Introduction
Alzheimer's disease (AD) is a neurodegenerative
disorder, accounting for 50–70% of all dementia cases and affecting
>12 million individuals worldwide. AD is an age-related
neurodegenerative illness characterized by a progressive decline in
cognitive functions, and histopathologically by the presence of
neuritic plaques containing amyloid-β (Aβ) peptide and
neurofibrillary tangles, mostly constisting of hyperphosphorylated
Tau protein (1). AD exists in
familial (early- and late-onset) forms, as well as a sporadic form,
which is very common, accounting for ~90% of all cases. AD has a
complex multietiological origin; however, its exact genetic and
environmental causative factors are unknown. Increasing evidence
indicates that different processes contribute to neurodegeneration,
including mitochondrial dysfunction, oxidative stress and
impairment of the autophagic/lysosomal/endosomal system (2,3).
Numerous studies have demonstrated that oxidative stress is one of
the initiating events and one of the molecular changes underlying
the pathogenesis of AD (4,5).
Oxidative stress may result from suppression of
mitochondrial function. Cytochrome oxidase (COX) is a mitochondrial
enzyme that plays an important role in aerobic energy metabolism
and mitochondrial function. Mitochondrial abnormalities,
particularly of COX, are found in the brains of subjects with AD
(6). The principal toxic action
of sodium azide (NaN3) is inhibiting the function of COX
in the mitochondrial electron transport chain (7). The model of brain mitochondrial COX
inhibition by NaN3 was described in rats in an attempt
to mimic the morphological and behavioral pathology of AD (8). The involvement of mitochondrial
dysfunction and the consequent overproduction of reactive oxygen
species (ROS) is increasingly recognized and widely accepted as an
etiopathological factor of AD. The tissue-specific inhibition of
COX by NaN3 may serve as a useful tool for the
evaluation of AD in vivo and in vitro.
Hydrogen sulfide (H2S), is considered as
the third most abundant endogenous signaling gasotransmitter
following nitric oxide (NO) and carbon monoxide, which affects
physiological and pathophysiological processes in a wide range of
biological systems (9).
H2S is produced endogenously in mammals, including
humans. In particular, cystathionine-β-synthase (CBS) in the
central nervous system (CNS) and cystathionine-γ-lyase in the
cardiovascular system are the key enzymes that are mainly
responsible for the endogenous generation of H2S
(10). 3-Mercaptopyruvate
sulfurtransferase (3- MST) is also known to be a significant
producer of endogenous H2S in the brain (11). Increasing evidence demonstrates
that H2S is associated with AD pathogenesis (12). The dysfunction of CBS in the
transsulfuration pathway may lead to a decrease in H2S
production in AD (13).
Interestingly, the levels of H2S are severely decreased
in AD patients; moreover, plasma H2S levels are
negatively correlated with the severity of the disease in AD
patients (14). Moreover, our
previous research demonstrated downregulation of the expression and
activity of CBS and 3-MST in neuron-like rat pheochromocytoma
(PC12) cells induced by NaN3 (data not shown). However,
it is not known whether H2S has any therapeutic benefits
in AD. Therefore, the present study was undertaken to assess the
beneficial effects of sodium hydro-sulfide (NaHS), which is an
exogenous H2S donor, on the underlying cellular and
molecular mechanisms in neuronal cells treated with
NaN3.
The characteristics of neuronal damage induced by
NaN3 treatment were first investigated. The
neuroprotective activity of H2S and its effect on
oxidative stress were also investigated in PC12 cells with neuronal
damage induced by NaN3. To the best of our knowledge,
this is the first study to demonstrate that H2S can
suppress NaN3-induced oxidative stress and apoptosis in
PC12 cells. This study was conducted to gain better insight into
the physiological functions of H2S under normal and
injury conditions, and its association with the cellular and
molecular mechanisms underlying nervous system disease.
Materials and methods
Cell culture
Rat pheochromocytoma PC12 cells were obtained from
the Shanghai Institute of Cell Biology, Chinese Academy of Sciences
(Shanghai, China). PC12 cells were grown on polystyrene tissue
culture dishes in DMEM containing 10% horse serum and 5% fetal
bovine serum (FBS; Sijiqing Biological Engineering Materials Co.,
Ltd., Hangzhou, China), supplemented with 2 mmol/l glutamine, 100
μg/ml streptomycin, and 100 U/ml penicillin (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) at 37°C with 95% air, 5%
CO2. Prior to differentiation, the medium was exchanged
twice a week and the cultures were subcultured at a ratio of 1:4
once a week. For differentiation, the cells were washed and
incubated in a fresh medium containing nerve growth factor (NGF;
final concentration, 50 ng/ml) for 48 h at 37°C in a cell
incubator. The concentration of NGF was maintained throughout all
experiments. All the experiments were performed on cells between
passages 3–8.
Cell injury model
The injury model was constructed as follows:
briefly, the DMEM was removed, PC12 cells were washed twice with
glucose-free Earle's balanced salt solution (EBSS) at pH 7.5, and
maintained in glucose-free DMEM without FBS. Subsequently,
neurotoxic damage was induced by adding the indicated concentration
of NaN3 to the cultured cells for different periods of
time. The cells were preincubated with the indicated concentrations
of NaHS (donor of H2S) for 30 min prior to
NaN3 treatment and maintained throughout the entire
experiment. NaHS was dissolved in saline and was freshly prepared
immediately prior to use. The stock solutions were directly added
into the bath solution to achieve the final concentration. Control
cultures were maintained in DMEM for the same duration under
normoxic conditions. The concentrations of all the reagents were
maintained throughout the injury period.
Determination of cell viability
The viability of PC12 cells was determined using the
Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies,
Inc., Kumamoto, Japan), according to the manufacturer's
instructions. PC12 cells were cultured in 96-well plates at 37°C
under an atmosphere of 5% CO2 and 95% air. At the end of
treatment, CCK-8 reagent (10 μl) was added to each well and
the plates were then incubated at 37°C for 3 to 4 h in the
incubator. Absorbance at a wavelength of 450 nm was measured with a
microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
The means optical density (OD) from 6 wells in the indicated groups
were used to calculate the cell viability, which was expressed as a
percentage of cell survival rate compared with the control. All the
experiments were performed in triplicate and repeated three
independent times.
Determination of mitochondrial membrane
potential (MMP)
MMP was examined by staining PC12 cells with JC-1.
Staining was performed using 2.5 mg/ml JC-1 at 37°C for 15 min.
After staining, cells were rinsed three times with
phosphate-buffered saline (PBS). A confocal laser scanning
microscope was used to measure MMP using the JC-1 assay kit (C2006;
Beyotime Institute of Biotechnology, Haimen, China). Under the
microscope, images of different color were obtained. Green
fluorescence indicated cells with low MMP (Δψm), revealing that
JC-1 maintains (or reacquires) monomeric form, while red
fluorescence indicated cells with high Δψm. The relative
proportions of red and green fluorescence were used to measure the
extent of mitochondrial depolarization.
Intracellular ROS measurement
Production of intracellular ROS was determined using
the fluorescent probe dichlorofluorescin diacetate (DCFH-DA), which
can cross cell membranes and is subsequently hydrolyzed by
intracellular esterase to non-fluorescent DCFH (Beyotime Institute
of Biotechnology). Following treatment with NaN3 for 12
h in the presence or absence of 200 mM H2S, the culture
medium was changed to fresh DMEM containing 10 μM DCFH-DA
for 30 min in an incubator at 37°C in the dark. After washing three
times with PBS, the cells were observed under a fluorescence
spectrophotometer with an excitation wavelength of 488 nm and an
emission wavelength of 535 nm.
Measurement of lipid peroxidation
Malondialdehyde (MDA), a terminal product of lipid
peoxidation, was measured to estimate the extent of lipid
peoxidation in PC12 cells. The cells were then homogenized in lysis
buffer [1% NP-40, 50 mmol/l Tris, pH 7.5, 5 mmol/l EDTA, 1% sodium
dodecyl sulphate (SDS), 1% sodium deoxycholate, 1% Triton X-100, 1
mmol/l phenylmethanesulfonylfluoride, 10 μg/ml aprotinin,
and 1 μg/ml leupeptin] and clarified by centrifuging for 20
min in a microcentrifuge at 4°C. MDA concentration in cell
homogenates was determined with an MDA assay kit (S0131; Beyotime
Institute of Biotechnology), using the thiobarbituric acid method.
The assay was based on the ability of MDA to form a conjugate with
thiobarbituric acid and create a red product, which has maximum
absorbance at 532 nm.
Western blot analysis
The cells were then homogenized in lysis buffer (1%
NP-40, 50 mmol/l Tris, PH 7.5, 5 mmol/l EDTA, 1% SDS, 1% sodium
deoxycholate, 1% Triton X-100, 1 mmol/l
phenylmethanesulfonylfluoride, 10 μg/ml aprotinin, and 1
μg/ml leupeptin) and clarified by centrifuging for 20 min in
a microcentrifuge at 4°C. Following determination of its protein
concentration with the Bradford assay (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), the resulting supernatant (50 μg of
protein) was subjected to SDS polyacrylamide gel electrophoresis.
The separated proteins were transferred to a polyvinylidine
difluoride membrane (Millipore, Billerica, MA, USA) by a transfer
apparatus at 90V for 1 h. The membrane was then blocked with 5%
non-fat milk and incubated with primary antibody against caspase-3
(1:500, cat. no. BS1518) and Bcl-2 (1:500, cat. no. BZ00479) (both
from Bioworld Technology, Inc., St. Louis Park, MN USA) or GAPDH
(1:1,000, cat. no. G8795; Sigma-Aldrich; Merck KGaA, St. Louis, MO,
USA). After incubating with an anti-rabbit or anti-mouse
horseradish peroxidase-conjugated secondary antibody, protein was
visualized using an enhanced chemiluminescence system (ECL; cat.
no. 32106; Pierce; Thermo Fisher Scientific, Inc., Bellefonte, PA,
USA).
Immunofluorescence analysis
Immunofluorescence analysis was performed as
follows: the PC12 cell in a 24-well plate were fixed with 4%
paraformaldehyde for 15 min at room temperature and washed with PBS
three times, for 10 min each time. After the cells were prepared,
they were blocked with 5% donkey serum (Gibco; Thermo Fisher
Scientific, Inc.) with 0.3% Triton X-100 and 5% bovine serum
albumin (BSA) for 2 h at room temperature and incubated with rabbit
polyclonal primary anti-caspase-3 antibodies (1:50, cat. no.
ab13847; Abcam, Cambridge, UK). Briefly, the cells were incubated
with the primary antibodies overnight at 4°C, followed by a mixture
of fluorescein isothiocyanate-conjugated secondary antibodies for 2
h at room temperature, then washed with PBS three times, for 10 min
each time. Finally, the samples were mounted with coverslips using
antifade mounting medium (Beyotime Institute of Biotechnology) and
observed under a fluorescence microscope (Eclipse Ti-S; Nikon,
Tokyo, Japan). At least three random slides from each group were
examined.
Statistical analysis
All statistical analyses were conducted with SPSS
statistical software 16.0 (SPSS, Inc., Chicago, IL, USA). All the
values are expressed as means ± standard error of the mean. The
statistical significance of differences between groups was
determined by one-way analysis of variance followed by Tukey's post
hoc multiple comparison tests or Student's t-test (two means
comparison). P<0.05 was considered to indicate statistically
significant differences. Each experiment consisted of at least
three replicates per condition.
Results
NaN3 is cytotoxic to PC12
cells
Cell cytoxicity was evaluated by the CCK-8 assay
following incubation of PC12 cells with increasing concentrations
of NaN3 from 5 to 100 mmol/l at 1, 3, 6, 12, 18 and 24
h. As shown in Fig. 1A, at
concentrations of 5–100 mmol/l, treatment of PC12 cells with
NaN3 for 12 h led to a concentration-dependent reduction
in cell viability. The results demonstrated that the general trend
of cell survival rate decreased with increasing treatment time
(Fig. 1B).
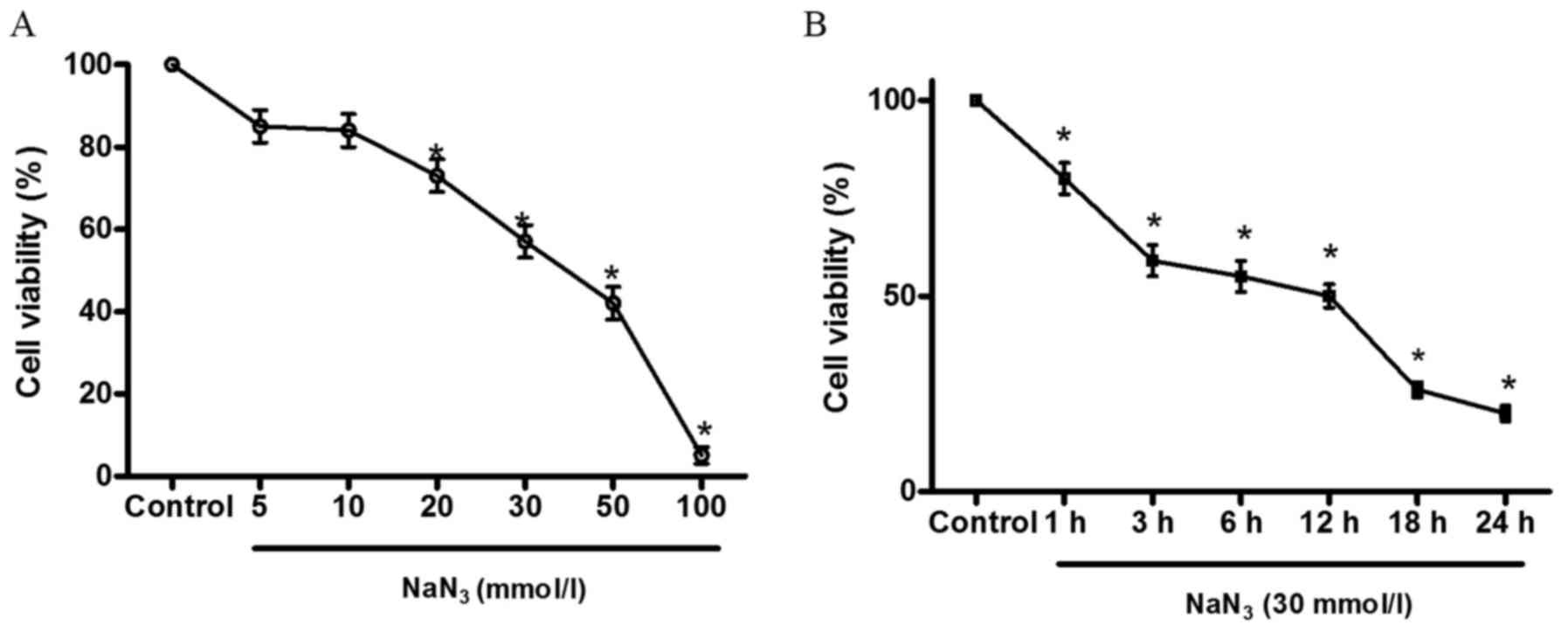 | Figure 1Concentration- and time-dependent
effect of sodium azide (NaN3) on cell viability in PC12
cells. (A) PC12 cells were treated with 5, 10, 20, 30, 50 and 100
mmol/l NaN3 for 12 h, and cell viability was determined
by the Cell Counting Kit-8 (CCK-8) assay. Exposure of PC12 cells to
NaN3 for 12 h induced a decrease in cell viability in a
concentration-dependent manner. (B) PC12 cells were treated for 1,
3, 6, 12, 18 and 24 h with 30 mmol/l NaN3, and cell
viability was determined by the CCK-8 assay. Exposure of PC12 cells
to 30 mmol/l NaN3 induced a decrease in cell viability
in a time-dependent manner. Columns represent means ± standard
error of the mean of viable cells (n=6), and expressed as
percentage of control values. *P<0.05 vs. control
group. |
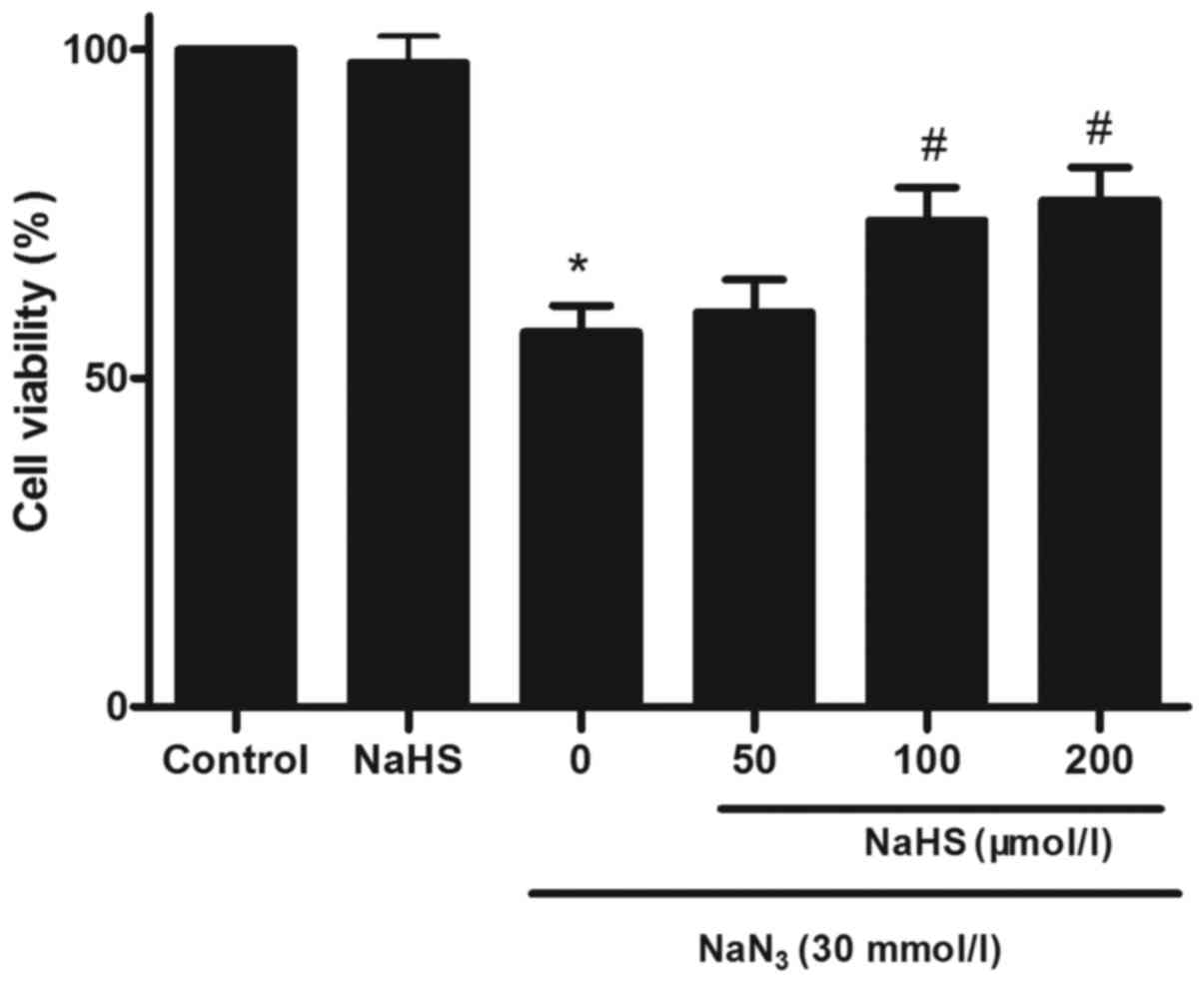
H2S protects PC12 cells
against NaN3-induced cytotoxicity
To investigate the effect of H2S on
NaN3-induced cytotoxicity, cell viability was analyzed
by determining the percentage of CCK-8 reduction. As shown in
Fig. 2, treatment with
NaN3 at concentrations of 30 mmol/l for 12 h attenuated
cell viability. The cytotoxic effects of NaN3 on PC12
cells were significantly prevented by pretreatment with NaHS at
100–200 μmol/l for 30 min. At 200 μmol/l, NaHS alone
did not measurably affect the viability of PC12 cells.
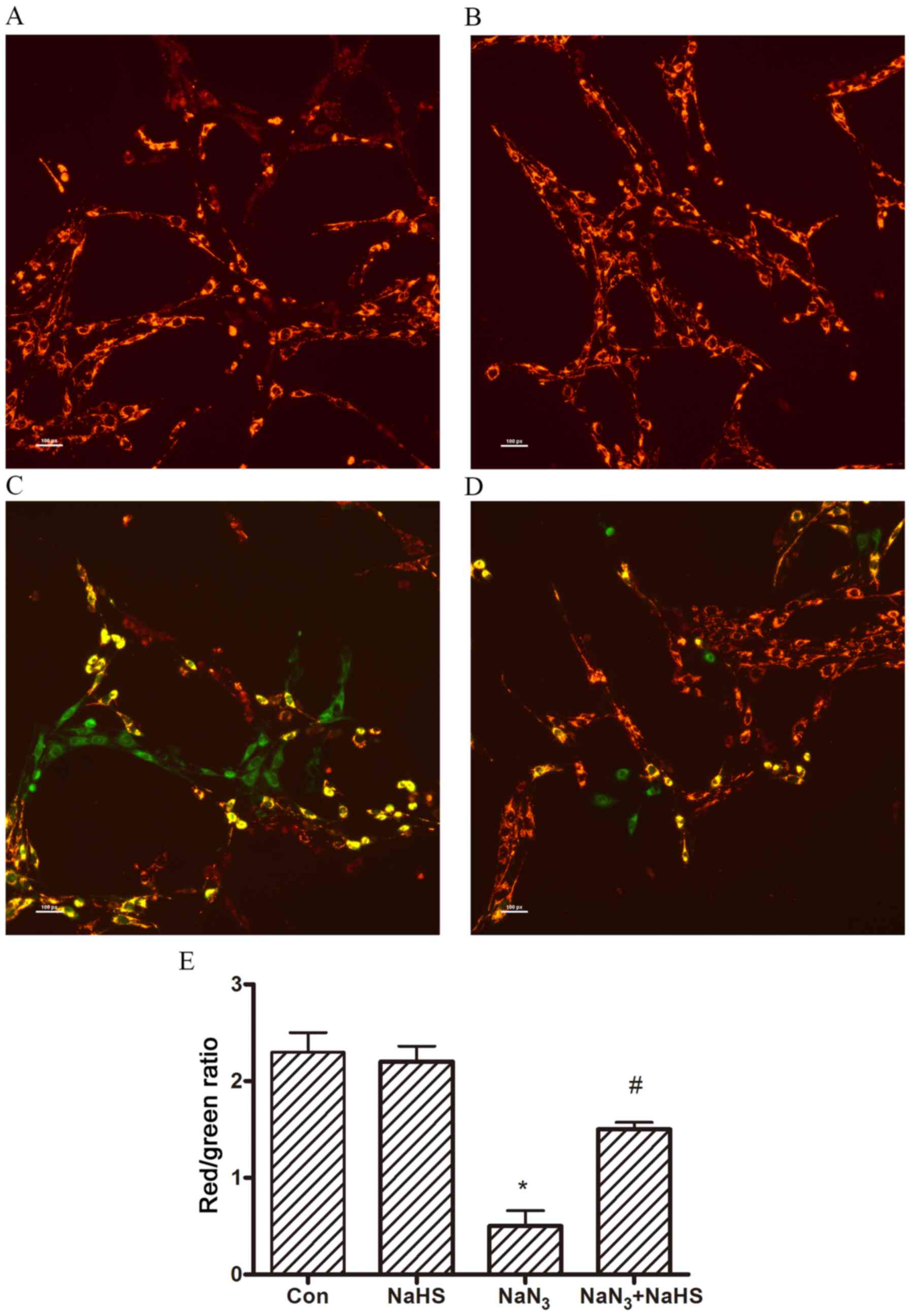
H2S exerts a protective effect
against NaN3-induced dissipation of the mitochondrial
membrane potential
To confirm that NaN3 induced a
mitochondrial membrane potential reduction in PC12 cells, laser
microscopy was used to visualize the fluorescence dye-stained
mitochondria. When PC12 cells were exposed to NaN3, the
mitochondrial membrane rapidly depolarized, as shown by the
increase in green fluorescence (Fig.
3C). Pretreatment with NaHS reduced the changes in
mitochondrial membrane potential, as indicated by repression of
green fluorescence and restoration of red fluorescence (Fig. 3D). The quantitative analysis of
the red/green ratios also demonstrated the protective role of
H2S in NaN3-induced a mitochondrial membrane
potential reduction (Fig.
3E).
H2S reduces
NaN3-induced intracellular ROS accumulation in PC12
cells
The mitochondrion is considered to be the main site
of ROS production, and an increased ROS level within the cell
reflects mitochondrial dysfunction. Therefore, the effect of
H2S on ROS levels was investigated.
N-acetyl-L-cysteine (NAC) is commonly used to identify and
test ROS inducers and to inhibit ROS as a positive control.
Intracellular ROS accumulation may be measured by the use of
DCF-DA, which freely crosses the cell membrane. Once inside the
cells, the compound is hydrolyzed by cellular esterase to DCF,
which interacts with peroxides forming fluorescent
2′,7′-dichlorofluorescin. PC12 cells treated with NaN3
displayed intense fluorescence after staining with DCF dye
(Fig. 4C). Intracellular ROS
accumulation resulting from NaN3 treatment was
significantly reduced when H2S (Fig. 4D) or NAC (Fig. 4E) was present in the medium.
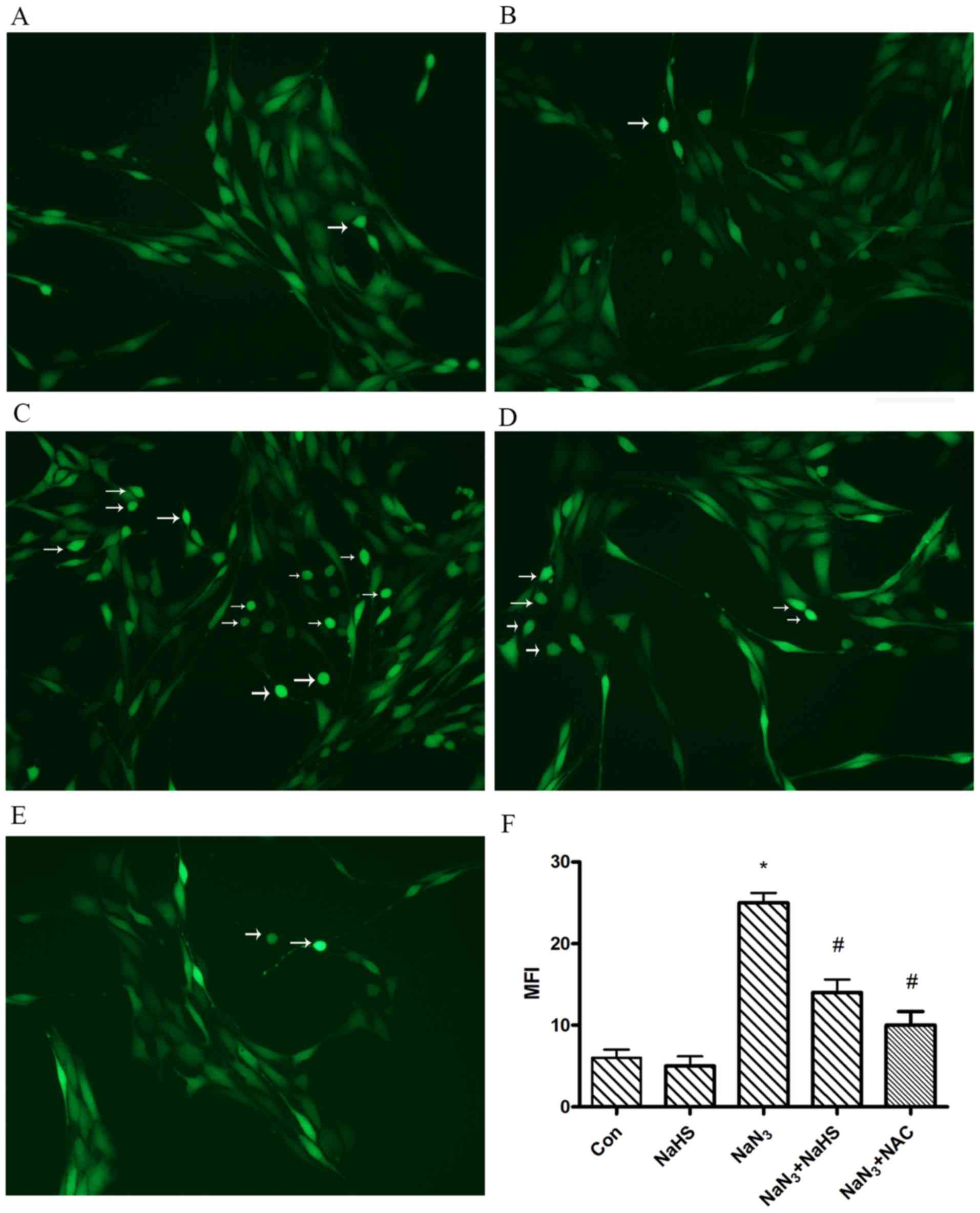 | Figure 4Protective effect of hydrogen sulfide
(H2S) on sodium azide (NaN3)-induced
intracellular reactive oxygen species (ROS) accumulation.
Intracellular peroxide levels were determined based on the
dichlorofluorescein (DCF) fluorescence, as described in Materials
and methods. Cells were plated and grown for 24 h and were then
exposed to NaN3 in the presence or absence of NaHS. (A)
Control, (B) 200 μmol/l NaHS, (C) 30 mmol/l NaN3,
(D) 30 mmol/l NaN3 + 200 μmol/l NaHS, (E) 30
mmol/l NaN3 + N-acetyl-L-cysteine (NAC), and (F)
the bar chart demonstrates the quantitative analysis of median
fluorescence intensity (MFI). NAC is commonly used to identify and
test ROS inducers, and to inhibit ROS. The data are expressed as
means ± standard error of the mean (n=6). *P<0.05,
significantly different from the control group;
#P<0.05, significantly different from the
NaN3 group. Arrows indicate DCF-positive cells. Scale
bars, 50 μm. |
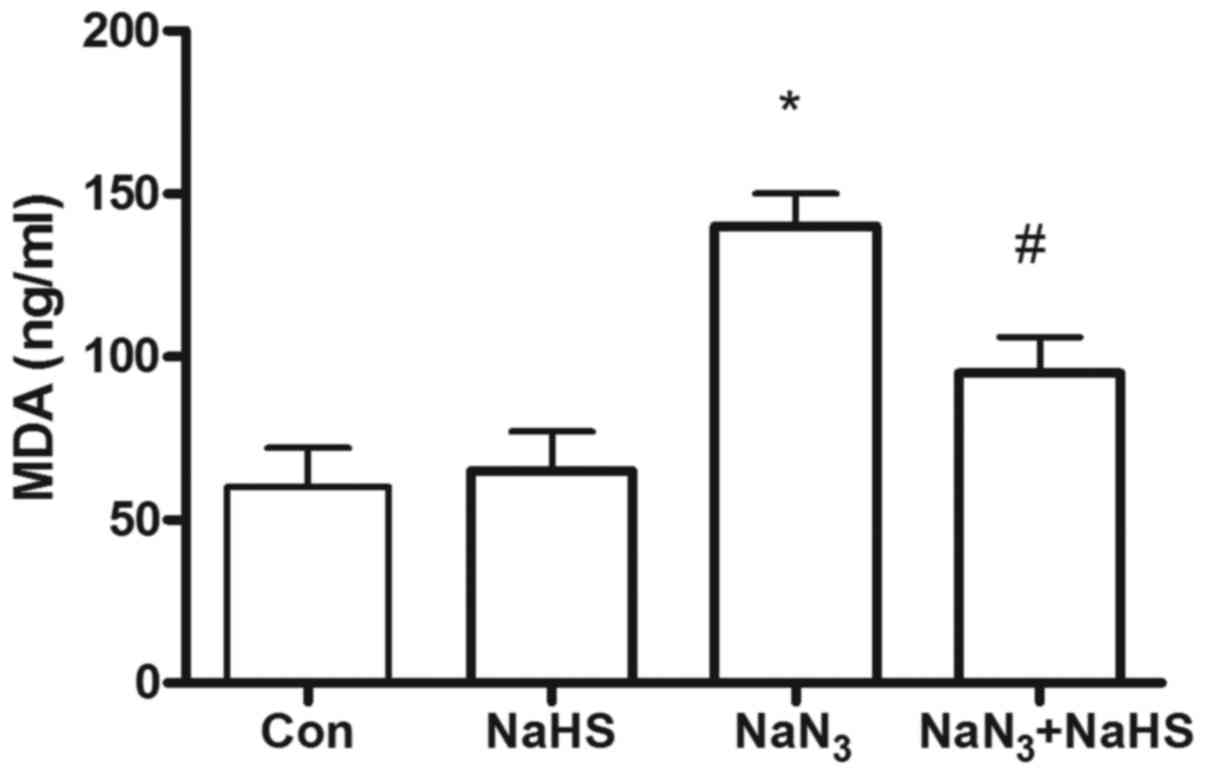
H2S attenuates
NaN3-induced lipid peroxidation increase in PC12
cells
When PC12 cells were exposed to NaN3 (30
mmol/l) for 12 h, an increase in the lipid peroxidation level, as
indicated by the excessive formation of MDA in PC12 cells, was
observed to 150% of control values (Fig. 5). Pretreatment with H2S
at concentrations of 200 μmol/l significantly decreased
lipid peroxidation (decrease in the formation of MDA) compared with
the levels observed in the NaN3 group.
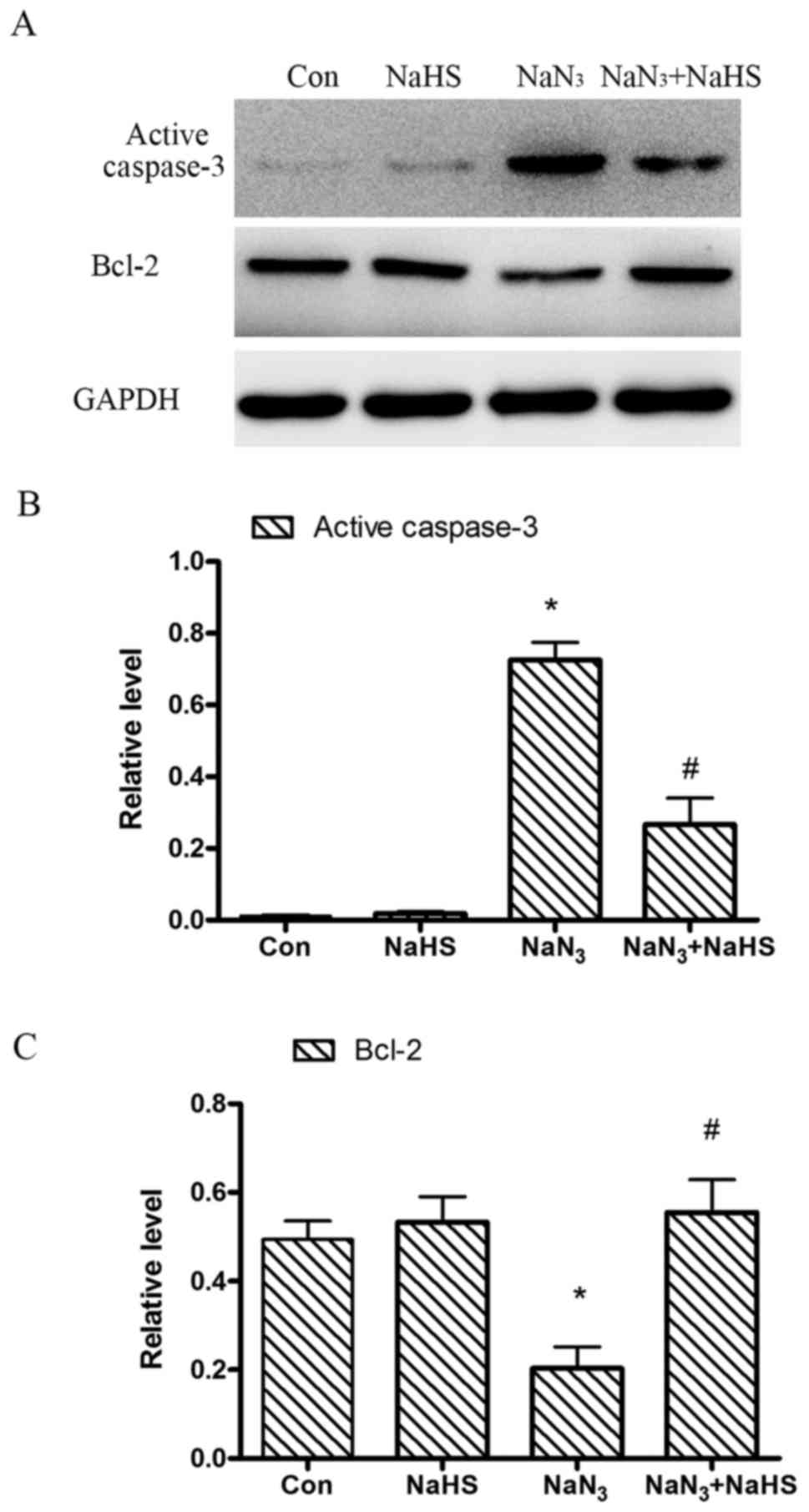
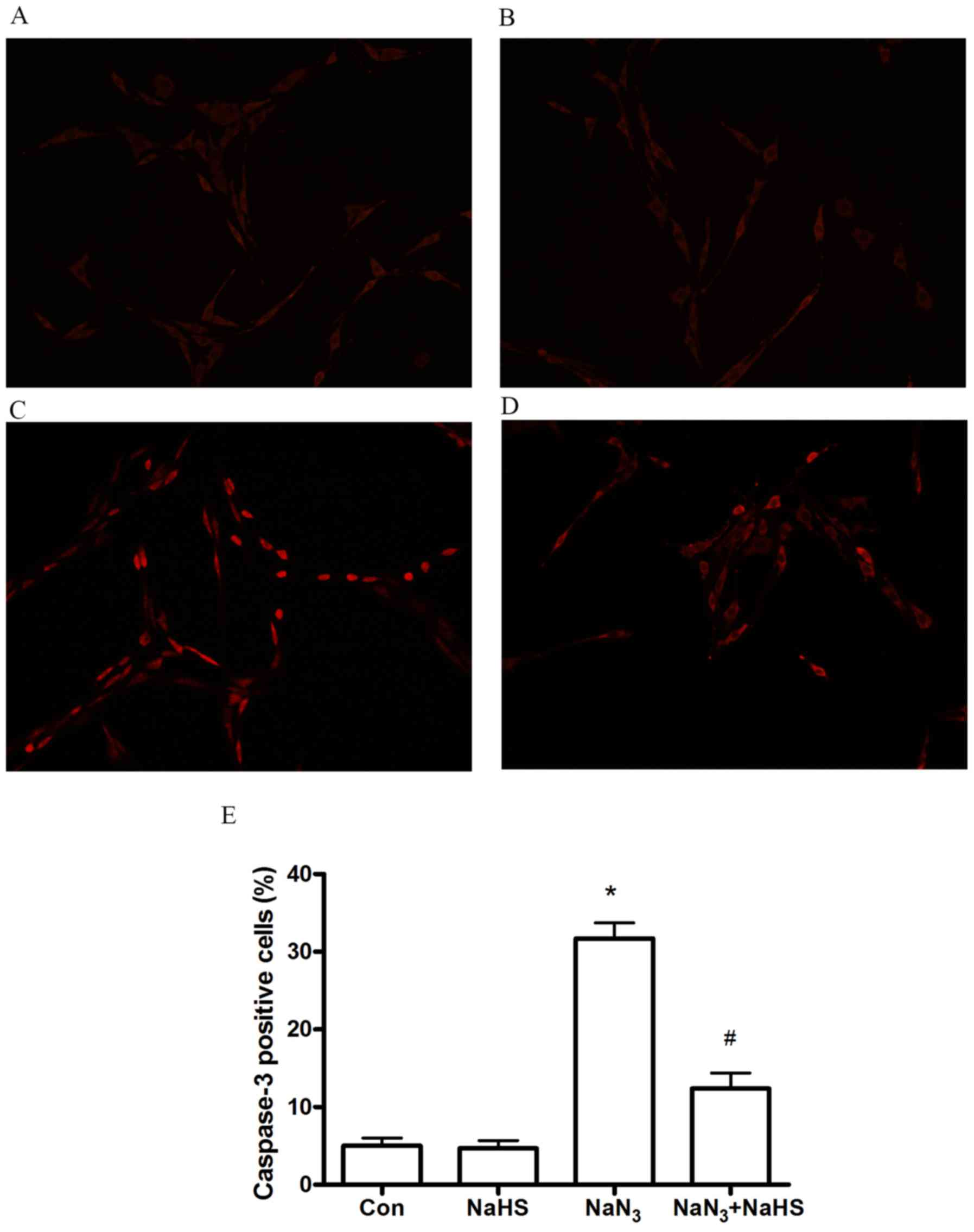
H2S suppresses
NaN3-induced apoptosis in PC12 cells
To certify the effects of H2S on
NaN3-induced apoptosis, the following studies were
performed: caspase-3 is a critical marker of apoptosis. To
determine the effects of NaN3 on cell apoptosis and the
response of H2S to the effects of NaN3, the
levels of caspase-3 and Bcl-2 expression were measured by western
blot analysis. As illustrated in Fig.
6, 12-h treatment with NaN3 (30 mmol/l)
significantly increased the amount of caspase-3 expression
(Fig. 6B), but decreased Bcl-2
expression (Fig. 6C). However,
co-treatment with 200 μmol/l NaHS for 12 h significantly
abolished the NaN3-induced decrease in Bcl-2 expression
and increase in caspase-3 expression. These results indicate that
H2S is able to block the NaN3-elicited
downregulation of Bcl-2 expression and upregulation of caspase-3
expression. In addition, we observed the effect of NaHS on
autophagic cell death in PC12 cells treated with NaN3 by
caspase-3 staining. As shown in Fig.
7, at 12 h after treatment with NaN3 (30 mmol/l),
the number of caspase-3-positive cells observably increased.
Co-treatment with NaHS (200 μmol/l) significantly
ameliorated the NaN3-induced increase of
caspase-3-positive cells. The control and NaHS-treated groups
displayed few caspase-3-positive cells.
Discussion
Endogenous H2S may have multiple
physiological functions in the brain. Our previous study
demonstrated that H2S improved spatial memory impairment
and alleviated cerebral edema in traumatic brain injury (TBI) mice
(15,16). A number of epidemiological studies
provided compelling evidence that sustaining a TBI is associated
with increased risk for degenerative conditions that may result in
dementia, including AD; however, several of the underlying
mechanisms have yet to be fully elucidated. A complex disease such
as AD involves multiple interwoven pathways leading to neuronal
damage, whereas increasing evidence suggests that oxidative stress
is one of the initiating events (17,18). The aim of the present study was to
examine the cell damage occurring in cultured PC12 cells when
exposed to NaN3. Due to its ability to induce oxidative
stress through inhibition of the electron transfer between COX and
oxygen, this model represents an interesting tool in neurotoxicity
studies. However, to the best of our knowledge, no study to date
has investigated whether H2S can prevent cytotoxicity
induced by NaN3 in PC12 cells, and whether oxidative
stress plays an important role in NaN3-induced
apoptosis. In the present study, the possible molecular mechanisms
underlying the neuroprotective effects of H2S against
NaN3-induced neuron cell injury were investigated.
Furthermore, we investigated the connection between the generation
of ROS and the activity of caspase-3 in NaN3-induced
apoptosis in PC12 cells and demonstrated a concentration- and
time-dependent reduction of cell viability induced by
NaN3. Our findings demonstrated that NaHS, a
H2S donor, attenuated NaN3-mediated
apoptosis, and the anti-apoptotic action of H2S was
partly dependent on suppressing the production of ROS and
inhibition of caspase-3 activity, and was associated with
increasing the expression of the anti-apoptotic protein Bcl-2.
AD is a multifactorial neurodegenerative disorder.
Among the numerous contributing factors, cellular stress and, in
particular, oxidative stress, have attracted considerable
attention, as several studies reported its involvement in AD
pathogenesis (19,20). The evidence of oxidative damage in
the postmortem AD brain is quite compelling, with significant
accumulation of markers of oxidative damage of lipids, proteins and
DNA, increased accumulation of transition metals, such as Fe, Cu
and Zn, as well as impaired antioxidant defense (21). The recent redox proteomics
analysis of the postmortem AD brain has demonstrated oxidative
damage to key enzymes involved in energy metabolism,
neurotransmitter-related proteins, mitochondrial proteins and
proteasomal components (22).
Multiple lines of evidence indicate that oxidative stress is an
early event in AD, occurring prior to cytopathological changes, and
may therefore play a key pathogenic role in the disease (20,23). Oxidative stress is the production
of ROS in amounts exceeding the ability of the body's antioxidant
systems to counteract their effects (24). These free radical species, which
contain one or more unpaired electrons, act as electron donors,
causing oxidation that potentially leads to damage of body
macromolecular polymers, such as lipids, proteins and nucleic acids
(25). The most important of all
cell targets of ROS are nervous system cells, particularly neurons,
which are highly susceptible to the harmful effects of ROS
(26).
NaN3, as a COX inhibitor, has been
extensively considered as a useful tool to study different
pathological conditions. Mitochondrial energy metabolism is
suggested to be a determining element for interpreting impaired
neuron function, reduced molecular turnover, and enhanced cell
death (27,28). Inhibition of mitochondrial COX has
been found to induce cell death in a variety of cells. Sato et
al reported that SCC131 cells died 48–72 h after
NaN3 treatment at concentrations more than 5 mM
(29). Lutton et al
reported that NaN3 treatment at a concentration of 1 mM
induced necrosis in rat osteoclasts (30). In those studies, the longest
treatment time required to induce cell death was more than 24 h.
The reason for this finding may be differences between the types of
cells, specifically differential sensitivity of the excretory
function or the detoxification function, and the quantity of the
mitochondria of the target cells. PC12 cells, which are generally
considered to display neuronal-like characteristics, appear to be
more sensitive to NaN3. To induce oxidative stress in
PC12 cells, NaN3 concentrations ranged from 1 to 10 mM
in several experiments (31,32). Wang et al reported that the
viability of PC12 cells treated with 64 mM NaN3 for 4 h
decreased by 47.8% (33). Zhang
et al reported that cultured PC12 cells was incubated with
NaN3 20 mM for 3–24 h to induce apoptosis (34). Increased autophagy was also
observed in multiple and distinct experimental injury models
(35,36). We tested the 5-mM concentration of
NaN3 at 36 h. Although the result of the cell viability
assay revealed that NaN3 induced cell death, autophagic
cell death was not observed under these conditions. However, it is
not known whether the role of autophagy is protective or
detrimental for neural cell injury. It is possible that the role of
autophagy after cell injury is dependent upon the cell's capacity
to respond to the cumulative burden of damaged or dysfunctional
macromolecules and organelles. If the increase in autophagic
capacity is insufficient, augmenting autophagy would likely be
beneficial. When there is excessive increase in autophagic
capacity, inhibiting autophagy may be beneficial. Thus, the role of
autophagy may be dictated by whether it is able to meet
intracellular demands. The cell viability data were important in
order to evaluate whether cells were still physiologically
responsive, or if they were likely to be entering the cell death
process. Therefore, the overall toxic effects of NaN3
was evaluated by monitoring cell viability in PC12 cells. In order
to induce cell death in PC12 cells, high concentrations of
NaN3 (30 mM) were applied in our experiments. Under
these more severe stress conditions, when PC12 cell viability is
already severely hampered, an accumulation of autophagic cell death
was observed (37). A future
study is planned to focus mainly on autophagic cell death in PC12
cells induced by NaN3.
Mitochondrial dysfunction induced by NaN3
provides a common platform for investigating the mechanisms of
neuronal injury, which may prove useful for screening potential
protective agents against neuronal death (38). Hyperoside has the neuroprotective
capacity to attenuate NaN3-induced apoptosis in PC12
cells (34). Wang et al
reported that aloe vera extract exerted a protective effect against
mitochondrial functional impairment induced by NaN3 in
PC12 cells (33). H2S
has increasingly been recognized as a gasotransmitter of comparable
importance to nitric oxide and carbon monoxide in mammalian
systems. Evidence suggests that these gasotransmitters are involved
in the origin of life and play key roles in the endosymbiotic
events that contribute to the biogenesis and development of
mitochondria. In addition to its function as a signaling molecule,
H2S also acts as a cytoprotectant in neurons and cardiac
muscle (11). The neuroprotective
properties of H2S have long been observed, leading to
extensive research that has been widely reported and continues to
attract interest (39). In a rat
model, it was demonstrated that H2S exerts a protective
effect and diminishes oxidative stress and homocysteine-induced
toxicity by its antioxidant properties in the adrenal medulla and
smooth muscle cells of the vesicles (40). This raises the possibility of
H2S being a possible therapeutic strategy in the
treatment of neurodegenerative disorders. To investigate whether
ROS are involved in NaN3-induced injury, PC12 cells were
pretreated with NAC (a ROS scavenger) prior to exposure to
NaN3. The cell viability data were important in order to
evaluate if cells remained physiologically responsive, or if they
were likely to be entering cell death. We observed that
NaN3 induced not only ROS production, but also initiated
injury of PC12 cells, including a decrease in cell viability, loss
of MMP and caspase-3 activation, as well as an increase in the
number of apoptotic cells. These effects were significantly
prevented by NAC pretreatment, indicating that
NaN3-induced neuronal injury is due to its induction of
ROS. Exogenously applied free H2S is immediately
absorbed in a sulfur store as bound sulfane sulfur (41). H2S may be transiently
stored and then released when the cells are stimulated.
H2S is absorbed in brain homogenates more slowly
compared with liver and heart homogenates, and the release from
brain homogenates is also slower compared with that from liver and
heart homogenates (41,42). Once H2S is released
from bound sulfane sulfur or from H2S-producing enzymes,
free H2S may remain longer in the brain compared with
the liver and the heart. Therefore, considering the slow absorption
of H2S in the brain, pretreatment with NaHS was
selected. Interestingly, it was observed that NaHS (a donor of
H2S) shared similar neuroprotective properties with NAC
with a comparable potency in this experimental model. This may
support the ability of H2S in: i) inhibiting
NaN3-mediated protein cytotoxicity; ii) inhibiting
NaN3-mediated oxidative damage; and iii) inhibiting
generation of ROS induced by NaN3.
H2S is synthesized in a number of
different cell types and can easily diffuse without involvement of
any transporters. H2S is involved in a number of
organ-specific functions, such as thermoregulation, modulating
myocardial activity and bronchodilation (43). H2S also exerts
organ-protective effects in ischaemia, acting as a vasodilator and
negative inotrope to reduce blood pressure (44). A number of studies have
investigated the possible benefit of H2S in
hypertension, and found that H2S donor administration
significantly reduced blood pressure and oxidative stress in
hypertensive mice (45,46). H2S has also been found
to play a role in the pathology and treatment of chronic
obstructive pulmonary disease. Exogenously supplied H2S
may counteract the oxidative stress-mediated lung damage that
occurs in allergic mice (47).
Low H2S levels have been observed in a number of
different diseases, while there is evidence that H2S may
be beneficial in a number of chronic organ degenerative conditions.
Chronic kidney disease is associated with a significant reduction
in plasma H2S concentration, and H2S may
ameliorate adenine-induced chronic renal failure in rats by
inhibiting apoptosis through ROS signaling pathways (48). Diabetes is a chronic metabolic
disease affecting the metabolism of carbohydrates and other
nutrients. H2S protects against the development of
hyperglycemia-induced endothelial dysfunction by attenuating the
hyperglycemia-induced enhancement of ROS formation (49). As regards degenerative diseases of
the CNS, H2S treatment can specifically inhibit
6-OHDA-evoked NADPH oxidase activation and oxygen consumption in
Parkinson's disease (50).
Moreover, H2S protects neurons against oxidative stress,
which is responsible for neuronal damage and degeneration in AD
(51). In conclusion,
H2S donors have consistently been shown to be beneficial
in acute organ injury and in chronic organ pathology through ROS
signaling pathways. More specifically, data available thus far
strongly suggest that H2S may be a potent preventive and
therapeutic agent used for the prevention and improvement of the
symptoms of oxidative stress-associated diseases, which is worthy
of further investigation in future studies.
Although H2S exhibited promising
efficacy in NaN3-mediated cell injury, research is still
underway to identify selective oxidative stress regulators as
potential treatment drugs. In summary, the evidence presented
herein indicates an actively protective role for H2S
against oxidative stress and apoptosis induced by NaN3.
Our data may provide a novel pathway to elucidate the underlying
molecular and cellular mechanisms in the CNS following inhibition
of COX, and a novel strategy for the treatment of CNS diseases.
Future studies attempting to characterize the functional
consequences of H2S under conditions of oxidative stress
and the identification of substrates and downstream signaling
targets are now possible. Other H2S donors, such as
drug-like H2S donor ATB-346 or orally active
H2S donor SG-1002, may be used to evaluate the
protective effect of H2S in injury models in the future.
Testing these drug-like H2S donors will not only
consolidate the protective effect of H2S, but also shed
light on the clinical application of H2S as a
therapeutic agent.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81601306, 81301039
and 81530062); the China Postdoctoral Science Foundation Funded
Project (grant no. 2015M570476); the Priority Academic Program
Development of Jiangsu Higher Education Institutions (PAPD); the
Jiangsu Talent Youth Medical Program (grant no. QNRC2016245); the
Key Laboratory of Evidence Science (China University of Political
Science and Law), Ministry of Education (grant no. 2016KFKT05); the
Shanghai Key Laboratory of Forensic Medicine (grant no. KF1502);
and the Suzhou Science and Technology Development Project (grant
no. SYSD2015119).
References
|
1
|
Takahashi RH, Nagao T and Gouras GK:
Plaque formation and the intraneuronal accumulation of β-amyloid in
Alzheimer's disease. Pathol Int. 67:185–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martinez-Vicente M: Neuronal mitophagy in
neurodegenerative diseases. Front Mol Neurosci. 10:642017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Menzies FM, Fleming A, Caricasole A, Bento
CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez
Sanchez M, Karabiyik C, et al: Autophagy and neurodegeneration:
pathogenic mechanisms and therapeutic opportunities. Neuron.
93:1015–1034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cai Q and Tammineni P: Alterations in
mitochondrial quality control in Alzheimer's disease. Front Cell
Neurosci. 10:242016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cai Q and Tammineni P: Mitochondrial
aspects of synaptic dysfunction in Alzheimer's disease. J
Alzheimers Dis. 57:1087–1103. 2017. View Article : Google Scholar
|
|
6
|
Cadonic C, Sabbir MG and Albensi BC:
Mechanisms of mitochondrial dysfunction in Alzheimer's disease. Mol
Neurobiol. 53:6078–6090. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bennett MC, Mlady GW, Kwon YH and Rose GM:
Chronic in vivo sodium azide infusion induces selective and stable
inhibition of cytochrome c oxidase. J Neurochem. 66:2606–2611.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weinstock M and Shoham S: Rat models of
dementia based on reductions in regional glucose metabolism,
cerebral blood flow and cytochrome oxidase activity. J Neural
Transm (Vienna). 111:347–366. 2004. View Article : Google Scholar
|
|
9
|
Huang CW and Moore PK: H2S
synthesizing enzymes: biochemistry and molecular aspects. Handb Exp
Pharmacol. 230:3–25. 2015. View Article : Google Scholar
|
|
10
|
Kimura H: Hydrogen sulfide and
polysulfides as biological mediators. Molecules. 19:16146–16157.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kimura H: Hydrogen sulfide: its
production, release and functions. Amino Acids. 41:113–121. 2011.
View Article : Google Scholar
|
|
12
|
Liu Y, Deng Y, Liu H, Yin C, Li X and Gong
Q: Hydrogen sulfide ameliorates learning memory impairment in
APP/PS1 transgenic mice: a novel mechanism mediated by the
activation of Nrf2. Pharmacol Biochem Behav. 150–151:207–216. 2016.
View Article : Google Scholar
|
|
13
|
Dwyer BE, Raina AK, Perry G and Smith MA:
Homocysteine and Alzheimer's disease: a modifiable risk. Free Radic
Biol Med. 36:1471–1475. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei HJ, Li X and Tang XQ: Therapeutic
benefits of H2S in Alzheimer's disease. J Clin Neurosci.
21:1665–1669. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang M, Shan H, Chang P, Wang T, Dong W,
Chen X and Tao L: Hydrogen sulfide offers neuroprotection on
traumatic brain injury in parallel with reduced apoptosis and
autophagy in mice. PLoS One. 9:e872412014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang M, Shan H, Wang T, Liu W, Wang Y,
Wang L, Zhang L, Chang P, Dong W, Chen X, et al: Dynamic change of
hydrogen sulfide after traumatic brain injury and its effect in
mice. Neurochem Res. 38:714–725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tayler H, Fraser T, Miners JS, Kehoe PG
and Love S: Oxidative balance in Alzheimer's disease: relationship
to APOE, Braak tangle stage, and the concentrations of soluble and
insoluble amyloid-β. J Alzheimers Dis. 22:1363–1373. 2010.
View Article : Google Scholar
|
|
18
|
Chauhan V and Chauhan A: Oxidative stress
in Alzheimer's disease. Pathophysiology. 13:195–208. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Henriques AG, Domingues SC, Fardilha M, da
Cruz e Silva EF and da Cruz e Silva OA: Sodium azide and 2-deoxy-
D-glucose-induced cellular stress affects phosphorylation-dependent
AbetaPP processing. J Alzheimers Dis. 7:201–212. 2005. View Article : Google Scholar
|
|
20
|
Zhu X, Raina AK, Lee HG, Casadesus G,
Smith MA and Perry G: Oxidative stress signalling in Alzheimer's
disease. Brain Res. 1000:32–39. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Butterfield DA, Swomley AM and Sultana R:
Amyloid β-peptide (1-42)-induced oxidative stress in Alzheimer
disease: importance in disease pathogenesis and progression.
Antioxid Redox Signal. 19:823–835. 2013. View Article : Google Scholar :
|
|
22
|
Butterfield DA, Di Domenico F, Swomley AM,
Head E and Perluigi M: Redox proteomics analysis to decipher the
neurobiology of Alzheimer-like neurodegeneration: overlaps in
Down's syndrome and Alzheimer's disease brain. Biochem J.
463:177–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Petersen RB, Nunomura A, Lee HG, Casadesus
G, Perry G, Smith MA and Zhu X: Signal transduction cascades
associated with oxidative stress in Alzheimer's disease. J
Alzheimers Dis. 11:143–152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dringen R: Metabolism and functions of
glutathione in brain. Prog Neurobiol. 62:649–671. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gadalla MM and Snyder SH: Hydrogen sulfide
as a gasotransmitter. J Neurochem. 113:14–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dumont M and Beal MF: Neuroprotective
strategies involving ROS in Alzheimer disease. Free Radic Biol Med.
51:1014–1026. 2011. View Article : Google Scholar
|
|
27
|
Ott M, Gogvadze V, Orrenius S and
Zhivotovsky B: Mitochondria, oxidative stress and cell death.
Apoptosis. 12:913–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yin F, Boveris A and Cadenas E:
Mitochondrial energy metabolism and redox signaling in brain aging
and neurodegeneration. Antioxid Redox Signal. 20:353–371. 2014.
View Article : Google Scholar :
|
|
29
|
Sato E, Suzuki T, Hoshi N, Sugino T and
Hasegawa H: Sodium azide induces necrotic cell death in rat
squamous cell carcinoma SCC131. Med Mol Morphol. 41:211–220. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lutton JD, Moonga BS and Dempster DW:
Osteoclast demise in the rat: physiological versus degenerative
cell death. Exp Physiol. 81:251–260. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Amador FC and Henriques AG: Monitoring
protein phosphatase 1 isoform levels as a marker for cellular
stress. Neurotoxicol Teratol. 26:387–395. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Satpute R, Bhattacharya R, S Kashyap R, J
Purohit H, Y Deopujari J, M Taori G and F Daginawala H: Antioxidant
potential of Fagonia arabica against the chemical ischemia-induced
in PC12 cells. Iran J Pharm Res. 11:303–313. 2012.PubMed/NCBI
|
|
33
|
Wang Y, Cao L and Du G: Protective effects
of Aloe vera extract on mitochondria of neuronal cells and rat
brain. Zhongguo Zhong Yao Za Zhi. 35:364–368. 2010.In Chinese.
PubMed/NCBI
|
|
34
|
Zhang L, Cheng X and Hu J: Neuroprotective
effects of hyperoside on sodium azide-induced apoptosis in PC12
cells. Chin J Nat Med. 9:450–455. 2011.
|
|
35
|
Smith CM, Chen Y, Sullivan ML, Kochanek PM
and Clark RS: Autophagy in acute brain injury: feast, famine, or
folly. Neurobiol Dis. 43:52–59. 2011. View Article : Google Scholar
|
|
36
|
Liu L, Sun T, Xin F, Cui W, Guo J and Hu
J: Nerve growth factor protects against alcohol-induced
neurotoxicity in PC12 cells via PI3K/Akt/mTOR pathway. Alcohol
Alcohol. 52:12–18. 2017. View Article : Google Scholar
|
|
37
|
Shan H, Chu Y, Chang P, Yang L, Wang Y,
Zhu S, Zhang M and Tao L: Neuroprotective effects of hydrogen
sulfide on sodium azide-induced autophagic cell death in PC12
cells. Mol Med Rep. Aug 25–2017.Epub ahead of print. View Article : Google Scholar
|
|
38
|
Selvatici R, Previati M, Marino S, Marani
L, Falzarano S, Lanzoni I and Siniscalchi A: Sodium azide induced
neuronal damage in vitro: evidence for non-apoptotic cell death.
Neurochem Res. 34:909–916. 2009. View Article : Google Scholar
|
|
39
|
Kimura H: Hydrogen sulfide: production,
release, and functions. Nippon Yakurigaku Zasshi. 139:6–8. 2012.In
Japanese. View Article : Google Scholar
|
|
40
|
Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH,
Liu LS and Jiang ZS: Hydrogen sulfide, the next potent preventive
and therapeutic agent in aging and age-associated diseases. Mol
Cell Biol. 33:1104–1113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ishigami M, Hiraki K, Umemura K, Ogasawara
Y, Ishii K and Kimura H: A source of hydrogen sulfide and a
mechanism of its release in the brain. Antioxid Redox Signal.
11:205–214. 2009. View Article : Google Scholar
|
|
42
|
Kimura H: Hydrogen sulfide: from brain to
gut. Antioxid Redox Signal. 12:1111–1123. 2010. View Article : Google Scholar
|
|
43
|
Yu XH, Cui LB, Wu K, Zheng XL, Cayabyab
FS, Chen ZW and Tang CK: Hydrogen sulfide as a potent
cardiovascular protective agent. Clin Chim Acta. 437:78–87. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jin Z, Chan H, Ning J, Lu K and Ma D: The
role of hydrogen sulfide in pathologies of the vital organs and its
clinical application. J Physiol Pharmacol. 66:169–179.
2015.PubMed/NCBI
|
|
45
|
Al-Magableh MR, Kemp-Harper BK and Hart
JL: Hydrogen sulfide treatment reduces blood pressure and oxidative
stress in angiotensin II-induced hypertensive mice. Hypertens Res.
38:13–20. 2015. View Article : Google Scholar
|
|
46
|
Meng G, Ma Y, Xie L, Ferro A and Ji Y:
Emerging role of hydrogen sulfide in hypertension and related
cardiovascular diseases. Br J Pharmacol. 172:5501–5511. 2015.
View Article : Google Scholar :
|
|
47
|
Benetti LR, Campos D, Gurgueira SA,
Vercesi AE, Guedes CE, Santos KL, Wallace JL, Teixeira SA,
Florenzano J, Costa SK, et al: Hydrogen sulfide inhibits oxidative
stress in lungs from allergic mice in vivo. Eur J Pharmacol.
698:463–469. 2013. View Article : Google Scholar
|
|
48
|
Wu D, Luo N, Wang L, Zhao Z, Bu H, Xu G,
Yan Y, Che X, Jiao Z, Zhao T, et al: Hydrogen sulfide ameliorates
chronic renal failure in rats by inhibiting apoptosis and
inflammation through ROS/MAPK and NF-κB signaling pathways. Sci
Rep. 7:4552017. View Article : Google Scholar
|
|
49
|
Suzuki K, Olah G, Modis K, Coletta C, Kulp
G, Gerö D, Szoleczky P, Chang T, Zhou Z, Wu L, et al: Hydrogen
sulfide replacement therapy protects the vascular endothelium in
hyperglycemia by preserving mitochondrial function. Proc Natl Acad
Sci USA. 108:13829–13834. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hu LF, Lu M, Tiong CX, Dawe GS, Hu G and
Bian JS: Neuroprotective effects of hydrogen sulfide on Parkinson's
disease rat models. Aging Cell. 9:135–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kimura Y and Kimura H: Hydrogen sulfide
protects neurons from oxidative stress. FASEB J. 18:1165–1167.
2004.PubMed/NCBI
|