Introduction
Androgen deprivation therapy (ADT, surgical or
chemical castration) has been the primary treatment for prostate
cancer (PCa) used to suppress the transcriptional activity of
androgen receptor (AR) since the 1940s; however, a proportion of
patients relapse within a median of 2-3 years with disease that is
usually more aggressive and is currently defined as
castration-resistant PCa (CRPC) (1–3).
It was well known that AR is actively involved in the development,
progression and metastasis of PCa (4). Over the past several years, studies
from many groups have proposed that AR is aberrantly re-activated
in CRPC (4–6). There are multiple mechanisms through
which AR is aberrantly re-activated to support cell growth in CRPC
in the androgen-depleted condition, including AR amplification (the
hypersensitive pathway), AR mutation and co-regulator alterations
(the promiscuous pathway), ligand-independent AR activation (the
outlaw pathway) (4).
In general, androgen is a driver which elicits PCa
initiation and progression (7).
However, it is contradictory that androgen plays a role as a
suppressor in LNCaP cell growth by inducing cell cycle arrest at
the G1 phase following treatment with high-dose dihydrotestosterone
(DHT), accompanied by p27 overexpression and the loss of the
phosphorylated retinoblastoma protein (8,9).
Intermittent androgen deprivation therapy (IADT) had been proposed
for many years, and preclinical models have demonstrated that
exposure to intermittent androgen leads to a delay in the time to
castration resistance in castrated animals bearing
androgen-dependent Shionogi carcinoma tumors (10). Nonetheless, in phase III clinical
trials, there is still insufficient evidence to support that IADT
prevents the long-term complications caused by ADT (11). Thus, the roles which androgen
plays in PCa are closely dependent on the context.
LNCaP was considered as a universally used cell line
derived from a human lymph node metastatic lesion of prostatic
adenocarcinoma (12). Previously,
we established a PCa progression model using LNCaP cells which were
continuously maintained in androgen-depleted conditions to mimic
patients subjected to androgen ablation therapy (13), terned the LNCaP-AI cell line.
LNCaP-AI cells have been previously reported (14); however, there are few studies
which completely explain the differences among the two cell lines.
In this study, we examined the alterations in cell biological
characteristics between the LNCaP and LNCaP-AI cells. We also
investigated the roles which androgen and AR play in regulating
LNCaP and LNCaP-AI cell growth, making a comparison between then
LNCaP and LNCaP cells as regards AR expression and PSA secretion
when androgen is absent or present. These findings shed more light
into the role of androgen in PCa and enhance our understanding of
PCa development and progression.
Materials and methods
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM), phenol
red-free DMEM, fetal bovine serum (FBS), charcoal-stripped FBS
(CS-FBS), penicillin, streptomycin, 0.25% trypsin and
phosphate-buffered saline (PBS) were purchased from Gibco
(Gaithersburg, MD, USA). DHT with a purity of >98%, was
purchased from Sigma-Aldrich (St. Louis, MO, USA). For all androgen
regulation experiments, the cells were maintained in phenol
red-free DMEM medium with 10% CS-FBS. The androgen DHT powder was
dissolved in ethanol and diluted into the required concentrations
with the culture medium. Antibodies against cyclin D1 (1:1,000;
ab134175), Cdk4 (1:1,000; ab199728), Cdk2 (1:1,000; ab32147),
retinoblastoma protein (pRb; 1:1,000; Ab173289) were purchased from
Abcam (Cambridge, MA, USA). Antibodies against AR (1:1,000; 3202S),
p21 (1:1,000; 2947S), p27 (1:1,000; 3686S), cyclin E1 (1:1,000;
4129S), E-cadherin (1:1,000; 14472S), N-cadherin (1:1,000; 14215S)
and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1000; 5174S)
were purchased from Cell Signaling Technology (Beverly, MA,
USA).
Cells and cell culture
LNCaP cells obtained from the Cell Bank of Shanghai
Institute of Biochemistry and Cell Biology, Chinese Academy of
Sciences (Shanghai, China) were cultured in a 5% CO2
incubator in DMEM with 10% FBS. The establishment of an
androgen-independent cell line (LNCaP-AI) was carried out as
previously described (13). The
LNCaP-AI cells were maintained in phenol red-free DMEM with 10%
CS-FBS.
Cell proliferation assay
Cell proliferation was measured using a cell
counting kit-8 (CCK-8; Dojindo Molecular Technologies, Kumamoto,
Japan) following the manufacturer's instructions. Briefly, the
cells were seeded in 96-well plate at a density of 5×103
cells/well. After 24 h, they were treated with 10 nM DHT or were
transfected with AR short hairpin RNA (shRNA) and scrambled shRNA
recombinant lentiviral vector for different periods of time. A
total of 10 µl CCK-8 solution was added to each well of the
plate and incubated at 37°C for 2 h in a humidified 5%
CO2 atmosphere. Cell viability was then determined by
testing the absorbance at a 450 nm wavelength with a microplate
reader (iMark™; serial no. 13492; Bio-Rad, Tokyo, Japan).
Transwell invasion assay
Transwells of 24-well coated with Matrigel
(8-µm pore size; BD Biosciences, Bedford, MA, USA) were used
in the cell invasion assay. The cells were cultured in serum-free
medium overnight before being trypsinized and resuspended at a
density of 5×104 cells/ml in serum-free medium. A total
of 1×104 cells (200 µl) were seeded into the
upper chambers. Subsequently, the lower chambers were supplemented
with 500 µl complete medium containing 10% FBS as a
chemoattractant. After 48 h of incubation, the Matrigel and the
cells remaining in the upper chamber were removed using cotton
swabs. The invaded cells were fixed in 4% paraformaldehyde and
stained with 0.5% crystal violet, counted using an inverted
microscope (Leica, Wetzlar, Germany).
Apoptosis analysis
Cell apoptosis was measured using the Annexin
V-PE/7-AAD kit (BD Biosciences, San Diego, CA, USA). In brief, the
cells were seeded in 6-well plates at a density of 2×105
cells/well and transfected with AR shRNA or scrambled shRNA
recombinant lentivirus vector for 72 h. The cells were typsinized
and resuspended in 100 µl 1× binding buffer, followed by the
addition of 5 µl of PE Annexin V and 5 µl 7-AAD, and
gently vortexing the cells and incubating for 15 min at room
temperature (25°C) in the dark. Subsequently, 400 µl 1×
binding buffer were replenished to each tube. Samples were analyzed
using a flow cytometer (FACSCalibur; BD Biosciences, San Jose, CA,
USA) within 1 h and CellQuest software (FACSCalibur).
Cell cycle analysis
Cell cycle distribution was measured by flow
cytometry with PI/RNase staining buffer (BD Biosciences). In brief,
2×105 cells were exposed to ethanol (vehicle) or 10 nM
DHT and transfected with AR shRNA or scrambled shRNA recombinant
lentivirus vector. The cells were then trypsinized, resuspended and
fixed with 70% ethanol/30% PBS at −20°C overnight. The cells were
then stained with PI/RNase staining buffer for 15 min in the dark
and analyzed using a flow cytometer and ModFit LT software
(FACSCalibur). For each measurement, 3×104 cells were
collected and analyzed.
Transfection with shRNA lentiviral
particles
shRNA lentiviral particles were purchased from
GenePharma Biotechnology, Inc. (Shanghai, China). The sequence of
the AR shRNA was as follows: 5′-GGAACTCGATCGTATCATTGC-3′; a
scrambled sequence was used as a negative control with the
following squence: 5′-TTCTCCGAACGTGTCACGT-3′. According to the
instructions provided with the lentiviral particles, the cells were
seeded at 2×105 cells/well in a 6-well plate prior to
lentiviral particles infection and incubated with 2 ml of complete
medium for 24 h. The cells were infected by the addition of the
shRNA lentiviral particles at a multiplicity of infection (MOI) of
50; the plates were then gently swirled for mixing followed by
incubation for 12 h. The virus-containing medium of infected wells
was removed and fresh complete medium was added. The transfection
efficiency was evaluated using a FACSCalibur (BD Biosciences) flow
cytometer detecting green fluorescence protein (GFP) expression.
Reverse transcription-quantitative PCR (RT-qPCR) and western blot
analysis were performed to determine the shRNA interference
efficiency.
RT-qPCR
Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) and dissolved in RNAase-free water
according to the manufacturer's instructions. Total RNA (1.0
µg) was used as a template for cDNA synthesis using the
PrimeScript™ RT reagent kit with gDNA Eraser (Takara, Dalian,
China). Real-time PCR was performed using FastStart Universal
SYBR-Green Master (Roche, Indianapolis, IN, USA). The GAPDH mRNA
level was used as an internal control. The sequences of primers
were used as follows: AR forward, 5′-GCCACTCAGACCCACTTAGC-3′ and
reverse, 5′-CCTCACTCTTCGTCCACATCG-3′; PSA forward,
5′-GCCCACTGCATCAGGAACAA-3′ and reverse,
5′-GCTGTGGCTGACCTGAAATACC-3′; GAPDH forward,
5′-AACAGCCTCAAGATCATCAGCA-3′ and reverse,
5′-CATGAGTCCTTCCACGATACCA-3′. The reactions were performed using
the ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City,
CA, USA). All data for each sample were collected in triplicate and
assessed using 2−ΔΔCq relative quantitative
analysis.
Determination of PSA secretion
The cells were seeded in a 6-well plate with
androgen-depleted medium at a density of 3×105
cells/well and then treated with ethanol (vehicle) or 10 nM DHT for
24 h, and were transfected with AR shRNA or scrambled shRNA
recombinant lentivirus vector for 72 h. The cell culture
supernatants were collected to measure the total PSA level on a
chemiluminescence apparatus (Architect i2000SR; Abbott Diagnostics,
Abbott Park, IL, USA) according to the manufacturer's instructions
by chemiluminescence immunoassay (CLIA).
Western blot analysis
The cells were lysed in pre-cold RIPA lysis buffer
containing protease inhibitor cocktail (Biotool, Houston, TX, USA),
and placed sample on ice vigorously vortex 15 sec every 10 min for
3 times. Cell lysates were centrifuged at 13,000 × g for 10 min and
the supernatants were collected. Protein concentrations were
measured using the Pierce BCA protein assay kit (Pierce, Rockford,
IL, USA) according to the manufacturer's instructions. Total
protein (40 µg) was loaded and separated by electrophoresis
on 10% SDS-PAGE at 100 V for 2 h and transferred onto 0.2 µm
polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA)
using wet blotter at 250 mA for 2 h. The membranes were blocked
with 5% defatted milk for 1 h at room temperature and then
incubated with primary antibody diluted in 5% bovine serum albumin
at 4°C overnight. After being washed 3 times for 15 min with 10%
TBST, the membranes were incubated with HRP-conjugated secondary
antibodies [anti-mouse IgG (1:5,000; 7076S) and anti-rabbit IgG
(1:5,000; 7074S); both from Cell Signaling Technology] for 2 h at
room temperature. The bound antibodies were visualized using an
enhanced chemiluminescence kit (Millipore).
Statistical analysis
All data are presented as the means ± standard
deviation. Two-tailed Student's t-test was used and a value of
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were performed using SPSS 13.0
software (SPSS Inc., Chicago, IL, USA).
Results
Prolonged androgen deprivation increases
the LNCaP-AI cell proliferation rate and invasiveness, and induces
epithelial-mesenchymal transition (EMT)
To examine the alterations in the cell biological
characteristics in the transition from LNCaP to LNCaP-AI cells, we
performed CCK-8 cell proliferation assay, cell invasion assay and
western blot analysis of EMT marker proteins in the LNCaP-AI cells
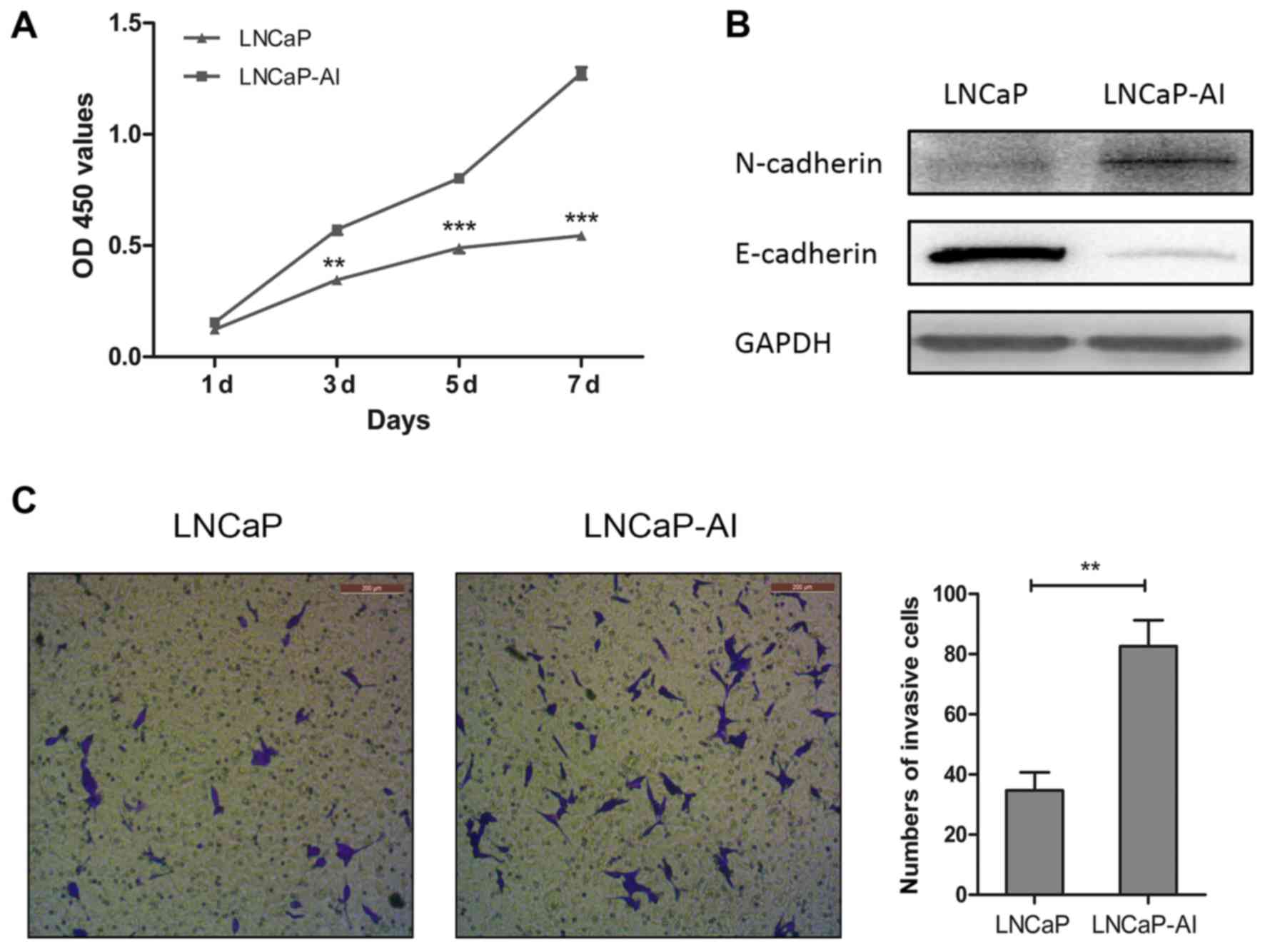
and LNCaP cells. CCK-8 proliferation assay revealed that the
LNCaP-AI cells grew adequately more rapidly than the LNCaP cells in
androgen-depleted medium (Fig.
1A). In the LNCaP-AI cells, western blot analysis demonstrated
a significant decrease in E-cadherin protein levels and a notable
increase in N-cadherin protein levels compared with the LNCaP
cells, suggesting that EMT had occurred in the transition from
LNCaP to LNCaP-AI cells (Fig.
1B). In addition, the numbers of invasive LNCaP-AI cells
significantly increased compared with the LNCaP cells,
demonstrating that prolonged androgen deprivation enhanced the
LNCaP-AI cell invasive ability (Fig.
1C).
Higher AR transcriptional activity in the
LNCaP-AI cells compared with the LNCaP cells
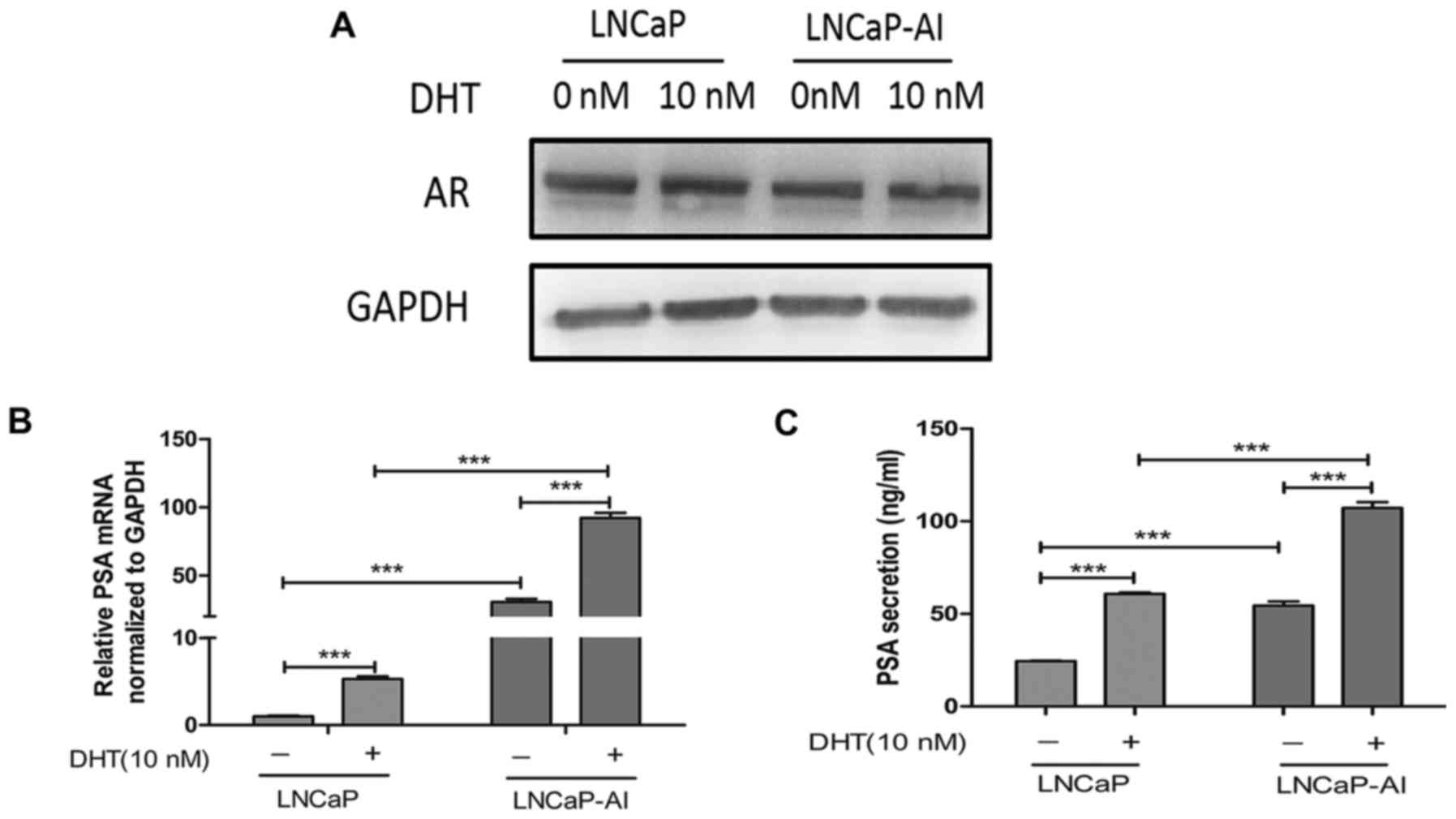
To examine the AR expression levels in the LNCaP-AI
cells and compare them to those of the LNCaP cells, and to
determine the effects of androgen on AR expression, western blot
analyses were performed using the LNCaP and LNCaP-AI cells
following treatment with ethanol (vehicle) or 10 nM DHT. The
results revealed that similar levels of AR protein were expressed
in the LNCaP and LNCaP-AI cells, and neither the LNCaP nor LNCaP-AI
cells expressed higher levels of AR protein following stimulation
with DHT (Fig. 2A). We then
examined the androgenic induction of mRNA expression and protein
secretion of PSA, one of the AR target genes, in order to assess
the transcriptional activity of AR in the LNCaP and LNCaP-AI cells.
The PSA mRNA and protein levels markedly increased with DHT
treatment in the LNCaP and LNCaP-AI cells. However, when DHT was
absent or present, the PSA protein level in the LNCaP-AI cells was
higher than that in the LNCaP cells (Fig. 2B and C). These findings
demonstrated that the LNCaP-AI cells retained a similar level of AR
as the LNCaP cells and were still sensitive to androgen
stimulation, and that the expression of AR are not affected by
androgen stimulation. The PSA protein levels in the LNCaP-AI cells
were significantly higher than those in the LNCaP cells in the
absence or presence of DHT.
Androgen suppresses the proliferation of
LNCaP-AI cells by inducing cell cycle arrest at the G1 phase,
contrary to the LNCaP cells
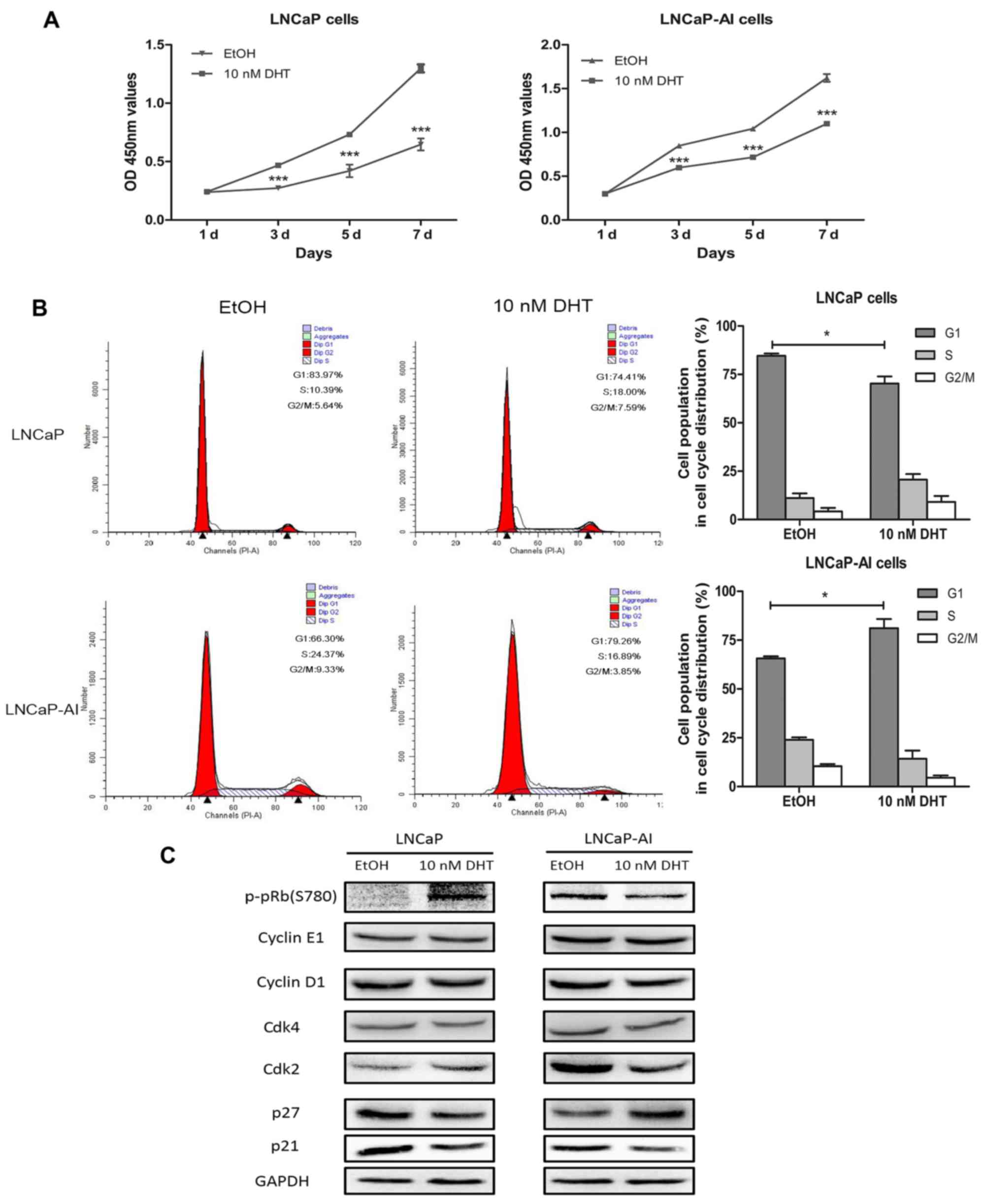
To determine the role that androgen plays in
LNCaP-AI cell growth compared with the LNCaP cells, the LNCaP-AI
and LNCaP cells were treated with DHT and cell viability was
examined. We found that the proliferation of the LNCaP-AI cells was
significantly suppressed by androgen (10 nM DHT) at a concentration
that is appropriate for LNCaP cell growth (Fig. 3A). Subsequently, we performed flow
cytometry to analyze the effects of androgen on the cell cycle
distribution of the LNCaP-AI and LNCaP cells. The results
demonstrated that androgen induced LNCaP-AI cell cycle arrest at
the G1 phase, but promoted LNCaP cell G1-S transition (Fig. 3B). To investigate the underlying
mechanisms, we measured the expression of G1 phase cell
cycle-related proteins. The results of western blot analysis
following treatment with 10 nM DHT revealed the loss of the
phosphorylated pRb (p-pRb) and Cdk2 protein in the LNCaP-AI cells,
in contrast to the LNCaP cells. This finding was in line with the
DHT-mediated arrest in the G1 phase in the LNCaP-AI cells. There
was no visible change in the protein levels of Cdk4, cyclin D1 and
cyclin E1. In addition, DHT induced the upregulation of p27 and the
downregulation of p21 in the LNCaP-AI cells, inconsistent with the
fact that p21 is a cyclin-dependent kinase inhibitor; however,
androgen deprivation (EtOH group) induced the upregulation of p27
and p21 in the LNCaP cells (Fig.
3C). Therefore, p21 may play different roles in mediating G1
arrest in the LNCaP-AI and LNCaP cells.
Assessment efficiency of shRNA lentiviral
vector transfection
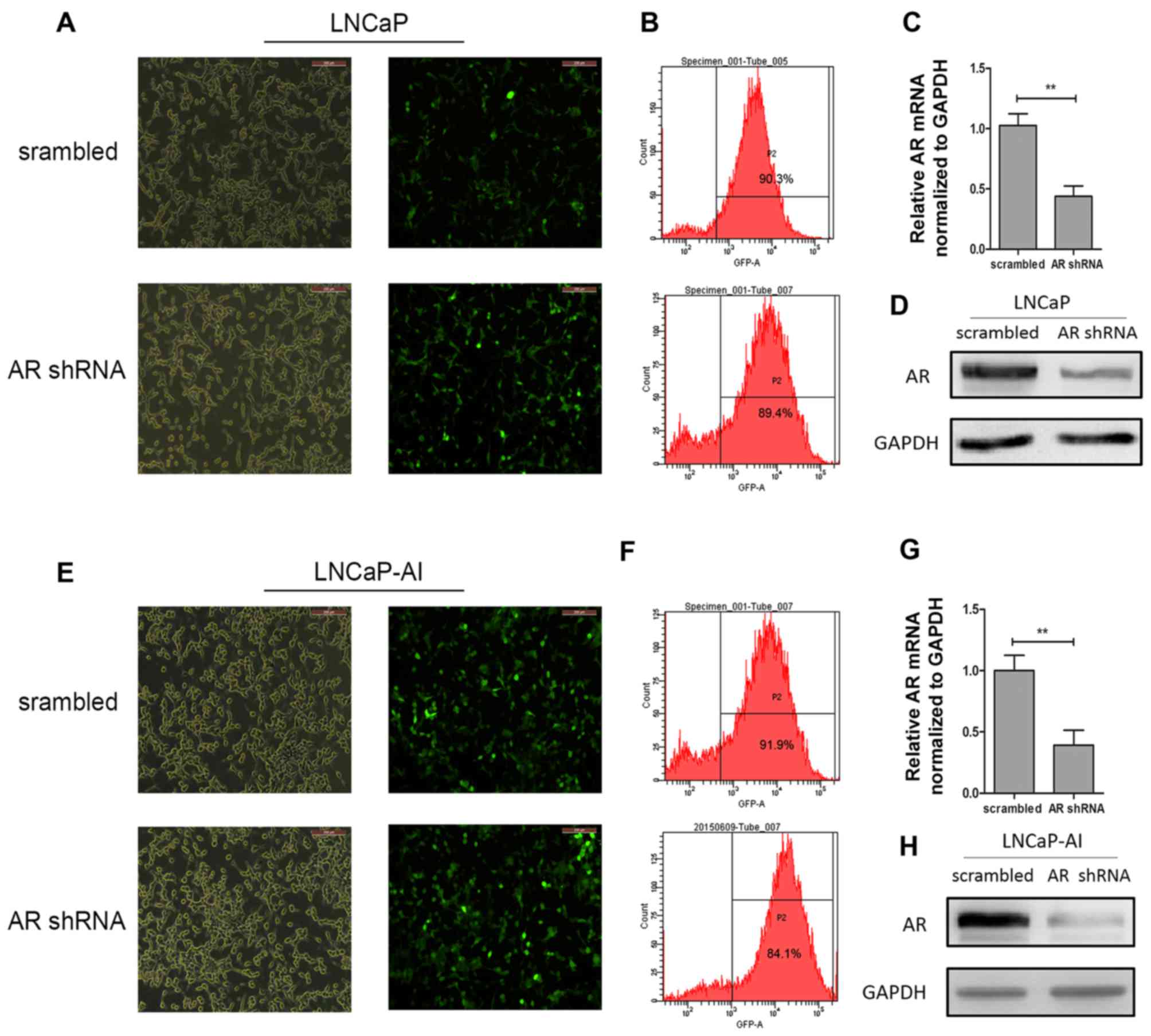
At 3 days after transfection, cells expressing GFP
were photographed under an inverted fluorescence microscope
(Fig. 4A and E). Moreover, a
FACSCalibur flow cytometer was used to detect GFP expression to
assess the efficiency. The efficiency of all groups was >80%
(Fig. 4B and F). RT-qPCR was used
to examine the mRNA level of AR after shRNA lentiviral vector
transfection. The results revealed that the AR levels in the LNCaP
cells in the AR shRNA group decreased by 56.2% compared with those
of the negative control group; the AR levels in the LNCaP-AI cells
in the AR shRNA group decreased by 60.7% compared with the negative
control group (Fig. 4C and G). As
expected, the AR protein levels were also significantly decreased
by transfection with AR shRNA lentiviral vector in the LNCaP and
LNCaP-AI cells (Fig. 4D and
H).
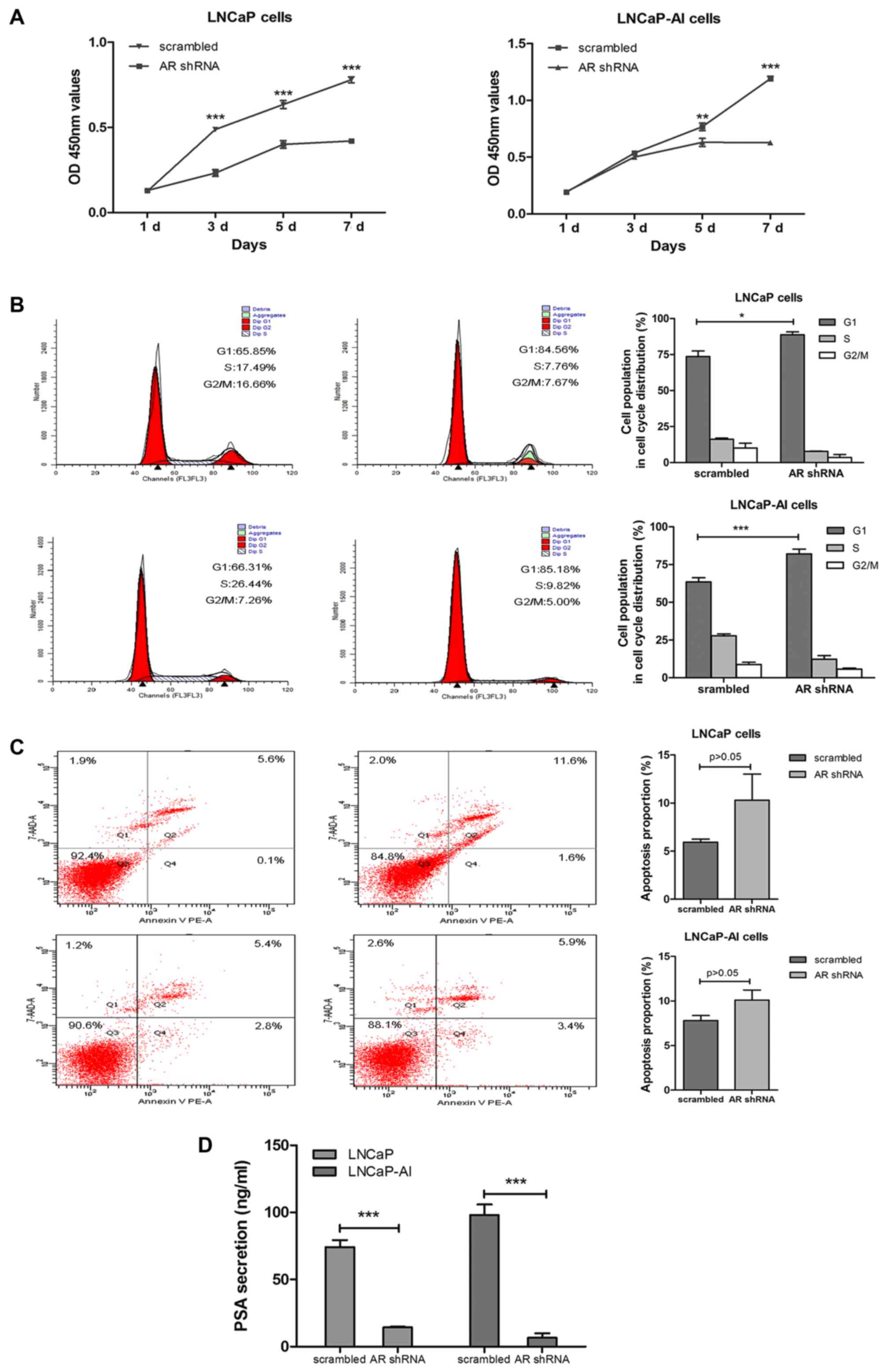
AR remains a critical factor in the
LNCaP-AI cells for cell cycle regulation and PSA secretion
To explore whether AR plays an important role in
LNCaP-AI cells, the cells were subjected to AR shRNA or scrambled
lentiviral particles transfection. CCK-8 assay, flow cytometry and
CLIA were used to analyze cell proliferation, cell cycle
distribution, the apoptotic rate and PSA secretion. The results
revealed that AR silencing inhibited LNCaP-AI and LNCaP cell growth
by inducing cell cycle arrest at the G1 phase (Fig. 5A and B); however, there was no
appreciable increase in the apoptotic rate of the LNCaP-AI and
LNCaP cells (Fig. 5C). Moreover,
AR silencing notably decreased PSA secretion in the LNCaP-AI and
LNCaP cells (Fig. 5D).
Discussion
Androgen deprivation therapy (ADT, surgical or
medical castration) remains the mainstay treatment for all patients
with PCa with metastatic disease (15,16). However, the effects of these
therapies are temporary, as within a median of 18-24 months, the
disease ultimately progresses to an androgen-independent stage that
is resistant to androgen ablation treatment, and is referred to as
CRPC (4,17). Therefore, to mimic the progression
of androgen-dependent PCa to androgen-independent PCa in clinical
practice, we established a subline of LNCaP cells (defined as
LNCaP-AI cells) by continuous cultured in the absence of androgen
(13). In this study, we found
that the LNCaP-AI cells represented a more aggressive phenotype
compared with the LNCaP cells, as regards the cell proliferation
rate, cell invasiveness, mesenchymal markers and PSA secretion.
Androgen suppressed LNCaP-AI cell growth by inducing G1 phase
arrest. Furthermore, we verified that AR remains a pivotal factor
in the LNCaP-AI cells for cell cycle governance and PSA
secretion.
To date, a number of studies have reported about
androgen-independent LNCaP cells, and have demonstrated that
androgen-independent LNCaP cells are characterized by an increased
rate of cell proliferation in androgen-depleted medium compared
with LNCaP cells (14,18,19), consistent with our observation.
However, there are few studies which have examined the effects of
ADT on EMT and the invasiveness of LNCaP-AI cells. Sun et al
reported that androgen deprivation caused EMT in PCa in
vivo, and partially reconstituted in vitro by treating
the LNCaP cell line with short-term androgen deprivation (20). This evidence further solidifies
our findings that long-term androgen deprivation renders the
LNCaP-AI cells to acquire a mesenchymal phenotype and stronger
invasiveness compared with the LNCaP cells. On the whole, the
present data suggest that LNCaP-AI cells, a subline of LNCaP cells,
have a more aggressive phenotype.
The aberrant AR re-activation in CRPC has been
reported in a variety of studies, and the mechanisms involved
include AR amplification, gain of function AR mutations,
ligand-independent AR activation and the overexpression of AR
co-factors (4,21–24). Kokontis et al revealed that
LNCaP-AI cells expressed a higher level of AR compared with LNCaP
cells, and androgen increased AR protein expression (25). However, Lu et al obtained
an opposite result that LNCaP-AI and LNCaP cells expressed similar
levels of AR protein, and independent of androgen stimulation
(14) consistent with our
finding. Thus, the underlying mechanisms of aberrant AR
re-activation are complex in CRPC. Of note, whether androgen is
absent or present, the levels of PSA expression and secretion in
LNCaP-AI cells were significantly higher than those in the LNCaP
cells (Fig. 2B and C).
Furthermore, Chuu et al found that the serum PSA level in
castrated mice bearing 104-R2 (an androgen-independent LNCaP cell
line) tumors was 8-fold higher than that of intact mice 104-S (an
androgen-dependent LNCaP cell line) tumors (19). This may indicate that the serum
levels of PSA in CRPC are also significantly higher than in
androgen-dependent PCa. In our study, as shown in Fig. 5D, there is overwhelming evidence
to indicate that aberrant AR re-activation occurs in CRPC, and
androgen induces PSA secretion in LNCaP-AI cells via the AR
signaling pathway.
Androgens are thought to be essential for LNCaP cell
growth and survival. Under conditions of androgen deprivation, we
found that LNCaP cell growth was suppressed by arrest in the G1
phase (14,26,27). However, the effects of androgen on
LNCaP-AI cells remain controversial. Lu et al demonstrated
that the growth of LNCaP-AI cells still progressed with androgen
stimulation (14). By contrast,
Kokontis et al emphasized that androgen suppressed LNCaP-AI
cell proliferation via the inhibition of Cdk2, Cyclin A and Skp2,
and an increase in p27 protein accumulation, giving rise to cell
cycle arrest at the G1 phase (25,28). Our study showed that androgen led
to pRb-dependent G1 phase LNCaP-AI cell cycle arrest through the
upregulation of p27, and the down-regulation of p21 and Cdk2,
ultimately resulting in the loss of Rb
phosphorylation/inactivation. This is in agreement with the fact
that p27 is a cyclin-dependent kinase inhibitor that binds to and
prevents the activation of cyclin E-Cdk2 or cyclin D-Cdk4
complexes, and thus blocks cell cycle progression at G1 (29). Surprisingly, this observation is
contrary to the role of p21 as a cyclin-dependent kinase inhibitor
(30), and it is totally
consistent with p21 as a positive regulator of cyclin-dependent
kinase activity by promoting the formation, activation and nuclear
enrichment of Cdk4/6-cyclin D complexes (31–34). Therefore, p21 may play a role as a
positive regulator to promote G1-S transition in the LNCaP-AI cells
in contrast to the LNCaP cells. Taken together, androgen exerts its
suppressive effects on LNCaP-AI cell growth via the upregulation of
p27 and the downregulation of p21 to inhibit CDK activity and cause
G1 cell cycle arrest.
In order to further examine the effects of AR on
LNCaP-AI cells, we designed AR-targeted shRNAs and used these to
infect the LNCaP-AI and LNCaP cells. Not surprisingly, it appeared
that the AR shRNA-transfected cells grew at a prominently slower
rate compared with the scrambled shRNA-transfected cells; this was
observed for both the LNCaP-AI and LNCaP cells. Our results are in
accordance with those of other studies, which have reported that AR
remains a critical factor for androgen-independent PCa cells
(35–37). In general, AR silencing suppressed
androgen-dependent PCa growth via a block of the G1-S transition
(38). Thus, in this study, we
investigated the underlying mechanisms through which AR inhibits
the proliferation of LNCap-AI cells. We found that AR played a
similar role in governing the cell cycle in both the LNCaP-AI and
LNCaP cells; that is, AR silencing elicited G1 cell cycle arrest.
In addition, AR silencing had no significant effect on the
apoptosis of both LNCaP-AI and LNCaP cells, which was contradictory
to the findings of previous studies showing that AR silencing leads
to PCa cell death in vitro (36,39,40). Nonetheless, Yuan et al
demonstrated that AR downregulation did not result in significant
apoptotic cell death in CWR22R3 cells (41). Perhpaps the reduction of AR
protein abundance by shRNA lentivral infection was insufficient to
trigger cell apoptosis. However, AR silencing decreased PSA
secretion in the LNCaP-AI and LNCaP cells.
In conclusion, our data demonstrate that LNCaP-AI
cells, a subline of LNCaP cells, have a more aggressive phenotype.
AR remains a crucial factor for cell growth and the transcriptional
activity of LNCaP-AI cells under androgen-depleted conditions.
Moreover, androgen suppressed LNCaP-AI cell growth via the
upregulation of p27 and the downregulation of p21, and reduced CDK
activity, causing cell cycle arrest in the G1 phase. Therefore,
LNCaP-AI cells can be defined as an
androgen-independent/AR-dependent cell line with a more aggressive
phenotype compared to the LNCaP cells.
Acknowledgments
This study was supported by a grant from the
National Natural Science Foundation of China (no. 81271917). We
would like to thank the Clinical Research Center of the Second
Affiliated Hospital of Zhejiang University School of Medicine for
providing technological support.
References
|
1
|
Attar RM, Takimoto CH and Gottardis MM:
Castration-resistant prostate cancer: locking up the molecular
escape routes. Clin Cancer Res. 15:3251–3255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sharifi N, Kawasaki BT, Hurt EM and Farrar
WL: Stem cells in prostate cancer: Resolving the castrate-resistant
conundrum and implications for hormonal therapy. Cancer Biol Ther.
5:901–906. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Damber JE and Aus G: Prostate cancer.
Lancet. 371:1710–1721. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feldman BJ and Feldman D: The development
of androgen-independent prostate cancer. Nat Rev Cancer. 1:34–45.
2001. View
Article : Google Scholar
|
|
5
|
Ryan CJ, Smith A, Lal P, Satagopan J,
Reuter V, Scardino P, Gerald W and Scher HI: Persistent
prostate-specific antigen expression after neoadjuvant androgen
depletion: An early predictor of relapse or incomplete androgen
suppression. Urology. 68:834–839. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tan MH, Li J, Xu HE, Melcher K and Yong
EL: Androgen receptor: structure, role in prostate cancer and drug
discovery. Acta Pharmacol Sin. 36:3–23. 2015. View Article : Google Scholar :
|
|
7
|
Heinlein CA and Chang C: Androgen receptor
in prostate cancer. Endocr Rev. 25:276–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsihlias J, Zhang W, Bhattacharya N,
Flanagan M, Klotz L and Slingerland J: Involvement of p27Kip1 in G1
arrest by high dose 5 alpha-dihydrotestosterone in LNCaP human
prostate cancer cells. Oncogene. 19:670–679. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hofman K, Swinnen JV, Verhoeven G and
Heyns W: E2F activity is biphasically regulated by androgens in
LNCaP cells. Biochem Biophys Res Commun. 283:97–101. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Akakura K, Bruchovsky N, Goldenberg SL,
Rennie PS, Buckley AR and Sullivan LD: Effects of intermittent
androgen suppression on androgen-dependent tumors. Apoptosis and
serum prostate-specific antigen. Cancer. 71:2782–2790. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sciarra A, Abrahamsson PA, Brausi M,
Galsky M, Mottet N, Sartor O, Tammela TL and Calais da Silva F:
Intermittent androgen-deprivation therapy in prostate cancer: A
critical review focused on phase 3 trials. Eur Urol. 64:722–730.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Horoszewicz JS, Leong SS, Chu TM, Wajsman
ZL, Friedman M, Papsidero L, Kim U, Chai LS, Kakati S, Arya SK, et
al: The LNCaP cell line - a new model for studies on human
prostatic carcinoma. Prog Clin biol Res. 37:115–132. 1980.
|
|
13
|
Xu G, Wu J, Zhou L, Chen B, Sun Z, Zhao F
and Tao Z: Characterization of the small RNA transcriptomes of
androgen dependent and independent prostate cancer cell line by
deep sequencing. PLoS One. 5:e155192010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu S, Tsai SY and Tsai MJ: Molecular
mechanisms of androgen-independent growth of human prostate cancer
LNCaP-AI cells. Endocrinology. 140:5054–5059. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Loblaw DA, Mendelson DS, Talcott JA, Virgo
KS, Somerfield MR, Ben-Josef E, Middleton R, Porterfield H, Sharp
SA, Smith TJ, et al American Society of Clinical Oncology: American
Society of Clinical Oncology recommendations for the initial
hormonal management of androgen-sensitive metastatic, recurrent, or
progressive prostate cancer. J Clin Oncol. 22:2927–2941. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sowery RD, So AI and Gleave ME:
Therapeutic options in advanced prostate cancer: Present and
future. Curr Urol Rep. 8:53–59. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Debes JD and Tindall DJ: Mechanisms of
androgen-refractory prostate cancer. N Engl J Med. 351:1488–1490.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J,
Chen Z, Beroukhim R, Wang H, Lupien M, et al: Androgen receptor
regulates a distinct transcription program in androgen-independent
prostate cancer. Cell. 138:245–256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chuu CP, Kokontis JM, Hiipakka RA, Fukuchi
J, Lin HP, Lin CY, Huo C, Su LC and Liao S: Androgen suppresses
proliferation of castration-resistant LNCaP 104-R2 prostate cancer
cells through androgen receptor, Skp2, and c-Myc. Cancer Sci.
102:2022–2028. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun Y, Wang BE, Leong KG, Yue P, Li L,
Jhunjhunwala S, Chen D, Seo K, Modrusan Z, Gao WQ, et al: Androgen
deprivation causes epithelial-mesenchymal transition in the
prostate: Implications for androgen-deprivation therapy. Cancer
Res. 72:527–536. 2012. View Article : Google Scholar
|
|
21
|
Chen CD, Welsbie DS, Tran C, Baek SH, Chen
R, Vessella R, Rosenfeld MG and Sawyers CL: Molecular determinants
of resistance to antiandrogen therapy. Nat Med. 10:33–39. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scher HI, Buchanan G, Gerald W, Butler LM
and Tilley WD: Targeting the androgen receptor: Improving outcomes
for castration-resistant prostate cancer. Endocr Relat Cancer.
11:459–476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Scher HI and Sawyers CL: Biology of
progressive, castration-resistant prostate cancer: Directed
therapies targeting the androgen-receptor signaling axis. J Clin
Oncol. 23:8253–8261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lonergan PE and Tindall DJ: Androgen
receptor signaling in prostate cancer development and progression.
J Carcinog. 10:202011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kokontis JM, Hay N and Liao S: Progression
of LNCaP prostate tumor cells during androgen deprivation:
Hormone-independent growth, repression of proliferation by
androgen, and role for p27Kip1 in androgen-induced cell cycle
arrest. Mol Endocrinol. 12:941–953. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Knudsen KE, Arden KC and Cavenee WK:
Multiple G1 regulatory elements control the androgen-dependent
proliferation of prostatic carcinoma cells. J Biol Chem.
273:20213–20222. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu Y, Chen SY, Ross KN and Balk SP:
Androgens induce prostate cancer cell proliferation through
mammalian target of rapamycin activation and post-transcriptional
increases in cyclin D proteins. Cancer Res. 66:7783–7792. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kokontis JM, Lin HP, Jiang SS, Lin CY,
Fukuchi J, Hiipakka RA, Chung CJ, Chan TM, Liao S, Chang CH, et al:
Androgen suppresses the proliferation of androgen receptor-positive
castration-resistant prostate cancer cells via inhibition of Cdk2,
CyclinA, and Skp2. PLoS One. 9:e1091702014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vidal A and Koff A: Cell-cycle inhibitors:
Three families united by a common cause. Gene. 247:1–15. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gartel AL, Serfas MS and Tyner AL: p21-
negative regulator of the cell cycle. Proc Soc Exp Biol Med.
213:138–149. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Labaer J, Garrett MD, Stevenson LF,
Slingerland JM, Sandhu C, Chou HS, Fattaey A and Harlow E: New
functional activities for the p21 family of CDK inhibitors. Genes
Dev. 11:847–862. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheng M, Olivier P, Diehl JA, Fero M,
Roussel MF, Roberts JM and Sherr CJ: The p21 (Cip1) and p27 (Kip1)
CDK 'inhibitors' are essential activators of cyclin D-dependent
kinases in murine fibroblasts. EMBO J. 18:1571–1583. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alt JR, Gladden AB and Diehl JA: p21(Cip1)
promotes cyclin D1 nuclear accumulation via direct inhibition of
nuclear export. J Biol Chem. 277:8517–8523. 2002. View Article : Google Scholar
|
|
35
|
Zegarra-Moro OL, Schmidt LJ, Huang H and
Tindall DJ: Disruption of androgen receptor function inhibits
proliferation of androgen-refractory prostate cancer cells. Cancer
Res. 62:1008–1013. 2002.PubMed/NCBI
|
|
36
|
Li TH, Zhao H, Peng Y, Beliakoff J, Brooks
JD and Sun Z: A promoting role of androgen receptor in
androgen-sensitive and -insensitive prostate cancer cells. Nucleic
Acids Res. 35:2767–2776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Snoek R, Cheng H, Margiotti K, Wafa LA,
Wong CA, Wong EC, Fazli L, Nelson CC, Gleave ME and Rennie PS: In
vivo knockdown of the androgen receptor results in growth
inhibition and regression of well-established, castration-resistant
prostate tumors. Clin Cancer Res. 15:39–47. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Comstock CE and Knudsen KE: The complex
role of AR signaling after cytotoxic insult: Implications for
cell-cycle-based chemotherapeutics. Cell Cycle. 6:1307–1313. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liao X, Tang S, Thrasher JB, Griebling TL
and Li B: Small-interfering RNA-induced androgen receptor silencing
leads to apoptotic cell death in prostate cancer. Mol Cancer Ther.
4:505–515. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang Q, Fung KM, Day WV, Kropp BP and Lin
HK: Androgen receptor signaling is required for androgen-sensitive
human prostate cancer cell proliferation and survival. Cancer Cell
Int. 5:82005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yuan X, Li T, Wang H, Zhang T, Barua M,
Borgesi RA, Bubley GJ, Lu ML and Balk SP: Androgen receptor remains
critical for cell-cycle progression in androgen-independent CWR22
prostate cancer cells. Am J Pathol. 169:682–696. 2006. View Article : Google Scholar : PubMed/NCBI
|