Introduction
Tuberculosis, caused by Mycobacterium
tuberculosis (MTB), has affected one-third of the world's
population; in 9 million individuals, this has developed into
active infection and 1.5 individuals succumbed to the disease in
2013 according to WHO reports (1,2).
Notwithstanding advances in the diagnosis and treatment of
tuberculosis, this disease remains a major cause of human morbidity
and mortality due to the limitations of currently available
therapies and vaccines, and the emergence and spread of
multi-drug-resistant strains (2,3).
MTB has evolved a diverse number of strategies to evade host
defense mechanisms, particularly in macrophages, by potentiating
bacterial invasion and survival, and subverting host cell-defending
signaling pathways (4,5). The successful survival strategies
within macrophages employed by MTB include the reduction of the
acidification and maturation of phagosomes that engulf the
mycobacteria (6), preventing
phagosome-lysosome fusion (7),
blocking autophagy-mediated killing (1), inducing inflammatory responses
(8) and promoting cholesterol
accumulation (9). Among the
specific mechanisms targeted by MTB are signaling pathways mediated
by mitogen-activated protein kinases (10), phosphatidylinositol 3-kinase
(11), calcium (Ca2+)
(12) and cytokines, such as
interleukin-1 and type I interferon (IFN) (13,14).
Pathogens can exploit host genes to enter, reproduce
in or exit from host cells or to circumvent host defenses (1,15).
Therefore, adjunctive host-directed therapies have the potential to
augment cellular immune responses, to modify inflammatory processes
and to enhance tuberculosis chemotherapy (16). Decreased interleukin-1 responses
and exaggerated type I interferon production in MTB-infected mice
have been shown to be associated with an eicosanoid imbalance,
leading to an increased mortality of the mice (13). A broad range of host-directed
candidate agents, such as autophagy inducers, protein kinase
inhibitors and antimicrobial peptide inducers, have been developed
for clinical evaluation; however, none of these therapeutic agents
have been approved for clinical use yet (16). The analysis of host genetic
variations, which can alter susceptibility, may provide novel
targets for host-directed therapies (15). These genetic variations can be
identified by genome-wide gene inactivation strategies, including
the use of siRNA/shRNA, miRNA and antisense/sense RNA libraries
(17–19).
In the present study, we established an
MTB-infection-based RAW264.7 macrophage screening model. First, we
non-selectively inactivated genes using lentivirus-based RNA
screening libraries, and isolated macrophage cell clones showing
increased survival following MTB infection. DNA sequencing and
bioinformatics analysis revealed several candidate host genes and
an unsuspected role of collagen α-5(IV) chain (Col4a5), a type IV
collagen subunit present on the cell surface, in mycobacterial
infection. In addition, the analysis of the molecular mechanisms
underlying the role of Col4a5 in MTB infection revealed the ability
of this collagen to alter phagosomal acidification mediated by
vacuolar-type H+-ATPase (V-ATPase) in murine
macrophages. Our findings indicate that host-directed therapy
targeting Col4a5 may represent an alternative therapeutic approach
for combating MTB infection.
Materials and methods
MTB and mycobacterium bovis
Calmette-Guérin (BCG) cultures
The MTB strain, H37Rv, and the BCG strain, Pasteur,
were grown to early log phase in Middlebrook 7H9 broth with BBL
Middlebrook OADC (oleic acid, albumin, dextrose and catalase)
enrichment medium (BD Pharminigen, San Diego, CA, USA) and 0.1%
(vol/vol) Tween-80 (Sigma-Aldrich, St. Louis, MO, USA).
Predominantly single-cell suspensions were collected by
centrifuging twice at 125 × g for 5 min, re-suspension in
phosphate-bovine serum (PBS) with 0.1% Tween-80, and
re-centrifugation at 125 × g for 10 min, and used for the infection
of macrophages.
Screening of macrophage host genes
affecting MTB infection
Lentiviruses were produced by transiently
transfecting 293T cells [American Type Culture Collection (ATCC),
Manassas, VA, USA] using a Random Homozygous Knock Out (RHKO) or an
EST library DNA along with DNAs for the G418 (both from
Sigma-Aldrich) resistance gene, packaging and VSVG envelope
constructs, as demonstrated by us and others previously (18,20,21). RAW264.7 macrophages (ATCC) were
infected with purified lentiviruses and selected with 0.7 mg/ml
G418 (Invitrogen, Carlsbad, CA, USA). Approximately
1×107 G418-resistant clones of RAW264.7 macrophages were
cultured at approximately 80% confluency, and exposed to MTB H37Rv
at multiplicities of infection (MOI) of 2–3, 10–17 or 52–77 for 3
days, until the cells appeared dead under microscopic inspection.
MTB was then killed with streptomycin (20 µg/ml), isoniazid
(5 µg/ml) and rifampicin (5 µg/ml) (Sigma-Aldrich).
Fresh DMEM (Invitrogen) plus 10% fetal bovine serum (FBS) (Atlanta
Biologicals, Flowery Branch, GA, USA) were added to support the
re-growth of surviving cells to approximately 80% confluency and
used for re-infection. Following 3 cycles of infection and
re-growth, cell colonies appeared. Single clones of cells were
obtained by limited dilution and expansion in 96-well plates.
Single clones stably transfected with antisense constructs were
confirmed by genomic PCR with primers ESTF and ESTR, and analyzed
for inserted sequences by DNA sequencing as described by us and
others previously (20,21).
In order to verify that the phenotypes of the
isolated clones are due to the inactivation of the genes targeted
by the RHKO or EST inserts, RNAi (shRNA-incorporated) and the
vector-only control RAW264.7 macrophages with target gene-specific
knockdowns were established. Briefly, the RAW264.7 macrophages were
seeded at approximately 5% confluency in fresh DMEM plus 10% FBS,
incubated for 24 h, suspended and infected with filtered lentivirus
in 5 mg/ml polybrene at 37°C for 6–18 h at a MOI retrospectively
calculated as approximately 0.1, followed by selection in puromycin
(Invitrogen) at 2.5 µg/ml for 72 h. A total of
1×107 puromycin-resistant clones was recovered after 10
days and the phenotype of target gene knockdown was confirmed using
reverse transcriptoin-quantitative polymerase chain reaction
(RT-qPCR) and/or western blot analysis.
RT-qPCR
RNA samples were prepared using Qiagen RNA
extraction kits (Quiagen, Hilden, Germany). Primers for Col4a5 were
AACGGGGGTTTCCAGGTTTAG (forward) and TTGGTTCCATTGCATCCAGG (reverse).
Primers for Wrn (Werner syndrome ATP-dependent helicase) were
GCCTGTTTACTTGGATCTGCACAG (forward) and TCATCCACAGCAATGAGAGTGATG
(reverse). Primers for glucuronidase beta (GUSB) were
CCTGCGGTTGTGATGTGGTC (forward) and CCTCCAAATGCCCATAGTCATG
(reverse). RNA was transcribed into cDNA with oligo-d(T) using
MulLV reverse transcriptase (Applied Biosystems, Carlsbad, CA,
USA). RT-qPCR reactions were performed using Bio-Rad SYBR kits and
Col4a5 mRNA levels were normalized to GUSB mRNA levels.
Preparation of murine bone marrow-derived
macrophages (BMMs)
Young adult male C57BL/6 mice were purchased from
the Zhejiang University animal facility. All animal experimental
protocols used in this study were approved by Zhejiang University's
Animal Care and Use Committee. BMMs were isolated and cultured by
the following procedures: L929 cells (ATCC) were cultured with an
initial 50% confluency in RPMI-1640 medium (Invitrogen) plus 10%
FBS for 5 days, and the conditioned media were collected, filtered
with a 0.22 µm filter and stored as L-cell conditioned
medium at −20°C. Bone Marrow Growth Medium containing RPMI-1640
(Invitrogen) plus 20% FBS, 30% L-cell conditioned medium and 1%
penicillin/streptomycin (P/S) (Invitrogen), was prepared, aliquoted
and stored at −20°C. The mice were sacrificed by cervical
dislocation, and the pelvic and femoral bones were separated,
immersed in 75% alcohol for 5 min followed by immersion in
Dulbecco's phosphate-buffered saline (Invitrogen) for 5 min, and
left in RPMI-1640 (Invitrogen) plus 1% P/S (Invitrogen). The ends
of the bones were cut and a 27 g needle/1 ml syringe filled with
bone marrow growth medium was injected to expel the bone marrows
from both ends of the bones and directed into a 15 ml cell culture
dish. The bone marrow cells were cultured and the medium was
changed every 3 days. Some of the cells became substrate attached
in culture, whereas most grew in suspension. The cells in
suspension were spun-down and re-cultured in new dishes. After 10
days, almost all cells became attached and the BMMs were ready for
further analysis. RNAi-treated BMMs were obtained by transient
siRNA-transfection as previously reported by us (18).
MTT assay
The mycobacteria were harvested, the OD600 of the
mycobacterial culture was measured, and the bacterial number was
calculated as 0.001 OD600 = 1×105/ml, as previously
described (22). The bacteria
were mixed vigorously using a vortex, and diluted in tissue-culture
medium to obtain the desired density to infect the macrophages at
an MOI of 3 or 9. Bacterial colony forming units (CFU) from
infected cultures were determined at the beginning and end of each
experiment in 7H10 medium supplemented with OADC. RAW264.7
monolayers infected with the MTB strains were incubated at 37°C and
5 % CO2 for up to 72 h. After supernatant removal, the
wells were incubated with
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide [MTT
(Sigma-Aldrich), 5 mg/ml in PBS] with RPMI-1640 medium for further
2 h. The resulting formation of formazan crystals in the cells was
documented, and the cultures were then dissolved in 100 µl
dimethyl sulfoxide. The absorbance of the purple formazan product
was measured at 490 nm with a reference wavelength of 620 nm using
a microplate reader (Tecan, Shanghai, China). All experiments were
performed in quadruplicate and the relative cell viability (%) was
expressed as percentage relative to the untreated control
cells.
CFU determination for intracellular
mycobacteria
The mycobacterial cultures were centrifuged at 125 x
g for 10 min at room temperature, pellets were re-suspended in
RPMI-1640, and then applied to macrophage cultures at an MOI of 3
or 9 for 6, 30 or 72 h at 37°C in a 0.5% CO2 incubator.
The monolayers were washed with warm PBS to remove extracellular
bacteria and re-cultured in RPMI-1640 (nutrient-enriched, 'GM'
condition) or PBS (starvation, 'ST' condition) containing 20
µg/ml gentamycin to inhibit the growth of
extracellular/released bacteria in infected wells. At various time
points, the medium was aspirated from infected macrophage wells and
100 µl of sterile lysis buffer (0.05% SDS in
ddH2O) were added to each well. The plates were
incubated at room temperature for 5 min, serial dilutions were
carried out, and 0.1 ml volumes were plated for 3 dilutions in
duplicate on 7H11 agar plates and further incubated at 37°C for 21
days. CFUs of intracellular mycobacteria were then counted.
Western blot analysis
The RAW264.7 cells were seeded in plates and
infected with or without BCG at the MOIs described above. At the
indicated time points, the macrophages were lysed and boiled in
reducing sodium dodecyl sulfate-poly-acrylamide gel electrophoresis
(SDS-PAGE) sample buffer, subjected to 4–15% gradient gel
electrophoresis (Bio-Rad, Hercules, CA, USA) and transferred onto a
0.2 µm nitrocellulose membrane (GE Healthcare, Piscataway,
NJ, USA) as previously described (23). The blots were probed with
anti-V-ATPase subunit B antibody (Cat. no. SC-271832; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), anti-Col4a5 antibody
(Cat. no. ABIN375285; Antibodies-online, Aachen, Germany), and
anti-tubulin (Cat. no. T3526), and anti-V-ATPase subunit E (Cat.
no. HPA016938) antibodies (Sigma-Aldrich). The detection of
immunoreactivity was performed using the ECL (New England
Bioscience, Ipswich, MA, USA) or Odyssey detection system (LI-COR
Biosciences, Lincoln, NE, USA).
Immunofluorescence
The macrophages were fixed with 4% paraformaldehyde
in PBS for 2 h and permeabilized with 0.2% saponin in PBS
containing 10% donkey serum (Sigma-Aldrich) for 30 min at room
temperature. The coverslips were incubated for 30 min at room
temperature with anti-Col4a5 antibody (dilution, 1:50; Cat. no.
ABIN375285), rinsed twice with PBS containing 0.2% saponin, and
stained for 1 h at room temperature with secondary antibody
(dilution, 1:500) conjugated with TexRed (Molecular Probes,
Carlsbad, CA, USA). The coverslips were washed twice with PBS
containing 0.2% saponin and once in ddH2O, and mounted
with DAPI on slides. Images were acquired with an inverted Laser
scanning confocal microscope (LSM 510; Leica Microsystems, Wetzlar,
Germany).
Measurement of the endosomal/lysosomal pH
in macrophages
Dextran-Oregon Green 488 is an endosomal and
lysosomal marker traveling in the cell via the endosomal/lysosomal
routes. The ratio of the fluorescence at excitation 490 to 440 nm
correlates with endosomal/lysosomal pH values. Using standard
curve-fitting, pH values can be calculated from these fluorescence
ratios (24). For fluorescence
determinations, the RAW264.7 cells were plated to 60–70% confluency
on fluorescence-proof microplates (Corning, New York, NY, USA). To
measure the endosomal/lysosomal pH values, the cells were incubated
with 0.5 µg/ml Dextran-Oregon Green 488 (Molecular Probes)
for 30 min to 5 h at 37°C. The fluorescence of Dextran-Oregon Green
488 was measured with the excitation wavelength set at 440 and 490
nm, respectively, and the fluorescence emission set at 520 nm using
a microplate reader (Tecan). The fluorescence ratio of 490/440 was
converted to pH values by standard curve-fitting as reported
previously (25). Briefly, the
cells in the plate were incubated with Dextran-Oregon Green 488 for
1 h and then incubated in isotonic K+-rich medium
buffered to predetermined pH values (between pH 7.0 and 3.5).
Alternatively, the membranes were permeabilized gently using a
solution of 0.1% Triton X-100 in PBS for 2 min, given direct access
to the Dextran-Oregon Green 488 particles. Calibration curves were
obtained by plotting the extracellular pH, which is assumed to be
identical to the internal pH, against the corresponding
fluorescence ratio.
Measurement of phagosomal pH in
macrophages
Phagosomal pH values were measured as previously
described (26). Briefly, BCG was
labeled by incubation in N-Hydroxysuccinimidyl ester 5- (and
-6)-carboxyfluorescein (NHS-CF, 0.1 mg/ml) (Sigma-Aldrich) and FITC
(1 mg/ml) (Sigma-Aldrich) on ice for 30 min. Unbound dye was
removed by repeated washing. The bacteria were heated at 100°C for
10 min; the heat-killed bacteria were used for comparison. The
RAW264.7 cells were incubated with FITC-BCG on fluorescence-proof
microplates (Corning) for 1–5 h. Measurements of phagosomal pH were
obtained through the fluorescence ratio reading method as described
for endosomal/lysosomal pH measurements. The calibration of the
fluorescence ratio versus the pH value was performed for each
experiment by equilibrating the cells in isotonic
K+-rich medium buffered to varying pH values (between pH
4.0 and 8.0) in the presence of the K+/H+
ionophores nigericin (5 µM; Sigma-Aldrich) and monensin (2
µM; Sigma-Aldrich). Calibration curves were obtained by
plotting the extracellular pH, which is assumed to be identical to
the internal pH, against the corresponding fluorescence ratio.
Measurement of microsomal V-ATPase
activity in macrophages
ATPase-mediated ATP hydrolysis is coupled
enzymatically to the oxidation of NADH, which can be followed by an
absorbance decrease at 340 nm; therefore, V-ATPase activity was
measured using a modification of the protocol previously described
(26). Briefly, microsomal
fractions were obtained by ultracentrifugation of a macrophage
homogenate using a sucrose gradient, and 10 µg of microsomal
protein were added to a master mix containing 37.5 mM MOPS, 4 mM
MgSO4, 50 mM KCl, 3 mM orthovanadate, 1 mM
phosphoenolpyruvate, 0.3 mM NADH, 10 mg/ml pyruvate kinase and 10
mg/ml lactate dehydrogenase (Sigma-Aldrich). Reactions with and
without the V-ATPase-specific inhibitor bafilomycin A1 (BA1; 100
nM; Sigma-Aldrich) were started by the addition of ATP to a final
concentration of 2 mM in a microtiter plate, and absorbance at 340
nm was monitored for 20 min in a Safire plate reader (Tecan). All
measurements were performed in triplicate, and V-ATPase activity
was calculated as BA1-inhibited ATP hydrolysis.
Statistical analysis
All experiments were repeated 3 times independently
and the data were presented as the means ± SEM. One-way analysis of
variance followed by Dunnett's test were used to determine
significant differences. Significant levels were set at P<0.05.
Statistical analyses were performed using SPSS statistical software
(version 21.0; SPSS, Inc., Chicago, IL, USA).
Results
Knockdown of host target genes increases
the viability of MTB-infected macrophages
RAW264.7 cell libraries in which cellular genes were
randomly inactivated by lentivirus-based antisense RNA methods were
constructed using the RHKO and EST DNA libraries, as previously
reported by us (18,21). These macrophage libraries were
screened for clones that survived multiple cycles of infection by
virulent MTB (H37Rv) at moderate to high MOI values. Individual
surviving macrophage cell clones were expanded, and DNA sequencing
identified 26 unique, inserted DNA sequences in a total of 81
macrophage clones (Table I).
Among these clones, the Wrn mutant survived in the highest
frequency from 3 cycles of the MTB infection and was the only
mutant surviving after 4 cycles of MTB infection. A series of
RT-qPCR assays confirmed the downregulation of the host target
genes in the isolated macrophage clones and additionally revealed
that the transcription of one of the 26 host target genes, Col4a5,
was downregulated in clones containing antisense inserts in several
of the host target genes, including Wrn, Runx1, Pkr, Pak1, Mllt3,
Rnf135 and Extl2 mutants (Fig.
1A). These results led us to speculate that the inserts
identified by our screening may affect a common pathway regulating
Col4a5 expression. Consistent with this notion, Col4a5 was
downregulated in Wrn mutant cells (Fig. 1B), while the expression of Wrn was
not altered in Col4a5 mutant cells (Fig. 1C). Moreover, the expression levels
of the Runx1, Pkr, Pak1, Mllt3, Rnf135 and Extl2 genes were not
significantly altered in the Col4a5 mutant cells (data not shown).
Col4a5 was also downregulated in 5 Wrn-shRNA-incorporated lines and
correlated with the decreased Wrn mRNA levels in those cell lines
(Fig. 1D and E), confirming that
Wrn is a positive regulator of Col4a5 expression at the
transcriptional level. Additionally, genes, such as IFNγ, known to
affect MTB infection may alter Col4a5 expression or function at the
transcriptional and/or post-transcriptional levels (27,28). These findings led us to explore a
possible role of Col4a5 as a target gene for the host-directed
therapy of mycobacterial infection.
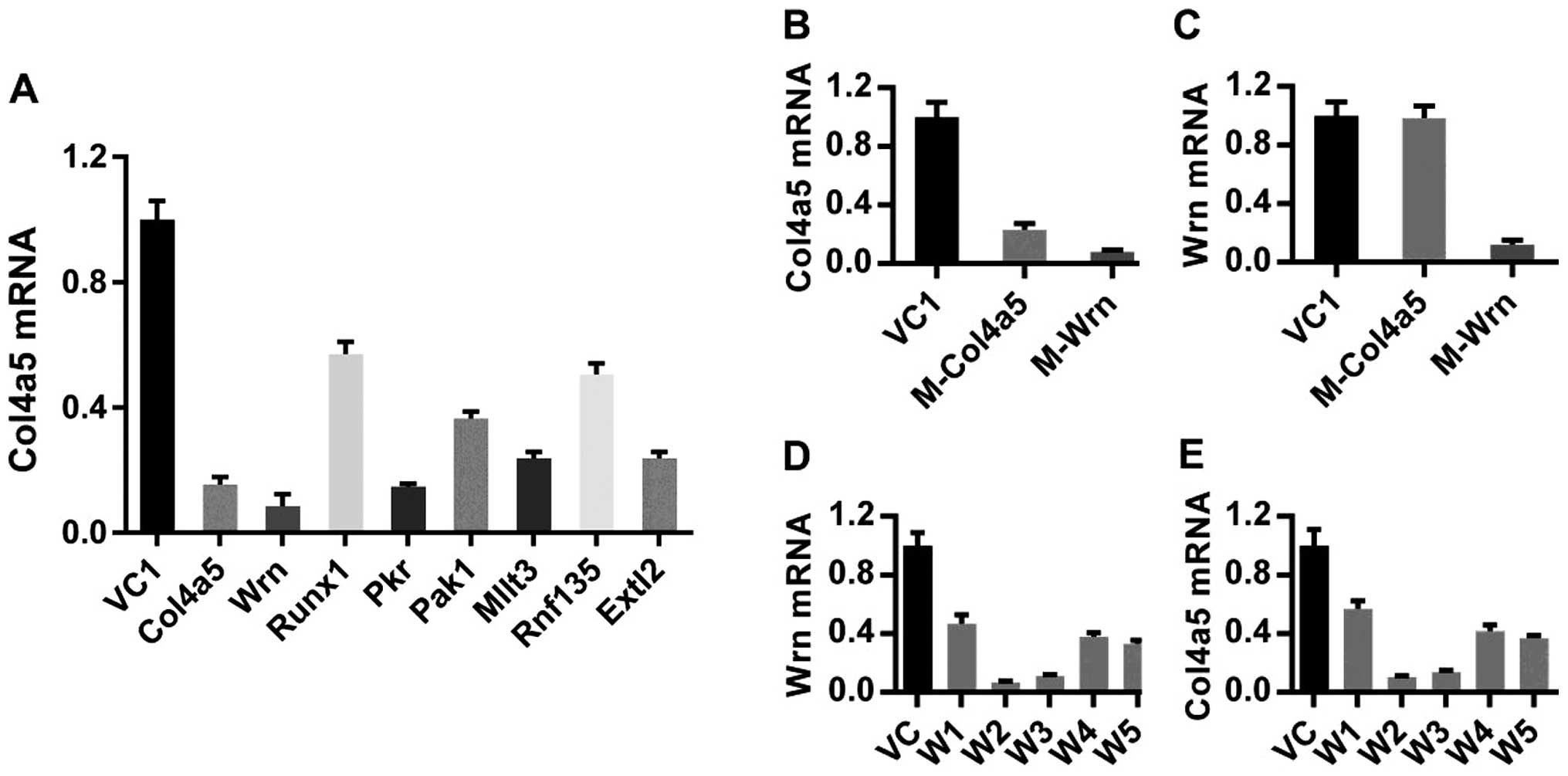 | Figure 1Identification and validation of host
target genes affecting Mycobacterium tuberculosis (MTB)
infection function as regulators of collagen α-5(IV) chain (Col4a5)
expression. (A) Relative quantification of Col4a5 mRNA expression
in vector control (VC1), Col4a5, Wrn, Runx1, Pkr, Pak1, Mllt3,
Rnf135 and Extl2 mutant cells was determined by RT-qPCR.
Quantification of relative (B) Col4a5 mRNA and (C) Wrn mRNA levels
determined by RT-qPCR in VC1, Col4a5-mutant (M-Col4a5) and
Wrn-mutant (M-Wrn) cell lines. Quantification of relative (D) Wrn
mRNA or (E) Col4a5 mRNA levels determined by RT-qPCR in VC1 and 5
Wrn-shRNA-transfected cell lines (W1, W2, W3, W4 and W5). |
 | Table IPutative host target genes involved
in the regulation of tuberculosis infection. |
Table I
Putative host target genes involved
in the regulation of tuberculosis infection.
| Gene |
Library/direction | MOI used | Annotation |
|---|
| Col4a5 | RHKO/antisense | 2–3 | Collagen IV
subunit |
| eIF4e | RHKO/antisense | 10–17 | Eukaryotic
translation initiation factor 4E |
| Mllt3 | RHKO/sense | 2–3,10–17 | Myeloid/lymphoid or
mixed-lineage leukemia (trithorax homolog, Drosophila) |
| Pgm2l1 | RHKO/sense | 2–3,10–17 | Phosphoglucomutase
2-like 1 |
| Wrn | RHKO/antisense | 2–3,10–17 | Werner syndrome,
RecQ helicase-like |
| Zfp52 | RHKO/antisense | 2–3 | Mouse zinc finger
protein 52 |
| NM_182745 | RHKO/sense | 2–3,10–17 | Hypothetical
protein, function unknown |
| PLBD1 | EST/antisense | 10–17 | Phospholipase B
domain containing 1 |
| F7 | EST/antisense | 10–17 | Coagulation factor
VII |
| EXTL2 | EST/antisense | 52–77 | Exostoses
(multiple)-like 2 |
| LMTK2 | EST/antisense | 52–77 | Lemur tyrosine
kinase 2 |
| DPEP1 | EST/antisense | 52–77 | Dipeptidase 1
(renal) |
| Sp100 | EST/antisense | 52–77 | SP100 nuclear
antigen |
| PKR | EST/antisense | 52–77 | EIF2AK2, eukaryotic
translation initiation factor 2-α kinase 2 |
| CST8 | EST/sense | 52–77 | Cystatin 8
(cystatin-related epididymal spermatogenic) |
| AL137616 | EST/sense | 52–77 | Unknown |
| ACADVL | EST/sense | 10–17 | Acyl-CoA
dehydrogenase, very long chain |
| PDE6B | EST/sense | 10–17 | Phosphodiesterase
6B, cGMP, rod receptor, beta polypeptide |
| CNKSR2 | EST/sense | 52–77 | Connector enhancer
of kinase suppressor of Ras |
| ARMC9 | EST/sense | 10–17 | Armadillo repeat
containing 9 |
| Hdac9 | EST/sense | 10–17 | Histone deacetylase
9 |
| Runx1 | EST/sense | 52–77 | Runt-related
transcription factor 1 |
| Spink1 | EST/sense | 52–77 | Serine peptidase
inhibitor, Kazal type 1 |
| Rnf135 | EST/sense | 10–17 | Ring finger protein
135 |
| TRAIP | EST/sense | 10–17 | TRAF interacting
protein |
| Pak1 | EST/sense | 10–17 | p21 protein
(Cdc42/Rac)-activated kinase 1 |
Col4a5 deficiency promotes macrophage
survival following mycobacterial infection
In order to further confirm the effects of Col4a5 on
mycobacterial infection, the knockdown of Col4a5 by shRNA was
employed in the RAW264.7 macrophages. To mitigate possible
off-target effects of the antisense RNA-based methods used to
identify Col4a5, 3 separate Col4a5-shRNA-transfected macrophage
lines, designated as C51, C52 and C53, were established, each of
which contained shRNA species targeting a different locus of the
Col4a5 mRNA. The results of RT-qPCR revealed that the Col4a5 mRNA
levels in the C51, C52 and C53 cells were decreased to
approximately 15–20% of the level observed for the vector-only
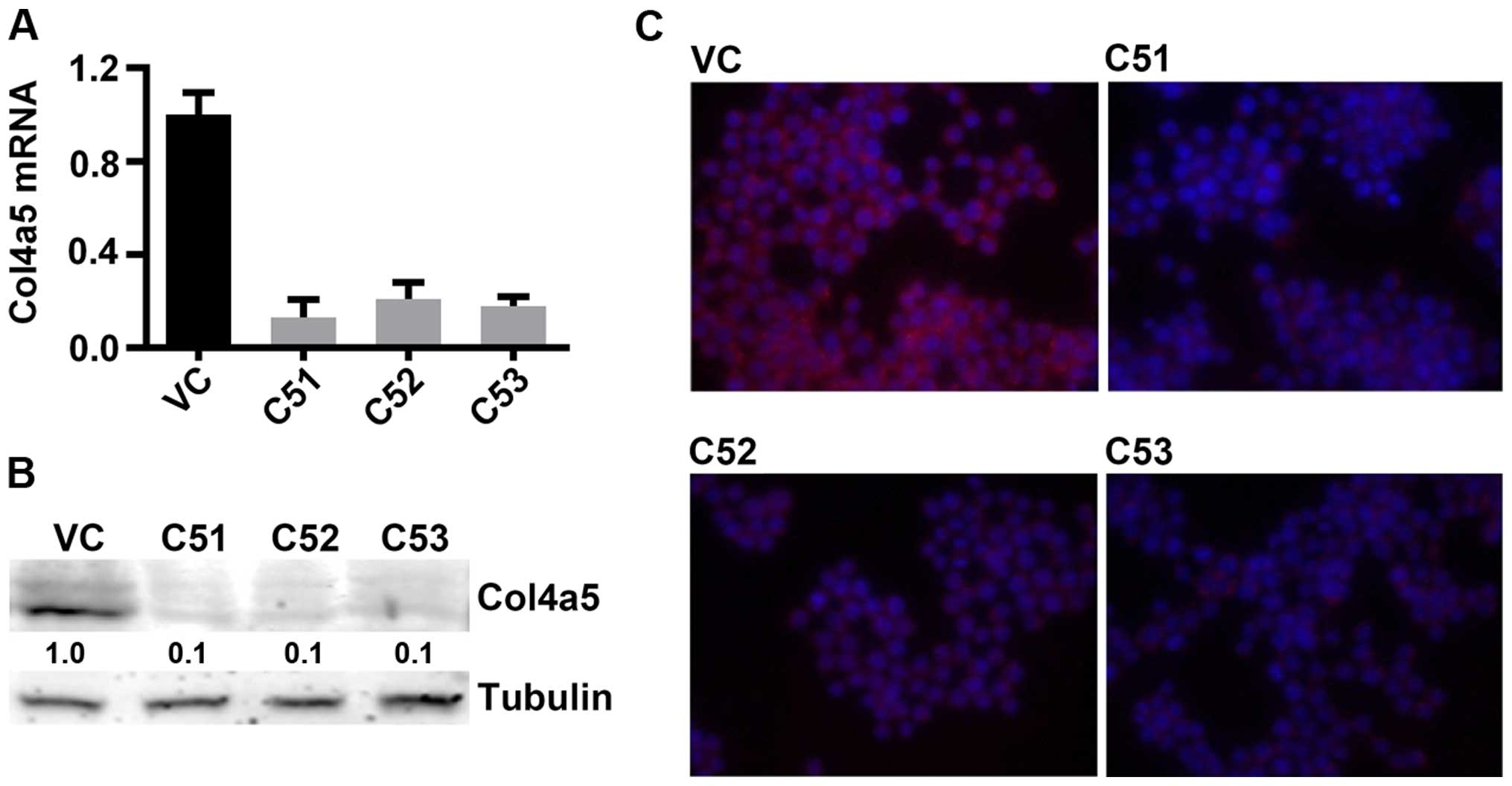
control (VC) cells (Fig. 2A).
Consistent with the reduction in mRNA expression, the results of
western blot analysis confirmed decreased Col4a5 protein levels in
the C51, C52 and C53 cells compared with the VC cells (Fig. 2B). Furthermore, western blot
analysis andd immunofluorescence staining revealed decreased Col4a5
protein staining in the cell membranes for all 3 cell lines
(Fig. 2B and C). Furthermore, the
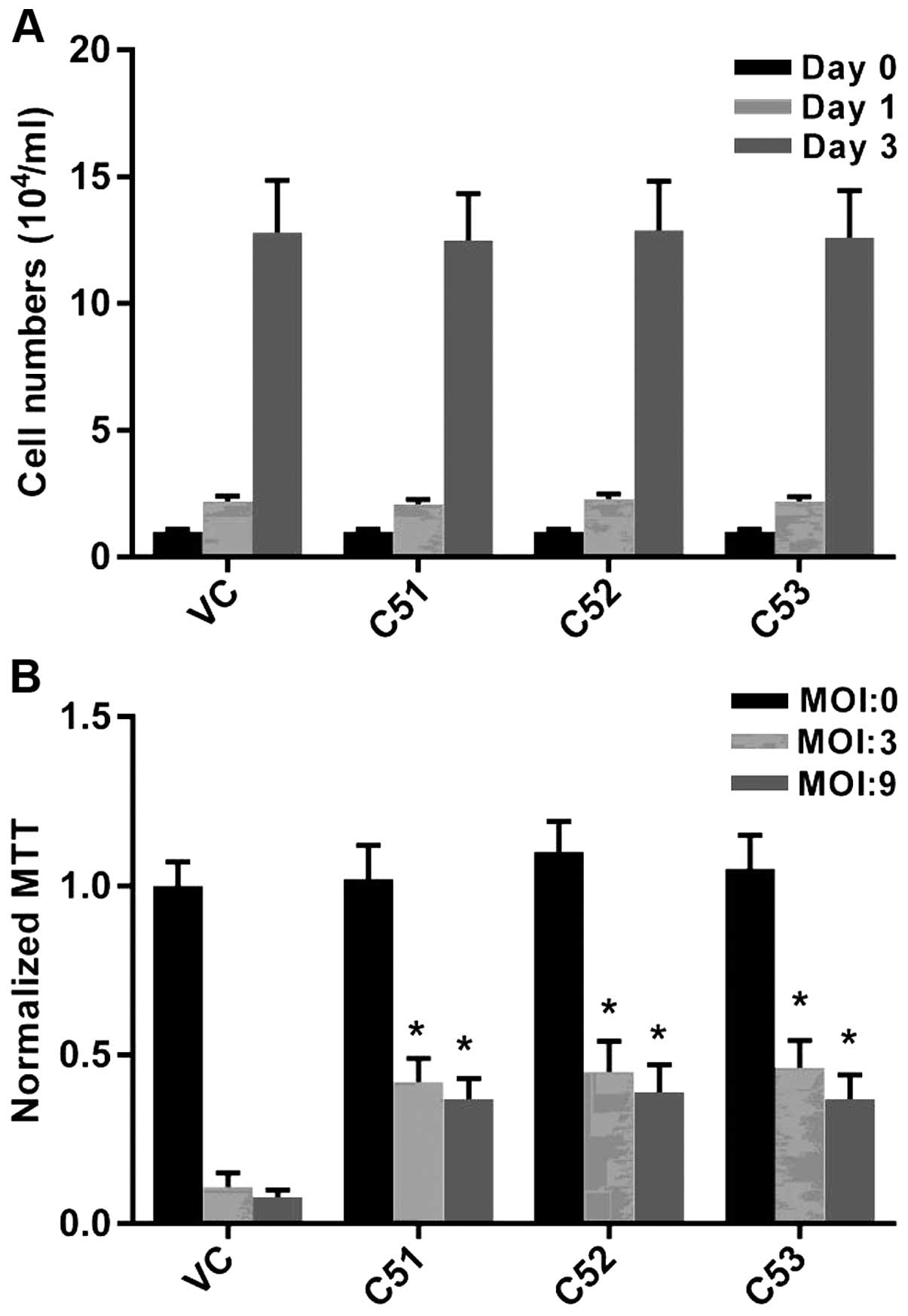
growth rates of Col4a5-deficient cell lines were similar to those
of the control cells without MTB infection, as indicated by the
cell numbers on days 1 and 3 (Fig.
3A). In addition, the deficiency of Col4a5 in the macrophages
was associated with an enhanced survival following infection by MTB
at either an MOI 3 or 9 on day 3. MTT assays consistently revealed
that after 3 days of MTB infection, only approximately 10% of the
vector-control cells survived, while >40% of the macrophages in
which Col4a5 was knocked down survived under the same culture
conditions (Fig. 3B). Taken
together, these results suggested that Col4a5 deficiency enhanced
macrophage viability following mycobacterial infection.
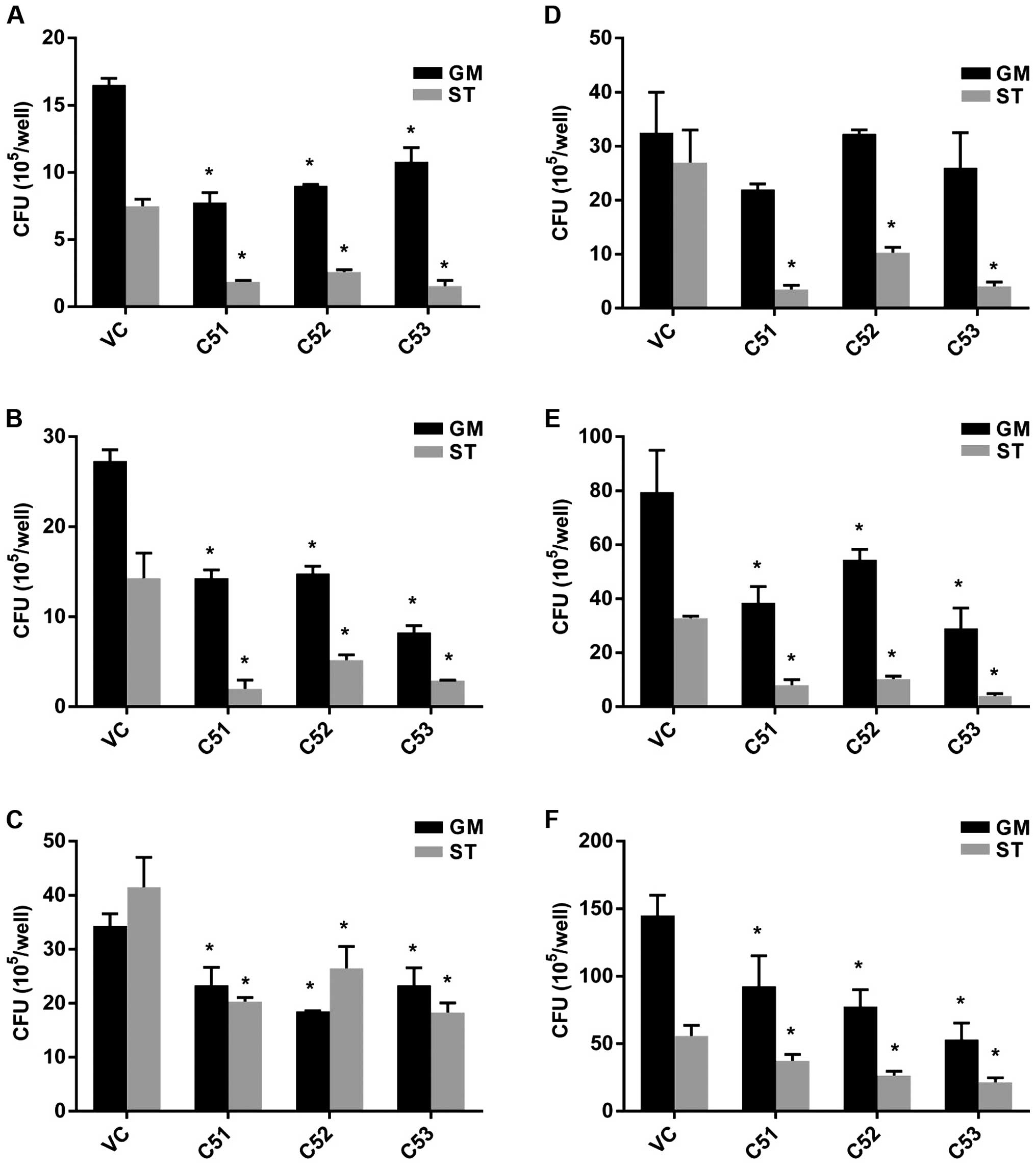
Col4a5 deficiency suppresses the
intracellular mycobacterial viability in macrophages
To further investigate the effects of Col4a5
deficiency on the viability of intracellular MTB, the intracellular
mycobacterial survival rate was estimated by CFU counting.
Macrophages were infected with MTB at an MOI of 3 in either
nutrient-enriched (GM) or starvation (ST) conditions. The knockdown
of Col4a5 led to an approximately 20–80% decrease in MTB numbers
recovered from the host cells incubated for 6, 30 or 72 h after
infection (Fig. 4A–C). When the
MOI of MTB was increased to 9, the knockdown of Col4a5 still led to
an approximately 10–80% decrease in MTB numbers recovered from the
host cells for 6, 30 or 72 h post-infection under nutrient-enriched
or starvation conditions (Fig.
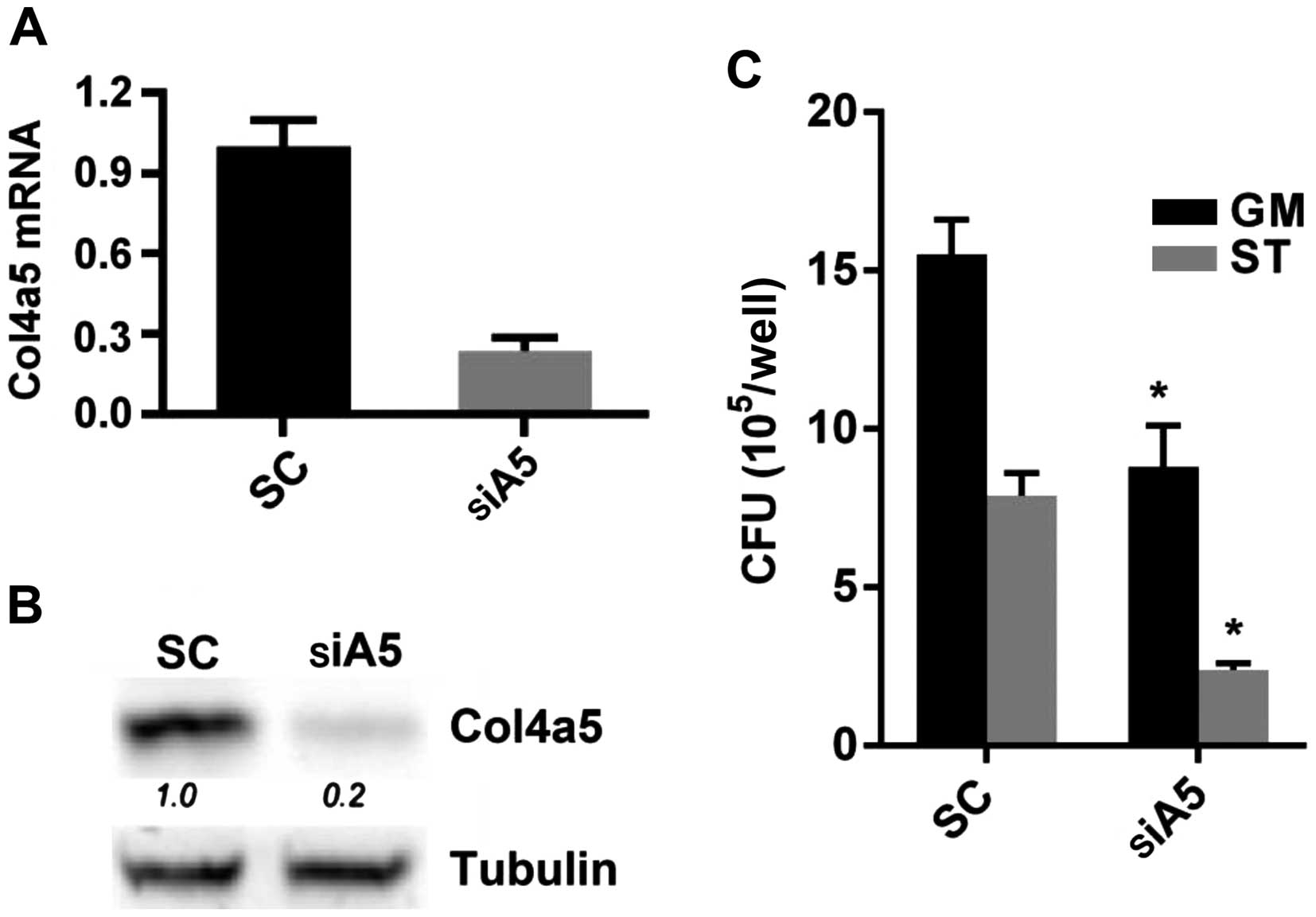
4D–F). Moreover, when siRNA transfection was employed to
knockdown Col4a5 expression in BMMs, which was confirmed by both
RT-qPCR and western blot analysis (Fig. 5A and B), Col4a5 knockdown led to
an approximately 40–80% decrease in MTB numbers recovered from the
host cells 6 h post-infection under nutrient-enriched or starvation
conditions (Fig. 5C).
Collectively, these results suggested that Col4a5 knockdown led to
the suppression of intracellular mycobacterial viability in
vitro under both nutrient-enriched or starvation
conditions.
Knockdown of Col4a5 enhances the
acidification of both culture media and intracellular vesicles
Macrophages represent the first line of defense
against MTB invasion by various mechanisms, a major one of which is
the acidification of phagosomes and lysosomes (29,30). During our studies, we observed
that Col4a5 knockdown in macrophages was associated with a
yellowing of the culture media, suggesting a possible decrease in
pH of the culture media (pHm). The direct measurement of
pHm values confirmed a 0.4–0.5 pH unit decrease (data
not shown) for media in which the C51, C52 or C53 cells were grown
for 3 days compared to the controls with the same growth rate as
Col4a5-deficient cells (Fig. 3A).
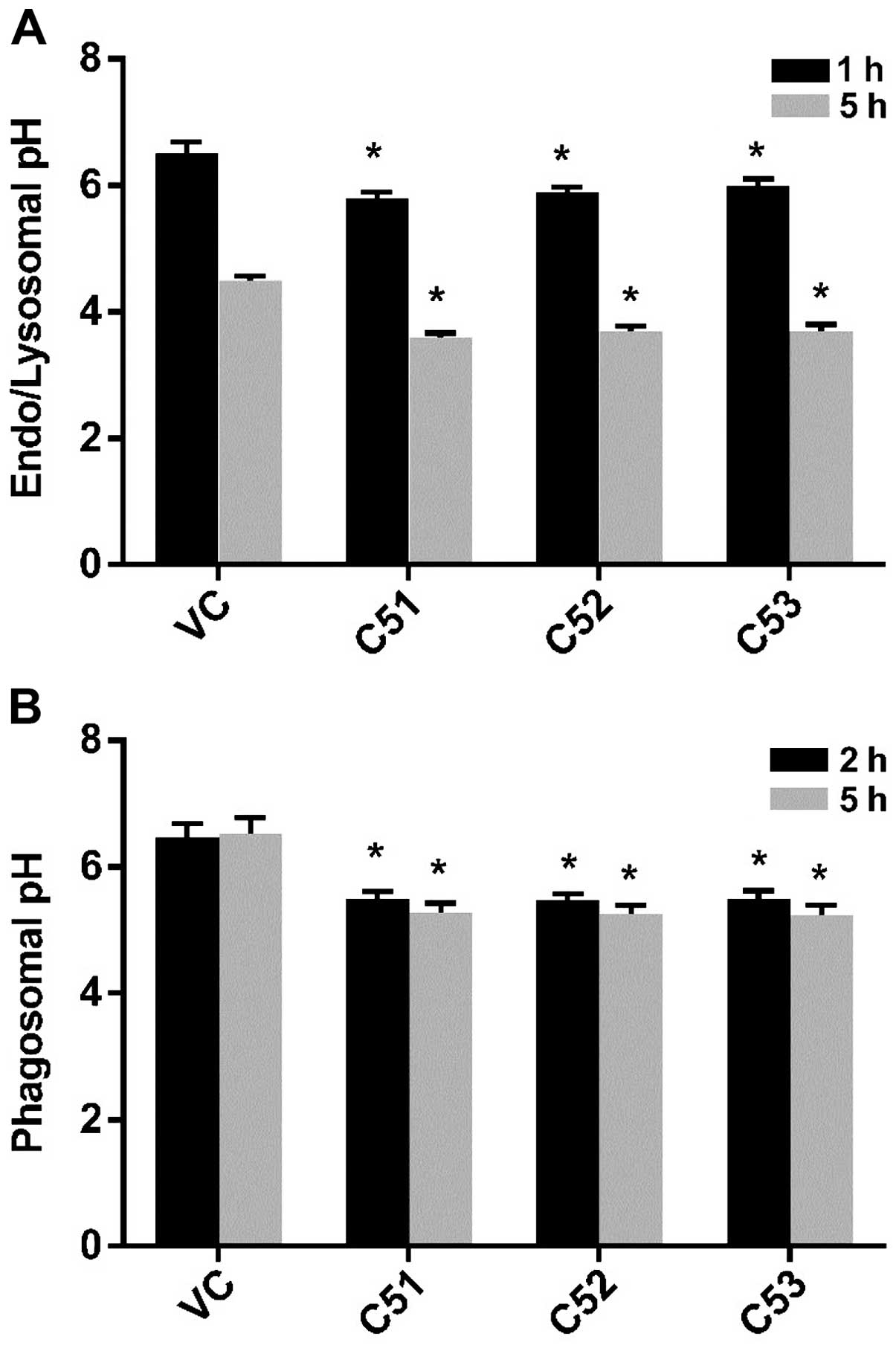
To further investigate the alterations in intravesicular pH
(pHi, i.e., in endosomes, lysosomes and phagosomes) in
macrophages, Dextran-Oregon Green 488, which is known to enter
cells via endocytosis within 1 h and enter the lysosome within
approximately 5 h, was used to determine whether the deficiency of
Col4a5 is associated with changes in pHi. The endosomal
pH was approximately 0.5 units lower and the lysosomal pH was
approximately 0.9 units lower in the macrophages in which Col4a5
had been knocked down compared with the controls (Fig. 6A). FITC-labeled BCG were used to
measure the phagosomal pH, which was approximately 0.9 units lower
in the cells in which Col4a5 had been knocked down compared with
the control macrophages (Fig.
6B). These results confirmed that Col4a5 knockdown enhances the
the acidification of intracellular vesicles in murine
macrophages.
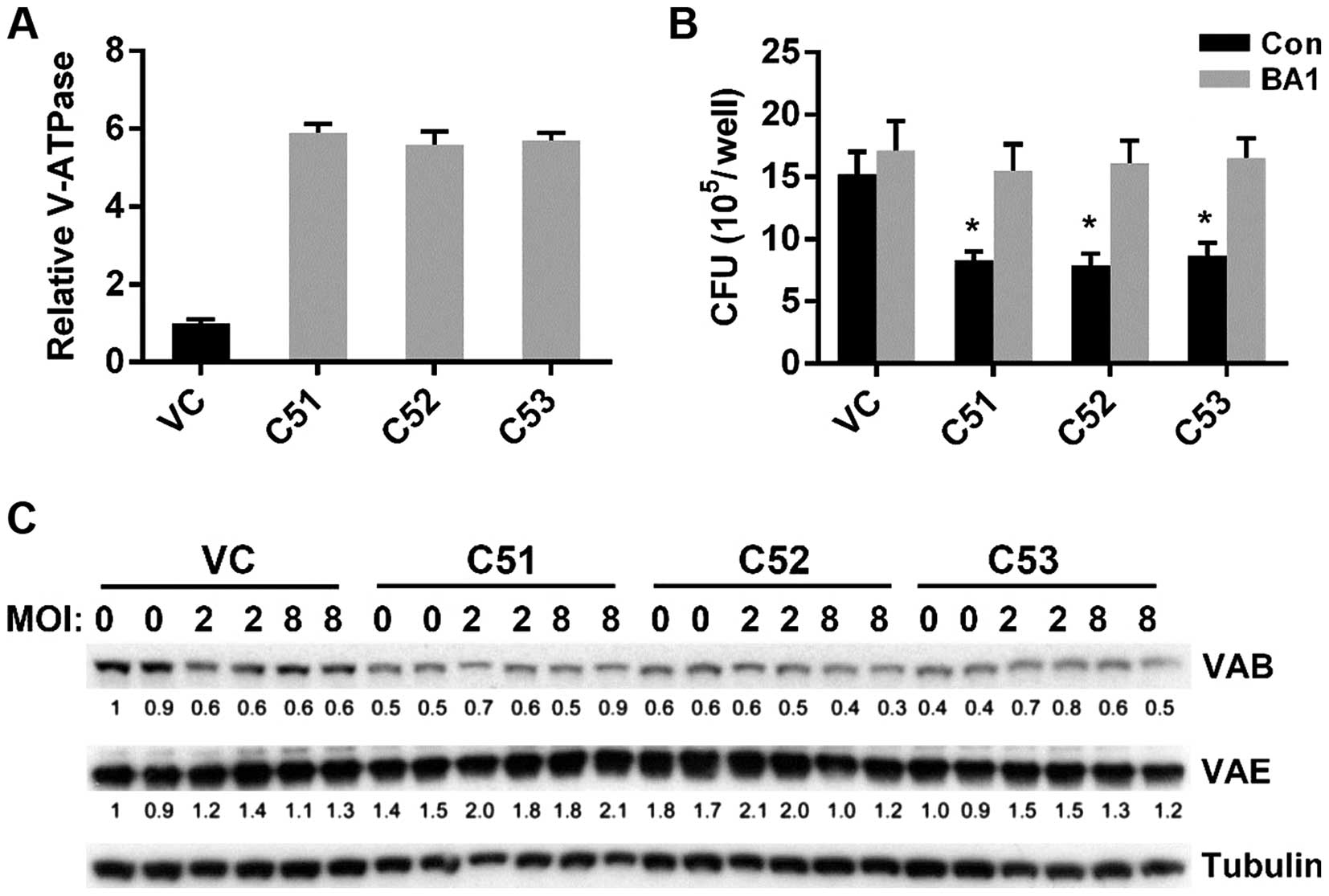
Increased microsomal V-ATPase activity
occurs post-translationally in cells in which Col4a5 is knocked
down
MTB is known to protect itself from digestion by
lysosomal enzymes by preventing the lowering of the pH necessary
for the activity of lysosomal enzymes (31). Moreover, macrophages are known to
actively secrete protons into the extracellular environment via the
V-ATPase, which is crucial for maintaining intravesicular
acidification (32). We
hypothesized that the lower pHm and pHi of
macrophages in which Col4a5 had been knocked down may be mediated
via the actions of V-ATPase. Treatment of the cell cultures with
BA1, a V-ATPase specific inhibitor, eliminated the pHm
difference in the media (data not shown), confirming the possible
role of V-ATPase for Col4a5-mediated acidification. Therefore, we
measured the total ATPase activity by a quantitative assay and
found that microsomal V-ATPase activity in the cells in which
Col4a5 had been knocked down was approximately 5.5-fold higher
compared with that in the control cells (Fig. 7A). Moreover, in the presence of
the V-ATPase specific inhibitor, BA1, the difference in BCG
infection between the cells in which Col4a5 had been knocked down
and the control cells was diminished (Fig. 7B). To further address whether the
upregulation of V-ATPase activity was due to the expression of
V-ATPase subunits, western blot analysis was performed detecting
the V-ATPase subunits B and E in controls and in the macrophages in
which Col4a5 had been knocked down without BCG infection (MOI 0) or
with BCG infection (MOI 2 or 8). Col4a5 knockdown did not
upregulate the protein expression of the ATPase subunits (Fig. 7C), suggesting that increased
microsomal V-ATPase activity occurs post-translationally in cells
in which Col4a5 is knocked down.
Discussion
In the present study, we screened for host genes
which are exploited by MTB during invasion and survival in murine
macrophages, and we identified Col4a5, a collagen IV subunit, as a
potential host target gene. In addition, we found 9 host genes as
positive regulators of Col4a5 gene expression. The knockdown of
Col4a5 reduced mycobacterial viability and increased the survival
of macrophages by promoting the acidification of intracellular
vesicles by increasing V-ATPase activity.
Tuberculosis remains one of the major causes of
mortality worldwide due to the lack of effective therapies and the
emergence of multidrug-resistant strains (2). In addition to conventional
chemotherapies, alternative host-directed therapies against
interleukin-1 and type I IFN have been explored to improve
treatment efficacies and to reduce the mortality rate associated
with the disease (13,33). Numerous host genes for MTB have
been identified by RNAi screens against all known kinases and
phosphatases in a mouse macrophage cell line (34) and against all genes in a human
macrophage cell line (19);
however, all these screens were based on reducing the intracellular
mycobacterial load. In this study, we investigated host genes
exploited by MTB during their invasion and survival in murine
macrophages and identified Col4a5 as a potential host target gene.
By contrast, the screening method we used to non-selectively
inactivate genes in RAW264.7 cells by lentivirus-based antisense
RNA methods, is the first one (to the best of our knowledge) to
identify host genes that regulate mycobacteria via a
macrophage-survival-based strategy. The death modality of
MTB-infected macrophages plays different roles in the pathogenesis
of tuberculosis and has extensive impacts on the outcome of the
disease (35). Virulent MTB
triggers necrosis and inhibits the apoptosis of host macrophages to
evade host defense by affecting eicosanoid biosynthesis, while the
attenuation of MTB induces apoptosis which then decreases bacterial
viability (36). Controversy
exists as to whether macrophage apoptosis is a self-protective
mechanism of the host or simply a consequence of bacterial
infection (37). Some
investigators believe that host cell apoptosis deprives the
pathogen of a protected intracellular environment, while others
argue that MTB triggers macrophage apoptosis in order to escape the
toxic intracellular environment and replicate in the extracellular
space (37). In any case, the
survival of virulent MTB-infected macrophages in vitro
provides a powerful selective tool for the identification of
macrophage mutants unable to take up MTB or to allow the bacterium
to replicate. Our results suggest that under the conditions we used
in this study, increased macrophage viability is consistent with
increased control of the intracellular mycobacterial load. The 26
host genes we identified in the present study are different from
those previously discovered, indicating that these host genes may
be condition-dependent, infection phase-dependent, and/or specific
for the macrophage survival-based strategy. Thus, it is not
surprising that Col4a5 was not previously identified as a host gene
reducing the MTB content in RNAi screens that targeted all kinases
(34) or all genes in macrophage
cell lines (19). However, in the
present study, we demonstrated that Col4a5 deficiency had similar
effect on the ability of macrophages to restrain the intracellular
mycobacterial load in vitro. Although, Col4a5 deficiency
enhanced two forms of anti-mycobacterial host defense in
vitro, i.e., apoptosis and macrophage activation in the present
study, it remains unclear whether it can lead to reduced MTB
numbers in vivo. The pathogenesis of infectious disease is
complex, and only limited aspects can be modeled in isolated cells.
In general, the macrophage control of MTB in vitro appears
to be far less effective than in the host. This presumably reflects
the shortcomings of in vitro models, such as the exclusion
of other cell types, differences in extracellular matrices,
supra-physiological levels of oxygen and the use of heterologous
sera (38).
Col4a5 encodes a 161 kDa type IV collagen protein
and its deficiency causes X-linked Alport syndrome, a hereditary
nephritis (39). It is known that
Col4a5 protein forms heterotrimers with other collagen IV subunits,
i.e., Col4a3, Col4a4 or Col4a6 in basement membranes. The
Col4a3/Col4a4/Col4a5 trimer is solid and rigid enough to enable the
glomerular basement membrane to protect cells against harsh
environments (39). Furthermore,
although Col4a5 is highly expressed in the kidneys, lungs and inner
ear, Col4a5-deficiency only causes diseases in the kidneys and
ears, but not in the lungs (39).
In this study, we describe a novel role of Col4a5 in negatively
regulating V-ATPase activity in murine macrophages. V-ATPase is an
essential cellular enzyme that acidifies intracellular vesicles,
thus providing a mycobactericidal micro-environment (32). It is well known that mycobacteria
attenuate phagosomal acidification, and initially this effect was
proposed to be mainly mediated through the exclusion of V-ATPases
from phagosomes, as a result of blocking of the fusion of
phagosomes with V-ATPase-rich vesicles, including late endosomes
and lysosomes (6). However, later
studies indicated that phagosomes containing mycobacteria still
acquire V-ATPases through interaction with early and recycling
endosomes, which are also sources of V-ATPases (40). Currently, it is still unclear as
to how Col4a5, an extracellular matrix protein, suppresses V-ATPase
activity in intracellular vesicles, but does not alter its protein
expression. There are at least two possible explanations. The first
possibility is that it may not be full-length Col4a5, but
internalized fragments that exert the inhibiting effects on
cellular V-ATPase. This is consistent with our observation that
Col4a5 fragments may accumulate in macrophages after 2 h or longer
incubation with mycobacteria (data not shown). Another possibility
is that Col4a5 may interact with cell surface receptors that
regulate the activity of V-ATPase-regulating protein kinases, which
in turn regulate V-ATPase activity through phosphorylation of
V-ATPase subunits (32).
There are several limitations to this study.
Firstly, it is not certain to which degree the observed in
vitro effects reflect the in vivo situation,
particularly due to the fact that no additional information is
available from Col4a5-knockout mice, which die shortly after birth
(41). Therefore, a conditional
knockout mouse model may help to elucidate the roles of Col4a5 in
mycobacterial infection. Secondly, although not available yet, the
screening and development of a specific Col4a5 inhibitor with high
bioavailability and low off-target toxicity would be invaluable for
further testing of Col4a5 functions in mouse models in vivo.
Thirdly, it would be also interesting to identify the functional
motifs in the Col4a5 protein, which are required for its effect on
inhibiting mycobacterial infection in macrophages. Finally, in
order to avoid the potential systematic toxicity, particularly the
toxicity to the kidneys, Col4a5-targeted therapy should be limited
to the lungs through pulmonary delivery. To this end, inhaled
formulations of Col4a5-targeted therapy could be developed in the
future.
In conclusion, we non-selectively inactivated genes
in RAW264.7 macrophages, screened for cells that survived infection
following virulent MTB infection, and identified 26 host genes, 9
of which were positive regulators of the expression of the Col4a5
gene, which encodes the dominant collagen IV subunit. In addition,
we demonstrated that the knockdown of Col4a5 reduced mycobacterial
viability and increased the survival of macrophages by promoting
the acidification of intracellular vesicles, including endosomes,
lysosomes and phagosomes. Finally, we demonstrated that Col4a5
knockdown may post-translationally increase microsomal vesicular
ATPase activity in macrophages, leading to the acidification of
both intracellular vesicles and culture media. Our findings reveal
a promising role for Col4a5 in the regulation of murine macrophage
responses to mycobacterial infection, identifying Col4a5 as a
potential target for anti-mycobacterial therapy, and providing
evidence of host-directed strategies that manipulate the survival
of macrophages.
Acknowledgments
This study was supported by the Zhejiang Provincial
Natural Science Foundation of China (grant no. LY14H010002) the Key
Personnel Grant of Zhejiang Medicine and Health Platform (no.
2012RCA025), the Grant for Returned Overseas Chinese Scholar of the
Personnel Department of Zhejiang Province (no. J20120565), the
Grant of Health and Family Planning Commission of Zhejiang province
(no. 2009B060) and the 5050 project of Binjiang district.
References
|
1
|
BoseDasgupta S and Pieters J: Striking the
right balance determines TB or not TB. Front Immunol. 5:4552014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zumla A, Raviglione M, Hafner R and von
Reyn CF: Tuberculosis. N Engl J Med. 368:745–755. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Horsburgh CR Jr, Barry CE III and Lange C:
Treatment of tuberculosis. N Engl J Med. 373:2149–2160. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dey B and Bishai WR: Crosstalk between
Mycobacterium tuberculosis and the host cell. Semin Immunol.
26:486–496. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fu X, Ding M, Zhang N and Li J:
Mycobacteriophages: An important tool for the diagnosis of
Mycobacterium tuberculosis (Review). Mol Med Rep. 12:13–19.
2015.PubMed/NCBI
|
|
6
|
Sturgill-Koszycki S, Schlesinger PH,
Chakraborty P, Haddix PL, Collins HL, Fok AK, Allen RD, Gluck SL,
Heuser J and Russell DG: Lack of acidification in Mycobacterium
phagosomes produced by exclusion of the vesicular proton-ATPase.
Science. 263:678–681. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ferrari G, Langen H, Naito M and Pieters
J: A coat protein on phagosomes involved in the intracellular
survival of mycobacteria. Cell. 97:435–447. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu L, Liu J, Niu G, Xu Q and Chen Q:
Mycobacterium tuberculosis 19-kDa lipoprotein induces Toll-like
receptor 2-dependent peroxisome proliferator-activated receptor γ
expression and promotes inflammatory responses in human
macrophages. Mol Med Rep. 11:2921–2926. 2015.
|
|
9
|
Brzostek A, Pawelczyk J,
Rumijowska-Galewicz A, Dziadek B and Dziadek J: Mycobacterium
tuberculosis is able to accumulate and utilize cholesterol. J
Bacteriol. 191:6584–6591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fratti RA, Chua J and Deretic V: Induction
of p38 mitogen-activated protein kinase reduces early endosome
autoantigen 1 (EEA1) recruitment to phagosomal membranes. J Biol
Chem. 278:46961–46967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lasunskaia EB, Campos MN, de Andrade MR,
Damatta RA, Kipnis TL, Einicker-Lamas M and Da Silva WD:
Mycobacteria directly induce cytoskeletal rearrangements for
macrophage spreading and polarization through TLR2-dependent PI3K
signaling. J Leukoc Biol. 80:1480–1490. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vergne I, Chua J and Deretic V:
Tuberculosis toxin blocking phagosome maturation inhibits a novel
Ca2+/calmodulin-PI3K hVPS34 cascade. J Exp Med.
198:653–659. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mayer-Barber KD, Andrade BB, Oland SD,
Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei
W, et al: Host-directed therapy of tuberculosis based on
interleukin-1 and type I interferon crosstalk. Nature. 511:99–103.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kindler V, Sappino AP, Grau GE, Piguet PF
and Vassalli P: The inducing role of tumor necrosis factor in the
development of bactericidal granulomas during BCG infection. Cell.
56:731–740. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meyer CG and Thye T: Host genetic studies
in adult pulmonary tuberculosis. Semin Immunol. 26:445–453. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wallis RS and Hafner R: Advancing
host-directed therapy for tuberculosis. Nat Rev Immunol.
15:255–263. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao HF, L'Abbé D, Jolicoeur N, Wu M, Li
Z, Yu Z and Shen SH: High-throughput screening of effective siRNAs
from RNAi libraries delivered via bacterial invasion. Nat Methods.
2:967–973. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen R, Liliental JE, Kowalski PE, Lu Q
and Cohen SN: Regulation of transcription of hypoxia-inducible
factor-1α (HIF-1α) by heat shock factors HSF2 and HSF4. Oncogene.
30:2570–2580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kumar D, Nath L, Kamal MA, Varshney A,
Jain A, Singh S and Rao KV: Genome-wide analysis of the host
intracellular network that regulates survival of Mycobacterium
tuberculosis. Cell. 140:731–743. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu Q, Wei W, Kowalski PE, Chang AC and
Cohen SN: EST-based genome-wide gene inactivation identifies ARAP3
as a host protein affecting cellular susceptibility to anthrax
toxin. Proc Natl Acad Sci USA. 101:17246–17251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu K, Koo J, Jiang X, Chen R, Cohen SN and
Nathan C: Improved control of tuberculosis and activation of
macrophages in mice lacking protein kinase R. PLoS One.
7:e305122012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Philips JA, Rubin EJ and Perrimon N:
Drosophila RNAi screen reveals CD36 family member required for
mycobacterial infection. Science. 309:1251–1253. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bao Z, Guan S, Cheng C, Wu S, Wong SH,
Kemeny DM, Leung BP and Wong WS: A novel antiinflammatory role for
andrographolide in asthma via inhibition of the nuclear
factor-kappaB pathway. Am J Respir Crit Care Med. 179:657–665.
2009. View Article : Google Scholar
|
|
24
|
Botelho RJ, Hackam DJ, Schreiber AD and
Grinstein S: Role of COPI in phagosome maturation. J Biol Chem.
275:15717–15727. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jankowski A, Scott CC and Grinstein S:
Determinants of the phagosomal pH in neutrophils. J Biol Chem.
277:6059–6066. 2002. View Article : Google Scholar
|
|
26
|
Hackam DJ, Rotstein OD, Zhang W, Gruenheid
S, Gros P and Grinstein S: Host resistance to intracellular
infection: Mutation of natural resistance-associated macrophage
protein 1 (Nramp1) impairs phagosomal acidification. J Exp Med.
188:351–364. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vivas JR, Regnault B, Michel V, Bussière
FI, Avé P, Huerre M, Labigne A, D'Elios MM and Touati E: Interferon
gamma-signature transcript profiling and IL-23 upregulation in
response to Helicobacter pylori infection. Int J Immunopathol
Pharmacol. 21:515–526. 2008.PubMed/NCBI
|
|
28
|
Ehrt S, Schnappinger D, Bekiranov S,
Drenkow J, Shi S, Gingeras TR, Gaasterland T, Schoolnik G and
Nathan C: Reprogramming of the macrophage transcriptome in response
to interferon-gamma and Mycobacterium tuberculosis: signaling roles
of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med.
194:1123–1140. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weiss G and Schaible UE: Macrophage
defense mechanisms against intracellular bacteria. Immunol Rev.
264:182–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vandal OH, Pierini LM, Schnappinger D,
Nathan CF and Ehrt S: A membrane protein preserves intrabacterial
pH in intraphagosomal Mycobacterium tuberculosis. Nat Med.
14:849–854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jayachandran R, BoseDasgupta S and Pieters
J: Surviving the macrophage: Tools and tricks employed by
Mycobacterium tuberculosis. Curr Top Microbiol Immunol.
374:189–209. 2013.
|
|
32
|
Jefferies KC, Cipriano DJ and Forgac M:
Function, structure and regulation of the vacuolar
(H+)-ATPases. Arch Biochem Biophys. 476:33–42. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nathan C: Fresh approaches to
anti-infective therapies. Sci Transl Med. 4:140sr22012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jayaswal S, Kamal MA, Dua R, Gupta S,
Majumdar T, Das G, Kumar D and Rao KV: Identification of
host-dependent survival factors for intracellular Mycobacterium
tuberculosis through an siRNA screen. PLoS Pathog. 6:e10008392010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Divangahi M, Behar SM and Remold H: Dying
to live: How the death modality of the infected macrophage affects
immunity to tuberculosis. Adv Exp Med Biol. 783:103–120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Behar SM, Divangahi M and Remold HG:
Evasion of innate immunity by Mycobacterium tuberculosis: Is death
an exit strategy? Nat Rev Microbiol. 8:668–674. 2010.PubMed/NCBI
|
|
37
|
Parandhaman DK and Narayanan S: Cell death
paradigms in the pathogenesis of Mycobacterium tuberculosis
infection. Front Cell Infect Microbiol. 4:312014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vogt G and Nathan C: In vitro
differentiation of human macrophages with enhanced
antimycobacterial activity. J Clin Invest. 121:3889–3901. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hudson BG, Tryggvason K, Sundaramoorthy M
and Neilson EG: Alport's syndrome, Goodpasture's syndrome, and type
IV collagen. N Engl J Med. 348:2543–2556. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Medina E and North RJ: Evidence
inconsistent with a role for the Bcg gene (Nramp1) in resistance of
mice to infection with virulent Mycobacterium tuberculosis. J Exp
Med. 183:1045–1051. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rheault MN, Kren SM, Thielen BK, Mesa HA,
Crosson JT, Thomas W, Sado Y, Kashtan CE and Segal Y: Mouse model
of X-linked Alport syndrome. J Am Soc Nephrol. 15:1466–1474. 2004.
View Article : Google Scholar : PubMed/NCBI
|