Colorectal cancer (CRC) is the second most common
type of cancer and the fourth leading cause of cancer-related
mortality worldwide, occupying approximately 9.7% of the total
cancer cases and 8.5% of the number of deaths in 2012 (1). CRC is composed of heterogeneous cell
populations differing in gene expression and tumorigenicity
(2,3). Sporadic CRC and hereditary CRC both
originate from the stem cell stage. CRC stem cells (CCSCs)
represent a small fraction of the CRC cell population with
self-renewal and multi-lineage differentiation potential and the
ability to drive tumorigenicity (4).
It is thought that CCSCs originate in three
different ways: first, they may be derived from the malignant
transformation of normal colorectal stem cells. Colorectal stem
cells have the ability to proliferate and self-repair. Gene
mutations can further accumulate during their long survival.
Evidence has demonstrated that colorectal stem cells become
tumorigenic more easily (4–6).
Barker et al suggested that only Leucine-rich
repeat-containing G protein-coupled receptor 5 (Lgr5)+
stem cells, in cooperation with APC-deficiency, may lead to
colorectal adenoma formation (7).
The stem-like Lgr5+ tumor initiating cells located in
the base of adenomas are similar to normal stem cells (8). In the initiation process of CRC,
normal colorectal stem cells acquire oncogenic mutations through
the interaction between internal and external factors.
Subsequently, in the evolution of CRC, the heterozygous loss of
APC, DCC and p53 occurs, accompanied by DNA
damage, DNA-repair mutations and altered methylation status
(9,10). Second, CCSCs may originate from
the dedifferentiation of common cancer cells. Cells with certain
differentiation characteristics, such as progenitor cells or mature
cells, acquire stemness by dedifferentiation. The successful
induction of induced pluripotent stem cells (IPS) has demonstrated
that differentiated cells, even in the stage of terminal
differentiation, can regain stemness through a reset by certain
specific regulation factors. Transducing transcription factor
Oct3/4, Sox2, c-Myc and Klf4 into mouse
fibroblast cells can drive cells to dediffer-entiate and acquire
stemness (6). Schwitalla et
al indicated that increasing nuclear factor-κB (NF-κB)
signaling in intestinal epithelial cells would activate the Wnt
signaling pathway, thus eliciting dedifferentiation and promoting
tumorigenicity (11). Third,
CCSCs may originate from cell malignant transformation through the
influence of the micro-environment. The transformation of
non-cancer stem cells to cancer stem cells is dependent on
transforming growth factor-β (TGF-β) signaling in the
micro-environment, and the process is most likely relevant to
epithelial-mesenchymal transition (EMT) (12,13). Mani et al found that
mammary gland cells undergoing EMT by Snail or Twist induction
regained stem cell markers and the ability to self-renew (14).
CCSCs are heterogeneous, as they contain various
subpopulations or are in different stages of stem cell development
(2). B-cell-specific Moloney
murine leukemia virus insertion site 1 (Bmi1)+ quiescent
cancer stem cells are insensitive to high-doses of radiation, while
Lgr5+ active cancer stem cells have a strong homeostatic
regeneration ability (15). If
the latter become injured or destroyed, the former can mobilize to
transform into an active status. Hence, quiescent cancer stem cells
most likely function as a reservoir to maintain the homeostasis of
stem cells. The micro-environment dictates the balance between them
(15,16). At present, therapy for CRC targets
mainly active cells, while quiescent stem cells can escape, leading
to relapse and resistance to treatment.
The characteristics of CCSCs are regulated by
different mechanisms. Self-renewal is a fundamental feature of the
malignant biological behaviors of CCSCs. Several pathways, cell
surface markers and micro-environmental factors are involved in
CCSC self-renewal.
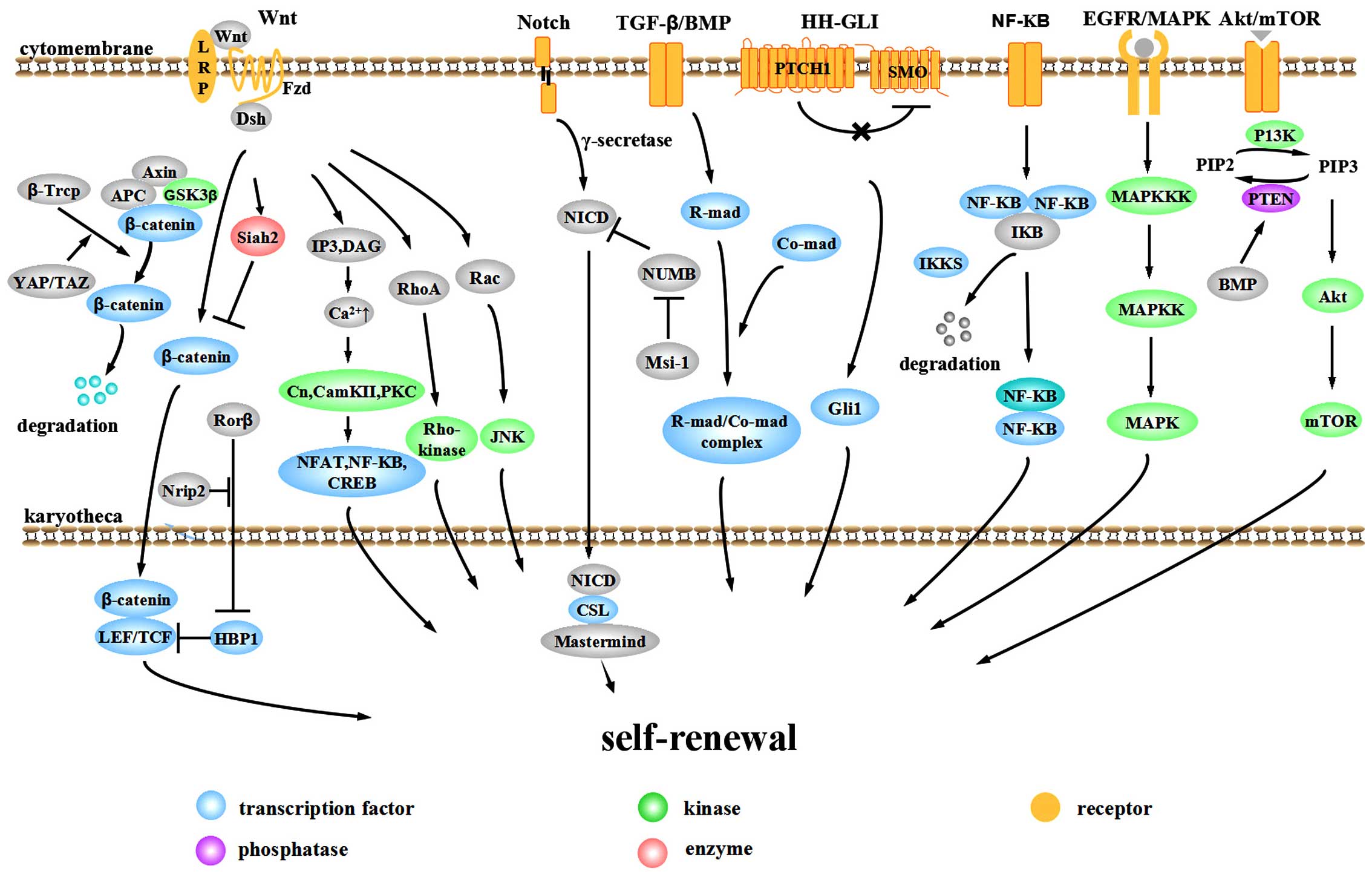
The aberrant activation of signaling pathways plays
important roles in the evolution and progression of CRC. The Wnt,
Notch, bone morphogenetic protein (BMP)/TGF-β, Hedgehog-Gli
(HH-GLI), epidermal growth factor receptor (EGFR)/mitogen-activated
protein kinase (MAPK), NF-κB and Akt/mTOR pathways are involved in
the self-renewal of CCSCs (Fig.
1).
The Wnt pathway is one of the most important
pathways in the tumorigenesis and progression of CRC. Over 90% of
CRC cases display an over-activation of Wnt signaling (26). According to whether this
activation is dependent on transcriptional regulation by
transporting β-catenin into the nucleus, the Wnt pathway is divided
into the canonical pathway (β-catenin-dependent) and the
non-canonical pathway (β-catenin-independent). The canonical
pathway mainly consists of extracellular signaling proteins (Wnt1,
Wnt3a, Wnt8, etc.), the transmembrane receptor Frizzled (Fzd),
co-receptor low-density lipoprotein-related receptor 5/6 (LRP5/6),
Dishevelled (Dsh), β-catenin, axis inhibitor (Axin) and the
intranuclear transcription factor T-cell factor (TCF)/lymphoid
enhancer factor (LEF). In the absence of Wnt, β-catenin interacts
with Axin, adenomatous polyposis coli (APC) and glycogen synthase
kinase-3β (GSK-3β) to form a destruction complex and is
phosphorylated by GSK-3β. Phosphorylated β-catenin recruits
ubiquitin E3 β-transducin repeat containing protein (β-TrCP) and is
then degraded by the proteasome, thus maintaining cytoplasmic
β-catenin at a relatively low level (27–29). With Wnt signaling, the Wnt protein
binds to the Fzd-LRP complex and recruits Dsh to the cytomembrane.
The induction of phosphorylation by Dsh separates GSK-3β from Axin
and inhibits the formation of the Axin-GSK-3β-APC complex,
inhibiting the phosphorylation and ubiquitination of β-catenin.
Free β-catenin accumulates in the cytoplasm and translocates to the
nucleus, targeting LEF and TCF (30). These proteins promote the
transcription and expression of downstream targets, such as
c-Myc, cyclin D1 and Axin2 (31). Disrupting the β-catenin/TCF-4
activity of CRC cells induces a rapid G1 arrest and blocks the
proliferative compartment in colon crypts from genetic programming.
The suppression by c-Myc on the promoter of the cell cycle
inhibitor p21 plays an important role in this process. Evidence
from conditional gene deletion of c-Myc suggests that
c-Myc(−/−) crypt cells are smaller in size and have a slower
cycle compared to wild-type cells and c-Myc-deficient crypts
are prone to loss and can be replaced by c-Myc-proficient
crypts within weeks (32,33).
In recent years, many other non-classical Wnt
proteins have been discovered, establishing a much more complex and
precise regulatory network for the canonical Wnt pathway. Yap/TAZ
appears to be an important component of the β-catenin destruction
complex. In the absence of Wnt, YAP/TAZ recruits β-TrCP to the
complex and degrades β-catenin, blocking pathway activity. When the
Wnt pathway is activated, YAP/TAZ is released from the complex so
that β-catenin can accumulate in the nucleus to stimulate
downstream effectors (34).
Polycomb group protein Bmi-1 activates Wnt signaling by
upregulating the transcription of Wnt factors Wnt3A, Wnt7A, Wnt10A
and Wnt4 or by repressing the DKK family. The Wnt downstream target
c-Myc in turn promotes the transcription and
trans-activation of Bmi-1, forming a positive feedback loop
(35). Oncogenic transcription
factor MYB cooperates with β-catenin to co-stimulate c-Myc
expression (36).
High Wnt activity can define the CCSC population
functionally. CRC cells with high Wnt activity upregulate the
expression of the stem cell-associated genes, Lgr5 and
achaete-scute family bHLH transcription factor 2 (ASCL2),
while ones with low Wnt activity upregulate the expression of the
epithelial differentiation-associated genes, mucin 2 (MUC2),
keratin 20 (KRT20) and fatty acid binding protein 2
(FABP2) (37).
CD133+, CD24+CD29+ or
CD44+CD166+ cells exhibit high Wnt activity
(37). High Wnt activity is
associated with high clonogenic cancer stem cell potential, while
low Wnt activity is not (37).
Similar evidence defines the association between Wnt activity and
CRC cell proliferation and EMT (38).
We recently found that nuclear receptor-interacting
protein 2 (Nrip2) is a novel interactor of the non-canonical Wnt
pathway. Nrip2 inhibits the transcription of HMG-box transcription
factor 1 (HBP1) through the arrest of retinoic acid-related orphan
nuclear receptor β (RORβ) in the cytoplasm and and its subsequent
degradation to promote the transcription of the downstream gene,
TCF/LEF, a process connected to activating the canonical Wnt
signaling pathway (unpublished data).
Notch can also be divided into canonical and
non-canonical pathways. Typical Notch ligands include Delta-like
(DLL)1, DLL3, DLL4, jagged (JAG)1 and JAG2 with a Delta-Serrate-Lag
2 (DSL) domain, while atypical ligands include DNER, F3/Contactin
and NB-3 without a DSL domain. When the ligand interacts with the
Notch1, Notch2, Notch3 or Notch4 receptor, continuous proteolysis
is triggered by γ-secretase, releasing the active Notch
intracellular domain (NICD). In the canonical Notch pathway, NICD
translocates to the nucleus and binds to the transcription factor,
CSL. Then CSL-NICD complex is activated by Mastermind family
co-activators for the transcriptional activation of targets
HES1 and HEY1 to suppress differentiation and
maintain stemness. Otherwise, NICD binds to nuclear p50 or c-Rel to
activate NF-κB activity 9 (non-canonical pathway). Another
non-canonical Notch pathway is triggered by an atypical Notch
ligand to form the CSL-NICD-Deltex complex, activating MAG
transcription and promoting differentiation (48). Which pathway is activated depends
on the interaction between ligands and receptors. NUMB regulates
intracellular Notch activity in the process of cell division,
inhibiting Notch transmission in the cytoplasm (49,50). Musashi-1 (Msi-1), a conservative
RNA-binding-protein (RBP), can upregulate Notch activity by
inactivating NUMB (51).
In general, an aberrantly activated Notch pathway is
oncogenic although anti-oncogenic partly in dermatoma and the
cervical uterus (48). Notch
activity in the CRC initiating cells is 10- to 30-fold higher than
in colon cancer cells. Notch inhibits apoptosis of CCSCs by
repressing the cell cycle inhibitor p27. In addition, Notch can
maintain self-renewal and inhibit differentiation through
repressing secretory cell lineage differentiation targets
MUC2 and atonal homolog 1 (ATOH1) (52).
The downstream targets of TGF-β are pivotal cell
cycle regulation proteins, including p21, p27 and p15. In most
situations, their activation leads to growth arrest (58). Mutated inactivation occurs in at
least one component of almost every CRC case, from frameshift
mutations caused by microsatellite instability to mutations of
Smad4 and Smad2 (59–62). Zubeldia et al injected
colon adenocarcinoma cells pre-treated with TGF-β into the spleens
of mice and found that it promoted primary tumor development and
liver metastasis (63). TGF-β can
promote EMT through inducing EMT-related transcription factors
Snail1/2, Twist and zinc finger E-box binding homeobox (ZEB)1/2 and
elicit cell dedifferentiation (64). Snail induces interleukin (IL)-8)
expression through binding to its E3/E4 E-boxes, maintaining
stemness through function of IL-8. ZEB2 activates the PI3K/AKT
pathway and induces cell transformation (65). ZEB2 attenuates the expression of
phosphatase and tensin homolog (PTEN) through microRNAs, such as
miR-181, miR-200b, miR-25 and miR-92a (66).
As regards the BMP signaling pathway, mutations in
the BMP receptor BMPR1A and Smad4 lead to juvenile intestinal
polyposis and Cowden disease, respectively (56). The downregulation of BMP3 occurs
in 89% of primary CRC cases. The early silencing and frequent
silencing of BMP3 is crucial in CRC progression (67). BMP4 is not expressed in CRC, while
it exists in differentiated cells. Recombinant BMP4 promotes the
differentiation and apoptosis of CCSCs (68). Inhibiting the BMP pathway with
Dorsomorphin causes mesenchymal stem cells to acquire
epithelial-like traits, including the expression of cytokeratin-18
and E-cadherin. The progress occurs through the downregulation of
Snail, Slug and COX2 to affect cell motility, invasiveness and
tumor growth in vitro (69). Moreover, in CRC initiation, BMP
signaling maintains the balanced control of stem cell self-renewal
by inhibiting the Wnt pathway. The zinc-finger transcription
factor, GATA binding protein 6 (GATA6) is a crucial regulation
factor connecting the Wnt and BMP pathways. Competing with
β-catenin/TCF4, GATA6 binds to a distal regulatory region of BMP4,
decreases the threshold of the BMP pathway for CCSC expansion and
inhibits stem cell differentiation (70).
It has been demonstrated that the self-renewal
ability of CCSCs is dependent on HH-GLI activity in vivo
(71). HH proteins initiate
signaling through binding to transmembrane receptor PATCHED1
(PTCH1) and relieve its inhibition to GPCR-like protein Smoothened
(SMO). SMO then localizes to primary cilia and the signaling
transduces through several parts to finally mediate the three GLI
zinc finger transcription factors, enhancing the GLI1 function and
inhibiting GLI repressors. GLI code (the sum of all functions of
the three GLI proteins) transforms into an activating state and
triggers the expression of target genes such as PTCH1,
GLI1, HIP, many of which are components of the HH-GLI
pathway. Varnat et al found that CRC cells exhibited a high
activity of HH-GLI signaling and located active HH-GLI signaling in
SHH+/PTCH1+/GLI1+ tumor cells
rather than the stroma. They inhibited HH-GLI signaling by RNAi or
cyclopamine treatment and tested the influence on CRC cell growth,
recurrence and metastases. The results suggested that the growth,
recurrence and metastases of CRC xenografts required HH-GLI
activity and cells with high HH-GLI activity owned stronger EMT
potentials. Furthermore, they injected CD133+ cells
infected with lentivirus encoding shSMO or shPTCH1
into nude mice, isolated tumor cells after 2–3 weeks and
subsequently analyzed the ratio of CD133+ subpopulation
to test stem cell behaviors in vivo. The results suggested
that the self-renewal of CCSCs relied on the direct function of
HH-GLI activity in vivo (71).
Components of the HH-GLI pathway may also influence
CRC progression by connecting to the Wnt pathway. The role is
controversial. GLI1 can downregulate the level of β-catenin, the
expression of c-Myc and proliferation ability (72). However, SMO significantly
increases in the intestinal adenoma of Apc(+/Delta716) mice and
knockdown of SMO in human CRC lines inhibites cell
proliferation. In Apc(+/Delta716)SMO(+/−) mice, the number of large
polyps decreases, polyp morphology recesses and proliferation of
cancer cells reduces. It is further demonstrated the decreased
expression of SMO attenuates the β-catenin-dependent transcription
instead of HH-responsive GLI-dependent transcription and SMO
promotes tumorigenesis through Wnt signaling (73).
The NF-κB pathway regulates immune-inflammatory
responses, cell survival and proliferation, playing an important
role in tumor formation. Members of the NF-κB family often display
as dipolymers. In a quiescent condition, NF-κB binds to IκB to form
a heteromultimer, residing in the cytoplasm in an inactive state.
With the stimulation of NF-κB activators, IκB kinase (IKKs) are
activated, leading to tripolymer phosphorylation, ubiquitylation of
the N-terminus of the IκB protein and degradation. Thus, the NF-κB
dipolymer is released and is further activated through
post-translational modifications. The NF-κB dipolymer then
translocates to the nucleus to interact with κB sequences and
regulates transcription (74).
The CCSC self-renewal mechanisms related to NF-κB
lie mainly in three areas. The first is related to
dedifferentiation. Schwitalla et al demonstrated that
increasing NF-κB recruits CREB-binding protein (CBP) and helps CBP
to bind RelA/p65 to β-catenin, thus activating Wnt and inducing the
dedifferentiation of non-cancer stem cells (11). The second is the induction of stem
cell gene expression. IκB kinase 2 (IKK2ca) is constitutively
activated in intestinal epithelial cells and increases stem-like
genes ASCL2, olfactomedin 4 (OLFM4), Delta-like 1
homolog (DLK1) and Bmi-1 in intestinal stem cells to
drive intestinal tumorigenesis (75). The third is that IKK2ca-expressing
intestinal epithelial cells can secrete cytokines and chemokines,
induce bone marrow cells, and activate fibroblasts, thus creating a
tumor micro-environment (75).
Epidermal growth factor (EGF) signals are essential
for the emergence and maintenance of CCSCs (76). The interaction between EGF and
EGFR promotes the dimerization and phosphorylation of receptors,
thus activating a downstream signaling cascade involving MAPK, AKT
and JAK-STAT (77). The MAPK
pathway passes through a three-step phosphorylation to further
activate downstream targets (78). EGF promotes the expression of high
levels of Musashi-1, Lgr5 and low levels of CK20 by CCSCs,
facilitating rapid tumor growth. The activation of EGFR signaling
promotes the expression of stem cell-related molecules Notch, Shh,
Oct3/4 and Wnt. EGFR inhibitors inhibit CCSC proliferation and
induce apoptosis through suppressing EGFR phosphorylation and
downstream signaling proteins, such as Akt kinase and ERK kinase
(76).
The Akt cascade regulates cell survival and
proliferation in many different tumors (79). Recently, a novel
kinase-independent Akt pathway was discovered (80). When the Akt kinase domain is
inactivated, Akt can resist a hostile environment through another
domain of the Akt protein, which is named PH domain. The process
may be related to the regulation of interacting protein partners.
The function of PH domain may be influenced by kinase domain to get
to a particular sub-cellular compartment and/or interact with
specific effector proteins (80).
Akt can be a bridge connecting the BMP pathway with Wnt pathway.
BMP positively regulates PTEN activity to inhibit Akt pathway. The
active serine/threonine kinase in the Akt pathway can activate
14-3-3zeta in the β-catenin complex. 14-3-3zeta contributes to the
stabilization and nuclear translocation of β-catenin, thus
facilitating CCSC self-renewal by activating Wnt (81).
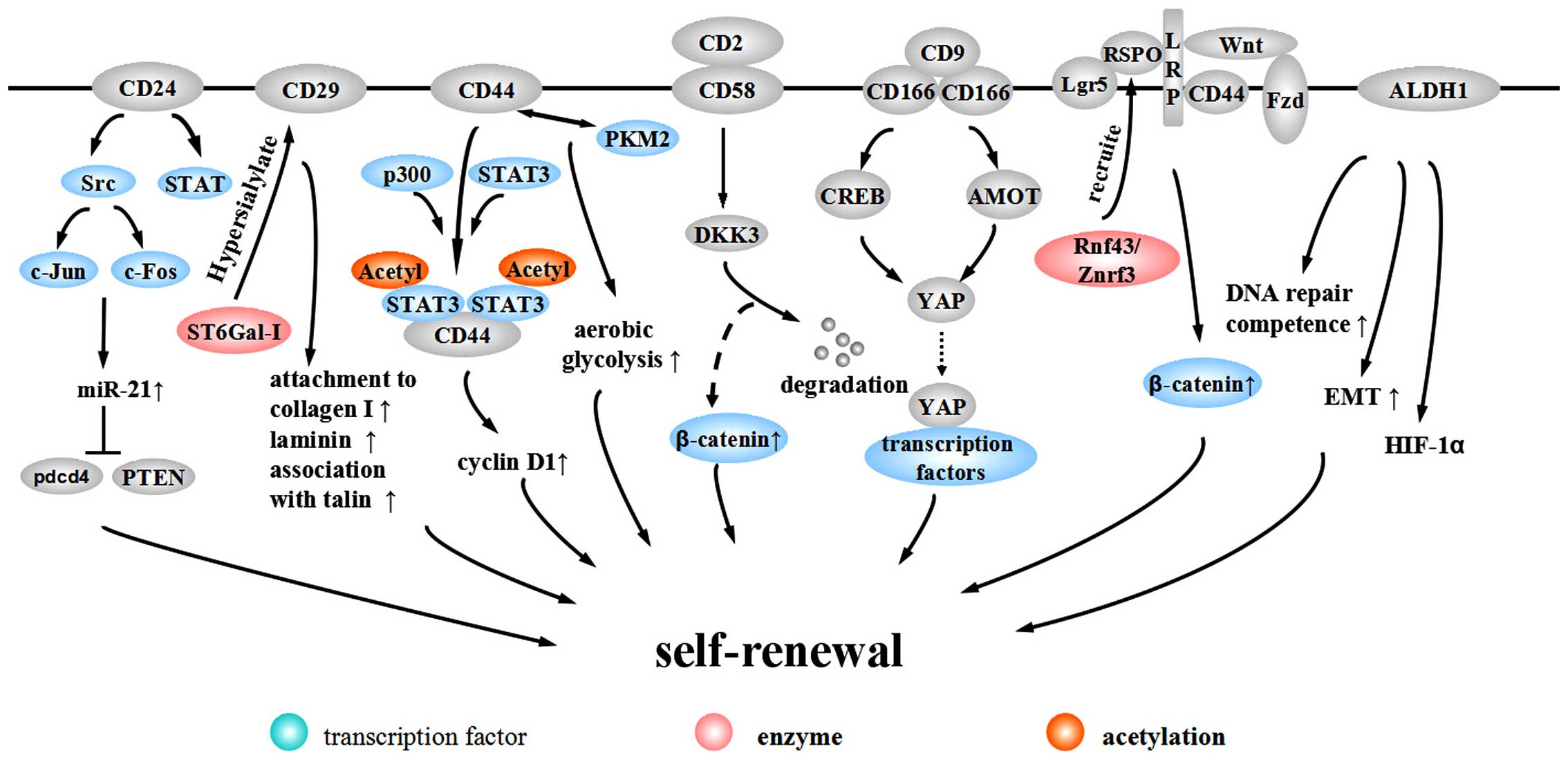
CD24 is a heavily glycosylated protein located on
the external membrane surface, promoting the renewal of the cell
cycle through increasing proliferation and suppressing apoptosis
(82). CD24+ cells
exhibit CCSC properties, including high chemotherapy resistance,
self-renewal ability and tumorigenic potential (83). The promoter of CD24 contains a
TCF/LEF sequence, suggesting that CD24 is a target of
the Wnt pathway (84). In
addition, CD24 activates the Src pathway and induces the expression
of c-Jun and c-Fos to promote the expression of miR-21, which
inhibits Pdcd4 and PTEN to facilitate CRC progression (85). Evidence from gastric cancer
suggests that CD24 can maintain cell survival and promote invasion
through activating STAT (86).
CD29, also known as β1-integrin, heterodimerizes
with one of 12 possible α subunits (87). β1-integrin is the major integrin
protein in normal cells and tumor-associated cells and is thought
to play a part in tumor invasion and metastasis. Alteration of
expression in integrin β1-subunit significantly correlates with
lymph node metastasis and depth of invasion in CRC (88). Song et al proposed
β1-integrin may induce proliferation and migration through HH-GLI
pathway (89).
The involvement of β1-integrin in tumor invasion
depends on the process of hypersialylation. Seales et al
discovered oncogenic ras expressed in 50% of colorectal
adenocarcinomas and it upregulated ST6Gal-I, a Golgi
glycosyltransferase. The enforced expression of ST6Gal-I in SW48
cells has been shown to induced the sialylation of β1-integrin to
upregulate attachment to collagen I and laminin and increase the
association with talin, a well-accepted marker for integrin
activation, which suggests that β1-integrin alters cell preference
for certain extracellular matrix milieus and stimulates cell
migration to promote CRC progression (87).
CD44 is type I transmembrane glycoprotein, comprised
of variable spliceosomes, generating different isoforms through
inserting one or more exons into a common framework region. These
variable isoforms may be a critical process in CRC metastasis
(90–92). CD44 expression is restricted to
the crypt of the epithelia in the normal intestinal mucosa of
APC-mutated mice while it is highly expressed in adenomas and
invasive carcinomas (93).
Although the specificity of CD44 to CCSCs is under debate, CD44 is
useful in isolating CCSCs combined with other surface markers
(20,21,94). The mechanism of the participation
of CD44 in maintaining stemness has mainly three aspects. First,
CD44 is a Wnt target gene and is involved in the Wnt-related
tumorigenesis. At the level of Wnt receptor LRP6, CD44
phosphorylates LRP6 on Wnt stimulation and assists LRP6 in correct
membrane localization (95).
Second, internalized CD44 interacts with STAT3 and
acetyltransferase p300 to form a complex, eliciting STAT3
acetylation and dimerization. The acetylated STAT3 dimer associated
with CD44 translocates to the nucleus dependent on a nuclear
localization signal (NLS) and binds to the promoter of cyclin
D1, increasing cyclin D1 expression and cell
proliferation (96). Further
research demonstrates that the C terminus of the CD44 molecule is
related to increased anoikis resistance in sphere-forming cultures
while the N terminus contributes to the interaction with STAT3 and
p300. Nuclear CD44/STAT3 signaling promotes the reprogramming of
cancer cells, with increasing expression of SOX2 and
OCT4. Additionally, CD44 is related to EMT (97). Third, Interaction between CD44 and
pyruvate kinase M2 (PKM2) facilitates synthesizing glutathione
through the pentose phosphate pathway to resist reactive oxygen
species (ROS) accumulation, thus promoting tumor cell aerobic
glycolysis to meet the requirements for tumor synthesis demands
(98).
CD58, also known as lymphocyte function-associated
antigen-3, is a heavily glycosylated protein on the cell surface
and a cell adhesion molecule belonging to the Ig superfamily. CD58
is expressed on the surface of most cells in the hematopoietic
system and non-hematopoietic system, including endothelial cells
and epithelial cells (20). Colon
cancer HT-29 and Caco-2 cells express CD58 at low levels (99). CD58 is a ligand for CD2 and their
interaction functions as a specific immunity co-stimulation to
promote T cells to secrete CXCL-8, which can promote the
self-renewal of CCSCs (100).
Our research demonstrated that CD58 promotes the maintenance of
self-renewal through activating the Wnt/β-catenin pathway (20). The DKK family is comprised of
inhibitors of the canonical Wnt signaling pathway. CD58 interacts
with downstream DKK3 and elicits DKK3 degradation to activate the
Wnt pathway (20).
ALCAM/CD166, belonging to the Ig super-family, is
highly expressed in endogenous intestinal stem cells and is related
to intercellular adhesion (101,102). CD166 is a target of the Akt
pathway regulated by TGF-β and NF-κB p50/p65 (103–105). Whether CD166 is a positive or
negative regulator of tumor development is still under debate.
Traditional opinions suggest that CD166 is associated with tumor
aggression and progression, anti-apoptosis and resistance to
autophagy (101,104). CD166 mediates its functions via
homophilic (CD166-CD166) or heterophilic (CD166-CD6/9) interactions
(102). A recent study indicated
that through the PI3K/AKT pathway, CD166 regulates the downstream
YAP protein to resist apoptosis, which can be promoted by the
interaction between CD9 and CD166. Additionally, the interaction
between PI3K/AKT signaling and CD166 can form a positive feedback
loop to further promote CD166 expression (105). PI3K/AKT/CD166 regulate the
expression and activity of YAP in at least two types of mechanisms.
The first utilizes the downstream target CREB to regulate YAP
transcription. The second mechanism is the post-transcriptional
control of YAP protein stability by suppressing AMOT130 (105).
Lgr5, a G-protein-coupled receptor with a leucine
rich repeat, is overexpressed in CRC cells and alters along with
CRC progression (32,106). Lgr5+ cells are
located in the crypt base and have the potential to generate all of
the intestinal epithelial cell lineages, maintaining self-renewal
and homeostasis of the intestinal mucosa (107,108). The mechanism of self-renewal
regulation by Lgr5+ is through the Wnt/β-catenin
pathway. With activated Wnt signaling, Lgr5+ forms a
complex with Frizzled/LRP. The complex can bind to Rspondin (RSPO),
a Wnt collaborative enhancer, to further enhance Wnt signaling
(109). Recently, it has been
indicated that after Lgr5 recruits RSPO, RSPO binds to Rnf43/Znrf3,
an E3 ligase that ubiquitinates Frizzled to protect Frizzled from
degradation and maintain Wnt signaling (110).
ALDH1 is reported to be a potential marker for
normal or malignant CCSCs. Immunostaining has demonstrated that
normal crypt bases only express a small amount of ALDH1. However,
in the transformation from normal epithelia to mutant epithelia,
and finally to adenomas, ALDH1+ cells increase in number
and distribute more widerly along the crypt axis (23). Patients with inflammatory bowel
diseases with atypical hyperplasia express higher levels of ALDH1,
while patients with simple inflammation exhibit no increase. This
finding suggests that ALDH1 may be a critical point distinguishing
atypical hyperplasia from inflammatory hyperplasia and a potential
marker for pre-cancerous lesions (111). Evidence from prostate cancer
suggests that ALDH1A1 activity is associated with increased DNA
repair competence and an induction of EMT. ALDH1A1+
cells express high level of hypoxia inducible factor-1α (HIF-1α).
The expression of ALDH1A1 is regulated by the Wnt pathway through
β-catenin/TCF-dependent transcription and co-occurs with the
expression of β-catenin (112).
The tumor micro-environment represents non-tumor
cells and adjacent tissues, including uncontrollable inflammation.
The micro-environment experiences pathological change and provides
cancer stem cells with soil, contributing to the complexity of CRC
biology. The interaction between CCSCs and the micro-environment
plays a role in regulating CCSC self-renewal.
Fibroblasts in the tumor micro-environment are
termed CAFs. This cell population is not stationary, but can be
transformed from mesenchymal cells, endothelial cells, adipose
cells and even cancer epithelial cells. CAFs promote tumor
progression and invasion by both mechanical forces and metabolic
machinery (113). On the one
hand, CAFs induce protease-mediated extracellular matrix
remodeling, serving to guide the structures that direct migration
(114). On the other hand, CAFs
secrete cytokines and growth factors to affect the proliferation,
survival, adhesion and migration of cancer cells. The secreted
mitogenic factors involve hepatocyte growth factor, EGF family
members, chemokine ligand 12, fibroblast growth factors and
stanniocalcin-1 (115). CAFs
promote invasion. One of the mechanisms is the secretion of matrix
metalloproteases or cytokines, such as tumor necrosis factor-α
(TNF-α) that promotes EMT. Activated CAFs by TGF-β can trigger
GP130/STAT3 signaling to express IL-11, contributing to CRC
metastasis (116).
Anoxia is recognized to promote tumor survival and
progression by inducing changes in tumor metabolism, angiogenesis,
invasion, metastasis and drug resistance to reduce the clinical
prognosis (117,118). Anoxia regulates gene expression
through the transcriptional control of HIF-1α and HIF-2α, which can
bind to hypoxia response elements (HRE) in the promoters of many
genes. The induction of these genes triggers aggressive tumor
growth, invasion and metastasis (117,118). HIFs are involved in the
canonical Wnt pathway. HIF-1α can increase the transcriptional
activity of β-catenin (119).
Moreover, Newton et al found that anoxia could inhibit
APC expression through the suppression of the HRE in
the APC promoter by HIF-1α (120). Shay et al treated
colitis-associated colon cancer with acriflavine, an inhibitor of
HIF transcription, and found that it could inhibit the hypoxic
induction of M-CSFR and angiogenic factors to inhibit tumor growth
and progression (121).
MDSCs are a heterogeneous cell population derived
from bone marrow, including immature neutrophils, immature
dendritic cells, immature monocytes and early myeloid progenitors
(122). The recruitment from the
circulation to the intestinal mucosa is associated with CXCR2
expression on CRC endothelial cells and immunocytes (123). With stimulation by cytokines
from the tumor stroma, such as cyclooxygenase 2 and
colony-stimulating factor, MDSCs expand and generate arginase 1,
ROS, and inducible nitric oxide synthase to inhibit the anticancer
function of NK cells and T cells. This process contributes to the
escape from immunological surveillance and promotes tumor
initiation and progression (123–125). Additionally, MDSCs can secrete
vascular endothelial growth factor A and matrix metalloproteinase
(MMP)9 to promote angiogenesis (126). Evidence from ovarian cancer
suggests that MDSCs can drive the expression of miRNA101 and
subsequently suppress repressor gene CtBP2. CtBP2 interacts with
nuclear genes, leading to increased stemness, tumorigenesis and
metastasis in cancer cells (127).
Inflammatory mediators and inflammatory effectors
are both important components of the tumor micro-environment. They
can be derived from the intrinsic pathway, such as the mutation of
an oncogene and chromosomal rearrangement, or the extrinsic
pathway, such as infection (128). The two pathways activate
transcription factors, mainly NF-κB and STAT3, to elicit the
release of downstream inflammatory factors, such as IL-1β, IL-6,
IL-8, IL-17, IL-23 and TNF-α (128,129). The inflammatory response has a
series of tumor facilitating effects, including tumor initiation,
survival, malignant transformation, invasion, metastasis,
destruction of the adaptive immune response, influencing immune
surveillance, and changing chemotherapy resistance (128,129). Activated STAT3 by IL-6 plays an
important role in the early stage of colitis-associated CRC. STAT3
upregulates the expression of cell cycle regulators cyclin
D1, cyclin D2, cyclin B, proto-oncogene
myc, and anti-apoptotic genes Bcl-2 and Bcl-2-like 1
to promote proliferation and survival (130). IL-23 can promote CRC cells to
generate TGF-β and induce the process of EMT to facilitate
malignant transformation (131).
Activated STAT5 by IL-23 with impaired Socs3 expression is
associated with the metastasis of CRC (132), and the function of the
IL-23/IL-17 axis on CRC initiation and progression has been
recently recognized (133).
IL-17A promotes the malignant progression of CRC through the
activation of ERK, p38 MAPK and NF-κB signaling while it also
regulates the production of IL-6 (134). Apart from activating multiple
signaling pathways to activate transcription factors, such as STAT3
and β-catenin to regulate stemness (135), IL-8 is related to EMT. In the
induction of EMT by Snail, Snail and IL-8 can form a positive
feedback loop and increase the expression of stemness genes
Sox2, Nanog and Oct4 (65). Leukotrienes D4 and prostaglandin
E2 can increase the ALDH+ subpopulation and enhance
sphere forming and tumor growth (136). The loss of TGF-β can induce the
secretion of chemokine CCL9 (CCL15 in humans), which recruits
immature myeloid cells that express the CCL9 receptor CCL1. These
cells secrete MMP9 and MMP2 to promote CRC invasion and metastasis
(137).
The components of the micro-environment are very
complex. In addition to the above mentioned, others, such as blood
vessels, microorganisms and normal cells, all exert an influence on
the micro-environment (113,138). All of these complex components
interact with each other to regulate CCSC self-renewal.
Because CCSCs possess a strong resistance to
therapy, the clinical effects on CRC are very limited. The
reactivation of signaling cascades, enhancement of DNA repair and
drug efflux by ABC transportation may be responsible for the
resistance (139). Considering
that the activation of Wnt is a dominating process in the evolution
and progression of CRC (26), the
targeted intervention of this pathway should be most reliable. How
to block this signaling pathway efficiently and specifically while
maintaining normal somatic function is the aim of future study.
Additionally, targets on other signaling pathways and
micro-environmental factors should be included to achieve the best
therapeutic effects.
This study was supported by grants from the Natural
Science of Foundation of Zhejiang Province (no. LY14H160023) and
the Foundation of Scientific Technology Bureau of Zhejiang Province
(no. 2013C33117). The English language editing was performed by
Elsevier Webshop.
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
De Sousa E, Melo F, Vermeulen L, Fessler E
and Medema JP: Cancer heterogeneity - a multifaceted view. EMBO
Rep. 14:686–695. 2013. View Article : Google Scholar
|
|
3
|
Zeuner A, Todaro M, Stassi G and De Maria
R: Colorectal cancer stem cells: From the crypt to the clinic. Cell
Stem Cell. 15:692–705. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vaiopoulos AG, Kostakis ID, Koutsilieris M
and Papavassiliou AG: Colorectal cancer stem cells. Stem Cells.
30:363–371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pardal R, Clarke MF and Morrison SJ:
Applying the principles of stem-cell biology to cancer. Nat Rev
Cancer. 3:895–902. 2003. View Article : Google Scholar
|
|
6
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barker N, Ridgway RA, van Es JH, van de
Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR,
Sansom OJ and Clevers H: Crypt stem cells as the cells-of-origin of
intestinal cancer. Nature. 457:608–611. 2009. View Article : Google Scholar
|
|
8
|
Schepers AG, Snippert HJ, Stange DE, van
den Born M, van Es JH, van de Wetering M and Clevers H: Lineage
tracing reveals Lgr5+ stem cell activity in mouse
intestinal adenomas. Science. 337:730–735. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang J, Papadopoulos N, McKinley AJ,
Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JK, Markowitz
SD, Morin P, et al: APC mutations in colorectal tumors with
mismatch repair deficiency. Proc Natl Acad Sci USA. 93:9049–9054.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Castets M, Broutier L, Molin Y, Brevet M,
Chazot G, Gadot N, Paquet A, Mazelin L, Jarrosson-Wuilleme L,
Scoazec JY, et al: DCC constrains tumour progression via its
dependence receptor activity. Nature. 482:534–537. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schwitalla S, Fingerle AA, Cammareri P,
Nebelsiek T, Göktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ,
Moreaux G, et al: Intestinal tumorigenesis initiated by
dedifferentiation and acquisition of stem-cell-like properties.
Cell. 152:25–38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scheel C, Eaton EN, Li SH, Chaffer CL,
Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, et al:
Paracrine and autocrine signals induce and maintain mesenchymal and
stem cell states in the breast. Cell. 145:926–940. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chaffer CL, Brueckmann I, Scheel C,
Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y,
Polyak K, et al: Normal and neoplastic nonstem cells can
spontaneously convert to a stem-like state. Proc Natl Acad Sci USA.
108:7950–7955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yan KS, Chia LA, Li X, Ootani A, Su J, Lee
JY, Su N, Luo Y, Heilshorn SC, Amieva MR, et al: The intestinal
stem cell markers Bmi1 and Lgr5 identify two functionally distinct
populations. Proc Natl Acad Sci USA. 109:466–471. 2012. View Article : Google Scholar :
|
|
16
|
Yeung TM, Chia LA, Kosinski CM and Kuo CJ:
Regulation of self-renewal and differentiation by the intestinal
stem cell niche. Cell Mol Life Sci. 68:2513–2523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ricci-Vitiani L, Lombardi DG, Pilozzi E,
Biffoni M, Todaro M, Peschle C and De Maria R: Identification and
expansion of human colon-cancer-initiating cells. Nature.
445:111–115. 2007. View Article : Google Scholar
|
|
18
|
O'Brien CA, Pollett A, Gallinger S and
Dick JE: A human colon cancer cell capable of initiating tumour
growth in immunodeficient mice. Nature. 445:106–110. 2007.
View Article : Google Scholar
|
|
19
|
Yeung TM, Gandhi SC, Wilding JL, Muschel R
and Bodmer WF: Cancer stem cells from colorectal cancer-derived
cell lines. Proc Natl Acad Sci USA. 107:3722–3727. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu S, Wen Z, Jiang Q, Zhu L, Feng S, Zhao
Y, Wu J, Dong Q, Mao J and Zhu Y: CD58, a novel surface marker,
promotes self-renewal of tumor-initiating cells in colorectal
cancer. Oncogene. 34:1520–1531. 2015. View Article : Google Scholar
|
|
21
|
Horst D, Kriegl L, Engel J, Kirchner T and
Jung A: Prognostic significance of the cancer stem cell markers
CD133, CD44, and CD166 in colorectal cancer. Cancer Invest.
27:844–850. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dalerba P, Dylla SJ, Park IK, Liu R, Wang
X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al:
Phenotypic characterization of human colorectal cancer stem cells.
Proc Natl Acad Sci USA. 104:10158–10163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang EH, Hynes MJ, Zhang T, Ginestier C,
Dontu G, Appelman H, Fields JZ, Wicha MS and Boman BM: Aldehyde
dehydrogenase 1 is a marker for normal and malignant human colonic
stem cells (SC) and tracks SC overpopulation during colon
tumorigenesis. Cancer Res. 69:3382–3389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miranda-Lorenzo I, Dorado J, Lonardo E,
Alcala S, Serrano AG, Clausell-Tormos J, Cioffi M, Megias D,
Zagorac S, Balic A, et al: Intracellular autofluorescence: A
biomarker for epithelial cancer stem cells. Nat Methods.
11:1161–1169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dotse E and Bian Y: Isolation of
colorectal cancer stem-like cells. Cytotechnology. 68:609–619.
2016. View Article : Google Scholar
|
|
26
|
Fevr T, Robine S, Louvard D and Huelsken
J: Wnt/beta-catenin is essential for intestinal homeostasis and
maintenance of intestinal stem cells. Mol Cell Biol. 27:7551–7559.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao C, Chen G, Romero G, Moschos S, Xu X
and Hu J: Induction of Gsk3β-β-TrCP interaction is required for
late phase stabilization of β-catenin in canonical Wnt signaling. J
Biol Chem. 289:7099–7108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aberle H, Bauer A, Stappert J, Kispert A
and Kemler R: beta-catenin is a target for the ubiquitin-proteasome
pathway. EMBO J. 16:3797–3804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sebio A, Kahn M and Lenz HJ: The potential
of targeting Wnt/β-catenin in colon cancer. Expert Opin Ther
Targets. 18:611–615. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Krausova M and Korinek V: Wnt signaling in
adult intestinal stem cells and cancer. Cell Signal. 26:570–579.
2014. View Article : Google Scholar
|
|
32
|
van de Wetering M, Sancho E, Verweij C, de
Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D,
Haramis AP, et al: The beta-catenin/TCF-4 complex imposes a crypt
progenitor phenotype on colorectal cancer cells. Cell. 111:241–250.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Muncan V, Sansom OJ, Tertoolen L, Phesse
TJ, Begthel H, Sancho E, Cole AM, Gregorieff A, de Alboran IM,
Clevers H, et al: Rapid loss of intestinal crypts upon conditional
deletion of the Wnt/Tcf-4 target gene c-Myc. Mol Cell Biol.
26:8418–8426. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Azzolin L, Panciera T, Soligo S, Enzo E,
Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V,
et al: YAP/TAZ incorporation in the β-catenin destruction complex
orchestrates the Wnt response. Cell. 158:157–170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cho JH, Dimri M and Dimri GP: A positive
feedback loop regulates the expression of polycomb group protein
BMI1 via WNT signaling pathway. J Biol Chem. 288:3406–3418. 2013.
View Article : Google Scholar :
|
|
36
|
Ciznadija D, Tothill R, Waterman ML, Zhao
L, Huynh D, Yu RM, Ernst M, Ishii S, Mantamadiotis T, Gonda TJ, et
al: Intestinal adenoma formation and MYC activation are regulated
by cooperation between MYB and Wnt signaling. Cell Death Differ.
16:1530–1538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vermeulen L, De Sousa E, Melo F, van der
Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M,
Merz C, Rodermond H, et al: Wnt activity defines colon cancer stem
cells and is regulated by the microenvironment. Nat Cell Biol.
12:468–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fodde R and Brabletz T: Wnt/beta-catenin
signaling in cancer stemness and malignant behavior. Curr Opin Cell
Biol. 19:150–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
De A: Wnt/Ca2+ signaling
pathway: A brief overview. Acta Biochim Biophys Sin (Shanghai).
43:745–756. 2011. View Article : Google Scholar
|
|
40
|
MacLeod RJ, Hayes M and Pacheco I: Wnt5a
secretion stimulated by the extracellular calcium-sensing receptor
inhibits defective Wnt signaling in colon cancer cells. Am J
Physiol Gastrointest Liver Physiol. 293:G403–G411. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gwak J, Cho M, Gong SJ, Won J, Kim DE, Kim
EY, Lee SS, Kim M, Kim TK, Shin JG, et al:
Protein-kinase-C-mediated beta-catenin phosphorylation negatively
regulates the Wnt/beta-catenin pathway. J Cell Sci. 119:4702–4709.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hernández-Maqueda JG, Luna-Ulloa LB,
Santoyo-Ramos P, Castañeda-Patlán MC and Robles-Flores M: Protein
kinase C delta negatively modulates canonical Wnt pathway and cell
proliferation in colon tumor cell lines. PLoS One. 8:e585402013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee JM, Kim IS, Kim H, Lee JS, Kim K, Yim
HY, Jeong J, Kim JH, Kim JY, Lee H, et al: RORalpha attenuates
Wnt/beta-catenin signaling by PKCalpha-dependent phosphorylation in
colon cancer. Mol Cell. 37:183–195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ishitani T, Kishida S, Hyodo-Miura J, Ueno
N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J and
Matsumoto K: The TAK1-NLK mitogen-activated protein kinase cascade
functions in the Wnt-5a/Ca(2+) pathway to antagonize
Wnt/beta-catenin signaling. Mol Cell Biol. 23:131–139. 2003.
View Article : Google Scholar :
|
|
45
|
Katoh M: WNT/PCP signaling pathway and
human cancer (Review). Oncol Rep. 14:1583–1588. 2005.PubMed/NCBI
|
|
46
|
Piazzi G, Selgrad M, Garcia M, Ceccarelli
C, Fini L, Bianchi P, Laghi L, D'Angelo L, Paterini P,
Malfertheiner P, et al: Van-Gogh-like 2 antagonises the canonical
WNT pathway and is methylated in colorectal cancers. Br J Cancer.
108:1750–1756. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sancho R, Nateri AS, de Vinuesa AG,
Aguilera C, Nye E, Spencer-Dene B and Behrens A: JNK signalling
modulates intestinal homeostasis and tumourigenesis in mice. EMBO
J. 28:1843–1854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Katoh M and Katoh M: Notch signaling in
gastrointestinal tract (Review). Int J Oncol. 30:247–251. 2007.
|
|
49
|
Couturier L, Mazouni K and Schweisguth F:
Inhibition of Notch recycling by Numb: Relevance and mechanism(s).
Cell Cycle. 12:1647–1648. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Giebel B and Wodarz A: Notch signaling:
Numb makes the difference. Curr Biol. 22:R133–R135. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pastò A, Serafin V, Pilotto G, Lago C,
Bellio C, Trusolino L, Bertotti A, Hoey T, Plateroti M, Esposito G,
et al: NOTCH3 signaling regulates MUSASHI-1 expression in
metastatic colorectal cancer cells. Cancer Res. 74:2106–2118. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sikandar SS, Pate KT, Anderson S, Dizon D,
Edwards RA, Waterman ML and Lipkin SM: NOTCH signaling is required
for formation and self-renewal of tumor-initiating cells and for
repression of secretory cell differentiation in colon cancer.
Cancer Res. 70:1469–1478. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Meng RD, Shelton CC, Li YM, Qin LX,
Notterman D, Paty PB and Schwartz GK: gamma-Secretase inhibitors
abrogate oxaliplatin-induced activation of the Notch-1 signaling
pathway in colon cancer cells resulting in enhanced
chemosensitivity. Cancer Res. 69:573–582. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gao F, Zhang Y, Wang S, Liu Y, Zheng L,
Yang J, Huang W, Ye Y, Luo W and Xiao D: Hes1 is involved in the
self-renewal and tumourigenicity of stem-like cancer cells in colon
cancer. Sci Rep. 4:39632014.PubMed/NCBI
|
|
55
|
Ghaleb AM, Aggarwal G, Bialkowska AB,
Nandan MO and Yang VW: Notch inhibits expression of the
Krüppel-like factor 4 tumor suppressor in the intestinal
epithelium. Mol Cancer Res. 6:1920–1927. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mishra L, Derynck R and Mishra B:
Transforming growth factor-beta signaling in stem cells and cancer.
Science. 310:68–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kamato D, Burch ML, Piva TJ, Rezaei HB,
Rostam MA, Xu S, Zheng W, Little PJ and Osman N: Transforming
growth factor-β signalling: Role and consequences of Smad linker
region phosphorylation. Cell Signal. 25:2017–2024. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yu M, Trobridge P, Wang Y, Kanngurn S,
Morris SM, Knoblaugh S and Grady WM: Inactivation of TGF-β
signaling and loss of PTEN cooperate to induce colon cancer in
vivo. Oncogene. 33:1538–1547. 2014. View Article : Google Scholar
|
|
59
|
Villanueva A, García C, Paules AB, Vicente
M, Megías M, Reyes G, de Villalonga P, Agell N, Lluís F, Bachs O,
et al: Disruption of the antiproliferative TGF-beta signaling
pathways in human pancreatic cancer cells. Oncogene. 17:1969–1978.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xu X, Brodie SG, Yang X, Im YH, Parks WT,
Chen L, Zhou YX, Weinstein M, Kim SJ and Deng CX: Haploid loss of
the tumor suppressor Smad4/Dpc4 initiates gastric polyposis and
cancer in mice. Oncogene. 19:1868–1874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Grady WM, Myeroff LL, Swinler SE, Rajput
A, Thiagalingam S, Lutterbaugh JD, Neumann A, Brattain MG, Chang J,
Kim SJ, et al: Mutational inactivation of transforming growth
factor beta receptor type II in microsatellite stable colon
cancers. Cancer Res. 59:320–324. 1999.PubMed/NCBI
|
|
62
|
Woodford-Richens KL, Rowan AJ, Gorman P,
Halford S, Bicknell DC, Wasan HS, Roylance RR, Bodmer WF and
Tomlinson IP: SMAD4 mutations in colorectal cancer probably occur
before chromosomal instability, but after divergence of the
microsatellite instability pathway. Proc Natl Acad Sci USA.
98:9719–9723. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zubeldia IG, Bleau AM, Redrado M, Serrano
D, Agliano A, Gil-Puig C, Vidal-Vanaclocha F, Lecanda J and Calvo
A: Epithelial to mesenchymal transition and cancer stem cell
phenotypes leading to liver metastasis are abrogated by the novel
TGFβ1-targeting peptides P17 and P144. Exp Cell Res. 319:12–22.
2013. View Article : Google Scholar
|
|
64
|
Moustakas A and Heldin CH: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:1512–1520.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH,
Chang SC, Teng HW, Yang SH, Lan YT, Chiou SH, et al: SNAIL
regulates interleukin-8 expression, stem cell-like activity, and
tumorigenicity of human colorectal carcinoma cells.
Gastroenterology. 141:279–291. 2912011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Karreth FA, Tay Y, Perna D, Ala U, Tan SM,
Rust AG, DeNicola G, Webster KA, Weiss D, Perez-Mancera PA, et al:
In vivo identification of tumor- suppressive PTEN ceRNAs in an
oncogenic BRAF-induced mouse model of melanoma. Cell. 147:382–395.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Loh K, Chia JA, Greco S, Cozzi SJ,
Buttenshaw RL, Bond CE, Simms LA, Pike T, Young JP, Jass JR, et al:
Bone morphogenic protein 3 inactivation is an early and frequent
event in colorectal cancer development. Genes Chromosomes Cancer.
47:449–460. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lombardo Y, Scopelliti A, Cammareri P,
Todaro M, Iovino F, Ricci-Vitiani L, Gulotta G, Dieli F, de Maria R
and Stassi G: Bone morphogenetic protein 4 induces differentiation
of colorectal cancer stem cells and increases their response to
chemotherapy in mice. Gastroenterology. 140:297–309. 2011.
View Article : Google Scholar
|
|
69
|
Garulli C, Kalogris C, Pietrella L,
Bartolacci C, Andreani C, Falconi M, Marchini C and Amici A:
Dorsomorphin reverses the mesenchymal phenotype of breast cancer
initiating cells by inhibition of bone morphogenetic protein
signaling. Cell Signal. 26:352–362. 2014. View Article : Google Scholar
|
|
70
|
Whissell G, Montagni E, Martinelli P,
Hernando-Momblona X, Sevillano M, Jung P, Cortina C, Calon A, Abuli
A, Castells A, et al: The transcription factor GATA6 enables
self-renewal of colon adenoma stem cells by repressing BMP gene
expression. Nat Cell Biol. 16:695–707. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Varnat F, Duquet A, Malerba M, Zbinden M,
Mas C, Gervaz P and Ruiz i Altaba A: Human colon cancer epithelial
cells harbour active HEDGEHOG-GLI signalling that is essential for
tumour growth, recurrence, metastasis and stem cell survival and
expansion. EMBO Mol Med. 1:338–351. 2009. View Article : Google Scholar
|
|
72
|
Akiyoshi T, Nakamura M, Koga K, Nakashima
H, Yao T, Tsuneyoshi M, Tanaka M and Katano M: Gli1, downregulated
in colorectal cancers, inhibits proliferation of colon cancer cells
involving Wnt signalling activation. Gut. 55:991–999. 2006.
View Article : Google Scholar
|
|
73
|
Arimura S, Matsunaga A, Kitamura T, Aoki
K, Aoki M and Taketo MM: Reduced level of smoothened suppresses
intestinal tumorigenesis by downregulation of Wnt signaling.
Gastroenterology. 137:629–638. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sakamoto K and Maeda S: Targeting
NF-kappaB for colorectal cancer. Expert Opin Ther Targets.
14:593–601. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Vlantis K, Wullaert A, Sasaki Y,
Schmidt-Supprian M, Rajewsky K, Roskams T and Pasparakis M:
Constitutive IKK2 activation in intestinal epithelial cells induces
intestinal tumors in mice. J Clin Invest. 121:2781–2793. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Feng Y, Dai X, Li X, Wang H, Liu J, Zhang
J, Du Y and Xia L: EGF signalling pathway regulates colon cancer
stem cell proliferation and apoptosis. Cell Prolif. 45:413–419.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Al Moustafa AE, Achkhar A and Yasmeen A:
EGF-receptor signaling and epithelial-mesenchymal transition in
human carcinomas. Front Biosci (Schol Ed). 4:671–684. 2012.
View Article : Google Scholar
|
|
78
|
Munshi A and Ramesh R: Mitogen-activated
protein kinases and their role in radiation response. Genes Cancer.
4:401–408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Moschetta M, Reale A, Marasco C, Vacca A
and Carratù MR: Therapeutic targeting of the mTOR-signalling
pathway in cancer: Benefits and limitations. Br J Pharmacol.
171:3801–3813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Vivanco I, Chen ZC, Tanos B, Oldrini B,
Hsieh WY, Yannuzzi N, Campos C and Mellinghoff IK: A
kinase-independent function of AKT promotes cancer cell survival.
eLife. 3:32014. View Article : Google Scholar
|
|
81
|
Tian Q, He XC, Hood L and Li L: Bridging
the BMP and Wnt pathways by PI3 kinase/Akt and 14-3-3zeta. Cell
Cycle. 4:215–216. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lim SC: CD24 and human carcinoma: Tumor
biological aspects. Biomed Pharmacother. 59(Suppl 2): S351–S354.
2005. View Article : Google Scholar
|
|
83
|
Ke J, Wu X, Wu X, He X, Lian L, Zou Y, He
X, Wang H, Luo Y, Wang L, et al: A subpopulation of
CD24+ cells in colon cancer cell lines possess stem cell
characteristics. Neoplasma. 59:282–288. 2012. View Article : Google Scholar
|
|
84
|
Shulewitz M, Soloviev I, Wu T, Koeppen H,
Polakis P and Sakanaka C: Repressor roles for TCF-4 and Sfrp1 in
Wnt signaling in breast cancer. Oncogene. 25:4361–4369. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Muppala S, Mudduluru G, Leupold JH, Buergy
D, Sleeman JP and Allgayer H: CD24 induces expression of the
oncomir miR-21 via Src, and CD24 and Src are both
post-transcriptionally downregulated by the tumor suppressor
miR-34a. PLoS One. 8:e595632013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang YC, Wang JL, Kong X, Sun TT, Chen HY,
Hong J and Fang JY: CD24 mediates gastric carcinogenesis and
promotes gastric cancer progression via STAT3 activation.
Apoptosis. 19:643–656. 2014. View Article : Google Scholar
|
|
87
|
Seales EC, Jurado GA, Brunson BA,
Wakefield JK, Frost AR and Bellis SL: Hypersialylation of beta1
integrins, observed in colon adenocarcinoma, may contribute to
cancer progression by upregulating cell motility. Cancer Res.
65:4645–4652. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Fujita S, Watanabe M, Kubota T, Teramoto T
and Kitajima M: Alteration of expression in integrin beta 1-subunit
correlates with invasion and metastasis in colorectal cancer.
Cancer Lett. 91:145–149. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Song J, Zhang J, Wang J, Wang J, Guo X and
Dong W: β1 integrin mediates colorectal cancer cell proliferation
and migration through regulation of the Hedgehog pathway. Tumour
Biol. 36:2013–2021. 2015. View Article : Google Scholar
|
|
90
|
Marhaba R and Zöller M: CD44 in cancer
progression: Adhesion, migration and growth regulation. J Mol
Histol. 35:211–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Du L, Wang H, He L, Zhang J, Ni B, Wang X,
Jin H, Cahuzac N, Mehrpour M, Lu Y, et al: CD44 is of functional
importance for colorectal cancer stem cells. Clin Cancer Res.
14:6751–6760. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Bánky B, Rásó-Barnett L, Barbai T, Tímár
J, Becságh P and Rásó E: Characteristics of CD44 alternative splice
pattern in the course of human colorectal adenocarcinoma
progression. Mol Cancer. 11:832012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wielenga VJ, Smits R, Korinek V, Smit L,
Kielman M, Fodde R, Clevers H and Pals ST: Expression of CD44 in
Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J
Pathol. 154:515–523. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang C, Xie J, Guo J, Manning HC, Gore JC
and Guo N: Evaluation of CD44 and CD133 as cancer stem cell markers
for colorectal cancer. Oncol Rep. 28:1301–1308. 2012.PubMed/NCBI
|
|
95
|
Schmitt M, Metzger M, Gradl D, Davidson G
and Orian-Rousseau V: CD44 functions in Wnt signaling by regulating
LRP6 localization and activation. Cell Death Differ. 22:677–689.
2015. View Article : Google Scholar :
|
|
96
|
Lee JL, Wang MJ and Chen JY: Acetylation
and activation of STAT3 mediated by nuclear translocation of CD44.
J Cell Biol. 185:949–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Su YJ, Lai HM, Chang YW, Chen GY and Lee
JL: Direct reprogramming of stem cell properties in colon cancer
cells by CD44. EMBO J. 30:3186–3199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Tamada M, Nagano O, Tateyama S, Ohmura M,
Yae T, Ishimoto T, Sugihara E, Onishi N, Yamamoto T, Yanagawa H, et
al: Modulation of glucose metabolism by CD44 contributes to
antioxidant status and drug resistance in cancer cells. Cancer Res.
72:1438–1448. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kvale D, Krajci P and Brandtzaeg P:
Expression and regulation of adhesion molecules ICAM-1 (CD54) and
LFA-3 (CD58) in human intestinal epithelial cell lines. Scand J
Immunol. 35:669–676. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ebert EC, Panja A and Praveen R: Human
intestinal intraepithelial lymphocytes and epithelial cells
coinduce interleukin-8 production through the CD2-CD58 interaction.
Am J Physiol Gastrointest Liver Physiol. 296:G671–G677. 2009.
View Article : Google Scholar
|
|
101
|
Levin TG, Powell AE, Davies PS, Silk AD,
Dismuke AD, Anderson EC, Swain JR and Wong MH: Characterization of
the intestinal cancer stem cell marker CD166 in the human and mouse
gastrointestinal tract. Gastroenterology. 139:2072–2082.e5. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Gilsanz A, Sánchez-Martín L,
Gutiérrez-López MD, Ovalle S, Machado-Pineda Y, Reyes R, Swart GW,
Figdor CG, Lafuente EM and Cabañas C: ALCAM/CD166 adhesive function
is regulated by the tetraspanin CD9. Cell Mol Life Sci. 70:475–493.
2013. View Article : Google Scholar
|
|
103
|
Hansen AG, Arnold SA, Jiang M, Palmer TD,
Ketova T, Merkel A, Pickup M, Samaras S, Shyr Y, Moses HL, et al:
ALCAM/CD166 is a TGF-β-responsive marker and functional regulator
of prostate cancer metastasis to bone. Cancer Res. 74:1404–1415.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Wang J, Gu Z, Ni P, Qiao Y, Chen C, Liu X,
Lin J, Chen N and Fan Q: NF-kappaB P50/P65 hetero-dimer mediates
differential regulation of CD166/ALCAM expression via interaction
with micoRNA-9 after serum deprivation, providing evidence for a
novel negative auto-regulatory loop. Nucleic Acids Res.
39:6440–6455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ma L, Wang J, Lin J, Pan Q, Yu Y and Sun
F: Cluster of differentiation 166 (CD166) regulated by
phosphatidylinositide 3-Kinase (PI3K)/AKT signaling to exert its
anti-apoptotic role via yes-associated protein (YAP) in liver
cancer. J Biol Chem. 289:6921–6933. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Barker N, van Es JH, Kuipers J, Kujala P,
van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H,
Peters PJ, et al: Identification of stem cells in small intestine
and colon by marker gene Lgr5. Nature. 449:1003–1007. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ritsma L, Ellenbroek SI, Zomer A, Snippert
HJ, de Sauvage FJ, Simons BD, Clevers H and van Rheenen J:
Intestinal crypt homeostasis revealed at single-stem-cell level by
in vivo live imaging. Nature. 507:362–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Gerbe F, van Es JH, Makrini L, Brulin B,
Mellitzer G, Robine S, Romagnolo B, Shroyer NF, Bourgaux JF,
Pignodel C, et al: Distinct ATOH1 and Neurog3 requirements define
tuft cells as a new secretory cell type in the intestinal
epithelium. J Cell Biol. 192:767–780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Schuijers J and Clevers H: Adult mammalian
stem cells: The role of Wnt, Lgr5 and R-spondins. EMBO J.
31:2685–2696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
de Lau W, Peng WC, Gros P and Clevers H:
The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength.
Genes Dev. 28:305–316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Toll AD, Boman BM and Palazzo JP:
Dysplastic lesions in inflammatory bowel disease show increased
positivity for the stem cell marker aldehyde dehydrogenase. Hum
Pathol. 43:238–242. 2012. View Article : Google Scholar
|
|
112
|
Cojoc M, Peitzsch C, Kurth I, Trautmann F,
Kunz-Schughart LA, Telegeev GD, Stakhovsky EA, Walker JR, Simin K,
Lyle S, et al: Aldehyde dehydrogenase is regulated by
Beta-catenin/TCF and promotes radioresistance in prostate cancer
progenitor cells. Cancer Res. 75:1482–1494. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Chen S and Huang EH: The colon cancer stem
cell microenvironment holds keys to future cancer therapy. J
Gastrointest Surg. 18:1040–1048. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Cirri P and Chiarugi P:
Cancer-associated-fibroblasts and tumour cells: A diabolic liaison
driving cancer progression. Cancer Metastasis Rev. 31:195–208.
2012. View Article : Google Scholar
|
|
115
|
Calon A, Tauriello DV and Batlle E:
TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer
Biol. 25:15–22. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Calon A, Espinet E, Palomo-Ponce S,
Tauriello DV, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung
P, Zhang XH, et al: Dependency of colorectal cancer on a
TGF-β-driven program in stromal cells for metastasis initiation.
Cancer Cell. 22:571–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
McIntyre A and Harris AL: Metabolic and
hypoxic adaptation to anti-angiogenic therapy: A target for induced
essentiality. EMBO Mol Med. 7:368–379. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Rebucci M and Michiels C: Molecular
aspects of cancer cell resistance to chemotherapy. Biochem
Pharmacol. 85:1219–1226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Santoyo-Ramos P, Likhatcheva M,
García-Zepeda EA, Castañeda-Patlán MC and Robles-Flores M:
Hypoxia-inducible factors modulate the stemness and malignancy of
colon cancer cells by playing opposite roles in canonical Wnt
signaling. PLoS One. 9:e1125802014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Newton IP, Kenneth NS, Appleton PL, Näthke
I and Rocha S: Adenomatous polyposis coli and hypoxia-inducible
factor-1{alpha} have an antagonistic connection. Mol Biol Cell.
21:3630–3638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Shay JE, Imtiyaz HZ, Sivanand S, Durham
AC, Skuli N, Hsu S, Mucaj V, Eisinger-Mathason TS, Krock BL,
Giannoukos DN, et al: Inhibition of hypoxia-inducible factors
limits tumor progression in a mouse model of colorectal cancer.
Carcinogenesis. 35:1067–1077. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Gabrilovich DI, Ostrand-Rosenberg S and
Bronte V: Coordinated regulation of myeloid cells by tumours. Nat
Rev Immunol. 12:253–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Katoh H, Wang D, Daikoku T, Sun H, Dey SK
and Dubois RN: CXCR2-expressing myeloid-derived suppressor cells
are essential to promote colitis-associated tumorigenesis. Cancer
Cell. 24:631–644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Gabrilovich DI and Nagaraj S:
Myeloid-derived suppressor cells as regulators of the immune
system. Nat Rev Immunol. 9:162–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Murdoch C, Muthana M, Coffelt SB and Lewis
CE: The role of myeloid cells in the promotion of tumour
angiogenesis. Nat Rev Cancer. 8:618–631. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Motz GT and Coukos G: The parallel lives
of angiogenesis and immunosuppression: Cancer and other tales. Nat
Rev Immunol. 11:702–711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Cui TX, Kryczek I, Zhao L, Zhao E, Kuick
R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG, et al:
Myeloid-derived suppressor cells enhance stemness of cancer cells
by inducing microRNA101 and suppressing the corepressor CtBP2.
Immunity. 39:611–621. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin
C and Flavell RA: Inflammation-induced cancer: Crosstalk between
tumours, immune cells and microorganisms. Nat Rev Cancer.
13:759–771. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Suzuki H, Ogawa H, Miura K, Haneda S,
Watanabe K, Ohnuma S, Sasaki H, Sase T, Kimura S, Kajiwara T, et
al: IL-23 directly enhances the proliferative and invasive
activities of colorectal carcinoma. Oncol Lett. 4:199–204.
2012.PubMed/NCBI
|
|
132
|
Zhang L, Li J, Li L, Zhang J, Wang X, Yang
C, Li Y, Lan F and Lin P: IL-23 selectively promotes the metastasis
of colorectal carcinoma cells with impaired Socs3 expression via
the STAT5 pathway. Carcinogenesis. 35:1330–1340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Grivennikov SI, Wang K, Mucida D, Stewart
CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung
KE, et al: Adenoma-linked barrier defects and microbial products
drive IL-23/IL-17-mediated tumour growth. Nature. 491:254–258.
2012.PubMed/NCBI
|
|
134
|
Wang K, Kim MK, Di Caro G, Wong J,
Shalapour S, Wan J, Zhang W, Zhong Z, Sanchez-Lopez E, Wu LW, et
al: Interleukin-17 receptor a signaling in transformed enterocytes
promotes early colorectal tumorigenesis. Immunity. 41:1052–1063.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Waugh DJ and Wilson C: The interleukin-8
pathway in cancer. Clin Cancer Res. 14:6735–6741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Bellamkonda K, Sime W and Sjolander A: The
impact of inflammatory lipid mediators on colon cancer-initiating
cells. Mol Carcinog. 54:1315–1327. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Taketo MM: Roles of stromal
microenvironment in colon cancer progression. J Biochem.
151:477–481. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Sun J: Enteric bacteria and cancer stem
cells. Cancers (Basel). 3:285–297. 2010. View Article : Google Scholar
|
|
139
|
Ischenko I, Seeliger H, Schaffer M, Jauch
KW and Bruns CJ: Cancer stem cells: How can we target them? Curr
Med Chem. 15:3171–3184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Fujimoto K, Beauchamp RD and Whitehead RH:
Identification and isolation of candidate human colonic clonogenic
cells based on cell surface integrin expression. Gastroenterology.
123:1941–1948. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
May R, Riehl TE, Hunt C, Sureban SM, Anant
S and Houchen CW: Identification of a novel putative
gastrointestinal stem cell and adenoma stem cell marker,
doublecortin and CaM kinase-like-1, following radiation injury and
in adenomatous polyposis coli/multiple intestinal neoplasia mice.
Stem Cells. 26:630–637. 2008. View Article : Google Scholar
|
|
142
|
Li D, Peng X, Yan D, Tang H, Huang F, Yang
Y and Peng Z: Msi-1 is a predictor of survival and a novel
therapeutic target in colon cancer. Ann Surg Oncol. 18:2074–2083.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Yu T, Chen X, Zhang W, Colon D, Shi J,
Napier D, Rychahou P, Lu W, Lee EY, Weiss HL, et al: Regulation of
the potential marker for intestinal cells, Bmi1, by β-catenin and
the zinc finger protein KLF4: Implications for colon cancer. J Biol
Chem. 287:3760–3768. 2012. View Article : Google Scholar
|
|
144
|
Sagiv E, Memeo L, Karin A, Kazanov D,
Jacob-Hirsch J, Mansukhani M, Rechavi G, Hibshoosh H and Arber N:
CD24 is a new oncogene, early at the multistep process of
colorectal cancer carcinogenesis. Gastroenterology. 131:630–639.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Haraguchi N, Ishii H, Mimori K, Ohta K,
Uemura M, Nishimura J, Hata T, Takemasa I, Mizushima T, Yamamoto H,
et al: CD49f-positive cell population efficiently enriches colon
cancer-initiating cells. Int J Oncol. 43:425–430. 2013.PubMed/NCBI
|
|
146
|
Gemei M, Mirabelli P, Di Noto R, Corbo C,
Iaccarino A, Zamboli A, Troncone G, Galizia G, Lieto E, Del Vecchio
L, et al: CD66c is a novel marker for colorectal cancer stem cell
isolation, and its silencing halts tumor growth in vivo. Cancer.
119:729–738. 2013. View Article : Google Scholar
|
|
147
|
Nakanishi Y, Seno H, Fukuoka A, Ueo T,
Yamaga Y, Maruno T, Nakanishi N, Kanda K, Komekado H, Kawada M, et
al: Dclk1 distinguishes between tumor and normal stem cells in the
intestine. Nat Genet. 45:98–103. 2013. View Article : Google Scholar
|
|
148
|
van der Flier LG, Haegebarth A, Stange DE,
van de Wetering M and Clevers H: OLFM4 is a robust marker for stem
cells in human intestine and marks a subset of colorectal cancer
cells. Gastroenterology. 137:15–17. 2009. View Article : Google Scholar : PubMed/NCBI
|