. 12:130–141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma S, Yang D, Li D, Tang B, Sun M and Yang
Y: Cardiac extracellular matrix tenascin-C deposition during
fibronectin degradation. Biochem Biophys Res Commun. 409:321–327.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Q, Ma S, Li D, Zhang Y, Tang B, Qiu
C, Yang Y and Yang D: Dietary capsaicin ameliorates pressure
overload-induced cardiac hypertrophy and fibrosis through the
transient receptor potential vanilloid type 1. Am J Hypertens.
27:1521–1529. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Andresen MC and Peters JH: TRPV1,
hypertension, and cardiovascular regulation. Cell Metab.
12:421author reply 422. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao JF, Ching LC, Kou YR, Lin SJ, Wei J,
Shyue SK and Lee TS: Activation of TRPV1 prevents OxLDL-induced
lipid accumulation and TNF-α-induced inflammation in macrophages:
role of liver X receptor α. Mediators Inflamm. 2013:9251712013.
View Article : Google Scholar
|
|
22
|
Ohanyan VA, Guarini G, Thodeti CK,
Talasila PK, Raman P, Haney RM, Meszaros JG, Damron DS and Bratz
IN: Endothelin-mediated in vivo pressor responses following TRPV1
activation. Am J Physiol Heart Circ Physiol. 301:H1135–H1142. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Suri A and Szallasi A: The emerging role
of TRPV1 in diabetes and obesity. Trends Pharmacol Sci. 29:29–36.
2008. View Article : Google Scholar
|
|
24
|
Motter AL and Ahern GP: TRPV1-null mice
are protected from diet-induced obesity. FEBS Lett. 582:2257–2262.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang L and Wang DH: TRPV1 gene knockout
impairs postischemic recovery in isolated perfused heart in mice.
Circulation. 112:3617–3623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhong B and Wang DH: TRPV1 gene knockout
impairs preconditioning protection against myocardial injury in
isolated perfused hearts in mice. Am J Physiol Heart Circ Physiol.
293:H1791–H1798. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang W, Rubinstein J, Prieto AR, Thang LV
and Wang DH: Transient receptor potential vanilloid gene deletion
exacerbates inflammation and atypical cardiac remodeling after
myocardial infarction. Hypertension. 53:243–250. 2009. View Article : Google Scholar
|
|
28
|
Huang W, Rubinstein J, Prieto AR and Wang
DH: Enhanced postmyocardial infarction fibrosis via stimulation of
the transforming growth factor-beta-Smad2 signaling pathway: role
of transient receptor potential vanilloid type 1 channels. J
Hypertens. 28:367–376. 2010. View Article : Google Scholar
|
|
29
|
Ren JY, Song JX, Lu MY and Chen H:
Cardioprotection by ischemic postconditioning is lost in isolated
perfused heart from diabetic rats: involvement of transient
receptor potential vanilloid 1, calcitonin gene-related peptide and
substance P. Regul Pept. 169:49–57. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y, Li L, Hua Y, Nunn JM, Dong F,
Yanagisawa M and Ren J: Cardiac-specific knockout of ET(A) receptor
mitigates low ambient temperature-induced cardiac hypertrophy and
contractile dysfunction. J Mol Cell Biol. 4:97–107. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao F, Liang Y, Wang X, Lu Z, Li L, Zhu S,
Liu D, Yan Z and Zhu Z: TRPV1 activation attenuates high-salt
diet-induced cardiac hypertrophy and fibrosis through PPAR-δ
upregulation. PPAR Res. 2014:4919632014. View Article : Google Scholar
|
|
32
|
Buckley CL and Stokes AJ: Mice lacking
functional TRPV1 are protected from pressure overload cardiac
hypertrophy. Channels (Austin). 5:367–374. 2011. View Article : Google Scholar :
|
|
33
|
Horton JS, Buckley CL and Stokes AJ:
Successful TRPV1 antagonist treatment for cardiac hypertrophy and
heart failure in mice. Channels (Austin). 7:17–22. 2013. View Article : Google Scholar
|
|
34
|
Ge W, Yuan M, Ceylan AF, Wang X and Ren J:
Mitochondrial aldehyde dehydrogenase protects against doxorubicin
cardiotoxicity through a transient receptor potential channel
vanilloid 1-mediated mechanism. Biochim Biophys Acta. 1862:622–634.
2016. View Article : Google Scholar
|
|
35
|
Bhuiyan MS, Shioda N and Fukunaga K:
Chronic beta-AR activation-induced calpain activation and impaired
eNOS-Akt signaling mediates cardiac injury in ovariectomized female
rats. Expert Opin Ther Targets. 13:275–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Belge C, Hammond J, Dubois-Deruy E,
Manoury B, Hamelet J, Beauloye C, Markl A, Pouleur AC, Bertrand L,
Esfahani H, et al: Enhanced expression of β3-adrenoceptors in
cardiac myocytes attenuates neurohormone-induced hypertrophic
remodeling through nitric oxide synthase. Circulation. 129:451–462.
2014. View Article : Google Scholar
|
|
37
|
Savergnini SQ, Ianzer D, Carvalho MB,
Ferreira AJ, Silva GA, Marques FD, Peluso AA, Beiman M, Cojocaru G,
Cohen Y, et al: The novel Mas agonist, CGEN-856S, attenuates
isoproterenol-induced cardiac remodeling and myocardial infarction
injury in rats. PLoS One. 8:e577572013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bharti S, Singh R, Chauhan SS, Hussain T,
Al-Attas OS and Arya DS: Phosphorylation of Akt/GSK-3β/eNOS
amplifies 5-HT2B receptor blockade mediated anti-hypertrophic
effect in rats. FEBS Lett. 586:180–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liao Y, Asakura M, Takashima S, Ogai A,
Asano Y, Shintani Y, Minamino T, Asanuma H, Sanada S, Kim J, et al:
Celiprolol, a vasodilatory beta-blocker, inhibits pressure
overload-induced cardiac hypertrophy and prevents the transition to
heart failure via nitric oxide-dependent mechanisms in mice.
Circulation. 110:692–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ching LC, Kou YR, Shyue SK, Su KH, Wei J,
Cheng LC, Yu YB, Pan CC and Lee TS: Molecular mechanisms of
activation of endothelial nitric oxide synthase mediated by
transient receptor potential vanilloid type s1. Cardiovasc Res.
91:492–501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ching LC, Chen CY, Su KH, Hou HH, Shyue
SK, Kou YR and Lee TS: Implication of AMP-activated protein kinase
in transient receptor potential vanilloid type 1-mediated
activation of endothelial nitric oxide synthase. Mol Med.
18:805–815. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Su KH, Lin SJ, Wei J, Lee KI, Zhao JF,
Shyue SK and Lee TS: The essential role of transient receptor
potential vanilloid s1 in simvastatin-induced activation of
endothelial nitric oxide synthase and angiogenesis. Acta Physiol
(Oxf). 212:191–204. 2014. View Article : Google Scholar
|
|
43
|
Guarini G, Ohanyan VA, Kmetz JG,
DelloStritto DJ, Thoppil RJ, Thodeti CK, Meszaros JG, Damron DS and
Bratz IN: Disruption of TRPV1-mediated coupling of coronary blood
flow to cardiac metabolism in diabetic mice: role of nitric oxide
and BK channels. Am J Physiol Heart Circ Physiol. 303:H216–H223.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kazakov A, Hall R, Jagoda P, Bachelier K,
Müller-Best P, Semenov A, Lammert F, Böhm M and Laufs U: Inhibition
of endothelial nitric oxide synthase induces and enhances
myocardial fibrosis. Cardiovasc Res. 100:211–221. 2013. View Article : Google Scholar : PubMed/NCBI
|