Introduction
Global cerebral ischemia (GCI) is a leading cause of
mortality and morbidity worldwide (1). It is essential to understand the
biological cascades that drive the delayed secondary phase
subsequent to GCI (2). In these
situations, a transient period of GCI causes selective cell death
in the vulnerable cortex pyramidal cell layer days after
reperfusion, which is known as delayed apoptotic neuronal death
(3). However, despite extensive
research, no effective treatment has been developed to repair the
damage resulting from GCI in the clinic.
Vitamin D is most commonly associated with the
regulation of calcium homeostasis (4). Vitamin D2 and D3 are two exogenous
forms of vitamin D, each of which is biologically inert. Their
activation requires a two-step hydroxylation reaction involving
25-hydroxylase in the liver and 1α-hydroxylase in the kidney
(5). The biologically active
metabolite of vitamin D (calcitriol) exerts its endocrinological
influence via a nuclear vitamin D receptor (VDR) (6). The wide distribution of VDR suggests
that vitamin D may regulate various physiological pathways, such as
brain development, inflammation, neurological function, cell-cycle
control, immune modulation and apoptosis (7–11).
In addition, vitamin D is currently being investigated as a
therapeutic strategy for various neurological disease, including
depressive disorder, traumatic brain injury, stroke and
neurodegenerative diseases (12–15).
The present study assessed whether post-injury
treatment with calcitriol has a therapeutic effect against ischemic
reperfusion-induced injury and alleviates neurological deficits.
The present study was also performed to further refine the
potential underlying mechanisms of calcitriol treatment. In
addition, through administering PD98059 as a tool to modulate
extracellular signal-regulated kinase (ERK)1/2 activation, it was
assessed whether the ERK1/2 pathway has a role in neuronal
apoptosis in the rat cortex.
Materials and methods
Animals
The Institutional Animal Care and Use Committee of
North China University of Science and Technology (Tangshan, China)
approved all experiments, which were performed according to the
guidelines of the National Institutes of Health (NIH) Guide for the
Care and Use of Laboratory Animals (NIH publication no. 80–23,
revised 1978). All efforts were made to minimize the number of
animals used and their suffering. A total of 145 male Sprague
Dawley rats (age, 10–16 weeks; weight, 350–380 g) were used in the
study. The rats were housed under a 12-h light/dark cycle at 20°C.
All of the rats were fasted for 12–16 h prior to the experiments,
but had free access to water.
Model of GCI
The transient GCI model was generated as previously
described (16). In brief,
ischemia was induced via bilateral carotid artery ligation under
conditions of arterial hypotension. A laser Doppler monitoring
system (VMS-LDF2; Moor Instruments, Axminster, UK) was used to
monitor cerebral blood flow (CBF) during the procedure. The sensor
of the monitor was placed (and fixed with bone cement) 1–2 mm
posterior and 4–5 mm lateral to the bregma on the left or right
skull hemisphere after a small midline skin incision had been made
on the same side. For blood withdrawal and re-infusion, the right
jugular vein was cannulated with a silicone catheter. After
heparinization (50 units), blood was quickly drawn through the
jugular vein until the mean arterial pressure (MAP) attained 25–30
mmHg and the reduction in regional CBF was 50% from baseline. The
two common carotid arteries were then clamped with vascular clips
for 10 min, and the MAP was maintained at 25–30 mmHg during the
ischemic period. The clips were carefully removed from both
arteries and the withdrawn blood was slowly re-infused. After
completion of the procedure, the rats were given 0.5% bupivacaine
injections around all of the incision sites and were allowed to
recover from anesthesia at room temperature.
Groups and drug administration
Experiment 1 was performed to determine the
neuroprotection effects of calcitriol after GCI. A total of 135
rats were randomly assigned to 3 groups as follows: Sham group
(n=45), GCI group (n=45) and GCI + calcitriol group (n=45). In the
calcitriol group, rats were intraperitoneally injected with
calcitriol (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
dissolved in 5% dimethyl sulfoxide (DMSO) and 0.9% normal saline at
a dose of 1 μg/kg at 30 min, 12 and 24 h after the GCI
insult. The Sham and GCI groups received equal volumes of 5% DMSO
by intraperitoneal injection at the same time. All investigations
were blind and group identities were revealed only at the end of
the behavioral and histological analyses.
Experiment 2 was performed to determine whether the
ERK1/2 pathway was involved in neuronal death in the rat cortex. A
total of 10 rats from the GCI + calcitriol group were randomly
divided into two groups: The vehicle group, which received an equal
volume of 20 μl 5% DMSO (n=5; vehicle group); and the
PD98059 group, which received 5 μg p-ERK1/2 inhibitor
PD98059 (Bio-Rad Laboratories, Inc., Hercules, CA, USA) dissolved
in 20 μl 5% DMSO (n=5; PD98059 group). Drug injection was
performed at 30 min prior to the GCI insult.
Infarct measurement by
2,3,5-triphenyltetrazolium chloride (TTC) staining
At 7 days after GCI, rats were anesthetized by
intraperitoneal injection with 300 mg/kg of a 10% chloral hydrate
solution (Beijing Sino fir Biological Engineering Co., Ltd.,
Beijing, China), and intracardially perfused with isotonic sodium
chloride solution. Following reperfusion, rat brains (n=5 from the
Sham, GCI and calcitriol groups) were removed and frozen at −80°C
for 5 min. Coronal slices (2 mm) were stained for 20 min at 37°C
with 2% TTC (Sigma-Aldrich; Merck KGaA) and were then fixed with 4%
formaldehyde. The sections were scanned and the infarct area in
each section was calculated by using ImageJ analysis software
(version 1.46; NIH, Bethesda, MD, USA).
Hematoxylin and eosin (H&E)
stain
At 3 days after GCI, rats were anesthetized and
perfused as described above, followed by perfusion with 4% (w/v)
paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4). The
brains were removed and fixed for 48 h in 4% (w/v)
paraformaldehyde. After fixation, brains were embedded in paraffin
and sliced into 4-μm coronal sections at the level of the
bregma and stained with H&E.
Transmission electron microscopy
(TEM)
Cortex region neurons of 9 animals (3 each from the
Sham, GCI and calcitriol group) were examined via TEM at 3 days
after GCI. Following perfusion, brains were removed, and the cortex
region was dissected and washed in 0.1 M phosphate buffer. Tissue
samples were immersed in 2% glutaraldehyde and 1% osmium tetroxide
(Sigma-Aldrich; Merck KGaA) for 2 h at 4°C, and then dehydrated in
a graded ethanol series. Following displacement of ethanol with
propylene oxide, the tissue was embedded in Epon (both from
Sigma-Aldrich; Merck KGaA) and sectioned along the coronal plane
with a diamond knife (FernAnclez-hIorln 1953; Ivan Sorvall, Inc.,
New York, NY, USA) at a thickness of 60 nm. The sections were
stained with lead citrate and observed using a CM-120 electron
microscope (Philips, Eindhoven, The Netherlands).
Evaluation of brain edema
At 1, 3 and 5 days following GCI, rat brains were
separated and weighed immediately with a chemical balance to
determine the wet weight (WW). Following drying in a desiccating
oven for 24 h at 100°C, dry tissues were weighed again to obtain
the constant dry weight (DW). The percentage of water in the
tissues was calculated according to the following formula: Brain
water content (%) = (WW-DW)/WW) ×100%.
Evaluation of neurological deficits
The neurological severity score (NSS) (17) was determined at 1, 3, 5 and 7 days
after surgery. The investigator who performed the test was blinded
to the groups. Neurological function was graded on a scale of 0–18
(normal score, 0; maximal deficit score, 18). In the severity score
of injury, a score of 0 was awarded for the ability to perform the
test, and the higher the score, the more severe the injury.
Terminal deoxynucleotidyl transferase
deoxyuridine triphosphate nick end-labelling (TUNEL) assay
TUNEL staining was performed using an Apoptag
Peroxidase in situ Apoptosis Detection kit S7100 (EMD
Millipore, Billerica, MA, USA) according to the manufacturer's
protocol. The nuclei of TUNEL-positive neurons that contained
apoptotic bodies stained blue and were identified as apoptotic. The
apoptotic cells were counted under high-power magnification (×400)
and the percentage of TUNEL-positive cells among the total cells
was determined.
Western blot analysis
Western blotting was performed at 3 days post-GCI.
Rat cortex tissues were lysed in Tissue Protein Lysis Solution
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented
with 5% proteinase inhibitor cocktail (Sigma-Aldrich; Merck KGaA),
incubated on ice for 30 min and centrifuged at 15,000 × g at
22–24°C for 15 min. Protein concentrations were determined using
the bicinchoninic acid protein assay (reagents obtained from
Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Protein samples (50 μg/lane) were subjected to 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to polyvinylidene difluoride membranes (Roche
Diagnostics GmbH, Mannheim, Germany) for 60 min. Nonspecific
binding sites were blocked with 5% bovine serum albumin (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) at 22–24°C for 1 h, and then
incubated with various antibodies: Rabbit anti-rat VDR (cat. no.
sc9164), caspase-3 (cat. no. sc7148), B-cell lymphoma-2 (Bcl-2;
cat. no. sc783), ERK (cat. no. sc292838), phosphorylated (p)-ERK
(cat. no. sc101760) and GAPDH (cat. no. sc25778) polyclonal
antibodies (dilution, 1:1,000; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) overnight at 4°C. The membranes were then
incubated with secondary antibodies (cat. no. sc2955; dilution,
1:5,000; Santa Cruz Biotechnology, Inc.) for 2 h at 4°C. The
immunoreactive bands were visualized using an enhanced
chemiluminescence reagent (Bio-Rad Laboratories, Inc.). Blots were
scanned and for densitometrical analysis, and the integrated
density of pixels was quantified using Image Quant software
(version 5.2; Molecular Dynamics; GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA).
Immunofluorescent staining
Immunofluorescent staining was performed at 3 days
post-GCI as described previously. After perfusion, brains were
removed, post-fixed in the same fixative for 1 day at room
temperature and subsequently soaked in 30% sucrose for 2–3 days.
Subsequently, the tissues were embedded in optimal cutting
temperature compound. Frozen cross sections (12 μm) were
prepared and examined. Following three washes with
phosphate-buffered saline (PBS), for 10 min each, the sections were
incubated with 5% donkey serum (Institute of Hematology, Chinese
Academy of Medical Sciences, Tianjin, China) and 0.4% Triton X-100
(Amresco, LLC, Solon, OH, USA) at room temperature for 2 h. The
sections were incubated with primary goat polyclonal antibody to
p-ERK1/2 (cat. no. sc7976; 1:200 dilution; Santa Cruz
Biotechnology, Inc.), rabbit polyclonal primary antibody to
caspase-3 (cat. no. sc7148; 1:200 dilution) and mouse polyclonal
primary antibody to neuronal nuclei (NeuN; cat. no. sc71667; 1:200
dilution) (both from Santa Cruz Biotechnology, Inc.), respectively,
overnight at 4°C, followed by incubation with a mixture of
secondary antibodies (cat. no. A-11055, A-11037 and A-32728; 1:200
dilution; Invitrogen; Thermo Fisher Scientific, Inc.) at room
temperature for 2 h. After washing with PBS 3 times for 10 min
each, the images were captured under a fluorescence microscope
(Olympus FluoView™ FV1000; Olympus, Tokyo, Japan).
Statistical analysis
SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA)
was used for statistical analysis. Statistical evaluation of the
data was performed using an independent Student's t-test for
comparison between two groups, and one-way analysis of variance for
comparison among three groups, followed by post-hoc comparisons
using the least significant differences or Kruskal-Wallis method.
Data from the mechanical allodynia and thermal hyperalgesia tests
were analysed via using repeated-measures tests. All experimental
data are expressed as the mean ± standard error and P<0.05 was
considered to indicates a statistically significant difference.
Results
Mortality of experimental rats
The mortality rate was low in rats following GCI.
Two rats died during the experiments, one of which was in the GCI
model group and the other was in the calcitriol treatment group. No
signs of unacceptable pain/stress/illness were observed in the two
rats prior to mortality.
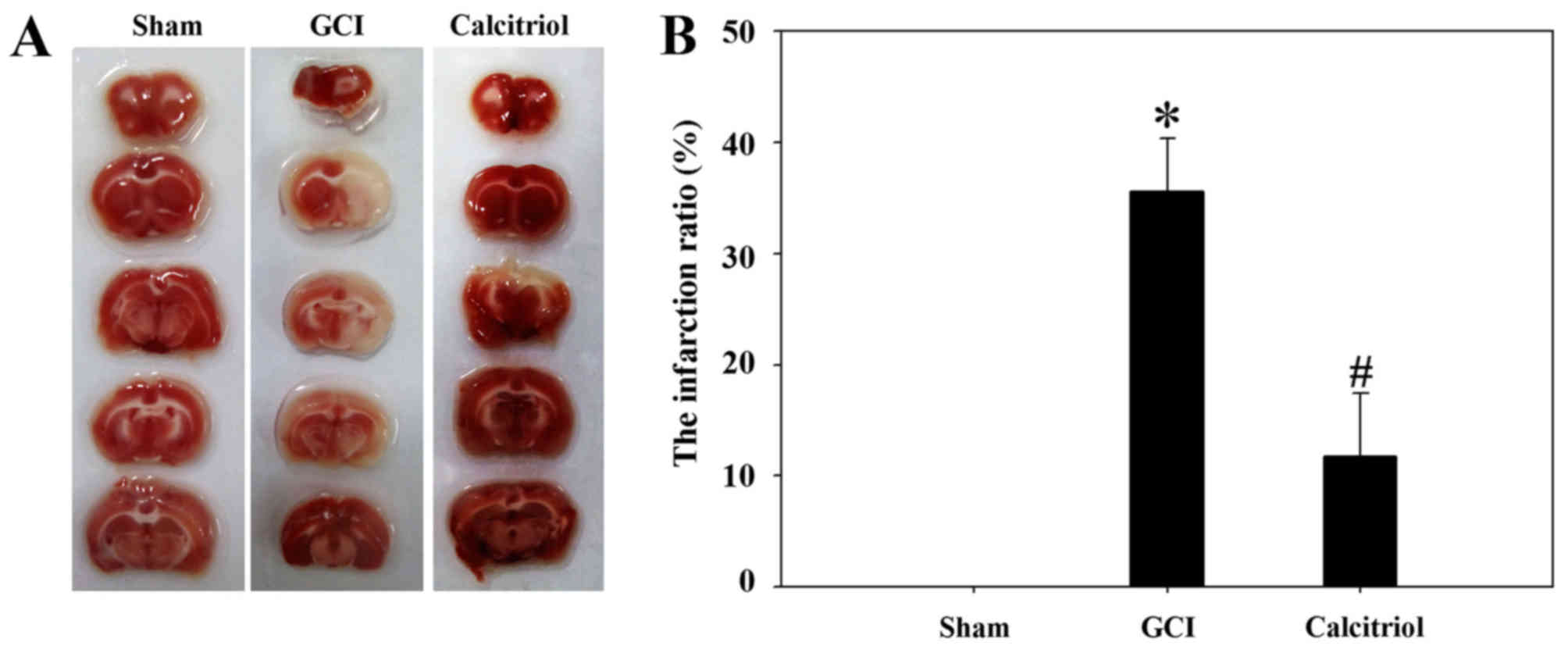
Calcitriol decreases the brain infarct
volume after GCI
As presented in Fig.
1, TTC staining revealed that the infarct ratio was
significantly increased after GCI compared with that in the
Sham-operated group (P<0.001), and intraperitoneal calcitriol
injection markedly decreased the infarct ratio (P<0.005). This
strongly indicated that vitamin D protects rats from
hypoxia-ischemia-induced brain cortex injuries.
Morphological analysis of cortical
regions of the brain
At 3 days after GCI, the cortical regions of the
brains were collected and morphological detection was performed via
H&E staining and TEM. As presented in Fig. 2A, the nuclei of normal neurons
were round and stained pale, whereas nuclei of dying neurons were
pyknotic and darkly stained after GCI. The pathological changes of
cortex neurons in the calcitriol-treated group were attenuated
compared with those in the GCI group. The ultrastructures of the
neurons in the cortex are depicted in Fig. 2B. In the Sham group, the neurons
in the cortex had large round nuclei, the double nuclear membranes
were clear and complete and the outer membrane enclosed the
periphery of the mitochondria. In the GCI model group, neurons were
irregular and exhibited chromatin condensation, cytoplasm
dissolution and vacuole formation. The nuclear membranes and cell
organelles were dissolved or absent. In the calcitriol treatment
group, the damage to the neurons was alleviated.
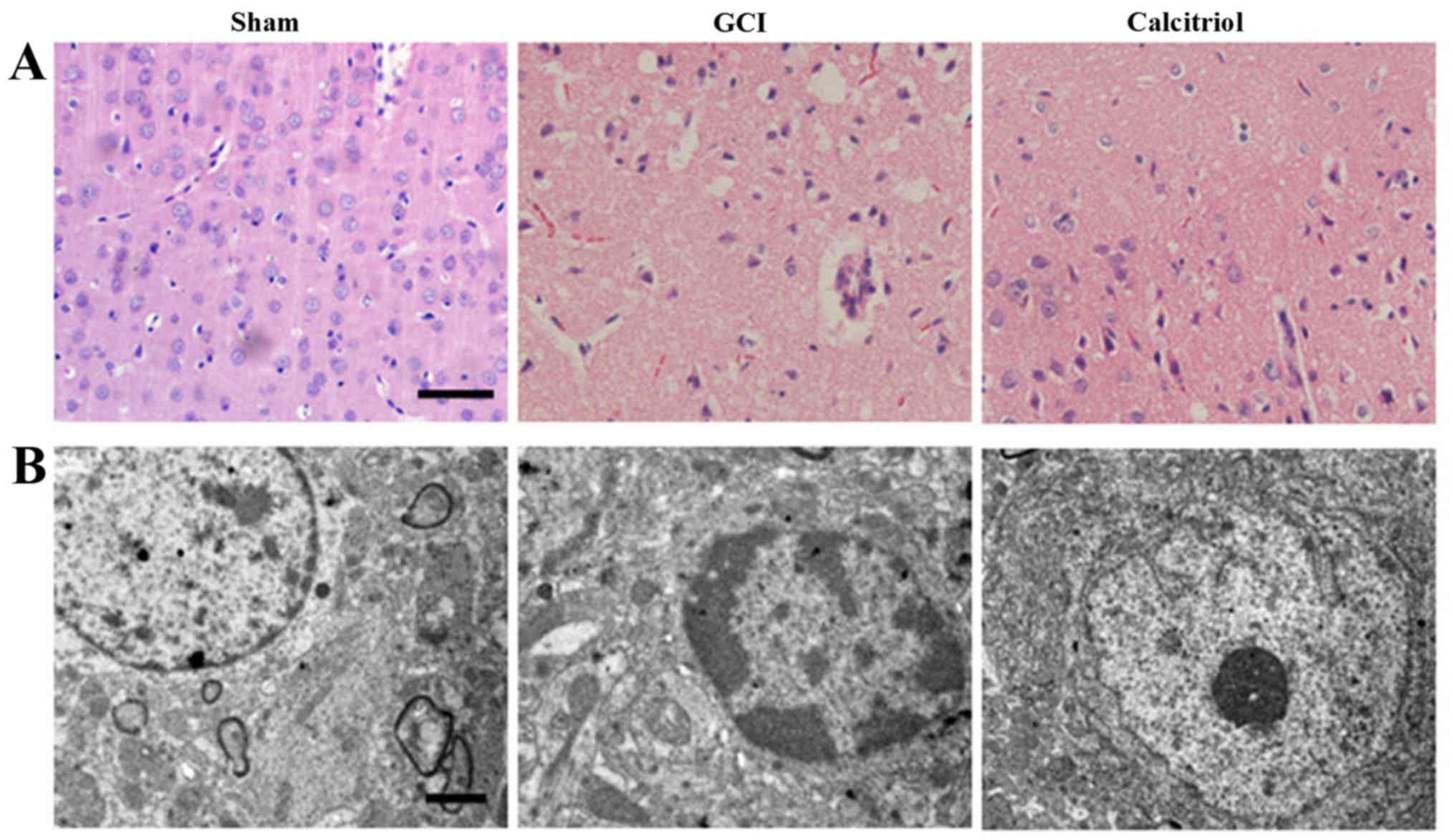 | Figure 2Morphological detection results in the
cortex region of the brain after GCI. (A) Assessment of
histopathologic changes in the experimental groups (hematoxylin and
eosin staining; scale bar, 50 μm). The nuclei of normal
neurons were round and stained pale, whereas nuclei of dying
neurons were pyknotic and darkly stained after GCI. The
pathological changes of cortex neurons in the calcitriol-treated
group were improved compared with those in the GCI group. (B)
Ultrastructures of neurons were observed via transmission electron
microscopy in the experimental groups (scale bar, 2 μm). In
the Sham group, the neurons in the cortex had large round nuclei,
the double nuclear membranes were clear and complete, and the outer
membrane enclosed the periphery of the mitochondria. In the GCI
group, neurons were irregular and exhibited chromatin condensation,
cytoplasm dissolution and vacuole formation. The nuclear membranes
and cell organelles were dissolved or absent. In the calcitriol
group, the damage to the neurons was alleviated. GCI, global
cerebral ischemia. |
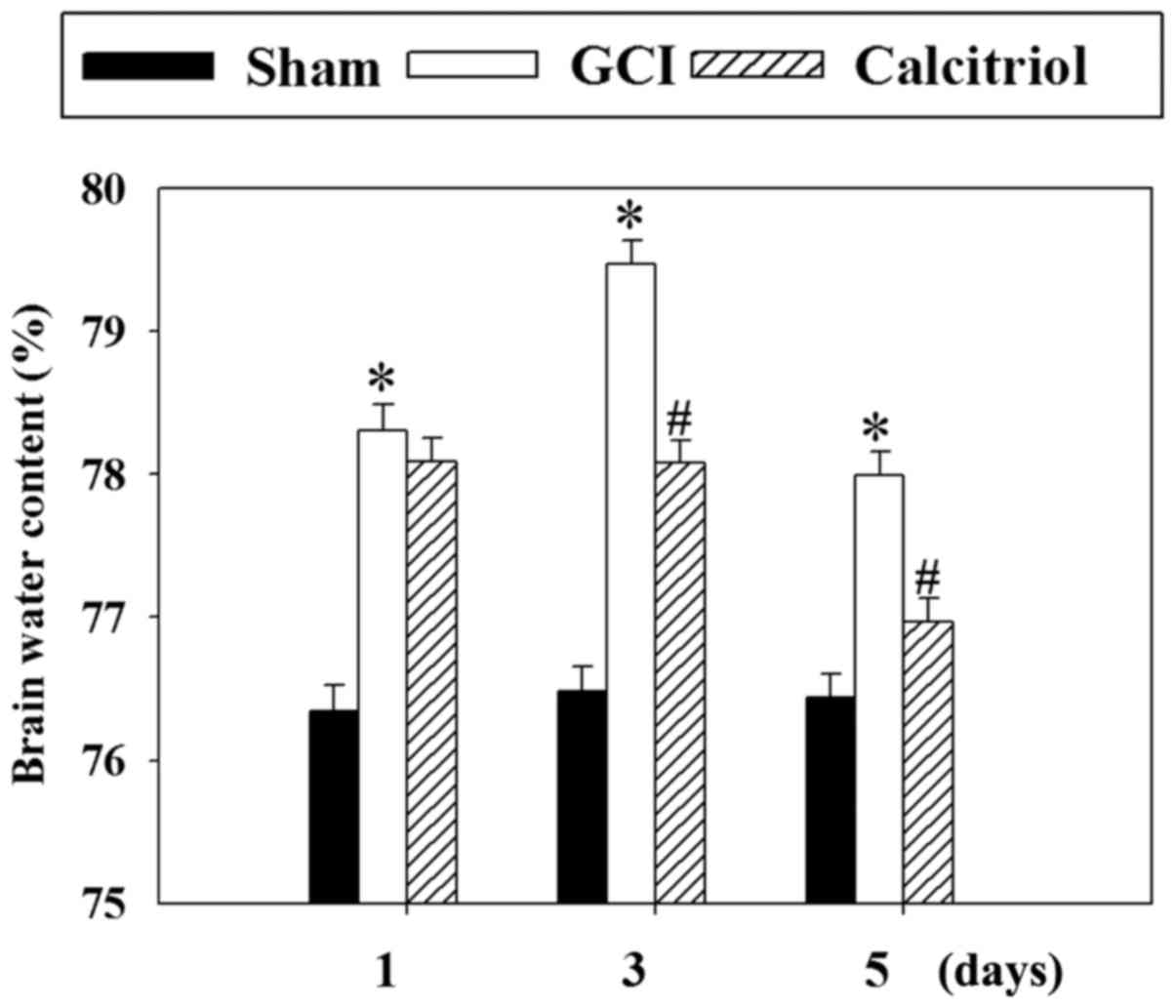
Calcitriol attenuates GCI-induced
cerebral edema
To evaluate the effects of calcitriol on brain
edema, the wet-dry weight method was employed at 1, 3 and 5 days
post-GCI. As presented in Fig. 3,
the cerebral water content was significantly increased at 1, 3 and
5 days after GCI (P<0.001 vs. Sham group). However, treatment
with calcitriol attenuated the cerebral water content at 3 and 5
days compared with that in the GCI model group (P<0.001 and
P=0.001, respectively).
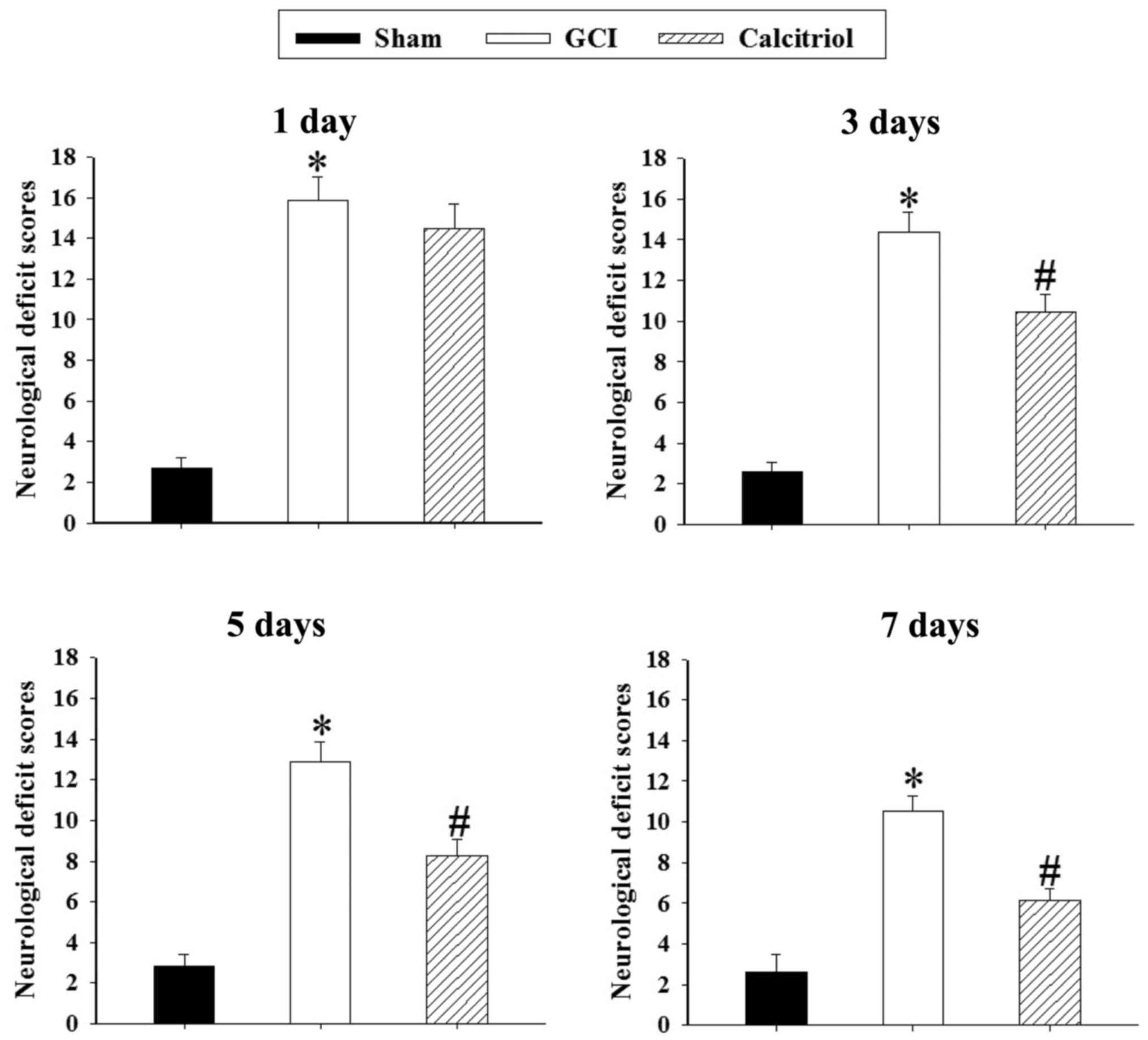
Calcitriol attenuates GCI-induced
neurological deficits
Neurological deficits after GCI were expressed as
the NSS in the present study. Changes in neurological deficits of
rats at 1, 3, 5 and 7 days are depicted in Fig. 4. The rats exhibited significant
neurological deficits after GCI (P<0.001). Post-injury
administration of calcitriol significantly improved the NSS at 3, 5
and 7 days after GCI (P=0.004, P=0.002 and P=0.001,
respectively).
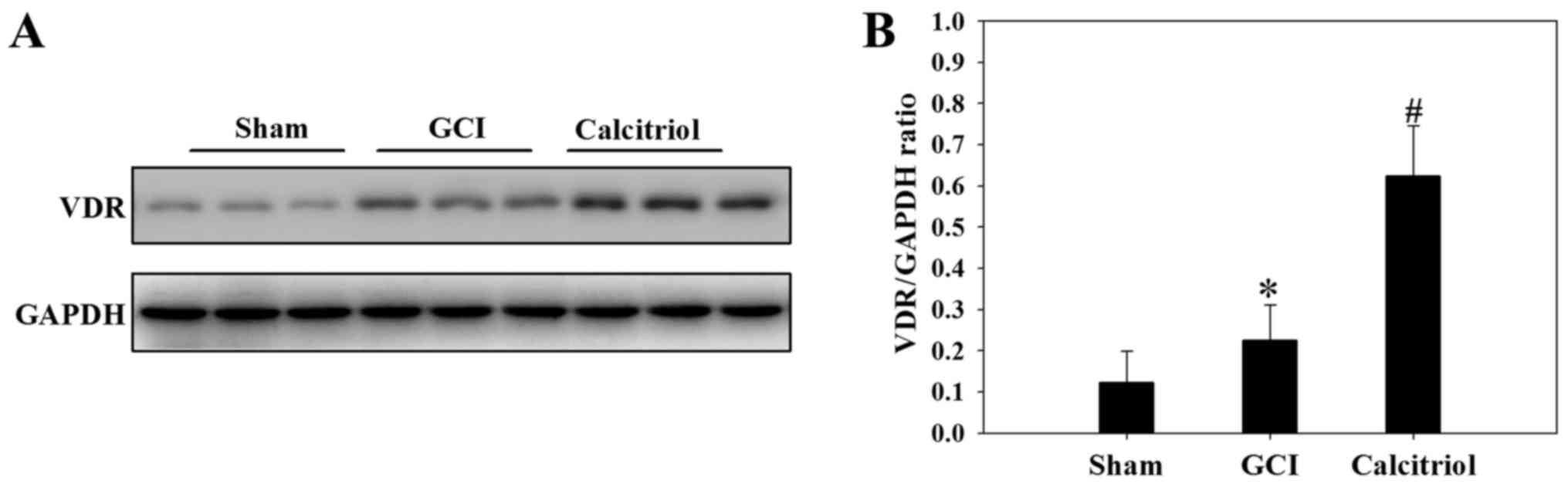
Increase of VDR expression after
treatment of calcitriol
To further investigate the underlying mechanisms of
the effect of calcitriol, the expression levels of VDR were
analyzed in the cortex tissue at 3 days following the GCI insult.
As demonstrated in Fig. 5, GCI
caused an upregulation of VDR protein expression compared with that
in Sham-operated rats (P<0.05), and supplementation with
calcitriol further increased VDR protein expression after GCI
(P<0.05).
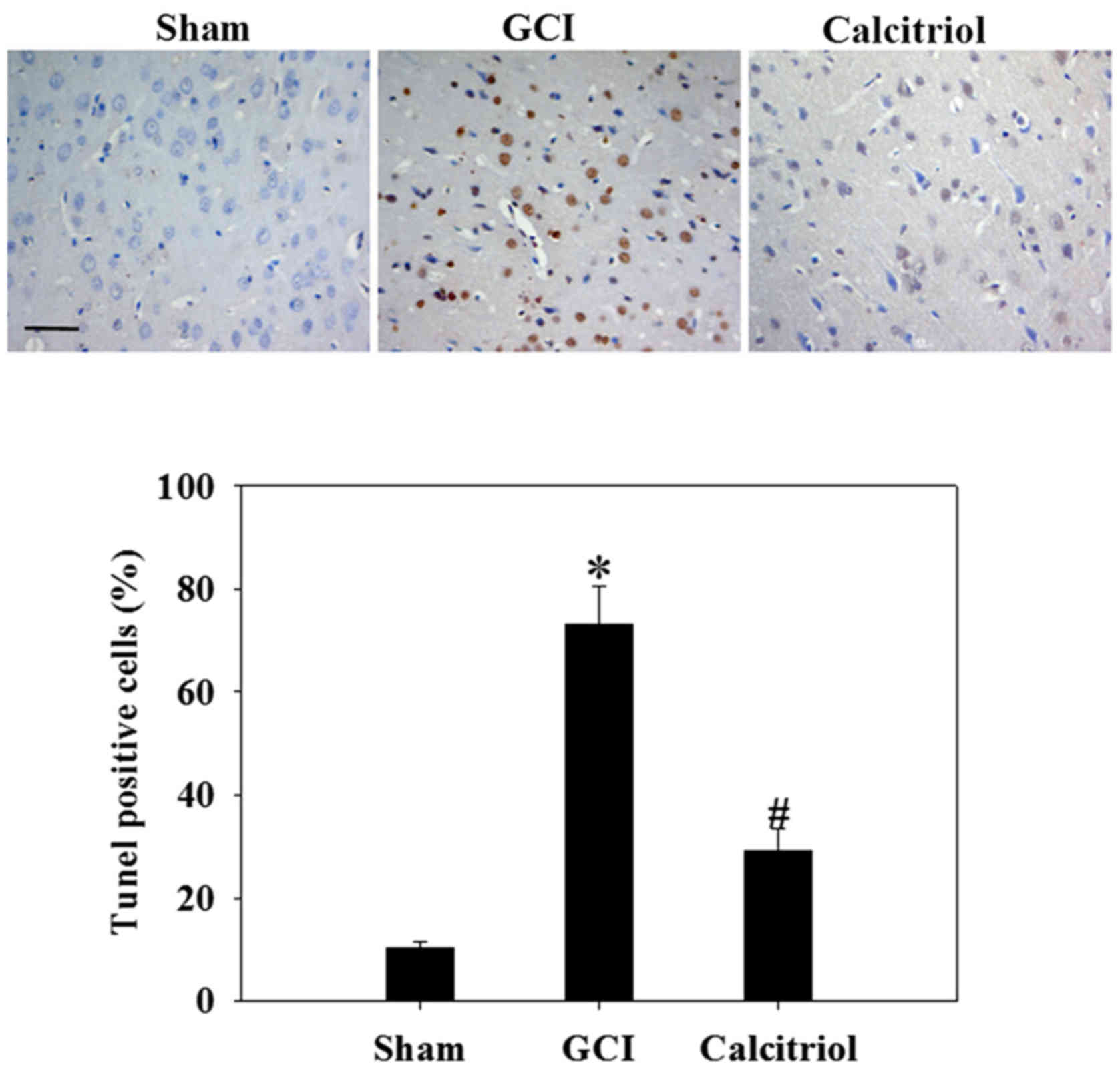
Calcitriol attenuates apoptotic death of
cortical neurons
To assess the effect of calcitriol on neuronal
apoptosis after GCI, the cortical region was subjected to a TUNEL
assay at 3 days following GCI insult (Fig. 6). The results revealed few
apoptotic cells in the Sham group. In the GCI model group, the
nerve cells were irregularly arranged and the cell bodies were
shrunken. The number of TUNEL-positive cells was significantly
increased after GCI compared with that in the Sham group
(P<0.001). However, calcitriol treatment significantly reduced
the number of TUNEL-positive cells compared with that in the GCI
group (P<0.001).
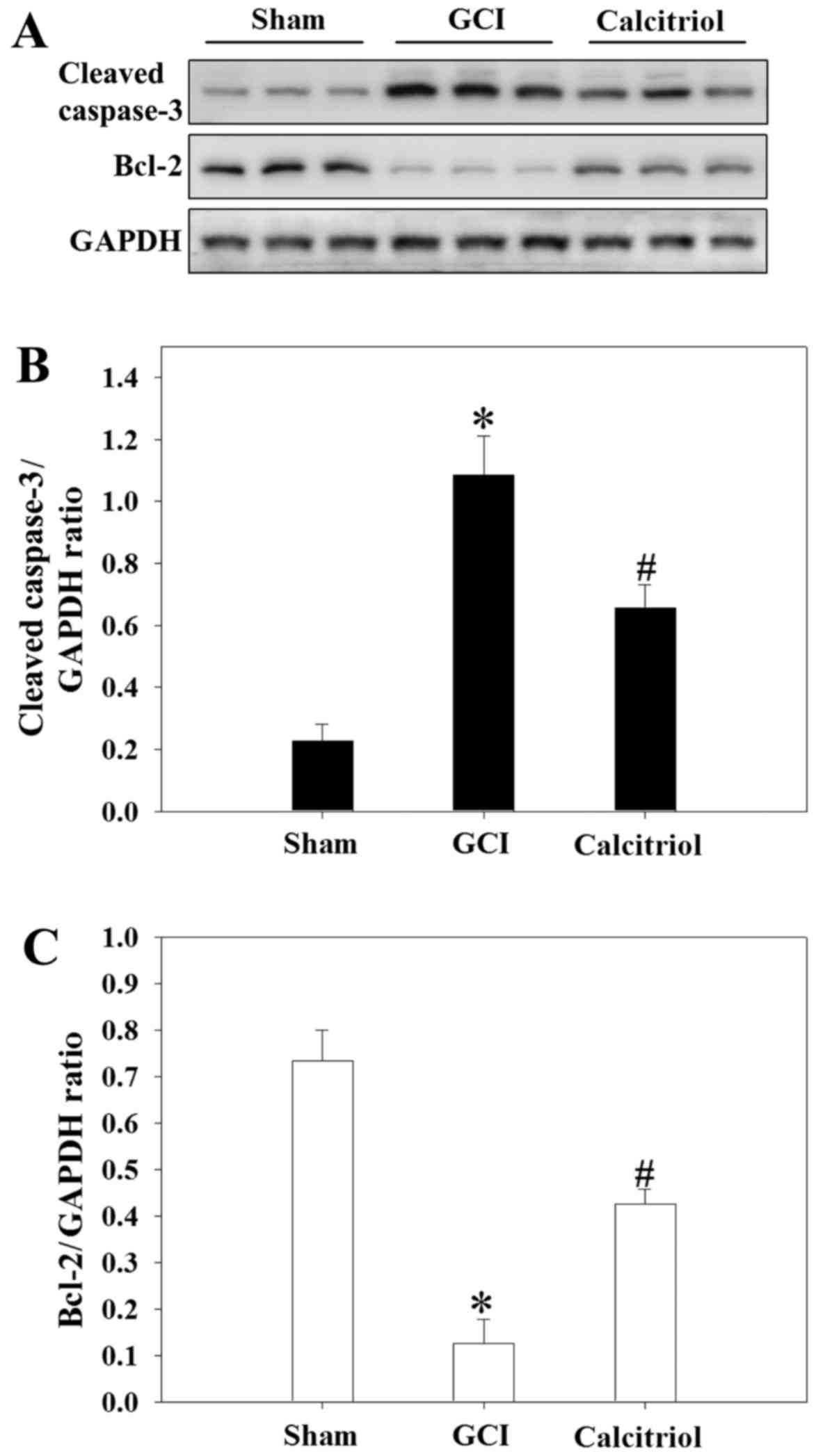
Calcitriol treatment inhibits GCI-induced
changes of caspase-3 and Bcl-2 protein expression
Western blot analysis was performed to detect the
expression of cleaved caspase-3 and Bcl-2 (markers of apoptosis) in
the cortex region of rats at 3 days. As presented in Fig. 7, GCI caused an increase in cleaved
caspase-3 expression, while attenuating Bcl-2 expressing compared
with that in the Sham-operated rats (P<0.001). However, compared
with those in the GCI group, the levels of cleaved caspase-3 were
significantly decreased in the calcitriol treatment group
(P=0.006), while Bcl-2 expression was markedly elevated
(P=0.002).
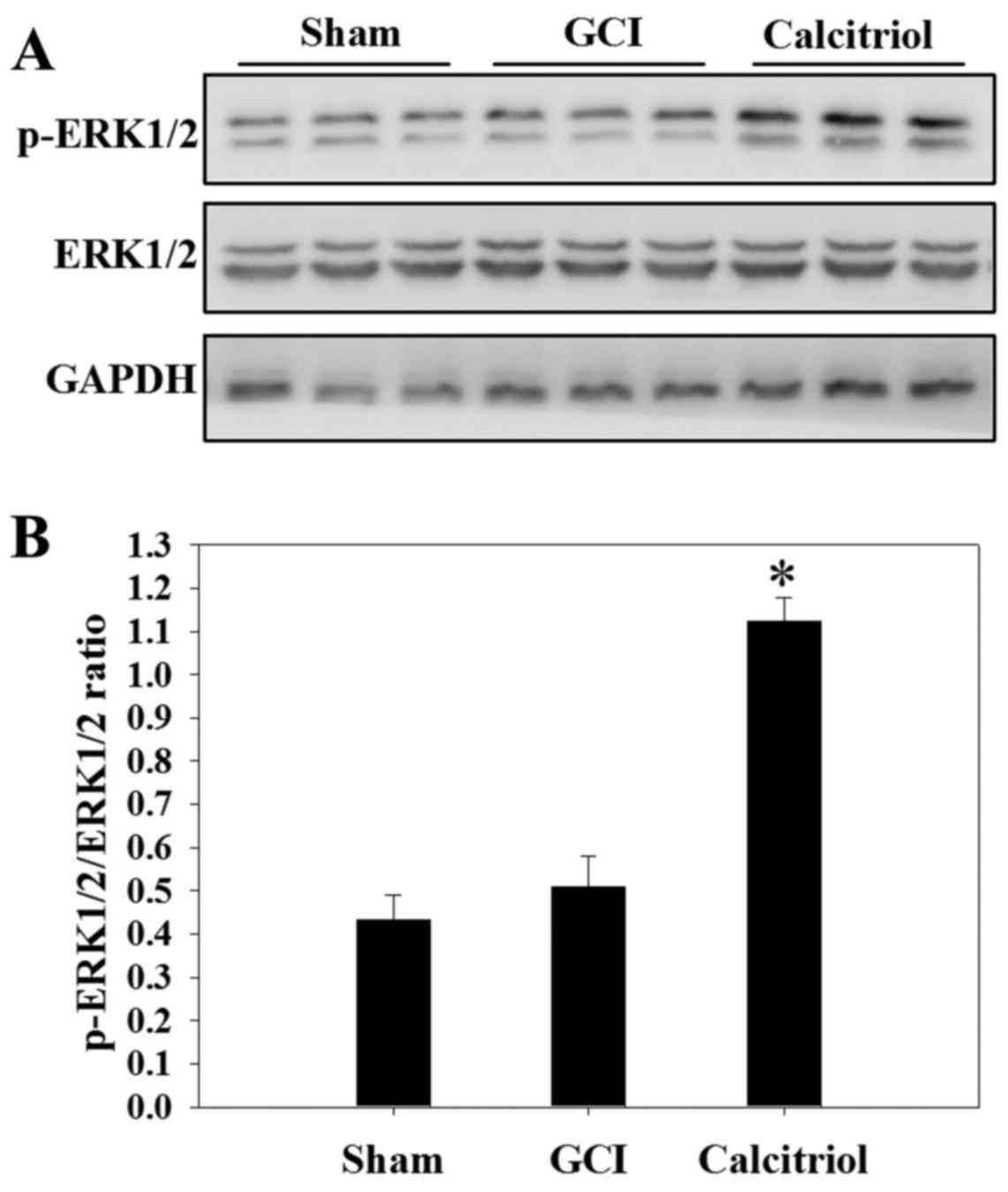
Calcitriol activates the ERK1/2 pathway
after GCI
Western blot analysis was performed to assess the
expression of ERK1/2 and the levels of p-ERK1/2 in the Sham, GCI
and calcitriol groups at 3 days. As depicted in Fig. 8, there was no significant
difference in the p-ERK1/2 to ERK1/2 ratio between the GCI group
and the Sham group. However, administration of calcitriol produced
a significant elevation of the phosphorylation of ERK1/2 (P=0.003
vs. GCI group).
The ERK1/2 pathway is involved in the
calcitriol-mediated inhibition of GCI-induced neuronal
apoptosis
To determine whether the ERK1/2 pathway was involved
in the effects of calcitriol on GCI-induced neuronal apoptosis,
immunofluorescence staining was performed at 3 days after GCI. As
depicted in Fig. 9, there was no
fluorescence observed for caspase-3 and p-ERK1/2 in the Sham group,
while staining for NeuN, a neuronal marker, was clearly visible.
GCI caused a significant decrease of cells staining for NeuN, while
the amount of p-ERK1/2 and caspase-3 expression was increased and
co-localized in numerous cells. However, administration of
calcitriol produced a marked elevation of p-ERK1/2 and attenuation
of caspase-3. The number of cells with co-localization of p-ERK1/2
and caspase-3 was significantly decreased. These co-staining
results indicated that the ERK1/2 pathway may participate in the
inhibition of GCI-induced neuronal apoptosis after calcitriol
treatment.
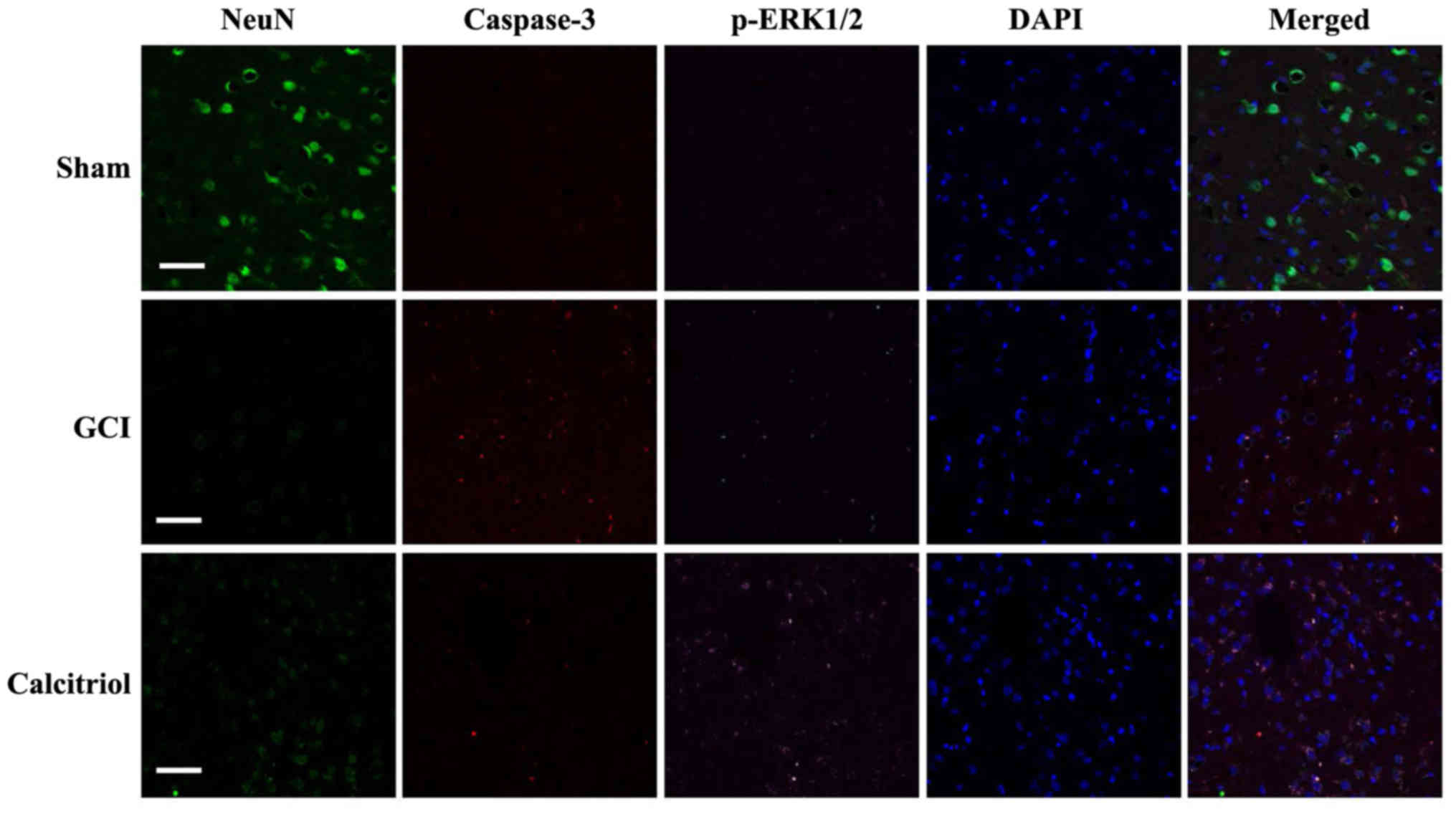 | Figure 9Fluorescence staining for NeuN,
caspase-3 and p-ERK1/2 in rat cortex, and the merged images.
Immunofluorescence staining was performed at 3 days in Sham, GCI
and calcitriol groups (n=5 for each group; scale bar, 50 μm;
green, NeuN; red, caspase-3; purple, p-ERK1/2; blue, DAPI). GCI,
global cerebral ischemia; p-ERK, phosphorylated extracellular
signal-regulated kinase. |
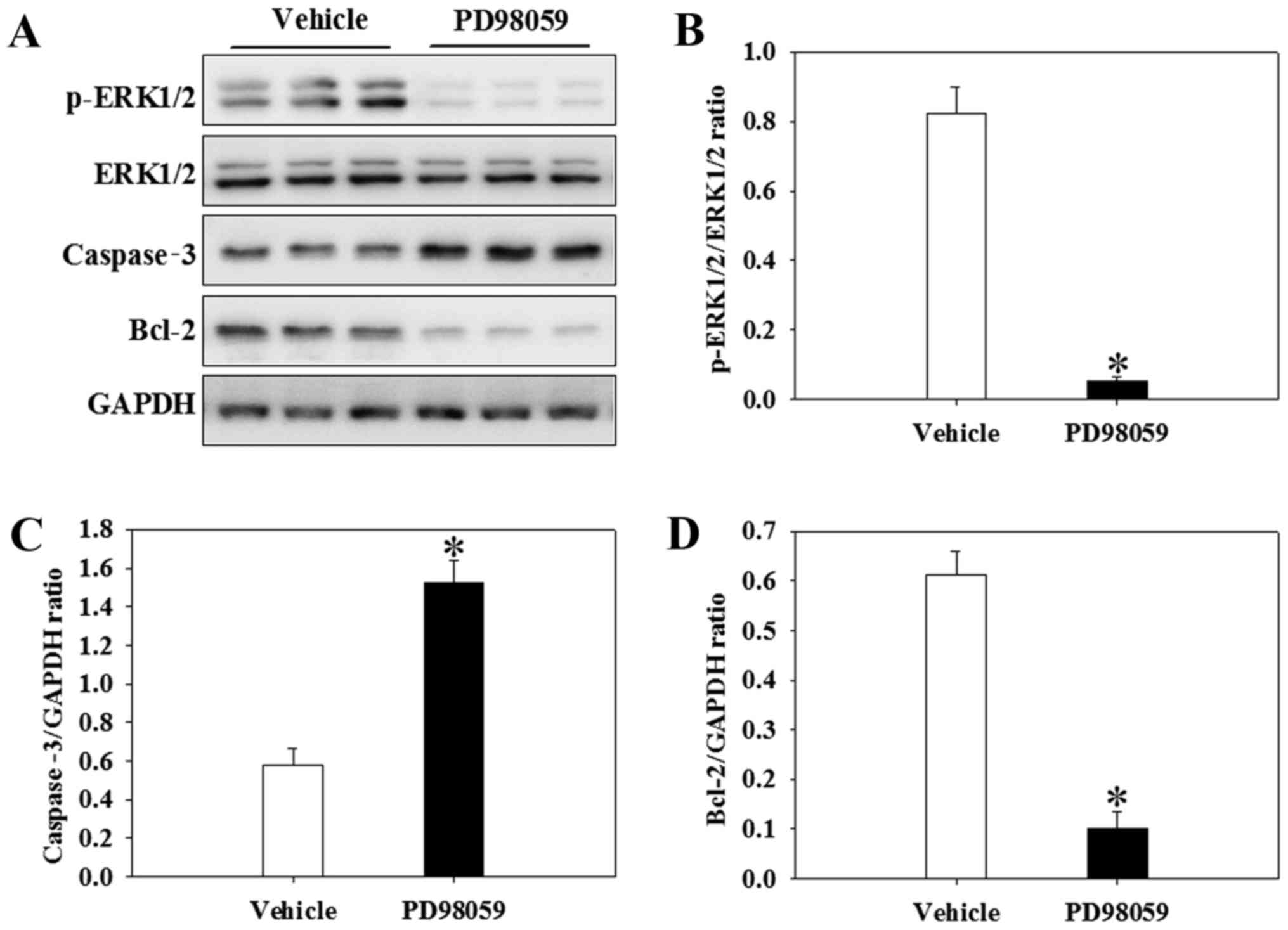
PD98059 inhibits Bcl-2 and p-ERK1/2 and
increases caspase-3 after GCI in the presence of calcitriol
To further investigate whether the attenuation of
apoptosis following calcitriol treatment was regulated by the
ERK1/2 signaling pathway, a p-ERK1/2 inhibitor, PD98059, was used
in the present study. The alterations in apoptotic cytokine levels
and cell signaling following intervention with PD98059 prior to the
GCI insult are depicted in Fig.
7. Of note, PD98059 inhibited the expression of p-ERK1/2 in the
presence of calcitriol after GCI (P<0.001) (Fig. 10A and B). Furthermore, the levels
of caspase-3 were significantly increased, whereas Bcl-2 was
significantly decreased in the PD98059 group compared with those in
the calcitriol and GCI-treated vehicle group (P=0.014 and
P<0.001, respectively) (Fig. 10C
and D).
Discussion
The purpose of the present study was to investigate
the neuroprotective effects of calcitriol on GCI. H&E staining
is a macroscopic and readily available method to assess
histopathological changes. Calcitriol treatment was revealed to
markedly attenuate injury. In the calcitriol group, the structure
of the brain tissue was better and the number of neurons was
greater than that in the GCI group. The ultrastructures of neurons
observed via TEM also indicated that calcitriol treatment
alleviated the damage in the cortex. In addition, GCI-induced
neurological deficits and brain edema were also suppressed by
calcitriol treatment. At the molecular level, VDR expression in the
cortex was significantly elevated following calcitriol treatment.
Calcitriol also inhibited GCI-induced activation of caspase-3 as
well as attenuation of Bcl-2. Furthermore, the effect of calcitriol
was associated with activation of the ERK pathway. The results were
consistent with those of previous studies reporting that calcitriol
exerted neuroprotective effects in various in vitro and
in vivo models (18–21). Therefore, calcitriol has the
potential to become a novel therapeutic for GCI.
The primary injury occurs simultaneously with GCI,
leading to a disruption of oxygen supply to the brain that
contributes to immediate (necrotic) cell death (2). Thereafter, oxygen free
radical-mediated lipid peroxidation and brain edema formation
appear to be fundamental mechanisms of secondary damage associated
with GCI (2). Sequential
activation of caspases, a family of proteases, has a pivotal role
in cellular apoptosis in the central nervous system (22). Apoptotic stimuli, including
ischemic injury, trigger the activation of initiator caspases and
subsequently the caspase cascade, finally leading to apoptotic cell
death (23). Of the various
subtypes of caspases, caspases-3 is the principal caspase actively
causing neuronal cell death (23). Numerous studies have demonstrated
that caspase-3 activity increases after cerebral
ischemia/reperfusion injury (24,25). In the present study, caspase-3 was
induced by GCI. However, treatment with calcitriol significantly
inhibited GCI-induced expression of cleaved caspase-3. The results
of the TUNEL assay revealed few apoptotic cells in the Sham group,
while a significant increase of apoptotic cells was noted in the
GCI model group. However, the apoptotic rate was significantly
lower in the calcitriol treatment group compared with that in the
GCI group. Taken together, reduction of apoptosis is an important
mechanism by which calcitriol exerts its neuroprotective
effects.
The intrinsic pathway of apoptosis is characterized
by mitochondrial outer membrane permeabilization, death-inducing
signaling complex formation, DNA fragmentation and caspase-3
activation (26). These events
have been demonstrated to be associated with ERK1/2 pathway
activation (27,28). In the present study, p-ERK1/2 and
caspase-3 were co-localized, which indicated that the ERK1/2
pathway may participate in the calcitriol-mediated inhibition of
neuronal apoptosis induced by GCI. Furthermore, PD98059 inhibited
the expression of p-ERK1/2 while significantly increasing the
expression levels of caspase-3, whereas Bcl-2 was significantly
decreased. Therefore, it is conceivable that the activation of the
ERK1/2 pathway induced by VDR activation may have an essential role
in the evasion of apoptosis after GCI.
In conclusion, the results of the present study
demonstrated that post-GCI administration of calcitriol attenuate
brain edema and improved neurological function in rats. Calcitriol
also caused a marked activation of the ERK1/2 pathway with
subsequent attenuation of neuronal apoptosis. The present study
provided novel clues for understanding the mechanisms by which
calcitriol exerts its neuroprotective effects in a rat model of
GCI.
References
|
1
|
Zhao P, Zhou R, Zhu XY, Hao YJ, Li N, Wang
J, Niu Y, Sun T, Li YX and Yu JQ: Matrine attenuates focal cerebral
ischemic injury by improving antioxidant activity and inhibiting
apoptosis in mice. Int J Mol Med. 36:633–644. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Han J, Sun L, Xu Y, Liang H and Cheng Y:
Activation of PPARγ by 12/15-lipoxygenase during cerebral
ischemia-reperfusion injury. Int J Mol Med. 35:195–201. 2015.
View Article : Google Scholar
|
|
3
|
Bao C, Wang Y, Min H, Zhang M, Du X, Han R
and Liu X: Combination of ginsenoside Rg1 and bone marrow
mesenchymal stem cell transplantation in the treatment of cerebral
ischemia reperfusion injury in rats. Cell Physiol Biochem.
37:901–910. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hellman P, Liu W, Westin G, Törmä H and
Akerström G: Vitamin D and retinoids in parathyroid glands
(Review). Int J Mol Med. 3:355–361. 1999.PubMed/NCBI
|
|
5
|
Hwang JH, Wang T, Lee KS, Joo JK and Lee
HG: Vitamin D binding protein plays an important role in the
progression of endometriosis. Int J Mol Med. 32:1394–1400. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bouillon R, Carmeliet G, Verlinden L, van
Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C and Demay M:
Vitamin D and human health: Lessons from vitamin D receptor null
mice. Endocr Rev. 29:726–776. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eyles DW, Burne THJ and McGrath JJ:
Vitamin D, effects on brain development, adult brain function and
the links between low levels of vitamin D and neuropsychiatric
disease. Front Neuroendocrinol. 34:47–64. 2013. View Article : Google Scholar
|
|
8
|
Pittas AG, Harris SS, Stark PC and
Dawson-Hughes B: The effects of calcium and vitamin D
supplementation on blood glucose and markers of inflammation in
nondiabetic adults. Diabetes Care. 30:980–986. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jorde R, Mathiesen EB, Rogne S, Wilsgaard
T, Kjærgaard M, Grimnes G and Schirmer H: Vitamin D and cognitive
function: The Tromsø Study. J Neurol Sci. 355:155–161. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Segaert S, Degreef H and Bouillon R:
Vitamin D receptor expression is linked to cell cycle control in
normal human keratinocytes. Biochem Biophys Res Commun. 279:89–94.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McGuire TF, Trump DL and Johnson CS:
Vitamin D(3)-induced apoptosis of murine squamous cell carcinoma
cells. Selective induction of caspase-dependent MEK cleavage and
up-regulation of MEKK-1. J Biol Chem. 276:26365–26373. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang H, Hua F, Wang J, Sayeed I, Wang X,
Chen Z, Yousuf S, Atif F and Stein DG: Progesterone and vitamin D:
Improvement after traumatic brain injury in middle-aged rats. Horm
Behav. 64:527–538. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang P, Xue Y, Li HD, Liu YP, Cai HL,
Tang MM and Zhang LH: Dysregulation of vitamin D metabolism in the
brain and myocardium of rats following prolonged exposure to
dexamethasone. Psychopharmacology (Berl). 231:3445–3451. 2014.
View Article : Google Scholar
|
|
14
|
Balden R, Selvamani A and Sohrabji F:
Vitamin D deficiency exacerbates experimental stroke injury and
dysregulates ischemia-induced inflammation in adult rats.
Endocrinology. 153:2420–2435. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nissou MF, Guttin A, Zenga C, Berger F,
Issartel JP and Wion D: Additional clues for a protective role of
vitamin D in neurodegenerative diseases: 1,25-dihydroxyvitamin D3
triggers an anti-inflammatory response in brain pericytes. J
Alzheimers Dis. 42:789–799. 2014.PubMed/NCBI
|
|
16
|
Lee H, Park YH, Jeon YT, Hwang JW, Lim YJ,
Kim E, Park SY and Park HP: Sevoflurane post-conditioning increases
nuclear factor erythroid 2-related factor and haemoxygenase-1
expression via protein kinase C pathway in a rat model of transient
global cerebral ischaemia. Br J Anaesth. 114:307–318. 2015.
View Article : Google Scholar
|
|
17
|
Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M
and Chopp M: Therapeutic benefit of intravenous administration of
bone marrow stromal cells after cerebral ischemia in rats. Stroke.
32:1005–1011. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kajta M, Makarewicz D, Ziemińska E, Jantas
D, Domin H, Lasoń W, Kutner A and Łazarewicz JW: Neuroprotection by
co-treatment and post-treating with calcitriol following the
ischemic and excitotoxic insult in vivo and in vitro. Neurochem
Int. 55:265–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Orme RP, Bhangal MS and Fricker RA:
Calcitriol imparts neuroprotection in vitro to midbrain
dopaminergic neurons by upregulating GDNF expression. PLoS One.
8:e620402013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fu J, Xue R, Gu J, Xiao Y, Zhong H, Pan X
and Ran R: Neuroprotective effect of calcitriol on
ischemic/reperfusion injury through the NR3A/CREB pathways in the
rat hippocampus. Mol Med Rep. 8:1708–1714. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alkharfy KM, Al-Daghri NM, Yakout SM and
Ahmed M: Calcitriol attenuates weight-related systemic inflammation
and ultrastructural changes in the liver in a rodent model. Basic
Clin Pharmacol Toxicol. 112:42–49. 2013. View Article : Google Scholar
|
|
22
|
Talanian RV, Quinlan C, Trautz S, Hackett
MC, Mankovich JA, Banach D, Ghayur T, Brady KD and Wong WW:
Substrate specificities of caspase family proteases. J Biol Chem.
272:9677–9682. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tamatani M, Ogawa S, Niitsu Y and Tohyama
M: Involvement of Bcl-2 family and caspase-3-like protease in
NO-mediated neuronal apoptosis. J Neurochem. 71:1588–1596. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chiarini A, Liu D, Armato U and Dal Prà I:
Bcl10 crucially nucleates the pro-apoptotic complexes comprising
PDK1, PKCζ and caspase-3 at the nuclear envelope of
etoposide-treated human cervical carcinoma C4-I cells. Int J Mol
Med. 36:845–856. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu YJ, Sun WY, Zhang S, Li XR and Wei W:
Targeted blockade of JAK/STAT3 signaling inhibits proliferation,
migration and collagen production as well as inducing the apoptosis
of hepatic stellate cells. Int J Mol Med. 38:903–911. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu LF, Wei BL, Guo YT, Ye YQ, Li GP, Pu ZJ
and Feng JL: Apoptosis induced by adenosine involves endoplasmic
reticulum stress in EC109 cells. Int J Mol Med. 30:797–804. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Y, Lu X, Qi H, Li X, Xiao X and Gao J:
Ursolic acid induces apoptosis through mitochondrial intrinsic
pathway and suppression of ERK1/2 MAPK in HeLa cells. J Pharmacol
Sci. 125:202–210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang H, Xiong J, Luo W, Yang J and Xi T:
8-Methoxypsoralen induces intrinsic apoptosis in HepG2 cells:
Involvement of reactive oxygen species generation and ERK1/2
pathway inhibition. Cell Physiol Biochem. 37:361–374. 2015.
View Article : Google Scholar : PubMed/NCBI
|