Introduction
Lung cancer accounts for over one million deaths
each year worldwide (1). Tumor
recurrence and metastasis are the most common events occurring
after resection and lead to mortality (2). The causes of tumor recurrence and
metastasis have not yet been completely elucidated. With
increasingly severe environmental pollution, high concentrations of
aerial irritants, such as asbestos or fine particulate matter, in
the outdoor air have resulted in an increase in the prevalence of
malignant pleural mesothelioma and lung cancer (3). This has focused our attention on the
association between inflammation and cancer.
Inflammation is a definite cancer-causing factor, as
has been revealed by clinical and epidemiological studies (3). Indeed, an estimated 20–25% of all
types of cancer worldwide are associated with inflammation
(4). Neutrophils are regarded as
playing principal roles in inflammation-associated tumor
development and progression; reactive oxygen species (ROS) and
nitric oxide (NO), which are derived from cells involved in the
inflammatory response such as neutrophils, are key molecules that
stimulate tumorigenesis. ROS are known to initiate and promote
cancer (5–7).
The epithelial-to-mesenchymal transition (EMT)
occurs during embryogenesis, tissue repair, fibrosis and tumor
metastasis (8). The early events
of metastasis begin with the EMT, which transforms epithelial cells
into elongated, motile and invasive phenotypes (9). The analysis of a large number of
lung tumor specimens has shown that the majority of primary lung
cancer and even premalignant lesions are characterized by the
mesenchymal phenotype (10).
Transforming growth factor-β1 (TGF-β1) is a cytokine with multiple
functions and is widely accepted to be a profibrogenic factor
(11). TGF-β1 also plays various
roles in the development of diseases, including cancer (12,13). The TGF-β1-induced EMT enables
cells to acquire migratory and invasive properties. ROS have been
considered as contributors to the TGF-β1-mediated pathology
(14). In view of this, we
suggest that ROS may also be associated with tumor metastasis.
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2)
plays a crucial role in the regulation of antioxidant gene
expression. One of its most important functions is to encode for
ROS scavenging enzymes, such as superoxide dismutase (SOD) and
glutathione (GSH) peroxidase (15).
A recent study (16) has shown that Kelch-like
ECH-associated protein 1 (Keap1)-Nrf2-GSH signaling, which is
regarded as an effective modulator of TGF-β1-induced EMT changes,
suppresses Nrf2 activity and increases the level of ROS following
treatment with TGF-β1 in normal cells. In addition, it has been
demonstrated that the nuclear translocation level of pyruvate
kinase M2 (Pkm2), an alternatively spliced variant of the pyruvate
kinase gene that results in the cancer-specific Warburg effect, is
enhanced by EMT, leading to the acquisition of invasive behavior
(17). ROS have also been
demonstrated to inhibit the expression of Pkm2 through the
oxidation of Cys358, which is essential for catalytic
activity (18).
In the present study, we examined the association
between oxidative stress and energy metabolism in TGF-β1-induced
EMT. We examined the expression of Pkm2 to reflect the changes in
energy metabolism. Furthermore, we demonstrated the functionality
of a pro-resolving lipid mediator, aspirin-triggered resolvin D1
(AT-RvD1), which is known to affect the resolution of inflammation
(19,20) by inhibiting TGF-β1-induced EMT.
This occurs through the inhibition of the mTOR pathway by reducing
the expression of Pkm2.
Materials and methods
Reagents
Recombinant human TGF-β1 was purchased from
Peprotech, Inc. (Rocky Hill, NJ, USA). AT-RvD1 was purchased from
Cayman Chemical Co. (Ann Arbor, MI, USA). Antibodies against Nrf2
(sc-722), β-actin (sc-7210 and Pkm2 (sc-135048) were obtained from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The mTOR
activator MHY1485 was purchased from Millipore Corp. (Bedford, MA,
USA). Fetal bovine serum (FBS) and Dulbecco's modified Eagle's
medium (DMEM) were obtained from Gibco (Carlsbad, CA, USA).
Cell culture
The human lung adenocarcinoma epithelial cell line
A549 was obtained from the Experimental Center of the Clinical
Medicine College of Yangzhou University (Yangzhou, China). These
cells were cultured in DMEM supplemented with 10% FBS, streptomycin
and penicillin (Beyotime Institute of Biotechnology, Shanghai,
China). The cells were grown at 37°C in a humidified 5%
CO2 atmosphere. The A549 cells were treated with
different concentrations of TGF-β1 (1,10 and 20ng/ml) for 48h. In
order to examine the effects of AT-RvD1, the cells were pre-treated
with AT-RvD1 for 30 min and then treated with TGF-β1. In
experiments using the mTOR activator, the cells were pre-treated
with MHY1485 (10μmol/ml) for 2 h and then treated with
AT-RvD1 or TGF-β1 as described above.
Measurement of cell proliferation
The cells were seeded in 100 μl serum-free
media with TGF-β1 or AT-RvD1 for 48 h in 96-well plates. At 48 h,
cell proliferation was measured with an MTT cell proliferation
assay kit (Beyotime Institute of Biotechnology) at an absorbance of
490 nm using a plate reader (3599; Corning, Corning, NY, USA). All
measurements were performed in triplicate and the experiments were
repeated three times.
Migration assay using scratch-wound
healing
The cell cultures were prepared with different
concentrations of TGF-β1 or AT-RvD1 in 6-well plates to 80–90%
confluence prior to performing the scratch-wound healing assay. The
cells were washed with phosphate buffered saline (PBS) three times
and cultured in serum-free medium. After incubating for 48 h at
37°C, images of the cells that had migrated onto the scratches were
captured and counted under a microscope (3516; Corning). All images
were randomly acquired with a 100X objective at consistent
intensity settings. We analyzed five fields of view for each
experimental condition. The experiment was repeated three
times.
Transwell invasion assay
Cell invasion was determined using 24-well plates
with Transwell chamber inserts (Chemicon International, Inc.,
Atlanta, GA, USA), which had membranes coated with Matrigel. In the
upper chambers, 3×104 A549 cells were seeded in FBS-free
culture media, and media with 10% serum were added to the lower
chambers. After a 24-h incubation, the cells on the upper surface
of the membrane were scraped off, and the cells that had migrated
onto the lower surface of the membrane were immersed in 500
μl media with 0.5 mg/ml MTT for enumeration.
Measurement of ROS
Cellular ROS levels were determined using a ROS
Assay kit (Beyotime Institute of Biotechnology). The cells in
96-well plates were treated with TGF-β1 or AT-RvD1 for 48 h. The
cells were then incubated with 200 μl DMEM and a
2′,7′-dichlorofluorescein diacetate (DCFH-DA) fluorescent probe for
30 min at 37°C. The fluorescence intensity was measured with a
microplate reader at 488 nm excitation and 525 nm emission
wavelengths. Moreover, a fluorescence microscope (Axio Observer A1,
Zeiss, Oberkochen, Germany) was used to capture images.
Total RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen, Carlsbad, USA) according to the manufacturer's
instructions. The reverse transcription of 1 μg RNA was
performed using qRT SuperMix (Vazyme, Nanjing, China). RT-qPCR was
performed using the ABI StepOne Plus system (Applied Biosystems
Life Technologies, Foster City, CA, USA) with the AceQ SYBR-Green
Master Mix (Vazyme). The primers were synthesized by Bioneer Corp.
(Daejeon, Korea), and the following primer sequences for the human
genes were used: E-cadherin, 5′-AAAGGCCCATTTCCTAAAAACCT-3′ and
5′-TGCGTTCTCTATCCAGAGGCT-3′; vimentin, 5′-TGCCGTTGAAGCTGCTAACTA-3′
and 5′-CCAGAGGGAGTGAATCCAGATTA-3′; Nrf2, 5′-TTCCCGGTCACATCGAGAG-3′
and 5′-TCCTGTTGCATACCGTCTAAATC-3′; Pkm2,
5′-ATGTCGAAGCCCCATAGTGAA-3′ and 5′-TGGGTGGTGAATCAATGTCCA-3′; and
β-actin, 5′-AAGACCTGTACGCCAACACAGT-3′ and
5′-AGAAGCATTTGCGGTGGACGAT-3′. The amplified expression of gene
transcriptions were normalized to β-actin expression. The following
PCR conditions were used: holding stage, 95°C for 5 min; cycling
stage, 95°C for 10 sec and 60°C for 30 sec 40 times; melt curve
stage, 95°C for 15 sec, 60°C for 60 sec and 95°C for 15 sec. Cycle
threshold (ΔΔCt) values were calculated for each experimental
group, which indicated the number of available template cDNA in
each reaction. The gene expression levels were compared using the
values of 2−ΔΔCt.
Total protein extraction and western blot
analysis
The total protein extracted from cells was analyzed
using a radioimmunoprecipitation assay (RIPA) and
phenylmethylsulfonyl fluoride (PMSF) (Beyotime Institute of
Biotechnology). Protein concentrations were measured using the
bicinchoninic acid (BCA) assay. The protein samples were separated
by electrophoresis on 6–10% sodium dodecyl sulfate
(SDS)-polyacrylamide gels and transferred to polyvinylidene
fluoride (PVDF) membranes (Invitrogen). The membranes were blocked
with 5% skim milk for 2 h and then incubated with the appropriate
antibodies against Nrf2, Pkm2 and β-actin (Santa Cruz
Biotechnology, Inc.) in 4% bovine serum albumin (BSA) at 4°C
overnight. The membranes were further incubated for 2 h with a
peroxidase-conjugated secondary antibody (Beyotime Institute of
Biotechnology) at room temperature, and the results were detected
using a ChemiDoc imaging system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and analyzed using ImageJ software.
Statistical analysis
All data are expressed as the means ± SEM and were
analyzed by one-way analysis of variance (ANOVA) followed by the
Newman-Keuls multiple comparison test (GraphPad Prism software;
GraphPad Software, Inc., La Jolla, CA, USA). Data were recorded
from at least three independent experiments. A p-value <0.05 was
considered to indicate a statistically significant difference.
Results
TGF-β1-induced EMT increases ROS levels
and the expression of Pkm2 in A549 cells
TGF-β1-induced EMT in the A549 cells enhanced the
mRNA expression of the mesenchymal marker vimentin, and reduced the
expression of the epithelial marker E-cadherin (Fig. 1D). These findings were consistent
with those of previous studies (13,21). The ROS levels were significantly
increased in the A549 cells following incubation with TGF-β1 in a
concentration-dependent manner, with 20 ng/ml TGF-β1 significantly
elevating the levels of ROS (Fig.
1A). The migration and invasion of A549 cells were enhanced by
TGF-β1 (Fig. 1C). It has been
previously demonstrated that Nrf2 plays a critical role in the
removal of cellular ROS, and TGF-β1 is capable of reducing Nrf2
expression in renal epithelial cells (16). The effect of TGF-β1 on Nrf2
expression in A549 cells was also confirmed by RT-qPCR and western
blot analyses (Fig. 1D and E). In
addition, the expression of Pkm2, which plays a crucial role in
tumor metabolism, was increased by exposure to TGF-β1 (Fig. 1D and E). These results suggest
that TGF-β1-induced EMT increased ROS levels and the expression of
Pkm2 in A549 cells.
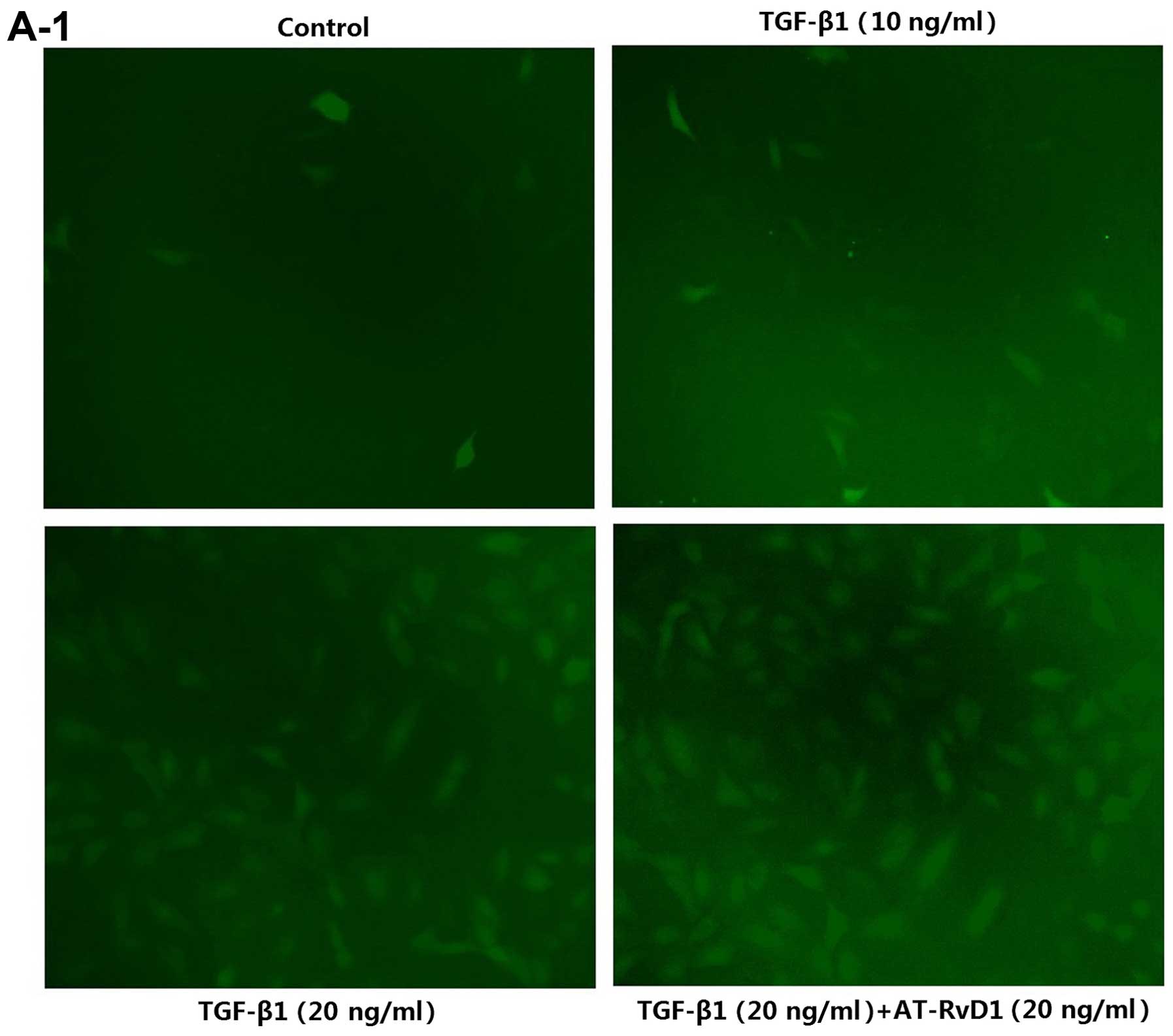 | Figure 1AT-RvD1 inhibition of TGF-β1-induced
EMT is associated with ROS and Pkm2. (A1 and 2) Determination of
ROS levels. Cells were pre-treated with AT-RvD1 for 30 min. The
A549 cells in 96-well plates were then treated with different
concentrations of TGF-β1 (1, 10 and 20 ng/ml) for 48 h. We used an
ROS assay kit, a microplate reader and a fluorescence microscope as
described in the Materials and methods. Images in A1, ×100
magnification. (B) Effects of AT-RvD1, TGF-β1 and mTOR activator on
the proliferation of A549 cells. The results showed that AT-RvD1
and TGF-β1 had no effect on cell proliferation. AT-RvD1,
aspirin-triggered resolvin D1; TGF-β1, transforming growth
factor-β1; EMT, epithelial-to-mesenchymal transition; ROS, reactive
oxygen species; Pkm2, pyruvate kinase M2. (C1-3) Effects of AT-RvD1
on TGF-β1-induced cell migration and invasion. The results showed
that AT-RvD1 in combinaton with TGF-β1 inhibited TGF-β1-induced
cell migration and invasion. AT-RvD1, aspirin-triggered resolvin
D1; TGF-β1, transforming growth factor-β1; EMT,
epithelial-to-mesenchymal transition; ROS, reactive oxygen species;
Pkm2, pyruvate kinase M2. (D) RT-qPCR of EMT markers in A549 cells
treated with TGF-β1 (0–20 ng/ml). (E) Western blot analysis of the
expression of Nrf2 and Pkm2. β-actin was used as a control. The
values were normalized to each control and the means ± SE from
three independent experiments are presented. *p<0.05
compared with control groups. AT-RvD1, aspirin-triggered resolvin
D1; TGF-β1, transforming growth factor-β1; EMT,
epithelial-to-mesenchymal transition; ROS, reactive oxygen species;
Pkm2, pyruvate kinase M2; Nrf2, nuclear factor (erythroid-derived
2)-like 2. |
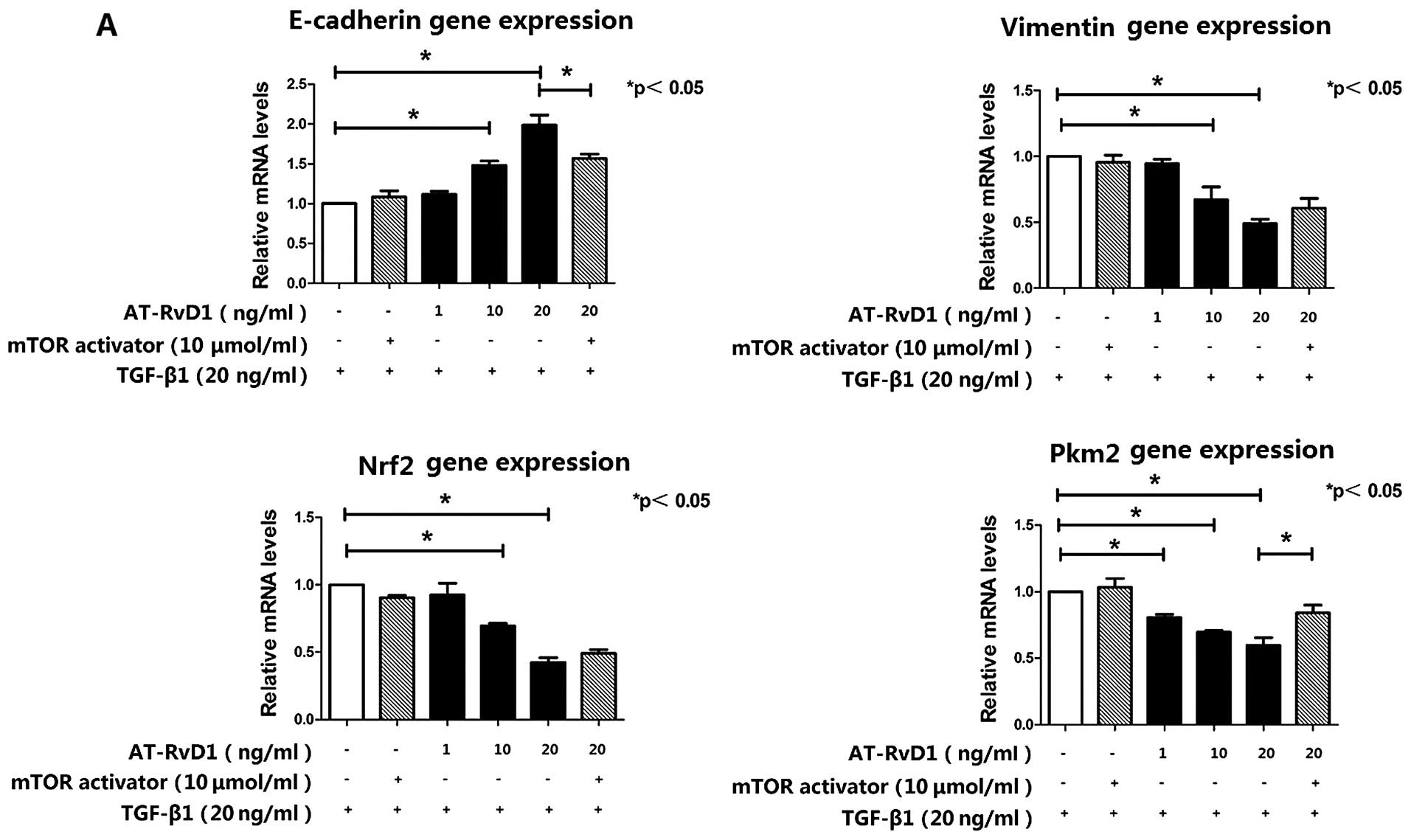
AT-RvD1 inhibits TGF-β1-induced EMT
through inhibition of the mTOR pathway by reducing the expression
of Pkm2
To elucidate the mechanism responsible for the
effects of AT-RvD1 on the TGF-β1-induced EMT process, the cells
were treated with different concentrations of AT-RvD1 30 min prior
to the addition of TGF-β1. AT-RvD1 treatment blocked the expression
of the mesenchymal marker vimentin and restored the expression of
E-cadherin. The effects of AT-RvD1 on the expression of E-cadherin
and vimentin in TGF-β1-treated A549 cells were confirmed using
RT-qPCR (Fig. 2A). However,
AT-RvD1 did not reduce the ROS levels in the TGF-β1-treated A549
cells (Fig. 1A). The reduced
expression of Nrf2 in the TGF-β1-treated A549 cells was further
lowered by AT-RvD1 (Fig. 1E).
However, AT-RvD1 clearly reduced the concentration-dependent
TGF-β1-induced migration and invasion (Fig. 1C) but did not affect the
proliferative properties of the TGF-β1-treated A549 cells (Fig. 1B). Our experiments have
conclusively shown that the mTOR activator had no effect on the
TGF-β1-induced EMT. However, the activation of mTOR signaling
restored the expression of Pkm2, which was suppressed by AT-RvD1,
and partly downregulated the expression of E-cadherin (Fig. 2). The inhibitory effect of AT-RvD1
on the TGF-β1-induced EMT was weakened by mTOR activator.
These results suggest that AT-RvD1 inhibited
TGF-β1-induced EMT through the inhibition of the mTOR pathway by
reducing the expression of Pkm2.
Discussion
Cancer, similar to chronic respiratory diseases and
diabetes, is a noncommunicable disease. It is characterized by a
long duration and slow development (3). The high mortality rates associated
with cancer are caused by tumor cell metastases. In fact,
metastases are the primary causes of 90% of cancer deaths (22). The EMT is involved in tumor cell
metastasis. The EMT process is directly associated with the
features of the primary tumor microenvironment (23). A molecular feature of the EMT is
the downregulation of epithelial markers and the upregulation of a
number of mesenchymal markers (24).
The tumor stroma is composed of an extracellular
matrix, vasculature, lymphatic systems and other cells. Changes to
these cell phenotypes are associated with tumorigenesis and
metastasis. The resultant cells are tumor-associated fibroblasts
and macrophages (21,25). Several inflammatory mediators are
released during these changes to the tumor microenvironment,
leading to the progression and metastasis of the tumor (21). Specialized pro-resolving lipid
mediators, such as lipoxin and resolvin, due to their involvement
in the inhibitory process of EMT (21,27), have the ability to prevent
excessive neutrophil trafficking to inflammation sites and thereby
promote the recovery of damaged tissue (20,26,27). However, the role of AT-RvD1
remains unclear. Thus, we examined the effects of the available
AT-RvD1 on the TGF-β1-induced EMT of lung cancer cells.
AT-RvD1 suppressed the TGF-β1-regulated expression
of vimentin and restored the expression of E-cadherin in A549 lung
cancer cells. However, the effects of AT-RvD1 are not associated
with cell proliferation, which is one of the characteristics of the
EMT. Moreover, AT-RvD1 also inhibited the TGF-β1-induced migration
and invasion of A549 cells. A previous study (21) has shown that resolvin (Rv)D1 and
RvD2 both suppressed TGF-β1-induced EMT, and RvD1 inhibited the
TGF-β1-induced EMT through the lipoxin A4 receptor/formyl peptide
receptor 2 (ALX/FPR2) and G protein-coupled receptor 32 (GPR32)
receptors by reducing the expression of ZEB1. Lipoxin A4 (LXA4),
another mediator of resolution, has also been reported to inhibit
the connective tissue growth factor (CTGF)-induced EMT process
(27). Our results have shown
that AT-RvD1 may also be useful in suppressing the EMT of lung
cancer cells. Our future studies will explore the association
between the EMT process and oxidative stress.
The effects of TGF-β1 and AT-RvD1 correlate with the
high levels of ROS in A549 lung cancer cells. ROS have been
reported to promote the TGF-β1-induced EMT genetic changes
(16). However, high
concentrations of ROS may also damage cellular components and
compromise cell viability (18).
Acute increases in the intracellular concentrations of ROS have
been found to inhibit the glycolytic enzyme Pkm2 in lung cancer
cells (18). Pkm2 enables
proliferating cells to divert glucose into anabolic pathways to
meet the increased biosynthetic demands of proliferation (18). Thus, TGF-β1-induced ROS may be
associated with cell proliferation. The inhibitory effect of TGF-β1
on cell proliferation is usually on normal cells and some types of
cancer cell, and is associated with the concentration of TGF-β1 and
some cancer-specific factors, so in our study TGF-β1 induced the
migration of the cells but had no effect on cell proliferation.
We also examined the expression of Nrf2, which is
one of the key proteins involved in antioxidant signaling pathways.
Both TGF-β1 and AT-RvD1 suppressed the expression of Nrf2. The
antioxidant signaling pathway Keap1-Nrf2 is known to have
protective effects in both normal and cancer cells (28). With the increase in ROS levels,
the activation of the antioxidant response element (ARE) reporter
has been found to lead to elevated levels of GSH, which enable
NADPH to sustain cell proliferation (20,29,30). The low expression of Nrf2 weakens
the cell protection and leads to increased ROS levels.
We also examined the elevated expression of Pkm2
during the TGF-β1-induced EMT. Treatment with AT-RvD1 reduced the
expression of Pkm2 in A549 cells. Glucose is also a major carbon
source for biosynthetic processes, and Pkm2 is known to partially
determine how glucose is metabolized in cancer cells (31). Pkm2 is commonly upregulated in
many types of human cancer and has been shown to play a crucial
role in reprogramming cellular metabolism, gene transcription and
cell cycle progression (26,32). A study (17) demonstrated that the EMT stimulates
the nuclear translocation of Pkm2 and decreases E-cadherin
transcription. Pkm2 interacts with transcription factor
TGF-β-induced factor homeobox 2 (TGIF2), which induces the
deacetylation of histone H3, resulting in the repressed expression
of E-cadherin. The discovery that AT-RvD1 reduces the expression of
Pkm2 is notable, and the the inhibition of TGF-β1-induced EMT by
AT-RvD1 is possibly due to the reduced expression of Pkm2. Further
studies are necessary in order to explore the associated
mechanisms.
The mTOR pathway is known to regulate the expression
of various glycolytic enzymes including Pkm2 (33,34). The inhibition of mTOR also reduces
Pkm2 expression (35). Thus, we
examined the effects of mTOR activator on changes made by AT-RvD1
to the TGF-β1-induced EMT and found that there was an association.
The mTOR activator did restore the expression of Pkm2, which had
been suppressed by AT-RvD1, and subsequently downregulated the
expression of E-cadherin. The inhibitory effect of AT-RvD1 on the
TGF-β1-induced EMT was weakened. However, increased oxidative
stress is known to inhibit the phosphoinositide 3-kinase
(P13K)/AKT/mTOR pathways through the disruption of mTORC1 formation
(36). This may potentially
explain the means through which increased oxidative stress affects
cellular energy metabolism. AT-RvD1 maintained a high level of
oxidative stress and reduced the expression of Pkm2 whereas it had
no effect on cell proliferation, thus, the mechanism may be
associated with a particular receptor (37). These results suggest that AT-RvD1
inhibited the TGF-β1-induced EMT through the inhibition of the mTOR
pathway by reducing the expression of Pkm2.
There are a few limitations to our experiments. We
illustrated the association between energy metabolism and oxidative
stress; however, the dynamics are complex and require further
investigation. In addition, the effect of AT-RvD1 has not yet been
confirmed in in vivo animal cancer models. In future
studies, we will focus our efforts on exploring other mechanisms
responsible for the low expression of Pkm2 mediated by AT-RvD1 and
also perform animal experiments.
Our results suggest the possibility that AT-RvD1
suppresses the TGF-β1-induced EMT by affecting cellular energy
metabolism and oxidative stress.
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Williams BA, Sugimura H, Endo C, Nichols
FC, Cassivi SD, Allen MS, Pairolero PC, Deschamps C and Yang P:
Predicting postrecurrence survival among completely resected
non-small-cell lung cancer patients. Ann Thorac Surg. 81:1021–1027.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Okada F: Inflammation-related
carcinogenesis: current findings in epidemiological trends, causes
and mechanisms. Yonago Acta Med. 57:65–72. 2014.PubMed/NCBI
|
|
4
|
Loomis D, Grosse Y, Lauby-Secretan B, El
Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Baan R, Mattock
H and Straif K; International Agency for Research on Cancer
Monograph Working Group IARC: The carcinogenicity of outdoor air
pollution. Lancet Oncol. 14:1262–1263. 2013. View Article : Google Scholar
|
|
5
|
Tazawa H, Okada F, Kobayashi T, Tada M,
Mori Y, Une Y, Sendo F, Kobayashi M and Hosokawa M: Infiltration of
neutrophils is required for acquisition of metastatic phenotype of
benign murine fibrosarcoma cells: implication of
inflammation-associated carcinogenesis and tumor progression. Am J
Pathol. 163:2221–2232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Okada F, Kobayashi M, Tanaka H, Kobayashi
T, Tazawa H, Iuchi Y, Onuma K, Hosokawa M, Dinauer MC and Hunt NH:
The role of nicotinamide adenine dinucleotide phosphate
oxidase-derived reactive oxygen species in the acquisition of
metastatic ability of tumor cells. Am J Pathol. 169:294–302. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Okada F, Tazawa H, Kobayashi T, Kobayashi
M and Hosokawa M: Involvement of reactive nitrogen oxides for
acquisition of metastatic properties of benign tumors in a model of
inflammation-based tumor progression. Nitric Oxide. 14:122–129.
2006. View Article : Google Scholar
|
|
8
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gemmill RM, Roche J, Potiron VA, Nasarre
P, Mitas M, Coldren CD, Helfrich BA, Garrett-Mayer E, Bunn PA and
Drabkin HA: ZEB1-responsive genes in non-small cell lung cancer.
Cancer Lett. 300:66–78. 2011. View Article : Google Scholar
|
|
10
|
Prudkin L, Liu DD, Ozburn NC, Sun M,
Behrens C, Tang X, Brown KC, Bekele BN, Moran C and Wistuba II:
Epithelial-to-mesenchymal transition in the development and
progression of adenocarcinoma and squamous cell carcinoma of the
lung. Mod Pathol. 22:668–678. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pohlers D, Brenmoehl J, Löffler I, Müller
CK, Leipner C, Schultze-Mosgau S, Stallmach A, Kinne RW and Wolf G:
TGF-β and fibrosis in different organs - molecular pathway
imprints. Biochim Biophys Acta. 1792:746–756. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kawata M, Koinuma D, Ogami T, Umezawa K,
Iwata C, Watabe T and Miyazono K: TGF-β-induced
epithelial-mesenchymal transition of A549 lung adenocarcinoma cells
is enhanced by pro-inflammatory cytokines derived from RAW 264.7
macrophage cells. J Biochem. 151:205–216. 2012. View Article : Google Scholar
|
|
13
|
Park MK, You HJ, Lee HJ, Kang JH, Oh SH,
Kim SY and Lee CH: Transglutaminase-2 induces N-cadherin expression
in TGF-β1-induced epithelial mesenchymal transition via
c-Jun-N-terminal kinase activation by protein phosphatase 2A
down-regulation. Eur J Cancer. 49:1692–1705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kashihara N, Haruna Y, Kondeti VK and
Kanwar YS: Oxidative stress in diabetic nephropathy. Curr Med Chem.
17:4256–4269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kobayashi M and Yamamoto M: Molecular
mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene
regulation. Antioxid Redox Signal. 7:385–394. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ryoo IG, Ha H and Kwak MK: Inhibitory role
of the KEAP1-NRF2 pathway in TGFβ1-stimulated renal epithelial
transition to fibroblastic cells: a modulatory effect on SMAD
signaling. PLoS One. 9:e932652014. View Article : Google Scholar
|
|
17
|
Hamabe A, Konno M, Tanuma N, Shima H,
Tsunekuni K, Kawamoto K, Nishida N, Koseki J, Mimori K, Gotoh N, et
al: Role of pyruvate kinase M2 in transcriptional regulation
leading to epithelial-mesenchymal transition. Proc Natl Acad Sci
USA. 111:15526–15531. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Anastasiou D, Poulogiannis G, Asara JM,
Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW,
Auld DS, et al: Inhibition of pyruvate kinase M2 by reactive oxygen
species contributes to cellular antioxidant responses. Science.
334:1278–1283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fadok VA, Bratton DL, Konowal A, Freed PW,
Westcott JY and Henson PM: Macrophages that have ingested apoptotic
cells in vitro inhibit proinflammatory cytokine production through
autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J
Clin Invest. 101:890–898. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Serhan CN, Hong S, Gronert K, Colgan SP,
Devchand PR, Mirick G and Moussignac RL: Resolvins: a family of
bioactive products of omega-3 fatty acid transformation circuits
initiated by aspirin treatment that counter proinflammation
signals. J Exp Med. 196:1025–1037. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee HJ, Park MK, Lee EJ and Lee CH:
Resolvin D1 inhibits TGF-β1-induced epithelial mesenchymal
transition of A549 lung cancer cells via lipoxin A4 receptor/formyl
peptide receptor 2 and GPR32. Int J Biochem Cell Biol.
45:2801–2807. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Steeg PS: Tumor metastasis: mechanistic
insights and clinical challenges. Nat Med. 12:895–904. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neil JR, Johnson KM, Nemenoff RA and
Schiemann WP: Cox-2 inactivates Smad signaling and enhances EMT
stimulated by TGF-β through a PGE2-dependent mechanisms.
Carcinogenesis. 29:2227–2235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Talbot LJ, Bhattacharya SD and Kuo PC:
Epithelial-mesenchymal transition, the tumor microenvironment, and
metastatic behavior of epithelial malignancies. Int J Biochem Mol
Biol. 3:117–136. 2012.PubMed/NCBI
|
|
25
|
Yang W and Lu Z: Pyruvate kinase M2 at a
glance. J Cell Sci. 128:1655–1560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spite M, Norling LV, Summers L, Yang R,
Cooper D, Petasis NA, Flower RJ, Perretti M and Serhan CN: Resolvin
D2 is a potent regulator of leukocytes and controls microbial
sepsis. Nature. 461:1287–1291. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu SH, Zhang YM, Tao HX and Dong L:
Lipoxin A(4) inhibits transition of epithelial to mesenchymal cells
in proximal tubules. Am J Nephrol. 32:122–136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Homma S, Ishii Y, Morishima Y, Yamadori T,
Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N,
et al: Nrf2 enhances cell proliferation and resistance to
anticancer drugs in human lung cancer. Clin Cancer Res.
15:3423–3432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brown SL, Sekhar KR, Rachakonda G, Sasi S
and Freeman ML: Activating transcription factor 3 is a novel
repressor of the nuclear factor erythroid-derived 2-related factor
2 (Nrf2)-regulated stress pathway. Cancer Res. 68:364–368. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dhakshinamoorthy S, Jain AK, Bloom DA and
Jaiswal AK: Bach1 competes with Nrf2 leading to negative regulation
of the antioxidant response element (ARE)-mediated NAD(P) H:quinone
oxidoreductase 1 gene expression and induction in response to
antioxidants. J Biol Chem. 280:16891–16900. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang
L, You MJ, Koh MY, Cote G, Aldape K, et al: EGFR-induced and PKCε
monoubiquitylation-dependent NF-κB activation upregulates PKM2
expression and promotes tumorigenesis. Mol Cell. 48:771–784. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Majumder PK, Febbo PG, Bikoff R, Berger R,
Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR,
et al: mTOR inhibition reverses Akt-dependent prostate
intraepithelial neoplasia through regulation of apoptotic and
HIF-1-dependent pathways. Nat Med. 10:594–601. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun Q, Chen X, Ma J, Peng H, Wang F, Zha
X, Wang Y, Jing Y, Yang H, Chen R, et al: Mammalian target of
rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is
critical for aerobic glycolysis and tumor growth. Proc Natl Acad
Sci USA. 108:4129–4134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iqbal MA and Bamezai RNK: Resveratrol
inhibits cancer cell metabolism by down regulating pyruvate kinase
M2 via inhibition of mammalian target of rapamycin. PLoS One.
7:e367642012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hambright HG, Meng P, Kumar AP and Ghosh
R: Inhibition of PI3K/AKT/mTOR axis disrupts oxidative
stress-mediated survival of melanoma cells. Oncotarget.
6:7195–7208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Al-Zaubai N, Johnstone CN, Leong MM, Li J,
Rizzacasa M and Stewart AG: Resolvin D2 supports MCF-7 cell
proliferation via activation of estrogen receptor. J Pharmacol Exp
Ther. 351:172–180. 2014. View Article : Google Scholar : PubMed/NCBI
|