Introduction
Breast cancer is a major worldwide threat to women's
health (1). It is the most common
malignancy in females resulting in ~40,000 deaths/year in the
United States (2). Breast cancer
has been reported to be associated with reproductive and hormonal
factors plus endogenous sex hormone levels (3). In addition, the metastasis of breast
cancer frequently presents in regional lymph nodes, bone marrow,
lung and liver, which primarily bypasses through the lymphatic
system (4,5). To prevent tumor growth and
metastasis, various treatments including surgery, radiotherapy,
chemotherapy, immunotherapy and combined strategies have been
employed (6,7). Advances in these therapies have
shown to have a beneficial effect on the quality of life of breast
cancer patients. However, no available therapy currently exists for
patients with advanced, invasive and metastatic breast cancer
(8). Hence, further exploration
of the molecular mechanisms involved in breast cancer development
and progression is still urgently needed. Human mitochondria are
considered as semi-autonomous cell organelles with their own genome
and are responsible for production of energy and regulation of
cellular processes (9,10). Immature colon carcinoma transcript
1 (ICT1) is a mitochondrial ribosome protein that belongs to a
family of human mitochondrial translation release factors, which
masters the termination stage of translation (11,12). In addition, it is also recruited
into the human mitochondrial ribosome as a functional peptidyl-tRNA
hydrolase (13). A previous study
showed that the ICT1 proteins are necessary for cell viability
(14). Mutations of the
Gly-Gly-Gln motif (GGQ motif) as well as knockdown of ICT1
were found to lead to loss of cell viability (15). In HeLa cells, depletion of
ICT1 caused apoptotic cell death (14). Lentiviral-mediated RNA
interference of ICT1 significantly blocked gliboblastoma
multiforme cell growth through inducing G2/M cell cycle arrest
(16). To date, the role of
ICT1 in breast tumor growth and spreading is unknown.
To uncover the biological function of ICT1 in
breast cancer, the ICT1 mRNA expression pattern was analyzed
using the Oncomine database. Furthermore, we conducted
loss-of-function experiments on breast cancer cells to further
confirm the biological function of ICT1. Our findings may
contribute to a better understanding of the role of ICT1 in
the regulation of breast cancer tumorigenesis.
Materials and methods
Oncomine dataset analysis
Microarray datasets for breast cancer were retrieved
from the Oncomine database (https://www.oncomine.org) to investigate ICT1
expression in breast cancer by the following definition: gene name,
ICT1; analysis type, cancer versus normal analysis. The cancer type
was defined as breast cancer, and data type was mRNA.
Cell lines and culture
Six breast cancer cell lines (ZR-75-30, T-47D,
MDA-MB-231, MDA-MB-435, MCF-7 and BT-474) and human embryonic
kidney cells 293T were provided by the Cell Bank of Shanghai
Institute of Biochemistry Cell Biology, Chinese Academy of
Sciences. ZR-75-30 and BT-474 cells were cultured in RPMI-1640
medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine
serum (FBS; S1810; Biowest, Nuaillé, France). MDA-MB-231,
MDA-MB-435, T-47D and HEK293T cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) (HyClone) with 10% FBS. MCF-7 cells
were maintained in Modified Eagle's medium (MEM; HyClone) with 10%
FBS (S1810; Biowest). All cell cultures were incubated in a
humidified 95% air atmosphere containing 5% CO2 at
37°C.
Construction of the lentivirus-based
vector to infect breast cancer cells
According to the ICT1 sequence (NM_001545),
two different shRNAs targeting ICT1 were designed using siRNA
Construct Builder (http://www.genscript.com/rnai.html). Primers of
shRNA(S1), shRNA(S2) and control shRNA were synthesized with the
following sequences:
5′-GCTGTTAATGCTTGTCTATAACTCGAGTTATAGACAAGCATTAACAGCTTTTTT-3′,
5′-GCAGAATGTGAACAAAGTGAACTCGAGTTCACTTTGTTCACATTCTGCTTT TTT-3′ and
5′-GATCCTTCTCCGAACGTGTCACGTCTCGAGACGACGCACTGGCGGAGAATTTTTG-3′,
respectively. Each shRNA was ligated between the NheI and
PacI sites downstream of the U6 promoter in the lentiviral
vector pFH-L (Shanghai Hollybio, Shanghai, China) to generate
Lv-shRNA (S1, S2 or Con). A mixture of the modified pFH-L plasmids
and packaging vectors (pVSVG-I and pCMVΔR8.92) were then
cotransfected into 293T cells using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA), as suggested by the manufacturer's
instructions.
For cell infection, human breast cancer ZR-75-30 and
T-47D cells (600,000 cells/well) were seeded in 6-well plates and
then infected with Lv-shICT1(S1), Lv-shICT1(S2), or Lv-shCon,
respectively. After incubation for 192 h, the infection efficiency
was determined by detecting green fluorescent protein (GFP)
expression under confocal fluorescence microscopy.
Real-time quantitative PCR (RT-qPCR)
analysis
Total RNAs were extracted from cells infected with
Lv-shICT1(S1), Lv-shICT1(S2), or Lv-shCon using TRIzol reagent
(Invitrogen). First-Strand complementary DNA (cDNA) was generated
from 1 µg of total RNA in the presence of SuperScript™
reverse transcriptase and oligo(dT)12–18 primer (both from
Gibco-BRL): ICT1 RT-qPCR primer set,
5′-CAGCCTGGACAAGCTCTACC-3′ and 5′-GGAACCTGACTTCTGCCTTG-3′;
actin RT-qPCR primers set, 5′-GTGGACATCCGCAAAGAC-3′ and
5′-AAAGGGTGTAACGCAACTA-3′. The reaction mixture contained 2X SYBR
Premix Ex Taq 10 µl, each primer (2.5 µM) 0.5
µl, cDNA 5 µl, and ddH2O 4.5 µl.
Samples were performed on Bio-Rad Connet Real-Time PCR platform and
initially denatured at 95°C for 1 min, denatured at 95°C for 5 sec,
40 cycles of 20 sec at 60°C, and 10 min at 72°C. The
2−ΔΔCt calculation method was used to analyze the
relative expression of ICT1 to actin.
Western blot analysis
The expression level of ICT1 protein along using
cell cycle and apoptosis regulatory proteins were analyzed in cells
with western blotting. Briefly, the cells were harvested, washed
with cold phosphate-buffered saline (PBS), and mixed with 2X sodium
dodecyl sulfate (SDS) sample buffer [100 mM Tris-HCl (pH=6.8), 10
mM EDTA, 4% SDS, 10% glycine]. Each sample of protein (30
µg/lane) was separated on 12% SDS-PAGE at 150 V for 1 h, and
then transferred to polyvinylidene fluoride) membrane (PVDF) at 300
mA for 2.0 h. After blocking with 5% non-fat milk, the blots were
incubated with primary antibodies: rabbit anti-ICT1 (1:1,000; Cat.
no. #AP20382b; Abgent, San Diego, CA, USA), rabbit anti-Bcl-2
(1:1,000; Cat. no. 2876), rabbit anti-caspase-3 (1:500; Cat. no.
9661) (both from Cell Signaling Technology, Danvers, MA, USA),
rabbit anti-CDK1 (1:500; Cat. no. 19532-1-AP; Proteintech, Chicago
IL, USA), rabbit anti-CDK2 (1:1,000; Cat. no. 2546; Cell Signaling
Technology), rabbit anti-cyclin B (1:1,000; sc-2005, Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) and rabbit
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:80,000 or
1:500,000; Cat. no. 10494-1-AP; Proteintech) at 4°C overnight.
Subsequently, the membrane was incubated with a goat anti-rabbit
horseradish peroxidase-conjugated secondary antibody (1:5,000;
Santa Cruz Biotechnology, Inc.) and visualized by Bio-Rad Gel Doc
XR+ Imaging system (Bio-Rad, Hercules, CA, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
cell viability assay
Cells were seeded onto 12-well culture plates at
2,000 cells/well and cultured for 1, 2, 3, 4 and 5 days before
adding MTT plus acidic isopropanol. After incubation for 4 h at
37°C, 100 µl acidic isopropanol containing 10% SDS, 5%
isopropanol and 0.01 mol/l HCl was then added to stop the reaction.
The absorbance of the reaction mixture was measured at 595 nm in a
UV/VIS spectrophotometer (U-2000; Hitachi, Tokyo, Japan).
Colony survival assay
To further assess the proliferation ability of the
human breast cancer cells, colony formation assay was performed in
ZR-75-30 and T-47D cells. On the third day following infection,
cells were seeded on 6-well culture plastic at an initial cell
density of 400 cells/well and allowed to incubate for 6 days at
37°C under 5% CO2 atmosphere. The cell pellet was washed
with ice-cold PBS, fixed with paraformaldehyde, and subsequently
stained with crystal violet for 20 min. Capability of colony
formation was examined under a fluorescence microscope. The number
of colonies consisting of >50 cells were counted and
quantified.
Cell cycle distribution analysis by flow
cytometry
The Lv-shCon- and Lv-shICT1(S1)-infected cells were
seeded at 40,000 cells in a 6-well plate and incubated for 96 h.
The cells were harvested, washed twice with pre-cold PBS, and fixed
in ice-cold 75% ethanol for 2 min. The samples were digested with
RNase, dyed with propidium iodide and analyzed on a flow cytometer
(BD Biosciences, Franklin Lakes, NJ, USA).
Cell apoptosis
Analysis of the cell apoptosis after 40 h of
infection was carried out using Annexin V-APC/7-AAD labeling
(Apoptosis Detection kit; KeyGEN, Nanjing, China) on a FACSCalibur
(BD Biosciences) according to the manufacturer's instructions. The
proportion of ZR-75-30 cells were categorized as viable (Annexin
V−/7-AAD−), necrotic (Annexin
V−/7-AAD+), early apoptotic (Annexin
V+/7-AAD−), and late apoptotic (Annexin
V+/7-AAD+).
Detectional intracellular signaling
pathways
To simultaneously evaluate the level of
phosphorylation or cleavage of 18 significant molecules, the
PathScan Intracellular Signaling array kit was utilized (Cell
Signaling Technology). ZR-75-30 cells were washed twice with
ice-cold PBS and lysed in 1 ml lysis buffer. After incubation with
the array blocking buffer, the mixtures were treated at room
temperature with lysate for 1 h. Reactions were incubated overnight
at 37°C, treated with monoclonal antibody cocktail, and reacted
with HRP-conjugated streptavidin in substrate buffer. The
chemiluminescence signal was captured by a ChemiDoc XRS imaging
system (Bio-Rad).
Statistical analysis
Each experiment was repeated at least three times
and the results are shown as mean ± standard deviation (SD). SPSS
19.0 (SPSS, Chicago, IL, USA) was used for statistical analyses.
Student's t-test was performed to compare differences between
groups. A p-value <0.05 indicates significant difference.
Results
ICT1 is overexpressed in human breast
cancers
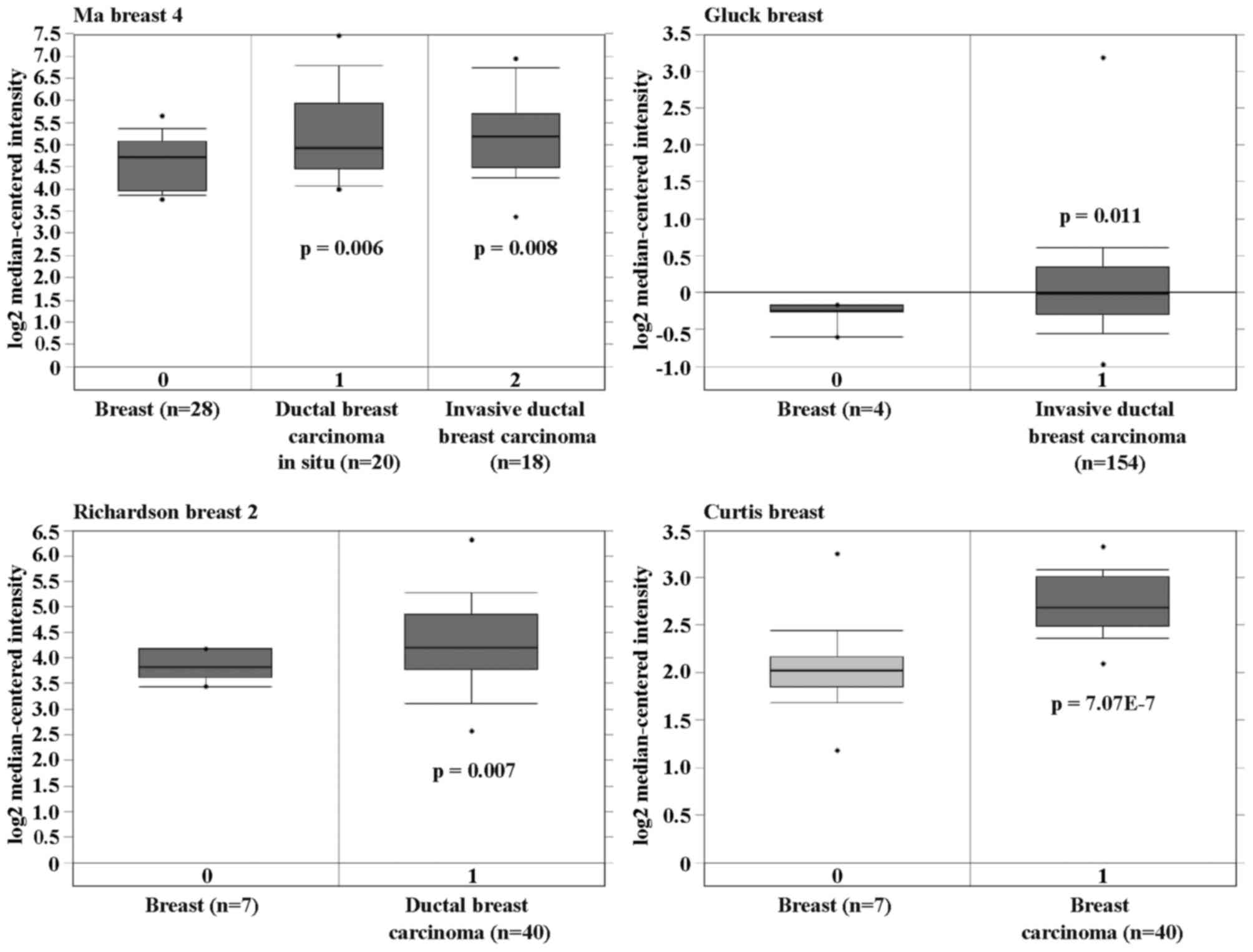
To investigate the correlation between ICT1
and human breast cancer, the ICT1 expression pattern in
normal and cancer samples were initially determined from the
publicly available Oncomine database (https://www.oncomine.org). Four data sets were
identified, including normal blood, breast carcinoma, male breast
carcinoma, tubular breast carcinoma and ductal breast carcinoma
in situ. Importantly, significant overexpression of
ICT1 was found in breast carcinoma compared with breast
(p=7.07E-7), in male breast carcinoma compared with blood and
breast (p=0.003), in tubular breast carcinoma compared with breast
(p=4.73E-24), and in ductal breast carcinoma compared with breast
(p=6.35E-4) (Fig. 1). Here, the
results demonstrated that ICT1 was highly overexpressed in
various human breast cancer subtypes.
Expression of ICT1 mRNA and protein
levels are clearly inhibited in breast cancer cells following
knockdown by shRNA
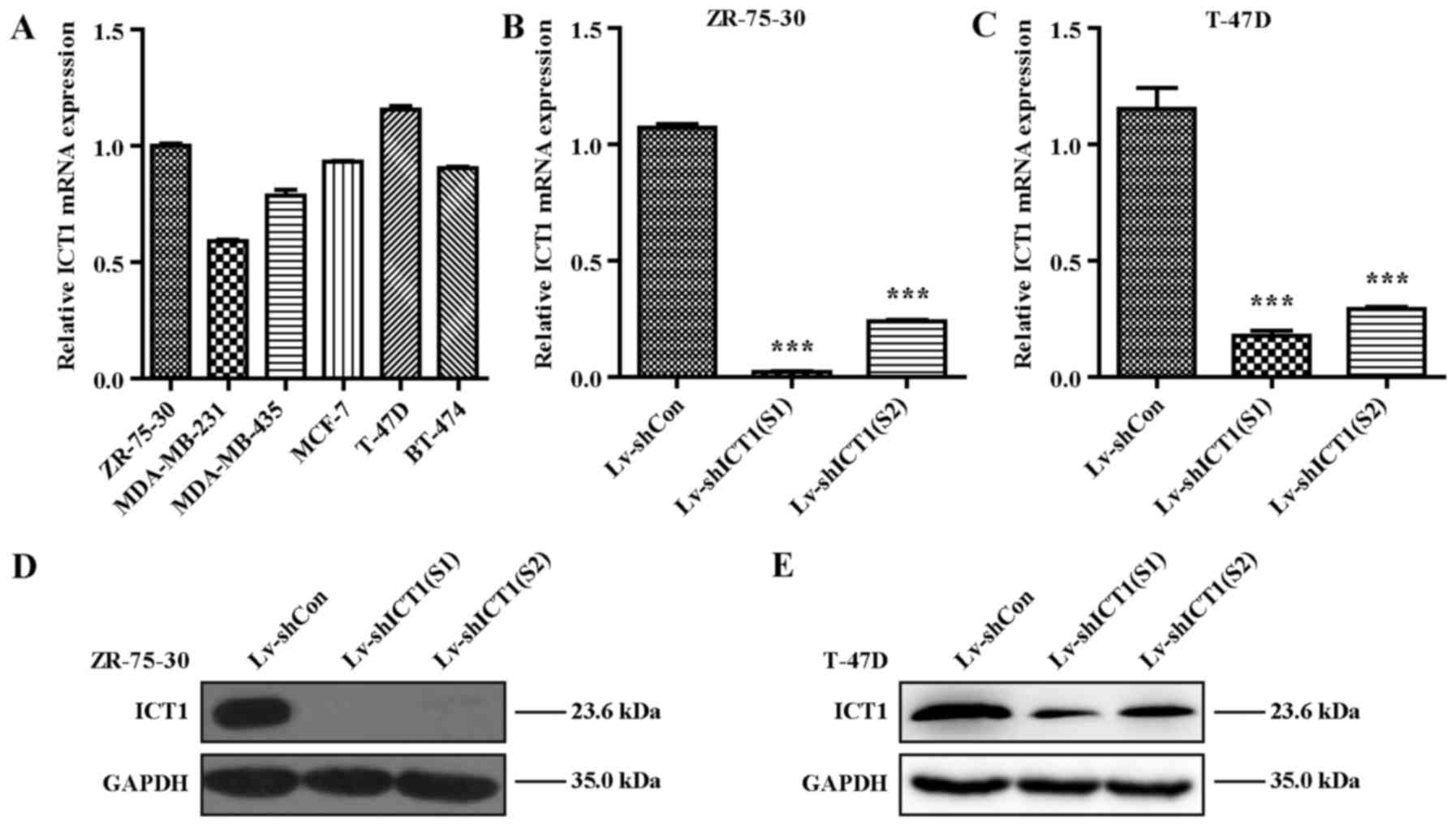
To further investigate the biological function of
ICT1 in breast cancer in vitro, we determined the expression
of ICT1 in several breast cancer cell lines using RT-qPCR analysis.
As shown in Fig. 2A, ZR-75-30 and
T-47D cells presented higher ICT1 expression, which thus were
chosen for the subsequent analysis. To conduct loss-of-function
analysis on breast cancer cells, ICT1 expression was specifically
knocked down in the ZR-75-30 and T-47D cells by lentivirus-mediated
RNA interference technology (shRNA). We evaluated the knockdown
efficiency using RT-qPCR and western blot analysis. These results
demonstrated that the ICT1 mRNA was significantly reduced in
the Lv-shICT1(S1) and the Lv-shICT1(S2) groups compared with that
in the negative controls in the ZR-75-30 (p<0.001) (Fig. 2B) and T-47D (p<0.01) (Fig. 2C) cells. Consistently, ICT1
protein expression was markedly decreased in the ZR-75-30 and T-47D
cells transduced with lentivirus carrying Lv-shICT1(S1) or
Lv-shICT1(S2) as determined by western blotting (Fig. 2D and E). These findings
demonstrated that the ICT1 transcriptional and translational
levels were successfully reduced by lentivirus-mediated delivery of
shRNA in breast cancer cells.
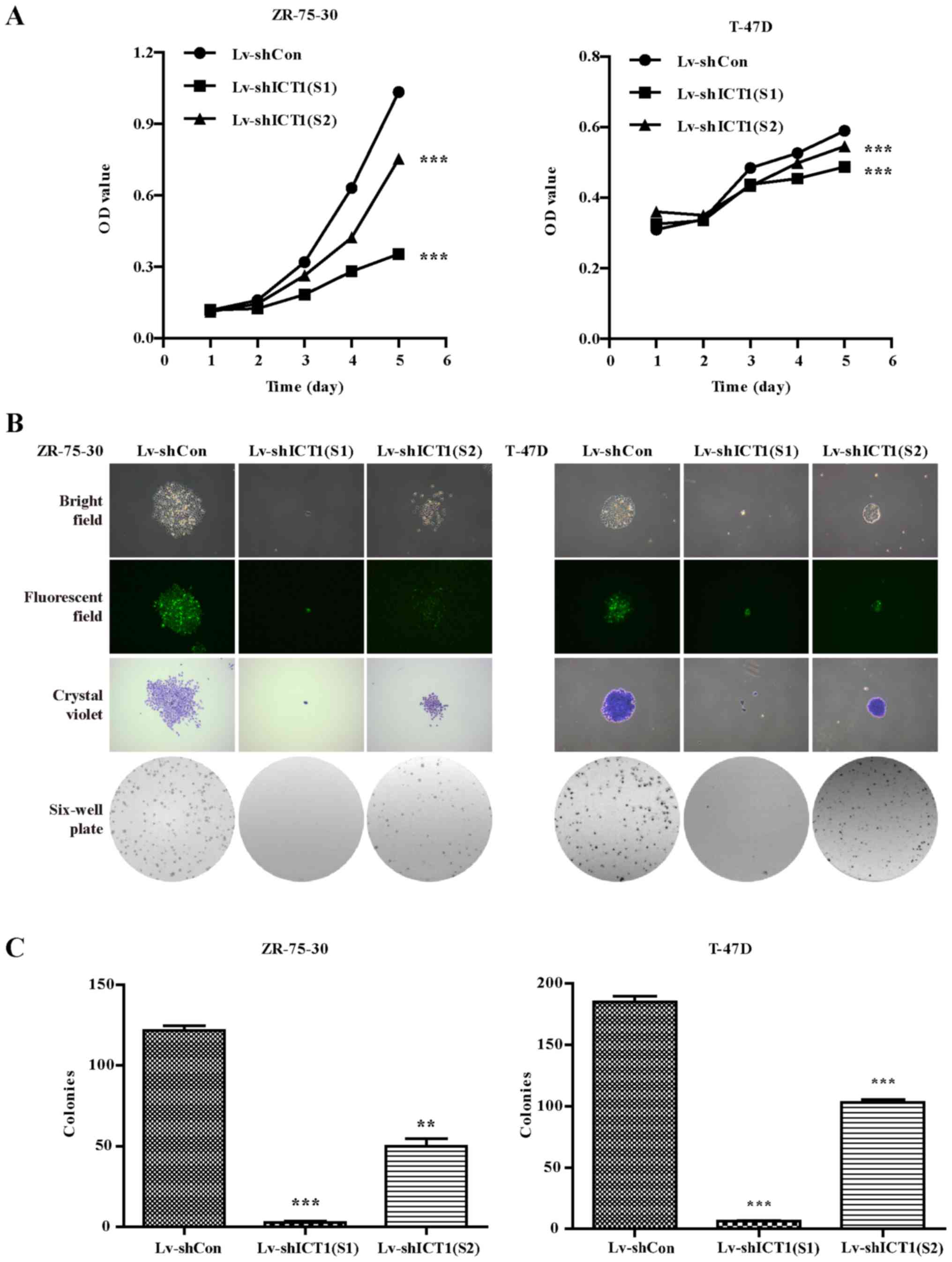
ICT1 knockdown suppresses the
proliferative and colony formation capabilities of human breast
cancer cells
MTT assays were performed to determine cell
proliferation 1, 2, 3, 4 and 5 days post-infection. On the 4th and
5th day, the cell viability was significantly decreased in the
Lv-shICT1(S1)- or Lv-shICT1(S2)-treated groups compared to the
control shRNA-treated ZR-75-30 and T-47D cells (Fig. 3A).
ZR-75-30 and T-47D cells were then used for colony
formation assay. Briefly, colonies were grown for 6 days and
stained with crystal violet. As shown in Fig. 3B, a smaller number of
colony-forming cells was observed under fluorescence microscope
after Lv-shICT1(S1) or Lv-shICT1(S2) infection. A further
examination of colony numbers showed that the number of colonies
was significantly reduced in the shICT1(S1)- and
shICT1(S2)-infected cells compared with the shCon-infected cells
(Fig. 3C). These findings
indicate that ICT1 knockdown inhibited human breast cancer
cell proliferation and colony formation ability and Lv-shICT1(S1)
displayed more powerful ability to inhibit cell proliferation.
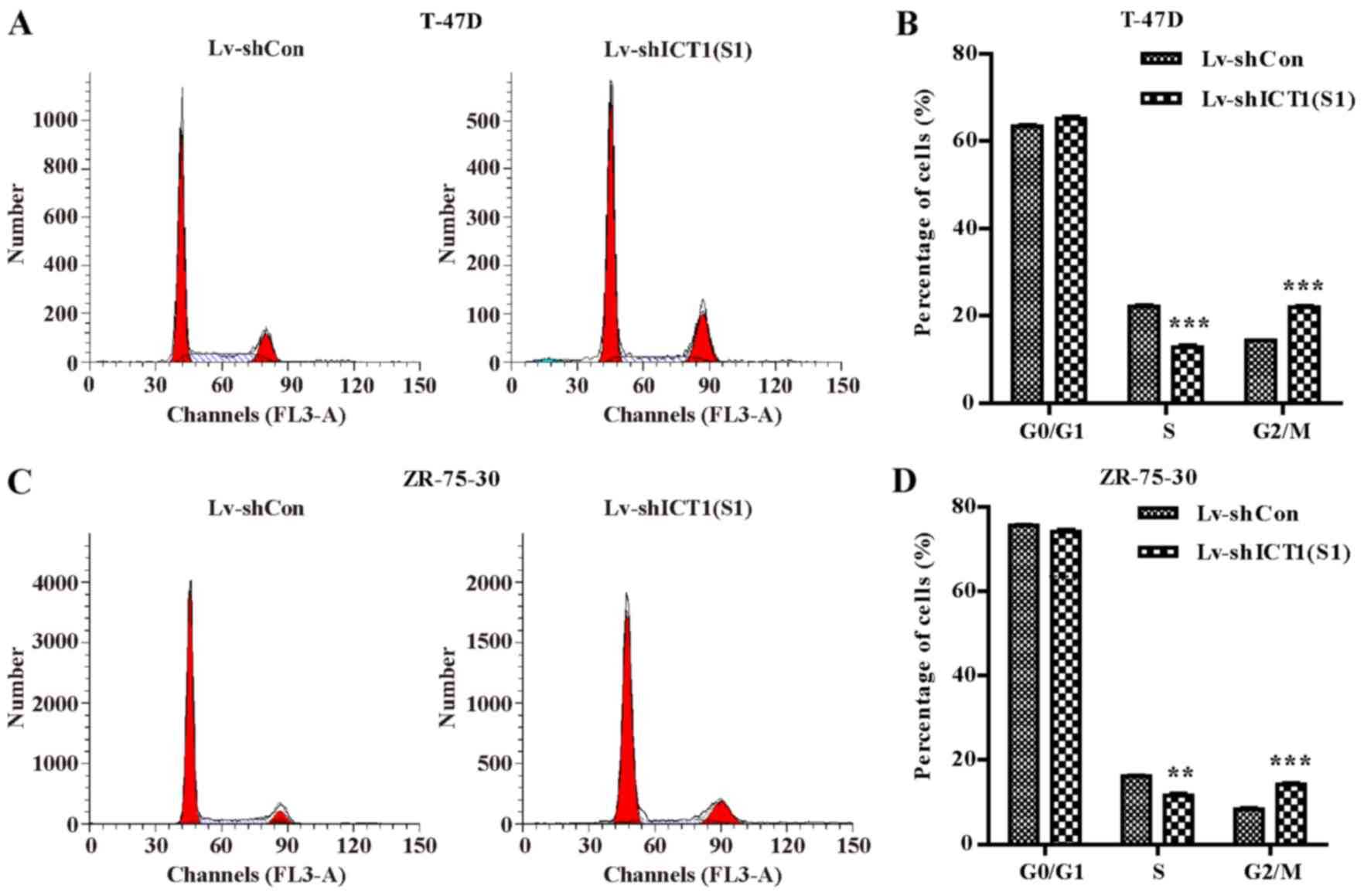
Knockdown of ICT1 causes cell cycle
arrest at G2/M phase in breast cancer cells
To investigate how ICT1 regulates cell
proliferation, flow cytometry was used to examine cell cycle
distribution in the ZR-75-30 and T-47D cells transduced with
Lv-shCon or Lv-shICT1(S1) (Fig. 4A
and C). Statistical analysis further demonstrated that the
percentage of cells in the G2/M phase were markedly increased in
the Lv-shICT1(S1) group in both the T-47D (p<0.001) (Fig. 4B) and ZR-75-30 (p<0.001)
(Fig. 4D) cells. These findings
suggest that down-regulation of ICT1 significantly arrested the
cell cycle at the G2/M phase in the breast cancer cells.
Knockdown of ICT1 promotes cell
apoptosis
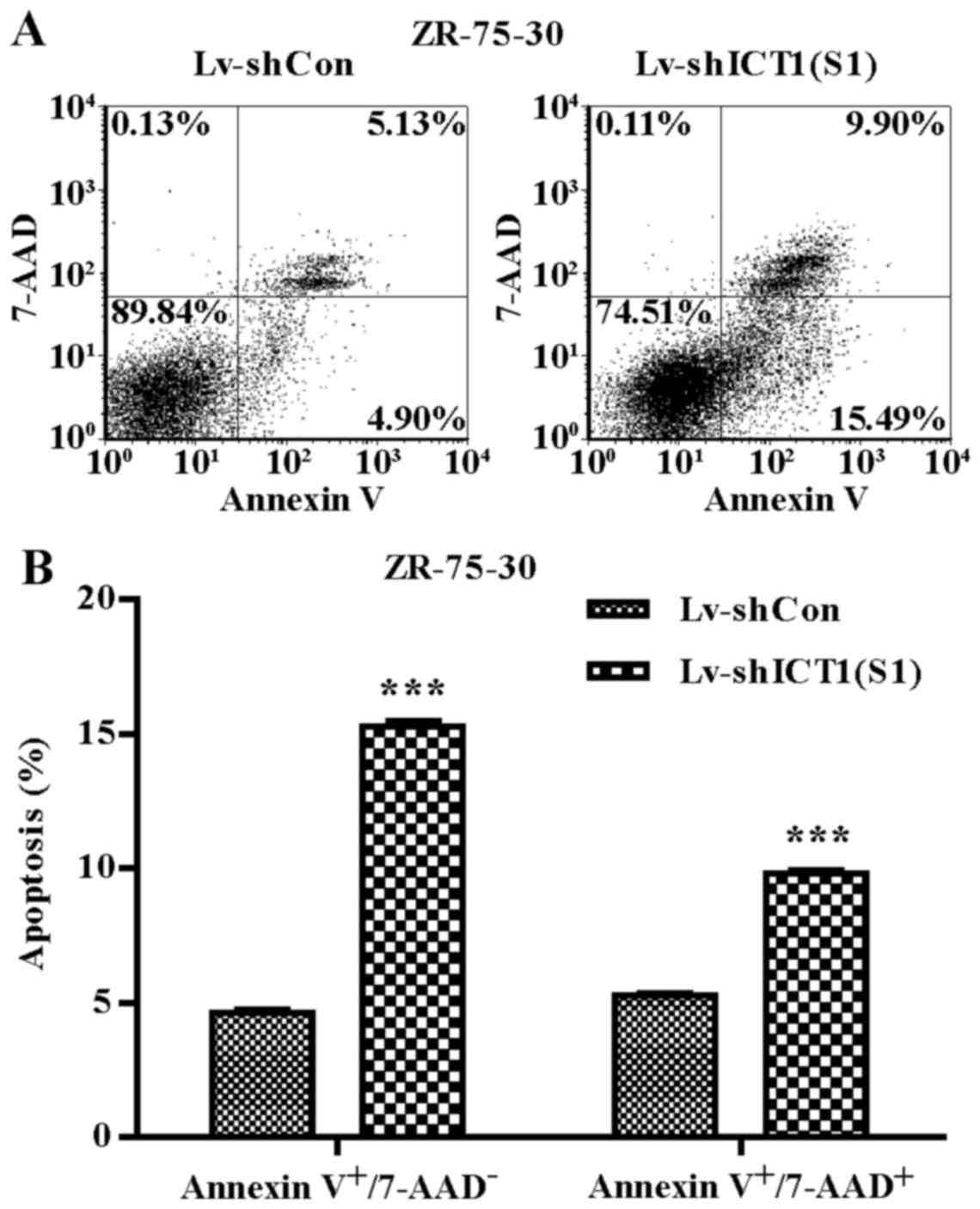
To verify whether ICT1 is involved in
regulating ZR-75-30 cell apoptosis, 7-AAD and Annexin V-APC
staining was performed (Fig. 5A).
The percentages of early apoptotic (Annexin
V-APC+/7-AAD−) and late apoptotic (Annexin
V-APC+/7-AAD+) cells were significantly
higher in the ICT1-knockdown group than that in the control
group (15.28±0.41 vs. 4.62±0.24 and 9.84±0.19 vs. 5.3±0.16)
(Fig. 5B). These results
indicated that knockdown of ICT1 significantly increased the
percentages of early and late apoptotic breast cancer cells.
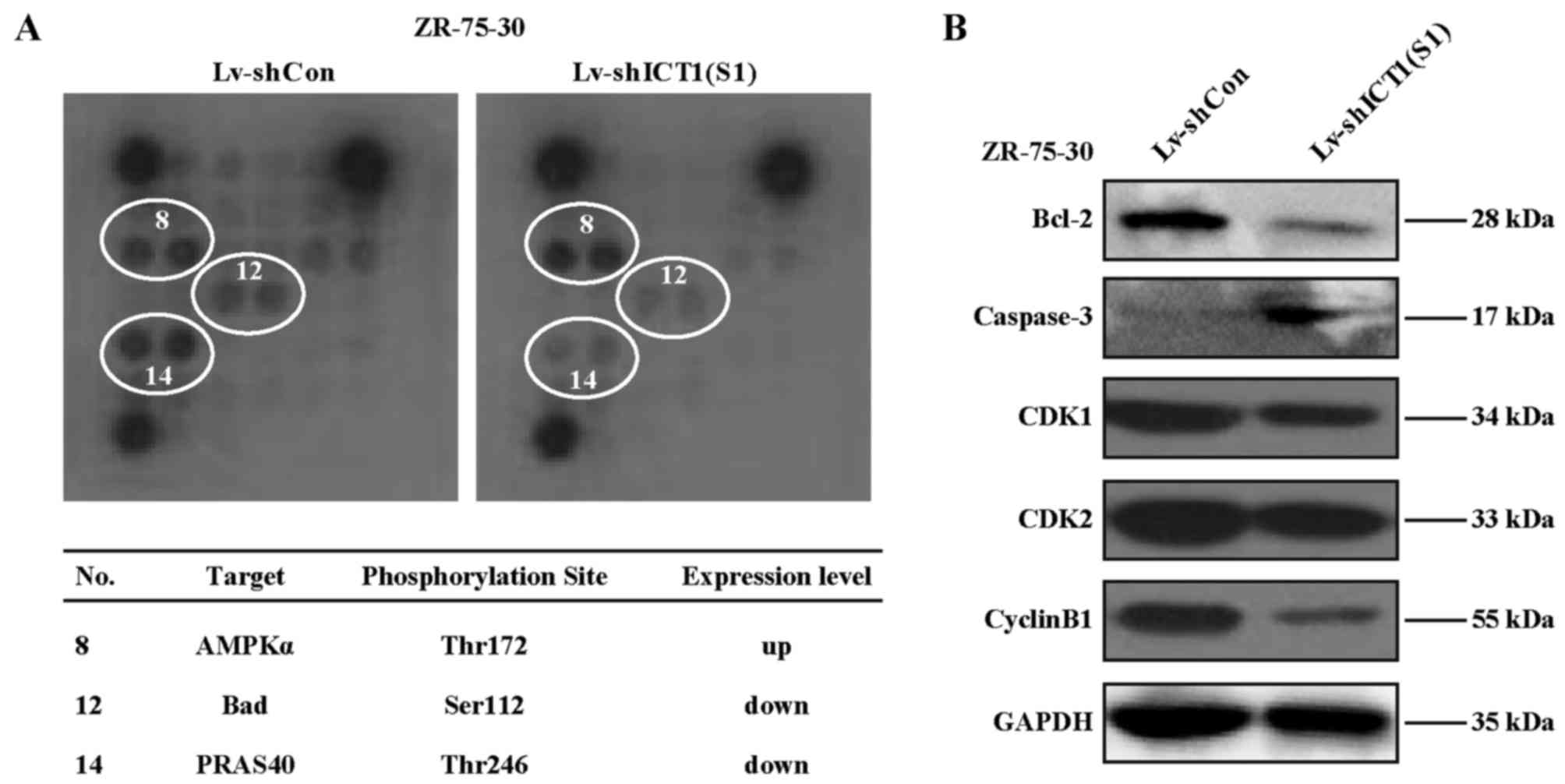
Depletion of ICT1 disrupts Bad (Ser112),
PRAS40 (Thr246), and AMP-activated protein kinase α (AMPKα)
(Thr172) phosphorylation in ZR-75-30 cells
To investigate the molecular mechanism of
ICT1-mediated cell proliferation and apoptosis regulation,
evaluation of ICT1 effects on 18 significant and
well-characterized signaling molecules in the ZR-75-30 cells was
detected by PathScan@ intracellular signaling array kit.
As a result (Fig. 6),
Lv-shICT1(S1)-transfected cells showed enhanced phosphorylation of
AMPKα at Thr172. In contrast, Bad phosphorylation at Ser112 and
PARS40 phosphorylation at Thr246 was decreased in the
Lv-shICT1(S1)-treated ZR-75-30 cells. Hence, it was suggested that
the signaling pathways mediated by Bad, AMPK or PRAS40 may be
altered after knockdown of ICT1 in ZR-75-30 cells.
Efeect of ICT1 knockdown on proteins
involved in cell apoptosis or cell cycle in ZR-75-30 cells
The above mentioned data suggested that the
regulation of cell apoptosis by ICT1 is closely correlated with
Bcl-2 family proteins. We further investigated the expression
pattern of Bcl-2 and caspase-3, two proteins that act as the
executor and modulator of cell apoptosis (17). As a result, Lv-shICT1(S1)
transduction significantly decreased the protein level of Bcl-2. In
contrast, the caspase-3 protein level was increased. Moreover, the
expression of cell cycle regulatory proteins including CDK1, CDK2
and cyclin B were all significantly downregulated in the
Lv-shICT1(S1)-infected cells.
Discussion
Human breast cancer is one of the most common
malignancies diagnosed worldwide and the overall incidence of
breast cancer has increased almost 2-fold over the past 20 years
(18). ICT1, a component
of the human mitoribosome, has been shown to be involved in the
proliferation of various types of cancer cells including HeLa,
hepatoblastoma HepG2 and glioblastoma multiforme cells (14,16). In the present study, the role of
ICT1 in breast cancer cells was explored. Our data revealed
that ICT1 deficiency led to the inhibition of proliferation
and colony formation, and the G2/M phase arrest. Consistently,
ICT1 knockdown caused cell cycle arrest at the G2/M phase in
prostate (19) and colon cancer
(20). But in lung cancer cells,
the cell cycle was arrested at the G0/G1 phase by ICT1
downregulation leading to cell growth inhibition (21). These differences in the different
cell cycle phases may be ascribed to the tumor type.
Moreover, intracellular signaling pathways,
apoptosis-related proteins, and cell cycle regulatory proteins were
partially altered in breast cancer cells following ICT1
knockdown. Cyclin-dependent kinase (CDK)/cyclin complexes play
central roles in the control of cell cycle progression in
eukaryotic cells (22). For
successful entry into mitosis, the activation of the CDK1/cyclin B
complex is essential for driving cells into the M phase (23). Meanwhile, the inactive form of
CDK1/cyclin B must occur for cell cycle arrest at G2/M transition
(24). CDK2 can form complexes
with cyclin A and B to participate in the cell cycle transition
from S phase to premitotic G2 phase and through the G2/M checkpoint
(25). p21Waf,
p27Kip1 and p57Kip2 are considered
cyclin-dependent kinase inhibitors that regulate cell cycle
distribution (26). In this
study, cell cycle arrest in G2/M phases occurred in the
ICT1-knockdown cells. It was suggested that the complexes
formed with CDK1, cyclin B or CDK2 were suppressed, as protein
levels of these proteins were all reduced in the ICT1-knockdown
cells. Therefore, the induction of G2/M arrest may be mediated
through the inactivation of CDK1-cyclin B and CDK2-cyclin B
complexes. Decreased activity of CDK2-cyclin A and CDK2-cyclin E
may have contributed to the S phase arrest in the ICT1
shRNA-transfected ZR-75-30 cells. It remain to be explored whether
ICT1 acts as negative regulator of p21Waf and/or
p27Kip1; targeting the downstream CDK1 and CDK2 in
breast cancer.
Phosphorylation of AMPK, Bad and PRAS40 has been
reported to be linked to cell apoptosis regulation (27,28). Intriguingly, the phosphorylation
of Bad Ser112, PRAS40 Thr246 and AMPKα-Thr172 could be regulated by
protein kinase A (PKA), PIM1 and AKT (29,30). Hurley et al demonstrated
that the phosphorylation of AMPKα at Thr172 is a determinant factor
for AMPK activation (27). Thus,
phosphorylation of AMPK, an energy and nutrient sensor, can
modulate its downstream signaling targets (31,32). In this investigation, the
phospho-AMPK (Thr172) was induced upon knockdown of ICT1,
indicating that the AMPK downsteam pathways may contribute to the
apoptosis of breast cancer cells.
Bad is a death-promoting member of the BCL-2 family
promoting apoptosis through interaction and inhibition of Bcl-xL
and Bcl-2 anti-apoptotic function (33). When Bad was phosphorylated at
Ser112, Ser136 and Ser155, the sequestration of Bad in the cytosol
occurs as a result of the tau form of 14-3-3 proteins that promotes
dissociation of Bcl-xL and Bcl-2 (34,35). Our data demonstrated that Bad
Ser112 phosphorylation was decreased in the ICT1-knockdown
cells, suggesting an increased binding of Bad to Bcl-xL and Bcl-2
and subsequent promotion of cell apoptosis.
PRAS40 has been identified as a proline-rich
substrate of Akt and is involved in cell survival, proliferation,
and mobility (28,36). Wang et al (36) showed that a decrease in PRAS40
(Thr246) phosphorylation is associated with tumor cell apoptosis.
During tumor cell growth progression in human melanocytes, the
phospho-Thr246 of PRAS40 was increased (36). In this study, reduced
phospho-Thr246 PRAS40 was observed following knockdown of
ICT1 contributing to the induction of apoptosis in breast
cancer cells. Moreover, caspase-3, a death protease that is usually
activated during cell death program (37) was found to be overexpressed in
ZR-75-30 cells with ICT1 knockdown.
In conclusion, our results demonstrated that
ICT1 may play a positive role in breast cancer cell
proliferation by regulating cell cycle progression and apoptosis.
Knockdown of ICT1 may be used as a new therapeutic approach for the
therapy for breast cancer. Furthermore, shRNA-mediated
ICT1-targeted therapy warrants further investigation in preclinical
and clinical studies.
Acknowledgments
The authors are thankful for the financial support
from the Natural Science Foundation of Zhejiang Province, China
(no. LY12H16030); the Foundation of Health Department of Zhejiang
Province, China (no. 2011BCA015) and the Traditional Chinese
Medical Research Foundation of Zhejiang Province, China (no.
2011ZA016).
References
|
1
|
Ferlay J, Héry C, Autier P and
Sankaranarayanan R: Global burden of breast cancer. Breast Cancer
Epidemiology. Li C: Springer; New York: pp. 1–19. 2010, View Article : Google Scholar
|
|
2
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Key T, Appleby P, Barnes I, Reeves G and
Endogenous H; Endogenous Hormones and Breast Cancer Collaborative
Group: Endogenous sex hormones and breast cancer in postmenopausal
women: Reanalysis of nine prospective studies. J Natl Cancer Inst.
94:606–616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Skobe M, Hawighorst T, Jackson DG, Prevo
R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K and Detmar
M: Induction of tumor lymphangiogenesis by VEGF-C promotes breast
cancer metastasis. Nat Med. 7:192–198. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Müller A, Homey B, Soto H, Ge N, Catron D,
Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al:
Involvement of chemokine receptors in breast cancer metastasis.
Nature. 410:50–56. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clarke M, Collins R, Darby S, Davies C,
Elphinstone P, Evans V, Godwin J, Gray R, Hicks C, James S, et al
Early Breast Cancer Trialists' Collaborative Group (EBCTCG):
Effects of radiotherapy and of differences in the extent of surgery
for early breast cancer on local recurrence and 15-year survival:
An overview of the randomised trials. Lancet. 366:2087–2106. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Slamon DJ, Leyland-Jones B, Shak S, Fuchs
H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M,
et al: Use of chemotherapy plus a monoclonal antibody against HER2
for metastatic breast cancer that overexpresses HER2. N Engl J Med.
344:783–792. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meraviglia S, Eberl M, Vermijlen D, Todaro
M, Buccheri S, Cicero G, La Mendola C, Guggino G, D'Asaro M,
Orlando V, et al: In vivo manipulation of Vgamma9Vdelta2 T cells
with zoledronate and low-dose interleukin-2 for immunotherapy of
advanced breast cancer patients. Clin Exp Immunol. 161:290–297.
2010.PubMed/NCBI
|
|
9
|
Das S, Ferlito M, Kent OA, Fox-Talbot K,
Wang R, Liu D, Raghavachari N, Yang Y, Wheelan SJ, Murphy E, et al:
Nuclear miRNA regulates the mitochondrial genome in the heart. Circ
Res. 110:1596–1603. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dames S, Eilbeck K and Mao R: A
high-throughput next-generation sequencing assay for the
mitochondrial genome. Methods Mol Biol. 1264:77–88. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Handa Y, Inaho N and Nameki N: YaeJ is a
novel ribosome-associated protein in Escherichia coli that can
hydrolyze peptidyl-tRNA on stalled ribosomes. Nucleic Acids Res.
39:1739–1748. 2011. View Article : Google Scholar
|
|
12
|
Akabane S, Ueda T, Nierhaus KH and
Takeuchi N: Ribosome rescue and translation termination at
non-standard stop codons by ICT1 in mammalian mitochondria. PLoS
Genet. 10:e10046162014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Richter R, Rorbach J, Pajak A, Smith PM,
Wessels HJ, Huynen MA, Smeitink JA, Lightowlers RN and
Chrzanowska-Lightowlers ZM: A functional peptidyl-tRNA hydrolase,
ICT1, has been recruited into the human mitochondrial ribosome.
EMBO J. 29:1116–1125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Handa Y, Hikawa Y, Tochio N, Kogure H,
Inoue M, Koshiba S, Güntert P, Inoue Y, Kigawa T, Yokoyama S, et
al: Solution structure of the catalytic domain of the mitochondrial
protein ICT1 that is essential for cell vitality. J Mol Biol.
404:260–273. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kogure H, Handa Y, Nagata M, Kanai N,
Güntert P, Kubota K and Nameki N: Identification of residues
required for stalled-ribosome rescue in the codon-independent
release factor YaeJ. Nucleic Acids Res. 42:3152–3163. 2014.
View Article : Google Scholar :
|
|
16
|
Xie R, Zhang Y, Shen C, Cao X, Gu S and
Che X: Knockdown of immature colon carcinoma transcript-1 inhibits
proliferation of glioblastoma multiforme cells through Gap
2/mitotic phase arrest. Onco Targets Ther. 8:1119–1127.
2015.PubMed/NCBI
|
|
17
|
Sparrow JR and Cai B: Blue light-induced
apoptosis of A2E-containing RPE: Involvement of caspase-3 and
protection by Bcl-2. Invest Ophthalmol Vis Sci. 42:1356–1362.
2001.PubMed/NCBI
|
|
18
|
Yan LX, Huang XF, Shao Q, Huang MY, Deng
L, Wu QL, Zeng YX and Shao JY: MicroRNA miR-21 overexpression in
human breast cancer is associated with advanced clinical stage,
lymph node metastasis and patient poor prognosis. RNA.
14:2348–2360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Z, Xu D, Gao Y, Liu Y, Ren J, Yao Y,
Yin L, Chen J, Gan S and Cui X: Immature colon carcinoma transcript
1 is essential for prostate cancer cell viability and
proliferation. Cancer Biother Radiopharm. 30:278–284. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lao X, Feng Q, He G, Ji M, Zhu D, Xu P,
Tang W, Xu J and Qin X: Immature colon carcinoma transcript-1
(ICT1) expression correlates with unfavorable prognosis and
survival in patients with colorectal cancer. Ann Surg Oncol.
23:3924–3933. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, He J, Zhang S, Yang Q, Wang B, Liu
Z and Wu X: Knockdown of immature colon carcinoma transcript-1
inhibits proliferation and promotes apoptosis of non-small cell
lung cancer cells. Technol Cancer Res Treat. Jul 13–2016.(Epub
ahead of print). doi:20161533034616657977. View Article : Google Scholar
|
|
22
|
Arellano M and Moreno S: Regulation of
CDK/cyclin complexes during the cell cycle. Int J Biochem Cell
Biol. 29:559–573. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji YB, Wu D, Dai QC, Guo L and Chen N: The
research on the medicinal value of Amaryllidaceae plants. Appl Mech
Mater. 411–414:3223–3226. 2013. View Article : Google Scholar
|
|
24
|
Cmielová J and Rezáčová M:
p21Cip1/Waf1 protein and its function based on a
subcellular localization [corrected]. J Cell Biochem.
112:3502–3506. 2011. View Article : Google Scholar
|
|
25
|
Caputi M, Russo G, Esposito V, Mancini A
and Giordano A: Role of cell-cycle regulators in lung cancer. J
Cell Physiol. 205:319–327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lim S and Kaldis P: Cdks, cyclins and
CKIs: Roles beyond cell cycle regulation. Development.
140:3079–3093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hurley RL, Anderson KA, Franzone JM, Kemp
BE, Means AR and Witters LA: The
Ca2+/calmodulin-dependent protein kinase kinases are
AMP-activated protein kinase kinases. J Biol Chem. 280:29060–29066.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morgensztern D and McLeod HL:
PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer
Drugs. 16:797–803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garcia-Haro L, Garcia-Gimeno MA, Neumann
D, Beullens M, Bollen M and Sanz P: Glucose-dependent regulation of
AMP-activated protein kinase in MIN6 beta cells is not affected by
the protein kinase A pathway. FEBS Lett. 586:4241–4247. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu J, Zhang T, Wang T, You L and Zhao Y:
PIM kinases: An overview in tumors and recent advances in
pancreatic cancer. Future Oncol. 10:865–876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meisse D, Van de Casteele M, Beauloye C,
Hainault I, Kefas BA, Rider MH, Foufelle F and Hue L: Sustained
activation of AMP-activated protein kinase induces c-Jun N-terminal
kinase activation and apoptosis in liver cells. FEBS Lett.
526:38–42. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shao JJ, Zhang AP, Qin W, Zheng L, Zhu YF
and Chen X: AMP-activated protein kinase (AMPK) activation is
involved in chrysin-induced growth inhibition and apoptosis in
cultured A549 lung cancer cells. Biochem Biophys Res Commun.
423:448–453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chiang CW, Kanies C, Kim KW, Fang WB,
Parkhurst C, Xie M, Henry T and Yang E: Protein phosphatase 2A
dephosphorylation of phosphoserine 112 plays the gatekeeper role
for BAD-mediated apoptosis. Mol Cell Biol. 23:6350–6362. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peso LD, González-García M, Page C,
Herrera R and Nuñez G: Interleukin-3 induced phosphorylation of BAD
through protein kinase Akt. Science. 278:687–689. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jin YP, Fishbein MC, Said JW, Jindra PT,
Rajalingam R, Rozengurt E and Reed EF: Anti-HLA class I
antibody-mediated activation of the PI3K/Akt signaling pathway and
induction of Bcl-2 and Bcl-xL expression in endothelial cells. Hum
Immunol. 65:291–302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang H, Zhang Q, Wen Q, Zheng Y,
Lazarovici P, Jiang H, Lin J and Zheng W: Proline-rich Akt
substrate of 40kDa (PRAS40): A novel downstream target of PI3k/Akt
signaling pathway. Cell Signal. 24:17–24. 2012. View Article : Google Scholar
|
|
37
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|