Introduction
Acute coronary syndrome (ACS) is a type of
cardiovascular disease (CVD) that occurs as a result of an acute
obstruction in the coronary arteries, and it represents a leading
cause of mortality and disability worldwide (1–3). A
previous epidemiological survey revealed that ~50% of
cardiovascular mortalities are due to ACS (4). Therefore, it is necessary to clarify
the underlying biological mechanisms of ACS to enable early
diagnosis and treatment of the disease.
The traditional diagnosis of ACS is achieved by a
combination of electrocardiogram (ECG) results and the detection of
specific serum markers (5).
However, the sensitivity of ECGs is generally <50% and the
application of serum markers, including creatine kinase isoenzyme
MB (CK-MB), cardiac troponin (cTn)I and cTnT, also has limitations
(6). Despite the high specificity
of cTnT and cTnI, they may only be detected 6–12 h following
myocardial ischemia, which is insufficient for early diagnosis and
treatment of ACS prior to myocardial injury (7). The development of novel, sensitive
and accurate ACS biomarkers are urgently required to enable the
early diagnosis and treatment of ACS.
Epigenetics are defined as stable, heritable
alterations in gene expression, which are not due to changes in the
DNA sequence (8). The role of
epigenetics has mainly been evaluated in relation to cancer
(9), but selected previous
studies have explored epigenetic involvement in CVD development and
progression (10–13). DNA methylation-induced genetic
repression is a major type of epigenetic modification (14). DNA methylation occurs at the C5
position of cytosine by the covalent modification of a methyl
group, which is most commonly found in 5′-C-phosphate-G-3′ (CpG)
dinucleotides (15). Alterations
in genome-wide and site-specific DNA methylation have been
described in CVD, although their specific functions are still
largely uninvestigated (16,17). Altered DNA methylation (hyper
methylation or hypomethylation) is one of the early events in the
pathogenesis of ACS (18,19). Hypermethylation of specific
promoter regions of crucial genes, such as transcription factor
genes, can repress the expression of these genes and alter
biological functions, which in turn increases the risk of ACS
(20). Furthermore, it has been
reported that numerous risk factors, including hypertension
(21,22), diabetes (23–26), atherosclerosis (27–30) and smoking (31,32), associated with the development of
ACS may cause epigenetic alterations. These previous studies
suggested that the detection of DNA methylation alterations may be
a promising tool for early ACS prediction and diagnosis. However,
systematic analyses of the changes to genome-wide DNA methylation
in patients with ACS are still lacking. In the present study, the
genome-wide aberrant DNA methylation patterns predominantly
associated with ACS were explored. Additionally, the differentially
methylated (DM) regions associated with ACS pathogenesis were
characterized, and novel DNA methylation biomarkers potentially
beneficial to the early diagnosis of ACS were identified.
Materials and methods
Study subjects
The present study recruited 81 patients with ACS, 74
patients with stable coronary artery disease (SCAD) and 53 control
subjects. The patients with CAD in the present study were admitted
to the Department of Cardiology of Jinling Hospital (Nanjing,
China) between January, 2014 and December, 2016, who were
undergoing clinically necessary coronary angiography. Angiograms of
all patients with CAD revealed ≥50% stenosis in a minimum of one
coronary artery. The 81 patients with ACS included patients with
acute myocardial infarction and unstable angina, with Braunwald
classification II or III who exhibited a positive cardiac biomarker
result (cTnI ≥0.090 ng/ml or cTnT ≥0.014 ng/ml), acute
ischemic-type chest pain (lasting for >15 min, duration from
symptom onset to emergency admission within 72 h) and
characteristic ECGs. The 74 patients with SCAD had a normal ECG and
documented normal left ventricular contractility, with the
exception of possible minor nonspecific ST-T features, and they had
a ≥1 year history without any cardiac events or procedures
suggestive of ACS. The exclusion criteria for the patients with CAD
included mild disease identified on angiography (a stenosis of
10–50% of the luminal diameter in all three coronary arteries),
prior coronary revascularization and the presence of renal disease.
The 53 control subjects were recruited to the study from routine
health examinations at Jinling Hospital over the same period. The
control patients had a normal physical examination, ECG and
laboratory tests, and were did not show signs of other diseases,
including hyperlipidemia, hypertension and diabetes mellitus, or
any clinically evident signs of atherosclerosis. The
clinicopathological features of all the patients and controls are
listed in Table I. The present
study was approved by the Ethics Committee of Jinling Hospital and
all the recruited subjects provided written informed consent prior
to the study.
 | Table IClinical and biochemical
characteristics of the participants included in the present
study. |
Table I
Clinical and biochemical
characteristics of the participants included in the present
study.
| Variable | Group
|
|---|
| Control (n=53) | SCAD (n=74) | ACS (n=81) |
|---|
| Age, years (mean ±
range) | 64.68±8.18 | 68.99±13.62 | 72.72±12.93 |
| Sex, n=m/f | 34/19 | 47/27 | 42/39 |
| History of
hypertension, n (%) | 0 (0) | 58 (78) | 61 (75) |
| History of
diabetes, n (%) | 0 (0) | 15 (20) | 37 (46) |
| TC, mmol/l | 4.43±0.47 | 4.16±0.95 | 4.50±0.73 |
| TG, mmol/l | 1.01±0.33 | 1.48±0.97a | 1.66±0.57b |
| HDL-C, mmol/l | 1.39±0.26 | 1.07±0.30b | 0.94±0.16b |
| LDL-C, mmol/l | 2.70±0.58 | 2.74±0.52 | 2.80±0.33 |
| Cardiac troponin I,
ng/ml | – | 0.03±0.01 | 6.61±17.31c |
| Cardiac troponin T,
ng/ml | – | 0.015±0.03 | 0.758±1.95d |
| Creatine kinase-MB,
U/l | – | 10.24±7.44 | 45.57±80.27d |
Blood sampling
Blood samples were collected from the patients with
81 ACS and 74 patients with SCAD in tubes containing EDTA
immediately following their hospitalization. The 53 control samples
were drawn from the elbow vein of a paired healthy individual on an
empty stomach. The samples were stored at −80°C prior to
analysis.
DNA isolation and sodium bisulfite
modification
Genomic DNA was isolated from 100 µl blood using the
DNeasy Blood and Tissue kit (Qiagen, Inc., Valencia, CA, USA)
according to the manufacturer's protocol. The quality and quantity
of the extracted genomic DNA was analyzed with a NanoDrop™ 2000
Spectrophotometer. Bisulfite modification of 1 μg DNA was
conducted using an EpiTect Bisulfite kit (Qiagen, Inc.) according
to the manufacturer's protocol.
Infinium HumanMethylation450 assay
A total of 3 patients with ACS were randomly
selected and paired with 3 control subjects for the Infinium
HumanMethylation450 assay (Illumina, Inc., San Diego, CA, USA). The
Infinium HumanMethylation450 assay was performed according to the
manufacturer's standard protocol. Processed methylation chips were
scanned using an iScan reader (Illumina, Inc.). Paired control
samples were processed on the same chip and all samples were
processed at the same time to avoid chip-to-chip variation. The
assays were conducted in the Beijing Genomics Institute (Beijing,
China).
Methylation-specific semi-quantitative
polymerase chain reaction (MSP) analysis
In the present study, MSP was developed for the
detection of mothers against decapentaplegic homolog 3 (SMAD3;
Chr1:67356838-Chr1:67356942) methylation in 81 patients with ACS,
74 patients with SCAD and 53 healthy individuals using two pairs of
primers, one was specific for the methylated alleles while the
other was specific for the unmethylated alleles. Primer sequences
are listed in Table II. The
thermocycling conditions were as follows: Complete denaturation of
the DNA at 95°C for 5 min; 35 cycles of denaturation at 95°C for 30
sec, annealing at 58°C for 30 sec, extension at 72°C for 45 sec;
and a final extension at 72°C for 10 min. The MSP products were
assessed by electrophoresis in 2% agarose gels with 0.5
μg/ml ethidium bromide and analyzed using a UVC1-1100
SmartView Pro 1100 Imaging system with version 1.0.0.2 software
(Major Science, Saratoga, CA, USA). Images demonstrating the
different methylation patterns were captured.
| Table IIPrimer sequences for the methylation
analysis of mothers against decapentaplegic homolog 3
(Chr1:67356838-Chr1:67356942). |
Table II
Primer sequences for the methylation
analysis of mothers against decapentaplegic homolog 3
(Chr1:67356838-Chr1:67356942).
| Method | Primer | Sequence
(5′-3′) | Amplicon size
(bp) |
|---|
| MSP | UF |
TTTAGATATTGGTTAGTTGGTTGT | 141 |
| UR |
ACACATTAAAAAACTCCACAACTT | |
| MF |
AGATATCGGTTAGTCGGTTGC | 141 |
| MR |
CATTAAAAAACTCCGCGACTT | |
| Sequenom | F |
AGGAAGAGAGGTTTTGTAATTTTTTATTTATTTGAGGG | 352 |
| MassARRAY | R |
CAGTAATACGACTCACTATAGGGAGAAGGCTATTTTATTTCCAAAACAATAAACTCACA | |
Sequenom MassARRAY
Primer pairs were designed using EpiDesigner
(epidesigner.com) and are presented in Table II. MassArray analysis was
performed in triplicate using matrix-assisted laser
desorption/ionization time-of-flight mass spectrometry (MassARRAY
Analyzer 4; Sequenom, San Diego, CA, USA) according to
manufacturer's protocol in 8 ACS patients, 8 SCAD patients and 8
healthy individuals randomly from each group. The experiments were
conducted at the Beijing Genomics Institute.
Differential methylation analysis
The Infinium HumanMethylation450 assay data were
normalized using internal controls (normalization control probe
pairs, which were designed to target the same region within
housekeeping genes and had no underlying CpG sites in the probe)
with GenomeStudio software (version 2011.1; Illumina, Inc.). For
quality control, methylation measures of P>0.05 and samples with
a CpG coverage <95% were removed. The methylation levels of CpG
sites were calculated as the β score [β score = intensity of the
methylated allele/(intensity of the unmethylated allele + intensity
of the methylated allele + 100)]. Limma software (version 3.24.15;
bioconductor. org/packages/release/bioc/html/limma.html) was used
to perform paired t-tests followed by Benjamini and Hochberg
correction for multiple testing in order to identify DM CpG sites
in the ACS group vs. the control group. Sites with a Benjamini and
Hochberg-corrected value of P<0.05 and a β score ≥0.1 were
considered to be significantly statistically different. A volcano
plot was used to display the mean DNA methylation differences for
the DM CpG sites. Hierarchical clustering of the methylation data
was performed when the β score was ≥0.2.
Functional annotation of DM CpG
sites
The enrichment analysis of the methylated regions
for Gene Ontology (GO) (33)
terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) (34) pathways was conducted using the
Genomic Regions Enrichment of Annotations Tool (35). Binomial statistics with false
discovery rate (FDR) correction were used to identify significantly
enriched ontologies. The most significant and replicable
corresponding genes were selected to be candidate markers for ASC
detection. These genes were chosen from the results of the
differential methylation tests at the site, gene region and gene
island region levels, and had a β score difference between the ACS
and control samples of >0.3.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 20.0; SPSS, Inc., Chicago, IL, USA). Normally
distributed values are expressed as the mean ± standard deviation.
A one-sample Kolmogorov-Smirnov test was used to evaluate the
normality of distribution of the variables. The significance of
differences between variants among groups was analyzed by one-way
analysis of variance, and the differences between groups were
subsequently determined using a post hoc Fisher's least significant
difference test. The differences in cTnI and CK-MB between two
groups were analyzed using the Student's t-test. The Kruskal-Wallis
H test was performed for non-parametric data. P<0.05 was
considered to indicate a statistically significant difference.
Results
Normalization of methylation data
For the methylated sites identified by Illumina
HumanMethylation450 assay, the original data of the 3 patients with
ACS and 3 paired controls were normalized to the internal controls
to eliminate the possibility of system error, and the β scores were
subsequently calculated. The β scores were generated based on the
intensity of the methylated and unmethylated probes, and were used
as an indicator of the degree of methylation at each CpG site.
Subsequently, a built-in detection score filter was used prior to
further calculations. This only left values with significantly
higher mean signal intensities from multiple probes for a given CpG
locus compared with those of the negative control in the same set
of chip data. The average number of loci detected for the three
pairs of ACS and control samples was 484,352 covering 42,343 genes
(Table III). The density plots
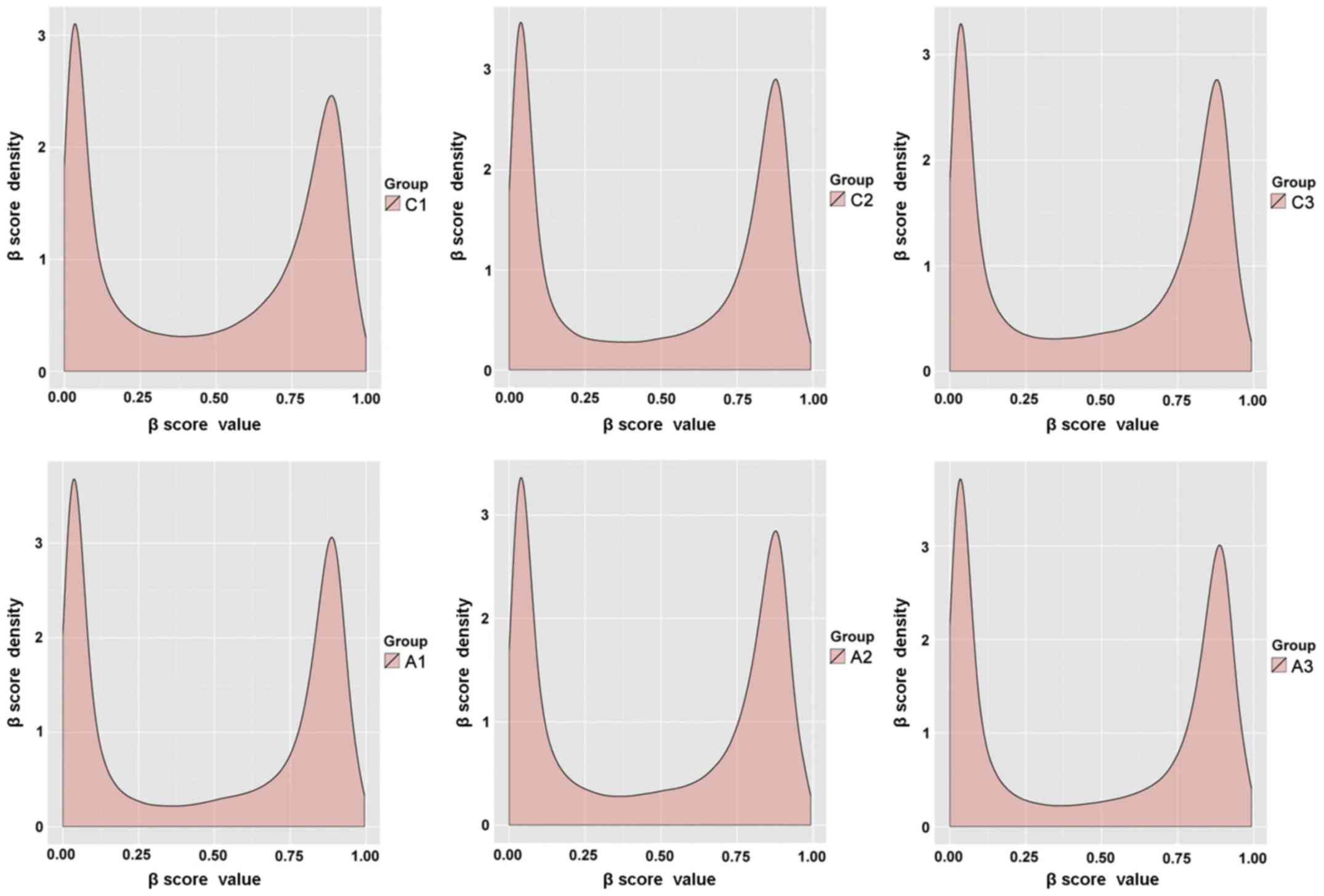
of the β score for the 6 samples exhibited a bimodal distribution
(Fig. 1). These results indicate
that the hybridization and amplification conditions were uniform
for all samples.
| Table IIIDM CpG sites and genes in patients
with acute coronary syndrome compared with the control group. |
Table III
DM CpG sites and genes in patients
with acute coronary syndrome compared with the control group.
| CpG site
methylation | No. of CpG
sites | No. of genes |
|---|
| Loci detected | 484,352 | 42,343 |
| DM | 11,342 | 5,073 |
|
Hypermethylated | 2,477 | 1,746 |
| Hypomethylated | 8,865 | 5,071 |
Epigenome-wide DNA methylation
analysis
Using Benjamini and Hochberg correction for multiple
comparisons, a total of 11,342 (2.34%) DM CpG sites covering 5,073
genes were identified in patients with ACS compared with the
control patients (Table III).
Among the 11,342 DM CpG sites, 8,865 (78.2%) covering 5,071 genes
were significantly hypomethylated, while 2,477 (21.8%) covering
1,746 genes were significantly hypermethylated in the ACS samples
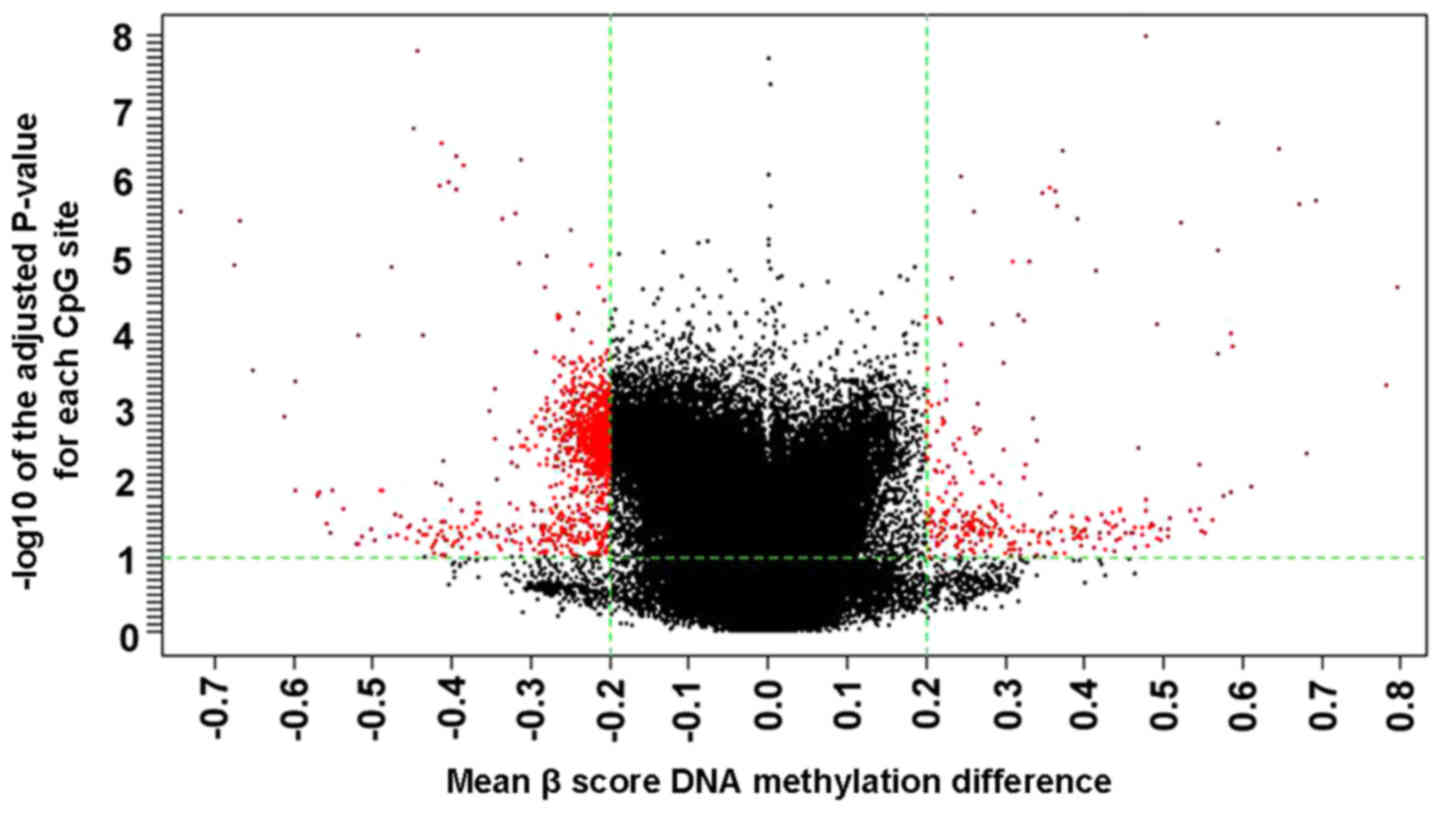
(Table III). A volcano plot of
the data exhibited the mean DNA methylation differences for all CpG
sites (Fig. 2). The frequencies
of all significant CpG sites between the ACS and control groups
were analyzed (Table IV). A
total of 9.04% (224) of the hypermethylated CpG sites and 11.8%
(982) of the hypomethylated CpG sites had an absolute ACS/control
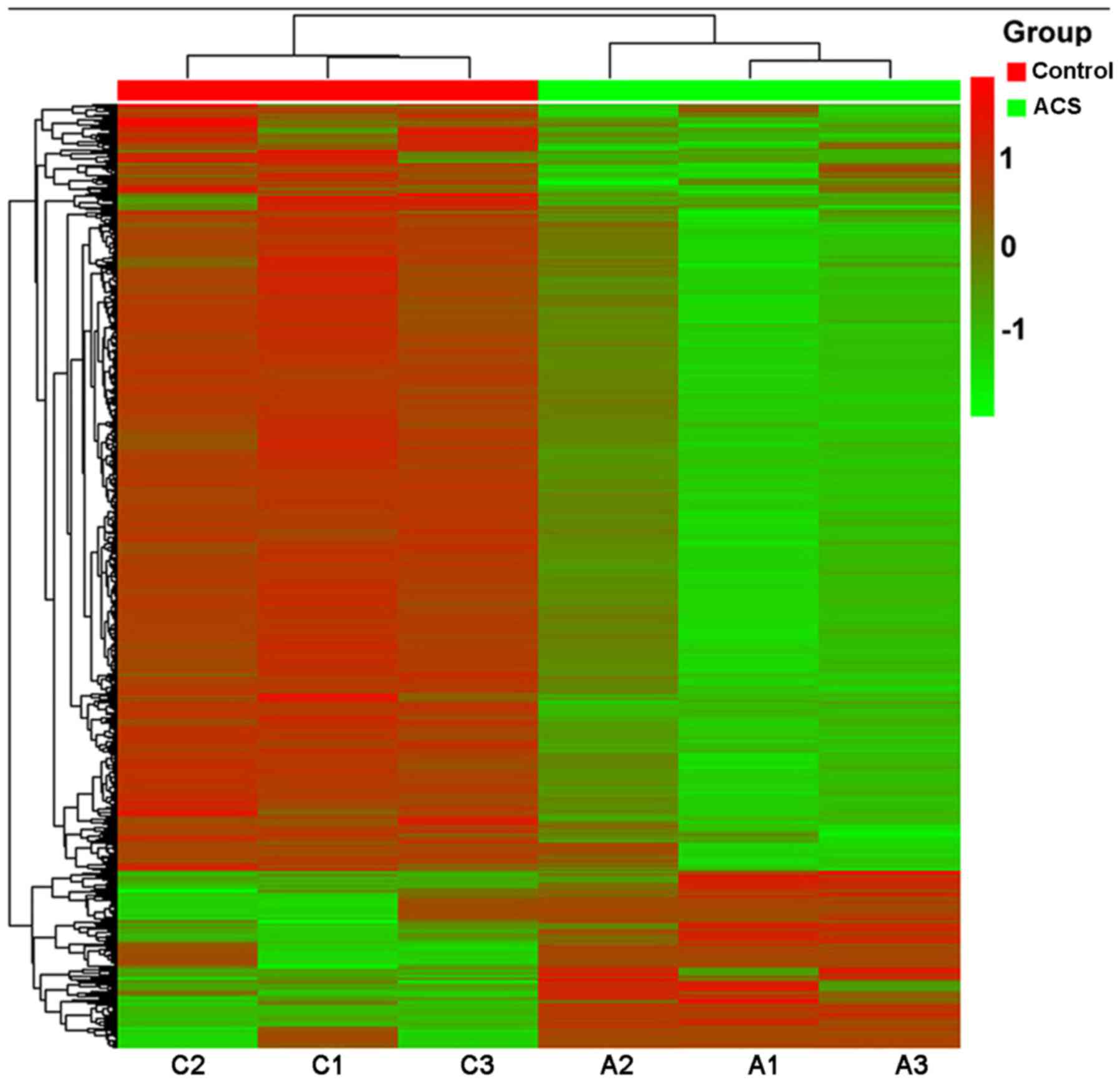
group methylation difference ≥20%. Fig. 3 presents a hierarchical cluster
analysis of the 1,206 CpG sites that distinguished the ACS samples
from the control samples.
| Table IVFrequency distribution of all
significantly differentially methylated CpG sites between acute
coronary syndrome and control samples by methylation status. |
Table IV
Frequency distribution of all
significantly differentially methylated CpG sites between acute
coronary syndrome and control samples by methylation status.
| ACS vs. control
group methylation β score difference (%) | Hypermethylated CpG
sites, n (%) | Cumulative (%) | Hypomethylated CpG
sites, n (%) | Cumulative (%) |
|---|
| ≥50 | 24 (0.97) | 0.97 | 16 (0.18) | 0.18 |
| ≥40–<50 | 32 (1.29) | 2.26 | 22 (0.25) | 0.43 |
| ≥30–<40 | 35 (1.41) | 3.67 | 40 (0.45) | 0.88 |
| ≥20–<30 | 133 (5.37) | 9.04 | 904 (10.20) | 11.08 |
| ≥10–<20 | 2,253 (90.96) | 100 | 7,883 (88.92) | 100 |
| Total | 2,477 | – | 8,865 | – |
Genomic distribution of DM CpG sites
Significant DM CpG sites were analyzed based on
their region and island region level throughout the genome
(Table V). There were 6
gene-based regions, including the transcription start site
(TSS)1500, TSS200 (within 200 base pairs of a TSS), 5′-untranslated
region (5′-UTR), 1stExon, body and 3′-UTR. In addition, there were
5 CpG island-based regions, including the island, N-Shore, S-Shore,
N-Shelf and S-Shelf sites. The gene and island regions were used to
calculate the methylation index. All the given region definitions
were based on the original Illumina methylation annotation for the
Human Methylation450 BeadChip array. The gene-based variant of the
region-level test resulted in the identification of 4,809
significant DM CpG sites, which covered 2,188 genes. This number
contained all unique genes, which resulted from six different,
separately analyzed gene categories: TSS1500 (349 genes), TSS200
(182 genes), 5′-UTR (246 genes), 1stExon (31 genes), body (1,250
genes) and 3′-UTR (130 genes). Similarly, the CpG island-based
variant of the region-level test resulted in 1,224 DM CpG sites
located in islands, and 1,593 and 1,247 DM CpG sites distributed
inside the N-Shore and S-Shore, respectively, while 751 and 685 DM
CpG sites were located in the N-Shelf and S-Shelf, respectively. In
total, 5,500 unique DM CpG sites covering 2,322 genes were
identified among the five CpG-related regions.
| Table VStatistics of DM CpG sites identified
by site, region and island region level. |
Table V
Statistics of DM CpG sites identified
by site, region and island region level.
| Test | Region | No. of DM CpG
sites | No. of genes |
|---|
| CpG site-level | Whole genome | 11,342 | 5,073 |
| Region-level gene
based | TSS1500 | 861 | 349 |
| TSS200 | 421 | 182 |
| 5′-UTR | 515 | 246 |
| 1st Exon | 73 | 31 |
| Body | 2,662 | 1,250 |
| 3′-UTR | 277 | 130 |
| Region-level island
based | Island | 1224 | 462 |
| N-Shore | 1593 | 644 |
| S-Shore | 1247 | 513 |
| N-Shelf | 751 | 372 |
| S-Shelf | 685 | 331 |
The genomic regions of the significantly
hypermethylated or hypomethylated CpG sites were distributed
similarly (Table VI). The
largest portion of hypermethylated CpGs (60.36%) were located in
the body of the genes and subsequently decreased in other
categories (15.17% in TSS1500, 6.88% in TSS200, 10.01% in the
5′-UTR, 1.72% in the 1stExon and 5.86% in the 3′-UTR). In addition,
over half (54.06%) of the significantly hypomethylated CpG sites
were located in the body, while the rest were distributed in the
TSS1500 (18.16%), TSS200 (9.24%), 5′-UTR (10.89%), 1stExon (1.47%)
and 3′-UTR (5.73%) regions. Furthermore, the distribution analysis
of DM CpG sites in island regions revealed that 18.5% (190) of the
significantly hypermethylated CpG sites were in CpG island regions,
with 47.81% (491) in CpG shores and 33.69% (346) in shelf regions.
As for the significantly hypomethylated CpG sites, 23.12% (1,034)
were located in islands, while the majority were identified in
shore (52.52%) or shelf (24.36%) regions (Table VII), suggesting they serve a
potential role in genomic instability.
| Table VIDistribution of the genomic region of
significantly differentially methylated CpG sites in the acute
coronary syndrome compared with the control samples. |
Table VI
Distribution of the genomic region of
significantly differentially methylated CpG sites in the acute
coronary syndrome compared with the control samples.
| Genomic region of
CpG sites | All CpG sites, n
(%) | Hypermethylated CpG
sites, n (%) | Hypomethylated CpG
sites, n (%) |
|---|
| TSS1500 | 861 (17.90) | 150 (15.17) | 711 (18.61) |
| TSS200 | 421 (8.75) | 68 (6.88) | 353 (9.24) |
| 5′-UTR | 515 (10.71) | 99 (10.01) | 416 (10.89) |
| 1st Exon | 73 (1.52) | 17 (1.72) | 56 (1.47) |
| Body | 2,662 (55.36) | 597 (60.36) | 2,065 (54.06) |
| 3′-UTR | 277 (5.76) | 58 (5.86) | 219 (5.73) |
| Total | 4,809 | 989 | 3,820 |
| Table VIIDistribution of island region of
significantly differentially methylated GpG sites in ACS vs.
control samples. |
Table VII
Distribution of island region of
significantly differentially methylated GpG sites in ACS vs.
control samples.
| Region level
island-based GpG sites | All GpG sites | Hypermethylated CpG
sites, no. (%) | Hypomethylated CpG
sites, no. (%) |
|---|
| Island | 1,224 (22.25) | 190 (18.50) | 1,034 (23.12) |
| N-Shore | 1,593 (28.96) | 304 (29.60) | 1,289 (28.82) |
| S-Shore | 1,247 (22.68) | 187 (18.21) | 1,060 (23.70) |
| N-Shelf | 751 (13.65) | 166 (16.16) | 585 (13.08) |
| S-Shelf | 685 (12.46) | 180 (17.53) | 505 (11.28) |
| Total | 5,500 | 1,027 | 4,473 |
GO term and KEGG pathway enrichment
analyses of the DM CpG sites
To identify the functions and pathways possibly
influenced by the genes nearest the DM CpG sites in patients with
ACS compared with the controls, GO term and KEGG pathway enrichment
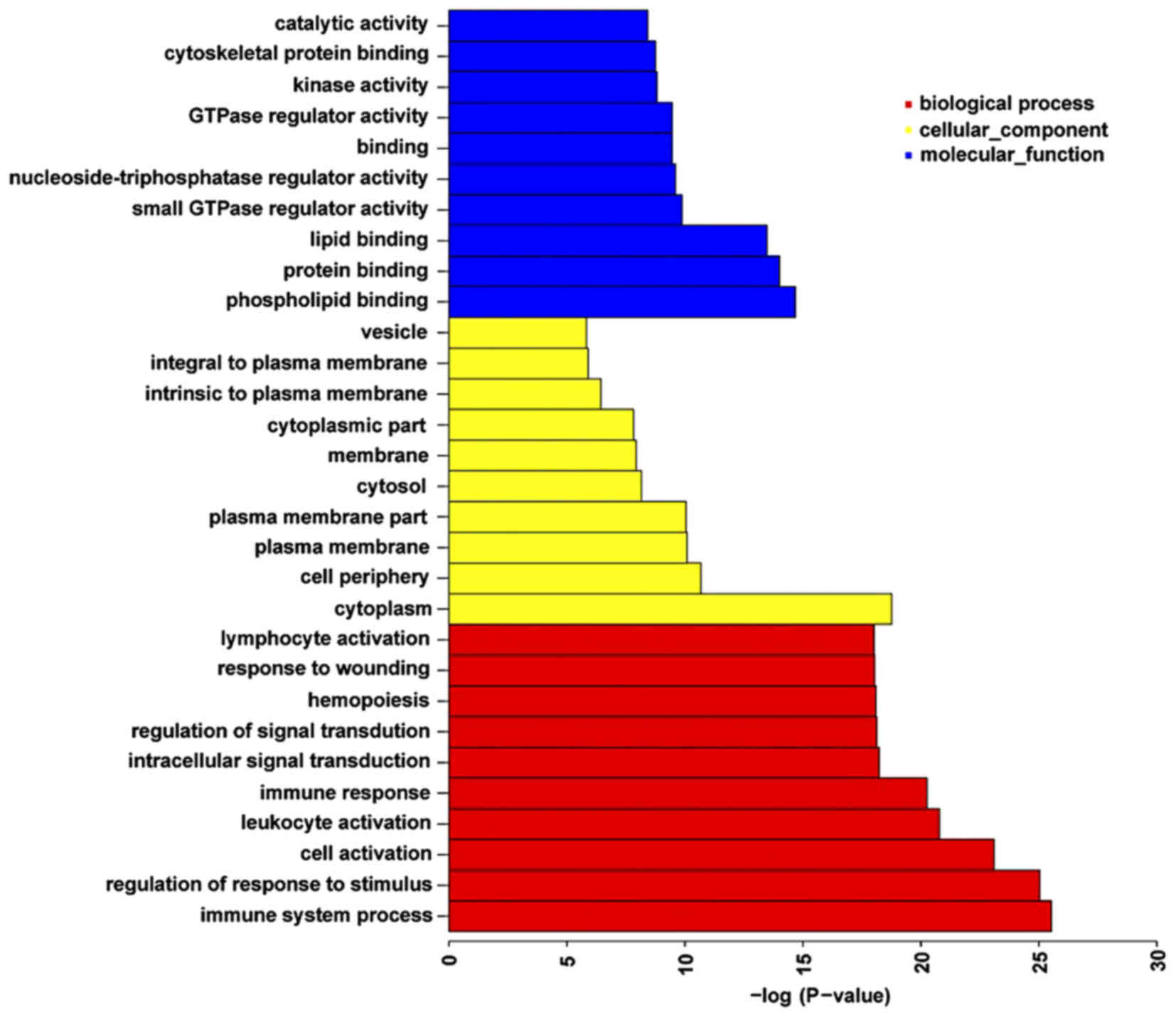
analyses were performed (Fig. 4).
There were 767 functional categories of DM genes (FDR <0.05),
the largest portion of the functional terms comprised of biological
processes (603), while the rest were cellular components (76) or
molecular functions (88). When the biological processes were used
for categorization, it was observed that the most enriched groups
included immune system processes, the regulation of responses to
stimuli and cell activation. Categorization by cellular components
indicated that proteins encoded by the analyzed genes were mainly
associated with the cytoplasm. The molecular function category
demonstrated significant enrichment for phospholipid, protein and
lipid binding activities. Fig. 4
presents the top 10 terms of the three GO categories ranked by
statistical significance.
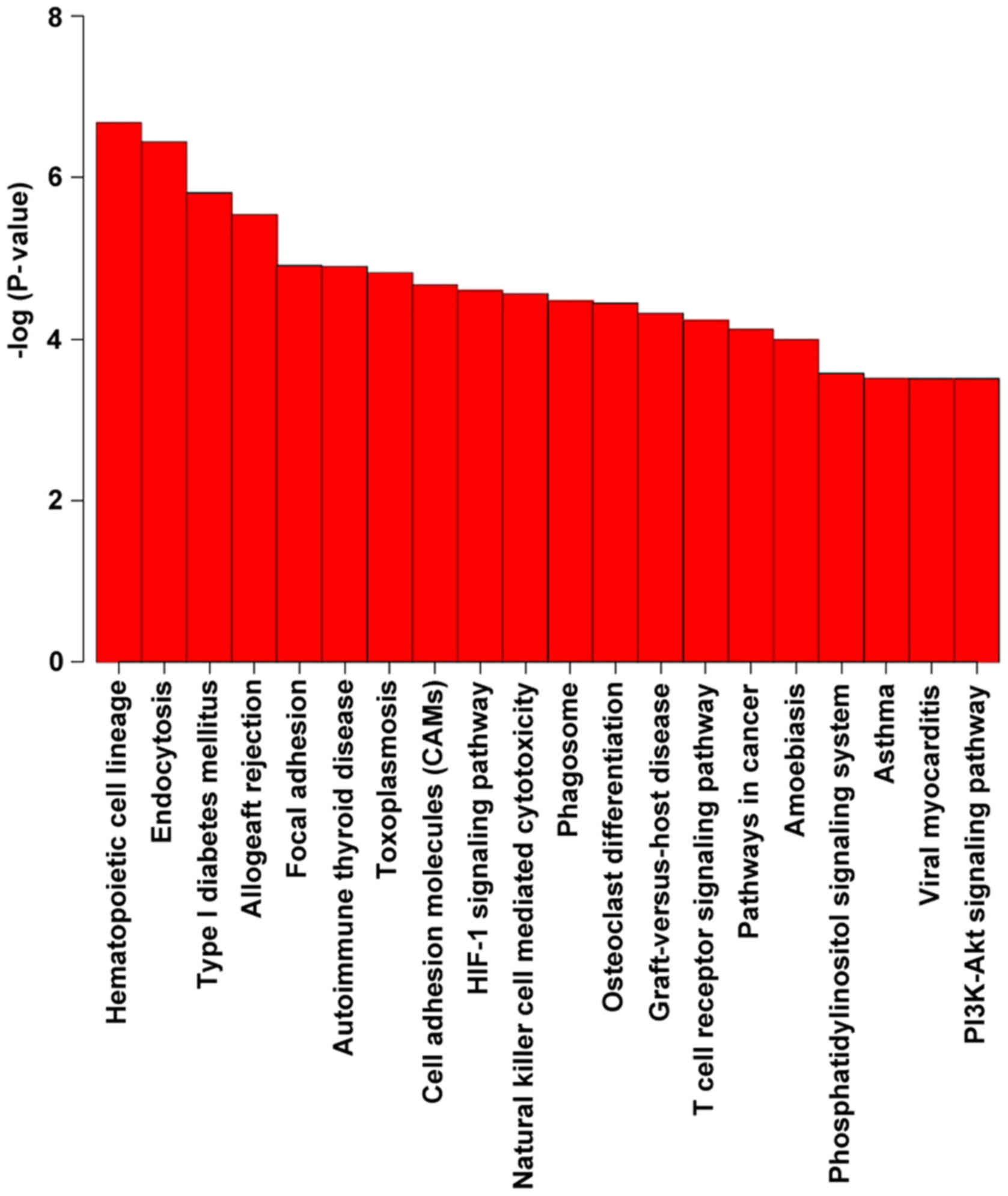
KEGG pathway analysis revealed a significant
enrichment of genes associated with the hematopoietic cell lineage
and endocytosis (Table VIII
and Fig. 5). Among them, the
immune system, and the transport and catabolism pathways attracted
were ranked highest in terms of significance.
| Table VIIIKEGG pathway enrichment analysis of
the hypermethylated and hypomethylated 5′-C-phosphate-G-3′ sites
between the acute coronary syndrome and control samples by
methylation status. |
Table VIII
KEGG pathway enrichment analysis of
the hypermethylated and hypomethylated 5′-C-phosphate-G-3′ sites
between the acute coronary syndrome and control samples by
methylation status.
| KEGG ID no. | Pathway name | Pathway class | No. of genes in
pathway | P-value | FDR |
|---|
| hsa04640 | Hematopoietic cell
lineage | Immune system | 43 |
2.13×10−7 |
4.72×10−5 |
| hsa04144 | Endocytosis | Transport and
catabolism | 83 |
3.63×10−7 |
4.72×10−5 |
| hsa04940 | Type I diabetes
mellitus | Endocrine and
metabolic diseases | 25 |
1.55×10−7 |
1.35×10−4 |
| hsa05330 | Allograft
rejection | Immune
diseases | 22 |
2.90×10−6 |
1.89×10−4 |
| hsa04510 | Focal adhesion | Cell
communication | 80 |
1.24×10−5 |
5.51×10−4 |
| hsa05320 | Autoimmune thyroid
disease | Immune
diseases | 24 |
1.27×10−5 |
5.51×10−4 |
| hsa05145 | Toxoplasmosis | Infectious
diseases: Parasitic | 50 |
1.51×10−5 |
5.63×10−4 |
| hsa04514 | Cell adhesion
molecules | Signaling molecules
and interaction | 58 |
2.12×10−5 |
6.90×10−4 |
| hsa04066 | HIF-1 signaling
pathway | Signal
transduction | 47 |
2.54×10−5 |
7.17×10−4 |
| hsa04650 | Natural killer cell
mediated cytotoxicity | Immune system | 52 |
2.76×10−5 |
7.17×10−4 |
| hsa04145 | Phagosome | Transport and
catabolism | 60 |
3.41×10−5 |
7.83×10−4 |
| hsa04380 | Osteoclast
differentiation | Development | 54 |
3.61×10−5 |
7.83×10−4 |
| hsa05332 | Graft vs. host
disease | Immune
diseases | 21 |
4.81×10−5 |
9.63×10−4 |
| hsa04660 | T cell receptor
signaling pathway | Immune system | 46 |
5.94×10−5 |
1.10×10−3 |
| hsa05200 | Pathways in
cancer | Cancers:
Overview | 113 |
7.67×10−5 |
1.33×10−3 |
| hsa05146 | Amoebiasis | Infectious
diseases: Parasitic | 46 |
1.03×10−4 |
1.67×10−3 |
| hsa04070 |
Phosphatidylinositol signaling system | Signal
transduction | 35 |
2.66×10−4 |
4.03×10−3 |
| hsa05310 | Asthma | Immune
diseases | 16 |
3.06×10−4 |
4.03×10−3 |
| hsa05416 | Viral
myocarditis | Cardiovascular
diseases | 31 |
3.10×10−4 |
4.03×10−3 |
| hsa04151 | PI3K-Akt signaling
pathway | Signal
transduction | 112 |
3.10×10−4 |
4.03×10−3 |
| hsa05221 | Acute myeloid
leukemia | Cancers: Specific
types | 27 |
3.34×10−4 |
4.14×10−3 |
| hsa05212 | Pancreatic
cancer | Cancers: Specific
types | 30 |
4.07×10−4 |
4.82×10−3 |
| hsa04670 | Leukocyte
transendothelial migration | Immune system | 46 |
5.73×10−4 |
6.28×10−3 |
| hsa04630 | Jak-STAT signaling
pathway | Signal
transduction | 53 |
5.80×10−4 |
6.28×10−3 |
| hsa05166 | HTLV-I
infection | Infectious
diseases: Viral | 89 |
7.26×10−4 |
7.55×10−3 |
| hsa05032 | Morphine
addiction | Substance
dependence | 38 |
8.19×10−4 |
8.01×10−3 |
| hsa05223 | Non-small cell lung
cancer | Cancers: Specific
types | 25 |
8.32×10−4 |
8.01×10−3 |
| hsa04062 | Chemokine signaling
pathway | Immune system | 67 |
9.16×10−4 |
8.32×10−3 |
| hsa04150 | mTOR signaling
pathway | Signal
transduction | 27 |
9.29×10−4 |
8.32×10−3 |
| hsa04662 | B cell receptor
signaling pathway | Immune system | 32 |
1.06×10−3 |
9.16×10−3 |
| hsa04915 | Estrogen signaling
pathway | Endocrine
system | 39 |
1.30×10−3 |
1.05×10−2 |
| hsa04612 | Antigen processing
and presentation | Immune system | 29 |
1.34×10−3 |
1.05×10−2 |
| hsa04664 | Fc epsilon RI
signaling pathwayc | Immune system | 30 |
1.36×10−3 |
1.05×10−2 |
| hsa04060 | Cytokine-cytokine
receptor interaction | Signaling molecules
and interaction | 83 |
1.37×10−3 |
1.05×10−2 |
| hsa05222 | Small cell lung
cancer | Cancers: Specific
types | 35 |
1.75×10−3 |
1.30×10−2 |
| hsa00562 | Inositol phosphate
metabolism | Carbohydrate
metabolism | 26 |
2.97×10−3 |
2.15×10−2 |
| hsa05140 | Leishmaniasisc | Infectious
diseases: Parasitic | 28 |
3.90×10−3 |
2.74×10−2 |
| hsa04666 | Fcγ R-mediated
phagocytosis | Immune system | 35 |
5.30×10−3 |
3.62×10−2 |
| hsa04360 | Axon guidance | Development | 47 |
5.58×10−3 |
3.72×10−2 |
| hsa04725 | Cholinergic
synapse | Nervous system | 41 |
7.36×10−3 |
4.78×10−2 |
Selection of significant genes as
possible diagnostic markers
The selection criteria used to select potential
diagnostic marker genes included CpG sites that were methylated in
the site, gene and gene island region, and a β score difference
between the ACS and control samples of >0.3. Following
filtering, a list of 19 hypermethylated and 17 hypomethylated CpG
sites were selected that matched these criteria (Table IX). These methylated CpG sites
were mapped to 37 known genes.
| Table IXList of the top 19 hypermethylated
and 17 hypomethylated corresponding genes selected as possible
diagnostic markers. |
Table IX
List of the top 19 hypermethylated
and 17 hypomethylated corresponding genes selected as possible
diagnostic markers.
| A, Hypermethylated
corresponding genes |
|---|
|
|---|
| CpG site | ACS vs. control
group methylation β score difference | Gene affected | P-value | FDR |
|---|
| cg03951394 | 0.31859 | DEAF1 |
4.61×10−2 | 0.43464 |
| cg11478607 | 0.33696 | GSTT1 |
1.43×10−3 | 0.24422 |
| cg01387905 | 0.34720 | ADAMTS2 |
1.35×10−6 | 0.03278 |
| cg13829625 | 0.35709 | SIN3B |
1.13×10−6 | 0.03160 |
| cg18424635 | 0.35947 | HLA-DRB5 |
2.83×10−2 | 0.39069 |
| cg26951705 | 0.38792 | ZNF787 |
4.81×10−2 | 0.43861 |
| cg00243527 | 0.39404 | LIF |
4.99×10−2 | 0.44222 |
| cg23782583 | 0.40248 | JAKMIP3 |
4.53×10−2 | 0.43270 |
| cg00248861 | 0.40500 | WHSC2 |
3.01×10−2 | 0.39564 |
| cg17005068 | 0.41609 | GSTT1 |
1.49×10−5 | 0.14324 |
| cg10662395 | 0.46909 | HCN2 |
3.48×10−3 | 0.24464 |
| cg19235645 | 0.47402 | FAM101A |
4.16×10−2 | 0.42548 |
| cg21829038 | 0.48534 | LRRK1 |
3.54×10−2 | 0.41016 |
| cg17713488 | 0.49270 | COL5A3 |
7.66×10−5 | 0.24422 |
| cg14594459 | 0.50806 | PKHD1L1 |
4.34×10−2 | 0.42957 |
| cg18443741 | 0.79680 | PYROXD1 |
2.48×10−5 | 0.18747 |
| cg19577958 | 0.54856 | SERPINB9 |
4.46×10−2 | 0.43166 |
| cg04234412 | 0.64833 | LOC391322 |
3.49×10−7 | 0.01914 |
| cg16836675 | 0.68159 | TOM1L1 |
4.18×10−3 | 0.24951 |
|
| B, Hypomethylated
corresponding genes |
|
| cg20592836 | −0.59591 | TP53INP2 |
1.33×10−2 | 0.32577 |
| cg03549208 | −0.55680 | ICA1L |
3.62×10−2 | 0.41253 |
| cg27141807 | −0.53542 | SPSB4 |
2.27×10−2 | 0.37233 |
| cg24851651 | −0.48666 | CCS |
1.31×10−2 | 0.32488 |
| cg06180910 | −0.43609 | GSTT1 |
1.11×10−4 | 0.24422 |
| cg10864200 | −0.41318 | PCGF3 |
1.12×10−2 | 0.31160 |
| cg24875593 | −0.40991 | PDXK |
5.26×10−3 | 0.26054 |
| cg04613734 | −0.39972 | TRPS1 |
4.07×10−2 | 0.42315 |
| cg19961153 | −0.38234 | ALPPL2 |
4.02×10−2 | 0.42219 |
| cg05141159 | −0.37248 | DEXI |
3.71×10−2 | 0.41476 |
| cg06049774 | −0.34306 | PLAC9 |
9.33×10−3 | 0.29687 |
| cg23231631 | −0.33541 | GABRB1 |
3.02×10−6 | 0.05052 |
| cg23731272 | −0.32429 | SMAD3 |
5.37×10−3 | 0.26137 |
| cg08253809 | −0.32072 | C14orf119 |
3.87×10−2 | 0.41857 |
| cg01655658 | −0.31142 | HLA-L |
4.82×10−7 | 0.02123 |
| cg13120108 | −0.31114 | PRKCH |
4.08×10−2 | 0.42353 |
| cg20433858 | −0.30584 | OR2L13 |
3.36×10−3 | 0.24464 |
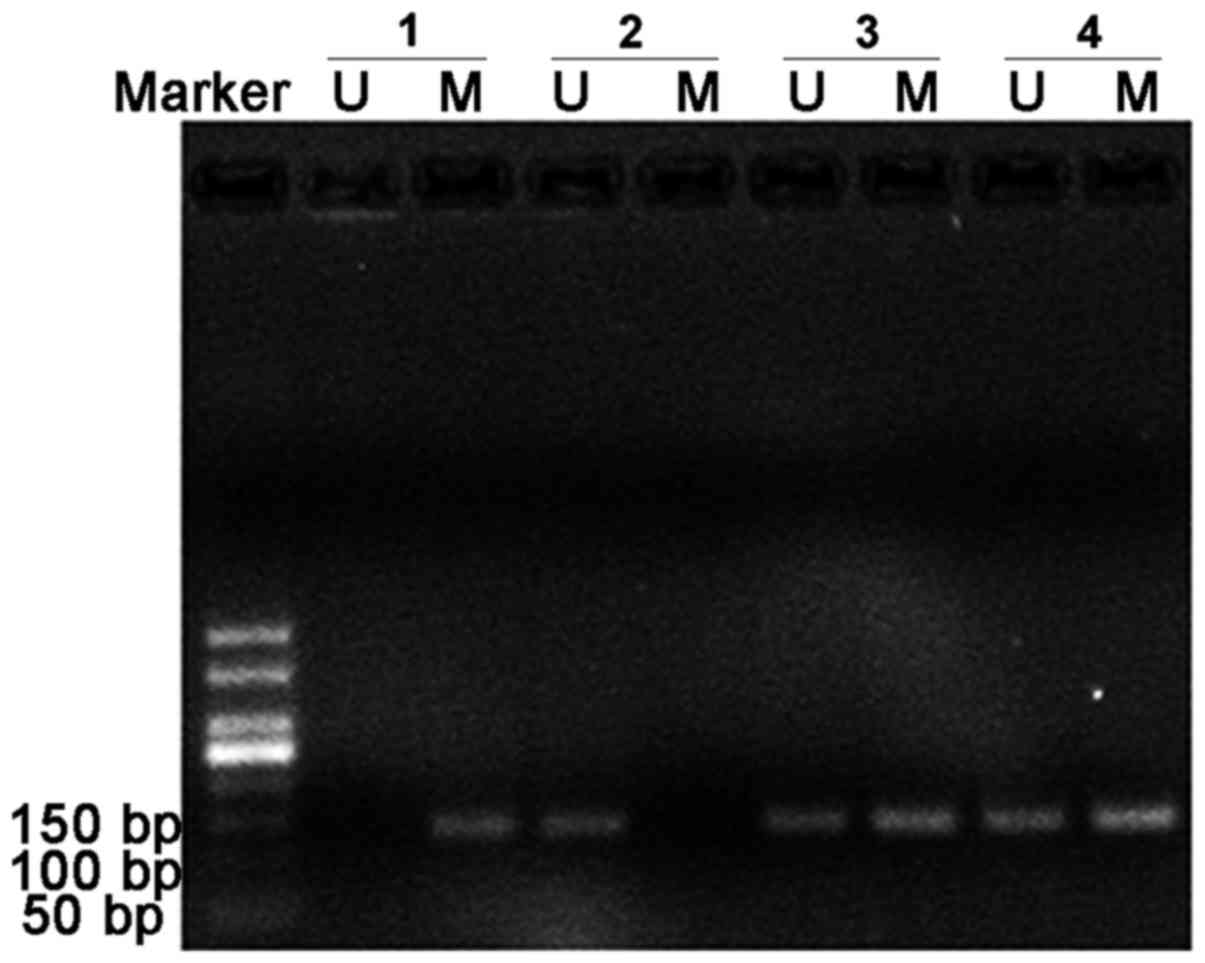
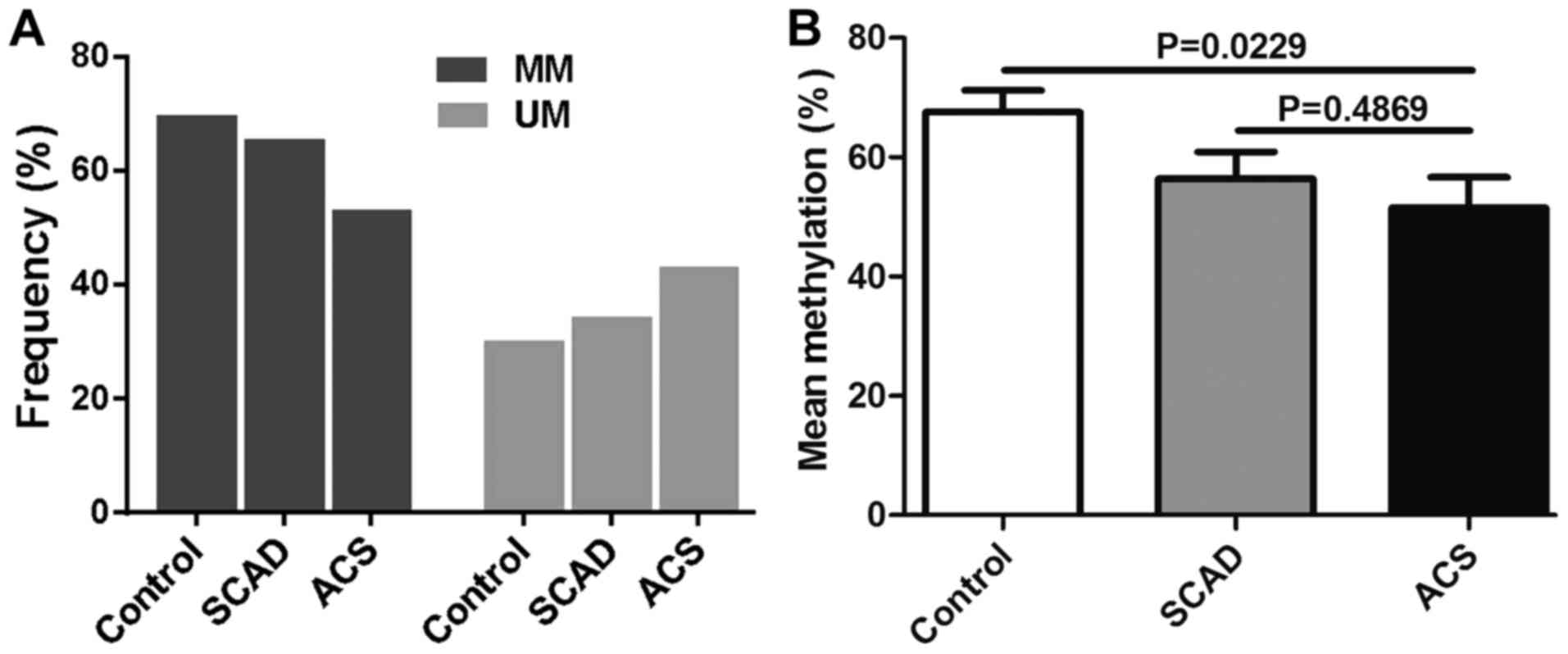
CpG methylation validation
The methylation state of the representative TSS1500
region (SMAD3; Chr1:67356838-Chr1:67356942) on the basis of DM
magnitude was detected by MSP (Figs.
6 and 7A) and Sequenom
MassARRAY (Fig. 7B). These
results were compared with the results from the HumanMethylation450
array. The frequency of semi-methylated sites
(ummethylated/methylated status) in patients with ACS was notably
higher than that in the patients with SCAD and the healthy donors
(Fig. 7A). The mean methylation
level was identified as being significantly lower in patients with
ACS compared with the SCAD and healthy control groups (Fig. 7B).
Discussion
To the best of our knowledge, the present study
presents the first genome-wide DNA methylation profiling of 3
paired ACS and normal human blood samples, using a high-throughput
DNA methylation microarray covering >450,000 CpG sites on the
human genome. The DM CpGs identified were enriched in genes
associated with immune system processes, the regulation of
responses to stimuli and cell activation, which suggests that they
are of potential mechanistic and therapeutic relevance.
The impact of epigenetic mechanisms in
cardiovascular pathophysiology is emerging as a key participant in
the interface between genotype and phenotype variability. Several
previous studies have focused on the role of DNA methylation in the
development of atherosclerosis and CVD. Hiltunen et al
(27) reported that a global
hypomethylation of DNA was present in advanced human, mouse and
rabbit atherosclerotic lesions. Notably, the results of Kim et
al (36) indicated that
elevated peripheral blood leukocyte DNA methylation was positively
associated with the prevalence of CVD predisposing conditions and
obesity in Chinese Singaporeans. A recent study also revealed that
lower global DNA methylation is associated with higher
cardiovascular risk in postmenopausal women (37). These studies led to the use of DNA
methylation as a novel biomarker for CAD. However, the majority of
their results were obtained from animal experiments or
gene-specific DNA methylation, which lack of the systematic
analysis of genome-wide changes to DNA methylation in patients with
ACS. Zaina et al (38)
identified an atherosclerosis-specific DNA methylation profile that
highlighted the contribution of different genes and pathways to the
disorder; however, this study did not take into account the
characteristics of acute onset in patients with atherosclerotic
CVD.
The present study analyzed the changes to
genome-wide DNA methylation in patients with ACS using the Illumina
HumanMethylation450 assay. A total of 11,342 CpG sites across the
whole genome demonstrated statistically significant DM in the ACS
samples compared with the control samples. The majority of the DM
CpG sites were hypomethylated (8,865; 78.2%) rather than
hypermethylated (2,477; 21.8%), which was consistent with the
results of a previous methylation study in atherosclerotic lesions
and CAD (28). In addition,
global hypomethylation, particularly in repeated sequences, serves
an important role in increasing chromosomal instability and may
lead to ACS (39). This suggests
that alteration of the DNA methylation profile, particularly
hypomethylation, is an important process in the pathogenesis of
ACS. Sharma et al (40)
proposed that genomic DNA methylation in patients with CAD is
significantly altered, as higher genomic DNA methylation changes
were observed in patients with CAD than in controls. Kim et
al (36) examined the
association between peripheral blood leukocyte global genomic DNA
methylation and the prevalence of CVD (myocardial infarction and
stroke) or its risk factors (hypertension and diabetes) to verify
the value of DNA methylation as a CVD risk biomarker. However, Kim
et al (36) only compared
differences in the overall methylation status between normal
subjects and patients with coronary heart disease; further details
on the differences in DNA methylation sites were not mentioned.
The genomic distribution of significant CpG sites
revealed that significantly DM CpG sites were more frequently
located at the body (55.3%) and shore (51.6%) sites. It has been
previously reported that methylation at the CpG island shore
regions regulates promoter activity (41), and that differential DNA
methylation in gene bodies and intergenic loci indicates more
complex mechanistic implications, as they are considered to promote
transcriptional elongation, and to regulate gene splicing and
enhancer activities (42).
The GO term and KEGG pathway enrichment analyses
results demonstrated a significant enrichment of the corresponding
methylated genes associated with the immune system processes,
regulation of responses to stimuli and cell activation, which are
all associated with the hematopoietic cell lineage and endocytosis
signaling pathways. The proteins encoded by the corresponding
methylated genes were mainly associated with the cytoplasm, and
were enriched in the molecular functions of phospholipid, protein
and lipid binding activities. These results are highly consistent
with data from a recent genome-wide systematic pathway analysis of
body mass index, which provided insights into the genetics of CVD
and its risk factors (43). The
results of the present study revealed a significant enrichment to
immune system processes in the ACS samples, which may be due to the
altered maturation status and possible immune exhaustion of immune
cells in the peripheral blood of patients with ACS (44). Additionally, the
neutrophil/lymphocyte ratio (NLR) has been identified to be
independently associated with the severity and 3-year outcomes of
CAD: Patients with higher NLR values had more advanced obstructive
CAD and a poorer prognosis (45).
CVD is often accompanied by disorders of lipid metabolism, and
previous studies have demonstrated that elevated concentrations of
oxidized lipoprotein were associated with the presence and severity
of ACS (46,47). In the present study, the network
analysis for GO term and KEGG pathway enrichment of the DM CpG
sites revealed significant enrichment change in patients with ACS,
which indicated that the methylation alterations at these sites
were biologically meaningful.
In the present study, 19 hypermethylated genes
(DEAF1, GSTT1, ADAMTS2, SIN3B, HLA-DRB5, ZNF787, LIF, JAKMIP3,
WHSC2, HCN2, FAM101A, LRRK1, COL5A3, PKHD1L1, PYROXD1, SERPINB9,
LOC391322 and TOM1L1) and 17 hypomethylated genes (TP53INP2, ICA1L,
SPSB4, CCS, GSTT1, PCGF3, PDXK, TRPS1, ALPPL2, DEXI, PLAC9, GABRB1,
SMAD3, C14orf119, HLA-L, PRKCH and OR2L13) demonstrated a potential
association with ACS. These genes may thus represent potential
novel candidate biomarkers for the early diagnosis of ACS. Three DM
genes were identified that have been deeply studied in CVD: SMAD3,
SERPINB9 and PRKCH. SMAD3, a key contributor to the transforming
growth factor-β pathway signaling, has been reported to serve an
essential role in CAD (48–51). Meta-analyses of genome-wide
association studies have identified that an intronic single
nucleotide polymorphism (SNP) in SMAD3 is associated with
protection from CAD (52).
SERPINB9 is most commonly known for its role in the control of
granzyme B, which can be applicable for prevention of ACS and
atherosclerotic plaque formation in patients with left ventricular
remodeling following acute myocardial infarction (53). However, SERPINB9 also serves a
role in suppressing interleukin-1β maturation through inhibiting
caspase-1 (54). Genome-wide
association and molecular mechanism studies have revealed that the
SNPs rs2230500 (55) and
rs2230500 (56,57) in PKRCH were significantly
associated with the risk of ischemic stroke. However, whether
SMAD3, SERPINB9 or PRKCH methylation is associated with the
pathogenesis of ACS is currently unknown. The validation
experiments performed for SMAD3 and TSS1500
(Chr1:67356838-Chr1:67356942) in the present study indicated that
the methylation degree was negatively associated with the
pathogenesis of ACS, and this was significantly corrected by the
reference results of the Illumina HumanMethylation450 assay.
In conclusion, the present study characterized the
genome-wide DNA methylation patterns occurring in ACS, and
identified significantly differentiated CpG sites and genes that
were correlated with ACS. Additionally, GO term and KEGG pathway
enrichment analyses provided information on the processes and
pathways that the methylated CpG sites and the participating genes
affected. These results may help clarify the role of aberrant
methylation in ACS pathogenesis, and provide the basis for future
research into potentially abnormally methylated genes as biomarkers
for the prevention and early diagnosis of ACS.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81271904, 81401742,
81572073 and 81572074), the Special-Funded Program on National Key
Scientific Instruments and Equipment Development of China (grant
no. 2012YQ030261) and Jinling Hospital Foundation (grant no.
2017060).
Glossary
Abbreviations
Abbreviations:
|
ACS
|
acute coronary syndrome
|
|
SCAD
|
stable coronary artery disease
|
|
GO
|
gene ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
DM
|
differentially methylated
|
|
MSP
|
methylation-specific polymerase chain
reaction
|
|
ECG
|
electrocardiogram
|
|
CK-MB
|
creatine kinase isoenzyme MB
|
|
cTn
|
cardiac troponin
|
|
CVD
|
cardiovascular disease
|
|
TSS
|
transcription start site
|
|
UTR
|
untranslated region
|
|
CAD
|
coronary artery disease
|
References
|
1
|
Falk E, Nakano M, Bentzon JF, Finn AV and
Virmani R: Update on acute coronary syndromes: The pathologists'
view. Eur Heart J. 34:719–728. 2013. View Article : Google Scholar
|
|
2
|
Corcoran D, Grant P and Berry C: Risk
stratification in non-ST elevation acute coronary syndromes: Risk
scores, biomarkers and clinical judgment. Int J Cardiol Heart Vasc.
8:131–137. 2015.
|
|
3
|
O'Gara PT, Kushner FG, Ascheim DD, Casey
DE Jr, MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin
BA, et al; American College of Cardiology Foundation/American Heart
Association Task Force on Practice Guidelines. 2013 ACCF/AHA
guideline for the management of ST-elevation myocardial infarction:
A report of the American College of Cardiology Foundation/American
Heart Association Task Force on Practice Guidelines. Circulation.
127:e362–e425. 2013. View Article : Google Scholar
|
|
4
|
Redfors B, Råmunddal T, Angerås O, Dworeck
C, Haraldsson I, Ioanes D, Petursson P, Libungan B, Odenstedt J,
Stewart J, et al: Angiographic findings and survival in patients
undergoing coronary angiography due to sudden cardiac arrest in
western Sweden. Resuscitation. 90:13–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hofer TP, Frankenberger M, Mages J, Lang
R, Meyer P, Hoffmann R, Colige A and Ziegler-Heitbrock L:
Tissue-specific induction of ADAMTS2 in monocytes and macrophages
by glucocorticoids. J Mol Med (Berl). 86:323–332. 2008. View Article : Google Scholar
|
|
6
|
Tomson T, Surges R, Delamont R, Haywood S
and Hesdorffer DC: Who to target in sudden unexpected death in
epilepsy prevention and how? Risk factors, biomarkers, and
intervention study designs. Epilepsia. 57(Suppl 1): 4–16. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mosleh W, Abdel-Qadir H and Farkouh M:
Biomarkers in the emergency workup of chest pain: Uses,
limitations, and future. Cleve Clin J Med. 80:589–598. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu SS, Lin X, Yuan LQ and Liao EY: The
role of epigenetics in arterial calcification. BioMed Res Int.
2015:3208492015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heyn H and Esteller M: DNA methylation
profiling in the clinic: Applications and challenges. Nat Rev
Genet. 13:679–692. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Handy DE, Castro R and Loscalzo J:
Epigenetic modifications: Basic mechanisms and role in
cardiovascular disease. Circulation. 123:2145–2156. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wallace RG, Twomey LC, Custaud MA, Moyna
N, Cummins PM, Mangone M and Murphy RP: Potential diagnostic and
prognostic biomarkers of epigenetic drift within the cardiovascular
compartment. BioMed Res Int. 2016:24657632016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Loscalzo J and Handy DE: Epigenetic
modifications: Basic mechanisms and role in cardiovascular disease
(2013 Grover Conference series). Pulm Circ. 4:169–174. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Turgeon PJ, Sukumar AN and Marsden PA:
Epigenetics of cardiovascular disease - A new 'Beat' in coronary
artery disease. Med Epigenet. 2:37–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kandi V and Vadakedath S: Effect of DNA
methylation in various diseases and the probable protective role of
nutrition: A mini-review. Cureus. 7:e3092015.PubMed/NCBI
|
|
15
|
Razin A, Webb C, Szyf M, Yisraeli J,
Rosenthal A, Naveh-Many T, Sciaky-Gallili N and Cedar H: Variations
in DNA methylation during mouse cell differentiation in vivo and in
vitro. Proc Natl Acad Sci USA. 81:2275–2279. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Afzali M, Nakhaee A, Tabatabaei SP,
Tirgar-Fakheri K and Hashemi M: Aberrant promoter methylation
profile of Niemann-pick type C1 gene in cardiovascular disease.
Iran Biomed J. 17:77–83. 2013.PubMed/NCBI
|
|
17
|
Nazarenko MS, Markov AV, Lebedev IN,
Freidin MB, Sleptcov AA, Koroleva IA, Frolov AV, Popov VA,
Barbarash OL and Puzyrev VP: A comparison of genome-wide DNA
methylation patterns between different vascular tissues from
patients with coronary heart disease. PLoS One. 10:e01226012015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Liu Y, Strickland FM and Richardson
B: Age-dependent decreases in DNA methyltransferase levels and low
transmethylation micronutrient levels synergize to promote
overexpression of genes implicated in autoimmunity and acute
coronary syndromes. Exp Gerontol. 45:312–322. 2010. View Article : Google Scholar :
|
|
19
|
Kim JM, Stewart R, Kang HJ, Bae KY, Kim
SW, Shin IS, Hong YJ, Ahn Y, Jeong MH and Yoon JS: BDNF methylation
and depressive disorder in acute coronary syndrome: The K-DEPACS
and EsDEPACS studies. Psychoneuroendocrinology. 62:159–165. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lü CX, Xu RD, Cao M, Wang G, Yan FQ, Shang
SS, Wu XF, Ruan L, Quan XQ and Zhang CT: FOXP3 demethylation as a
means of identifying quantitative defects in regulatory T cells in
acute coronary syndrome. Atherosclerosis. 229:263–270. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang LN, Liu PP, Wang L, Yuan F, Xu L,
Xin Y, Fei LJ, Zhong QL, Huang Y, Xu L, et al: Lower ADD1 gene
promoter DNA methylation increases the risk of essential
hypertension. PLoS One. 8:e634552013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim JD, Lee A, Choi J, Park Y, Kang H,
Chang W, Lee MS and Kim J: Epigenetic modulation as a therapeutic
approach for pulmonary arterial hypertension. Exp Mol Med.
47:e1752015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Z, Zheng Y, Hou C, Yang L, Li X, Lin
J, Huang G, Lu Q, Wang CY and Zhou Z: DNA methylation impairs TLR9
induced Foxp3 expression by attenuating IRF-7 binding activity in
fulminant type 1 diabetes. J Autoimmun. 41:50–59. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pasquier J, Hoarau-Véchot J, Fakhro K,
Rafii A and Abi Khalil C: Epigenetics and cardiovascular disease in
diabetes. Curr Diab Rep. 15:1082015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Babu M, Durga Devi T, Mäkinen P, Kaikkonen
M, Lesch HP, Junttila S, Laiho A, Ghimire B, Gyenesei A and
Ylä-Herttuala S: Differential promoter methylation of macrophage
genes is associated with impaired vascular growth in ischemic
muscles of hyperlipidemic and type 2 diabetic mice: Genome-wide
promoter methylation study. Circ Res. 117:289–299. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yip L, Fuhlbrigge R, Taylor C, Creusot RJ,
Nishikawa-Matsumura T, Whiting CC, Schartner JM, Akter R, von
Herrath M and Fathman CG: Inflammation and hyperglycemia mediate
Deaf1 splicing in the pancreatic lymph nodes via distinct pathways
during type 1 diabetes. Diabetes. 64:604–617. 2015. View Article : Google Scholar :
|
|
27
|
Hiltunen MO, Turunen MP, Häkkinen TP,
Rutanen J, Hedman M, Mäkinen K, Turunen AM, Aalto-Setälä K and
Ylä-Herttuala S: DNA hypomethylation and methyltransferase
expression in atherosclerotic lesions. Vasc Med. 7:5–11. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Castillo-Díaz SA, Garay-Sevilla ME,
Hernández-González MA, Solís-Martínez MO and Zaina S: Extensive
demethylation of normally hypermethylated CpG islands occurs in
human atherosclerotic arteries. Int J Mol Med. 26:691–700.
2010.PubMed/NCBI
|
|
29
|
Jiang YZ, Manduchi E, Stoeckert CJ Jr and
Davies PF: Arterial endothelial methylome: Differential DNA
methylation in atherosusceptible disturbed flow regions in vivo.
BMC Genomics. 16:5062015. View Article : Google Scholar
|
|
30
|
Eden A, Gaudet F, Waghmare A and Jaenisch
R: Chromosomal instability and tumors promoted by DNA
hypomethylation. Science. 300:4552003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Virani S, Rentschler KM, Nishijo M,
Ruangyuttikarn W, Swaddiwudhipong W, Basu N and Rozek LS: DNA
methylation is differentially associated with environmental cadmium
exposure based on sex and smoking status. Chemosphere. 145:284–290.
2016. View Article : Google Scholar :
|
|
32
|
Zhu X, Li J, Deng S, Yu K, Liu X, Deng Q,
Sun H, Zhang X, He M, Guo H, et al: Genome-wide analysis of DNA
methylation and cigarette smoking in a chinese population. Environ
Health Perspect. 124:966–973. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: The Gene Ontology Consortium: Gene ontology: Tool for the
unification of biology. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kanehisa M and Goto S: KEGG: Kyoto
Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar
|
|
35
|
McLean CY, Bristor D, Hiller M, Clarke SL,
Schaar BT, Lowe CB, Wenger AM and Bejerano G: GREAT improves
functional interpretation of cis-regulatory regions. Nat
Biotechnol. 28:495–501. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim M, Long TI, Arakawa K, Wang R, Yu MC
and Laird PW: DNA methylation as a biomarker for cardiovascular
disease risk. PLoS One. 5:e96922010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ramos RB, Fabris V, Lecke SB, Maturana MA
and Spritzer PM: Association between global leukocyte DNA
methylation and cardiovascular risk in postmenopausal women. BMC
Med Genet. 17:712016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zaina S, Heyn H, Carmona FJ, Varol N,
Sayols S, Condom E, Ramírez-Ruz J, Gomez A, Gonçalves I, Moran S,
et al: DNA methylation map of human atherosclerosis. Circ
Cardiovasc Genet. 7:692–700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li J, Chen L, Du L and Li M: Cage the
firefly luciferin! - a strategy for developing bioluminescent
probes. Chem Soc Rev. 42:662–676. 2013. View Article : Google Scholar
|
|
40
|
Sharma P, Kumar J, Garg G, Kumar A,
Patowary A, Karthikeyan G, Ramakrishnan L, Brahmachari V and
Sengupta S: Detection of altered global DNA methylation in coronary
artery disease patients. DNA Cell Biol. 27:357–365. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bockmühl Y, Patchev AV, Madejska A,
Hoffmann A, Sousa JC, Sousa N, Holsboer F, Almeida OF and Spengler
D: Methylation at the CpG island shore region upregulates Nr3c1
promoter activity after early-life stress. Epigenetics. 10:247–257.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao X, Gu J, Li M, Xi J, Sun W, Song G
and Liu G: Pathway analysis of body mass index genome-wide
association study highlights risk pathways in cardiovascular
disease. Sci Rep. 5:130252015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zidar DA, Mudd JC, Juchnowski S, Lopes JP,
Sparks S, Park SS, Ishikawa M, Osborne R, Washam JB, Chan C, et al:
Altered maturation status and possible immune exhaustion of CD8 T
lymphocytes in the peripheral blood of patients presenting with
acute coronary syndromes. Arterioscler Thromb Vasc Biol.
36:389–397. 2016. View Article : Google Scholar :
|
|
45
|
Arbel Y, Finkelstein A, Halkin A, Birati
EY, Revivo M, Zuzut M, Shevach A, Berliner S, Herz I, Keren G, et
al: Neutrophil/lymphocyte ratio is related to the severity of
coronary artery disease and clinical outcome in patients undergoing
angiography. Atherosclerosis. 225:456–460. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang JJ, Zhang CN, Meng Y, Han AZ, Gong JB
and Li K: Elevated concentrations of oxidized lipoprotein(a) are
associated with the presence and severity of acute coronary
syndromes. Clin Chim Acta. 408:79–82. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang JJ, Han AZ, Meng Y, Gong JB, Zhang
CN, Li K and Liu YX: Measurement of oxidized lipoprotein (a) in
patients with acute coronary syndromes and stable coronary artery
disease by 2 ELISAs: Using different capture antibody against
oxidized lipoprotein (a) or oxidized LDL. Clin Biochem. 43:571–575.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee AM, Shimizu C, Oharaseki T, Takahashi
K, Daniels LB, Kahn A, Adamson R, Dembitsky W, Gordon JB and Burns
JC: Role of TGF-β signaling in remodeling of noncoronary artery
aneurysms in kawasaki disease. Pediatr Dev Pathol. 18:310–317.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Turner AW, Nikpay M, Silva A, Lau P,
Martinuk A, Linseman TA, Soubeyrand S and McPherson R: Functional
interaction between COL4A1/COL4A2 and SMAD3 risk loci for coronary
artery disease. Atherosclerosis. 242:543–552. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Peng Q, Deng Y, Yang X, Leng X, Yang Y and
Liu H: Genetic variants of ADAM17 are implicated in the
pathological process of Kawasaki disease and secondary coronary
artery lesions via the TGF-β/SMAD3 signaling pathway. Eur J
Pediatr. 175:705–713. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lusis AJ: Cardiovascular disease genes
come together. Atherosclerosis. 242:630–631. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Turner AW, Martinuk A, Silva A, Lau P,
Nikpay M, Eriksson P, Folkersen L, Perisic L, Hedin U, Soubeyrand
S, et al: Functional analysis of a novel genome-wide association
study signal in SMAD3 that confers protection from coronary artery
disease. Arterioscler Thromb Vasc Biol. 36:972–983. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Saito Y, Kondo H and Hojo Y: Granzyme B as
a novel factor involved in cardiovascular diseases. J Cardiol.
57:141–147. 2011. View Article : Google Scholar
|
|
54
|
van der Burgh R, Meeldijk J, Jongeneel L,
Frenkel J, Bovenschen N, van Gijn M and Boes M: Reduced
serpinB9-mediated caspase-1 inhibition can contribute to
autoinflammatory disease. Oncotarget. 7:19265–19271. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hata J, Kubo M and Kiyohara Y: Genome-wide
association study for ischemic stroke based on the Hisayama study.
Nihon Eiseigaku Zasshi. 66:47–52. 2011.In Japanese. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wu L, Xi B, Hou D, Zhao X, Liu J, Cheng H,
Zhou X, Shen Y, Wang X and Mi J: The SNP (rs2230500) in PRKCH
decreases the risk of carotid intima-media thickness in a Chinese
young adult population. PLoS One. 7:e406062012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wu L, Shen Y, Liu X, Ma X, Xi B, Mi J,
Lindpaintner K, Tan X and Wang X: The 1425G/A SNP in PRKCH is
associated with ischemic stroke and cerebral hemorrhage in a
Chinese population. Stroke. 40:2973–2976. 2009. View Article : Google Scholar : PubMed/NCBI
|