Introduction
Pulmonary arterial hypertension (PAH) aggressively
threatens human health with high morbidity and mortality due to its
diverse etiologies. Unfortunately, current strategies are
ineffective to prevent and cure PAH. In recent years, multipotent
stem cells, such as adipose-derived stem cells (ADSCs), bone marrow
mesenchymal stem cells, and endothelial progenitor cells (EPCs),
have been employed as a promising strategy for PAH therapy
(1–3). These studies have revealed that stem
cells play crucial roles in the prevention and treatment of PAH by
repairing endothelial cells and angiogenesis and inhibiting
pulmonary arterial remodeling (1–3).
Moreover, our previous study also demonstrated that ADSCs
attenuated PAH and ameliorated pulmonary arterial remodeling in
rats with PAH (4).
Generally, due to the complex pathogenesis of PAH,
cell transplantation alone cannot solve all the issues. Cell-based
gene therapy has been found to be effective in preclinical models
of PAH (5,6). Several studies have indicated that
prepro calcitonin gene-related peptide (CGRP)-transfected EPCs,
vascular endothelial growth factor-transfected EPCs, and hypoxia
inducible factor 1α-transfected EPCs could effectively attenuate
PAH and reverse pulmonary vascular remodeling (7–9). A
PHACeT clinical trial reported that short-term hemodynamic
improvement and the associated long-term benefits in functional and
quality of life were evidenced in patients with PAH by delivering
endothelial NO-synthase (eNOS) gene-enhanced EPCs (10). Therefore, combination treatment is
more effective than single stem cell transplantation. Stem cell
transplantation combined with gene therapy may be an extremely
promising treatment for PAH.
Adiponectin (APN), a 30-kDa multimeric protein
produced by fat cells, has multiple biological functions including
anti-oxidation, anti-atherosclerosis, anti-inflammation and
anti-proliferation activity (11–13). It can protect blood vessels
through various mechanisms. APN-deficient mice (APN−/−)
suffer from PAH accompanied by infiltration of proinflammatory
cells and overexpression of endothelial E-selectin (14–16). Nakagawa et al reported that
upregulation of APN significantly suppressed pulmonary arterial
wall thickening and right ventricular (RV) hypertrophy induced by
chronic hypoxia in mice (17).
Nevertheless, APN treatment for PAH and the underlying mechanisms
have not been well probed.
Abnormal proliferation of pulmonary artery smooth
muscle cells (PASMCs) is a common pathologic feature of
pathological pulmonary vasculature (18). Bone morphogenetic protein (BMP)
receptor type 2 (BMPR2) is a member of the transforming growth
factor-β (TGF-β) receptor superfamily. Its mutations were confirmed
in the majority of patients with heritable PAH (19,20). In PAH models induced by
monocrotaline (MCT) and hypoxia, BMPR2 expression was downregulated
and the BMP signaling pathway was disordered (21,22). Enhancement of the BMPR2 pathway
could inhibit the proliferation and promote the apoptosis of PASMCs
(23), which may be ultilized for
the treatment of pathological pulmonary vasculature.
Therefore, this study aimed to probe the therapeutic
efficacy of APN gene-modified ADSCs for PAH in rats and elucidate
the underlying cellular and molecular mechanisms.
Materials and methods
Materials and animals
Adenosine monophosphate activated protein kinase
(AMPK) inhibitor compound C (CC) was purchased from Merck
(Darmstadt, Germany). APN, rabbit anti-α-smooth muscle actin
(α-SMA) antibody (ab5694), rabbit anti-AMPK antibody (ab32047), and
mouse anti-BMP2 antibody (ab6285) were obtained from Abcam
(Cambridge, UK). BCA protein assay kit and rabbit anti-Smad1/5/8/9
antibody (PA1-41238) were from Pierce (Rockford, IL, USA). BMP2
inhibitor noggin was purchased from R&D Systems Inc.
(Minneapolis, MN, USA). Cell Counting Kit-8 (CCK-8) was from
Dojindo (Kumamoto, Japan). CD29 (102205), CD45 (202205) and CD90
(202517) antibodies were from BioLegend (San Diego, CA, USA). CD31
antibody (ab119339), rat APN enzyme-linked immunosorbent assay
(ELISA) kit and adiponectin antibody were from Abcam. Collagenase
type I and MCT were purchased from Sigma-Aldrich (St. Louis, MO,
USA). 3,3′-Diaminobenzidine tetrahydrochloride hydrate (DAB) was
from Zhongshan Goldenbridge (Beijing, China).
4′,6-Diamidino-2-phenylindole (DAPI) was obtained from Beyotime
Institute of Biotechnology (Jiangsu, China). Dulbecco's modified
Eagle's medium (DMEM), DMEM-F12 medium, and fetal bovine serum
(FBS) were obtained from HyClone (Logan, UT, USA). Enhanced
chemiluminescent (ECL) reagent was from Amersham (Piscataway, NJ,
USA). Goat anti-rabbit IgG was obtained from ZSGB-Bio (Beijing,
China). Optimal cutting temperature compound (OCT) was obtained
from Miles Scientific (Naperville, IL, USA). Rabbit anti-p-AMPK
(2535S) and rabbit anti-p-Smad1/5/8/9 (9511S) antibodies were
purchased from Cell Signaling Technology (Danvers, CO, USA). Rabbit
anti-β-actin (sc-10731) and CD34 (sc-65261) antibodies were
obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). SD
rat ADSC functional identification kit was bought from Cyagen
Biosciences, Inc. (Guangdong, China).
Male Sprague-Dawley (SD) rats (8-weeks of age) were
obtained from Slaccas Co. Ltd. (Shanghai, China) (certificate no.
20120003). All animals were raised with food and water ad
libitum. The study protocol was reviewed and approved by the
Institutional Animal Care and Use Committee, The First Affiliated
Hospital of Fujian Medical University, China and was in accordance
with the guidelines established by the Chinese Council of Animal
Care.
Isolation and characterization of
ADSCs
ADSCs were isolated from the rats as described in
our previous study (4). Briefly,
bilateral inguinal subcutaneous adipose tissue was excised, minced,
and digested with 0.1% collagenase type I for 60 min at 37°C. The
mixture containing the ADSC fraction was filtered through a mesh
and centrifuged for 10 min at 1,000 × g. The resultant pellet was
resuspended and cultured in DMEM-F12 media containing 15% FBS at
37°C in 5% CO2. After 3 to 4 days, the culture medium
was completely changed. All cells were cultured for another 14 days
with a change of the culture media every 3 days. The multipotency
of ADSCs was assessed via adipogenic and osteogenic differentiation
assays by using the SD rat ADSC functional identification kit
according to the manufacturer's instructions for use. ADSCs were
incubated with CD31, CD29, CD34, CD45 and CD90 antibodies and then
assayed by using flow cytometry (BD FACSCalibur; BD Biosciences,
Franklin Lakes, NJ, USA).
Infection of ADSCs with the APN
lentiviral vector
After being cultured for 14 days, ADSCs were
infected with green fluorescent protein (GFP)-empty (ADSCs-V) or
APN-expressing lentiviral vector (ADSCs-APN) at a multiplicity of
infection (MOI) of 10. The GFP-empty lentivirus served as a
negative control. Rat APN-GFP cDNA was cloned in the GV287 vector
from GenePharma Co, Ltd. (Shanghai, China). Primers for rat APN
were forward, ACTCAGCATTCAGCGTAGGG and reverse,
CGTCGCCGTCCAGCTCGACCAG. After incubation for 12 h, the medium was
replaced by fresh DMEM and the cells were cultured for another 72
h. Supernatants were collected for the quantification of APN
protein using the rat APN ELISA kit according to the manufacturer's
instruction for use. The absorbance was determined at 450 nm with a
sensitivity of 1.5 ng/ml.
Animal treatments
Forty SD rats were randomly divided into five groups
(n=8): normal control (Ctr), PAH, ADSCs, ADSCs infected with the
GFP empty lentiviral vector (ADSCs-V), and APN-GFP gene-modified
ADSCs (ADSCs-APN). Rats in the PAH, ADSCs, ADSCs-V and ADSCs-APN
groups were intraperitoneally administered 40 mg/kg MCT, while
those in the Ctr group were injected with an equal volume of
saline. After 2 weeks, 1.0×106 ADSCs, ADSCs infected
with the GFP empty lentiviral vector, and APN-GFP gene-modified
ADSCs were injected into the left jugular vein of the rats,
respectively. Three weeks later, mean pulmonary arterial pressure
(mPAP) was measured as described below. Then the rats were
euthanized, and the lung and heart tissues were collected for
following analyses.
ADSCs-APN in the lung
To determine the location of the ADSCs in the lung,
GFP-labeled ADSCs were evaluated by confocal laser scanning
microscopy. The right upper lobe of the lung was dissected and
washed with phosphate-buffered saline (PBS). The tissue was
perfused with a mixture of formalin and OCT, embedded in OCT, and
quickly frozen in liquid nitrogen. Then the frozen tissue was
sectioned. Cryostat sections were fixed with formalin for 10 min
and incubated with DAPI for 10 min. The sections were examined
using FlowView software (V2.0c) on a confocal laser scanning
microscope (FV1000) (both from Olympus, Tokyo, Japan) for
determining the distribution of the GFP-labeled ADSCs in lung
tissue. Images were captured in the XYZ scan mode from top to
bottom of the section. GFP was excited at 488 nm and DAPI was
excited at 405 nm.
The expression of APN in the lung was detected by
western blot analysis. Lung tissue was homogenized and lysed in a
protein extraction buffer containing the protease inhibitor
phenylmethylsulfonyl fluoride. The protein concentration was
determined using a BCA protein assay kit. Proteins were separated
using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), and then transferred to a polyvinylidene fluoride
(PVDF) membrane. The membrane was incubated with the adiponectin
antibody (1:1,000) and the subsequent secondary antibody. The
membrane was analyzed on a gel imaging system (ChemiDoc™ XRS;
Bio-Rad, Hercules, CA, USA). The optical densities of protein
strips were evaluated with a Gel-Pro analyzer software (Media
Cybernetics, Rockville, MD, USA).
Measurement of mPAP and RV hypertrophy
index (RVHI)
After treatments, rats were anesthetized with 10%
chloral hydrate (400 mg/kg). mPAP was recorded as described
previously (4,24). In brief, polyethylene
micro-catheters (Chinese Peking Union Medical Physiology) were
inserted into the pulmonary artery via the right external jugular
vein and connected to a transducer. The mPAP data were collected
and analyzed by Powerlab-ML221 (AD Instruments, New South Wales,
Australia). RVHI was assessed by weighing the RV separately from
the left ventricle (LV) with the septal wall (SW).
Pulmonary vascular remodeling
analysis
Paraffin-embedded sections were prepared and stained
with hematoxylin and eosin (H&E). Pulmonary arteriolae between
50 and 200 μm in external diameter (ED) were chosen for
morphological analysis. Wall thickness (WT) and ED of the pulmonary
arteries were measured using IPP 6.0 image analysis software (Media
Cybernetics). The remodeling of pulmonary arterioles was calculated
as WT% = 2 × WT/ED ×100% (4,24).
For immunohistochemical analysis, sections were incubated with
α-SMA antibody at 4°C overnight followed by the secondary antibody.
Subsequently, they were incubated with DAB as a chromogen to
visualize the antigen-antibody reaction and with hematoxylin for
the counterstain of the nuclei.
RV function
The rats were sedated and laid in the supine
position. Transthoracic echocardiography was performed using a GE
Vivid-E9 ultrasound device (General Electric Co., Fairfield, CT,
USA) with a 12.0 MHz linear array transducer. During the ultrasonic
examination, the lead electrocardiogram was recorded and the heart
rate (HR) was monitored. Pulmonary artery acceleration time (PAAT)
was measured by a pulsed-wave Doppler in the parasternal short-axis
view and defined as the time interval from onset to the maximal
velocity of pulmonary artery forward flow. Since PAAT is
HR-dependent, we corrected the PAAT using the revised formula
PAAT/HR. Right ventricular WT (RVWT), right ventricular end
systolic diameter (RVESD), and right ventricular end diastolic
diameter (RVEED) were obtained from the apical 4-chamber view.
Tricuspid annular plane systolic excursion (TAPSE) was measured by
M-mode tracings. The echocardiographic parameters were analyzed
with Echo software by a sonographer who was blinded to the
hemodynamic and morphological data. Three representative cycles
were conducted.
Prepartion of PASMCs
Pulmonary artery tissues were isolated from the rats
in the PAH group. The pulmonary arteries were separated from the
connective tissues, minced to a size of ~1×1 mm, placed into a
dish, and incubated with DMEM supplemented with 15% FBS. PASMCs
were identified as having a smooth muscle phenotype by positive
immunofluorescence with the anti-α-SMA rabbit antibody. After 3–5
cell passages, the cells were used for the following
experiments.
Cell proliferation assay
The anti-proliferative effect of APN on PASMCs was
evaluated by the CCK-8 assay. PASMCs (2×104 cells/well)
were seeded in 96-well plates containing 100 μl DMEM
supplemented with 15% FBS per each well and cultured for 24 h.
Cells were washed twice with PBS. Subsequently, cells were starved
for 24 h in serum-free medium and treated with 200 ng/ml noggin
and/or 10 μg/ml APN. Cells without any treatments served as
the control. Following a 24-h incubation, 10 μl of CCK-8 was
added to each well for 2 h. The plates were submitted to a
microplate reader (Bio-Rad 550; Leading Biological Technology Co.
Ltd., Shanghai, China) for absorbance determination at 450 nm.
APN intervention
To investigate the effect of APN on the expression
of BMP2 in PASMCs, the cells (1×105/ml) were seeded in
6-well plates. After confluence reached ~70–80%, the cells were
exposed to different concentrations (0, 1.0, 5.0, 10.0 and 20.0
μg/ml) of APN for 1 h. In addition, cells were exposed to
APN at 10 μg/ml for 0, 5, 15 and 30 min, 1, 6, 24 and 36 h
(15,25). The expression of BMP2 was detected
by western blot analysis. To probe the effect of APN on the
expression of Smad, phosphorylated (p)-Smad (p-Smad), AMPK, and
p-AMPK in PASMCs, before exposure to APN (10 μg/ml), the
cells were starved in serum-free medium for 24 h, washed twice with
PBS, and treated with 200 ng/ml BMP2 inhibitor noggin and AMPK
inhibitor CC for 4 h, respectively.
Western blot analysis
After treatments, PASMCs were lysed with ice-cold
RIPA lysis buffer for 10 min and centrifuged for 15 min at 12,000 ×
g and 4°C. The supernatants were collected. The protein
concentration was determined by using a BCA protein assay kit.
Proteins were separated on 6 or 10% SDS-PAGE and then transferred
to a PVDF membrane. The membrane was blocked with 5% nonfat milk
for 1 h at room temperature and then incubated with rabbit
anti-p-AMPK (1:1,000), rabbit anti-AMPK (1:1500), mouse anti-BMP2
(1:1,000), rabbit anti-p-Smad1/5/8/9 (1:500), rabbit
anti-Smad1/5/8/9 (1:1,000), and rabbit anti-β-actin (1:1,000)
antibodies at 4°C overnight. After washing, the membrane was
incubated with horseradish peroxidase (HRP)-conjugated secondary
antibody (X-Y Biotechnology Co., Ltd., Shanghai, China) in TBS-T
for 1 h and then incubated with ECL reagent at room temperature.
The membrane was analyzed on a gel imaging system (ChemiDoc™ XRS;
Bio-Rad). The optical densities of protein strips were assessed
with a Gel-Pro analyzer software (Media Cybernetics).
Statistical analysis
Data are expressed as mean ± standard error of the
mean (SEM). Comparisons among groups were made using one-way ANOVA
followed by the Newman-Keuls test for post hoc testing on SPSS 20.0
software. A significant difference was defined as P<0.05.
Results
Characterization of ADSCs and
transfection of APN into ADSCs
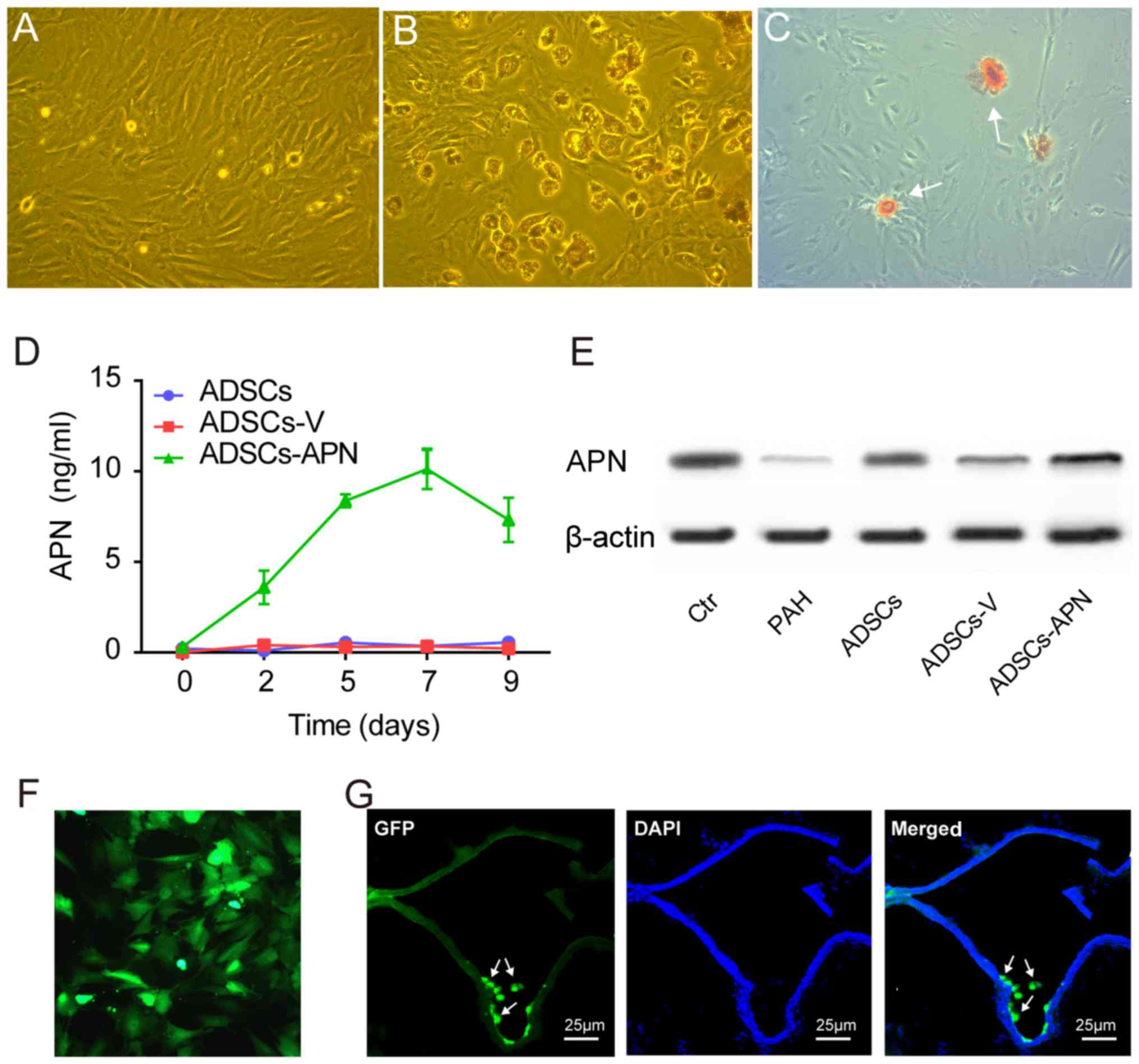
ADSCs isolated from rats exhibited spindle-shape and
fibroblast-like morphological features (Fig. 1A). ADSCs were able to
differentiate into adipogenic and osteogenic cells after
appropriate induction (Fig. 1B and
C). Flow cytometry showed that these ADSCs were positive for
mesenchymal stem cell markers CD90 (97.9%) and CD29 (87.6%), but
negative for hematopoietic/endothelial cell markers CD45 (6.2%),
CD31 (0.9%) and CD34 (37.9%) (data not shown).
After transfection of the GFP empty vector (ADSCs-V)
or APN-GFP vector (ADSCs-APN) into ADSCs, the APN concentration in
the ADSC supernatant was measured by ELISA for determining the
secretion rate of APN. We found that the APN concentration was
invariably not elevated in the ADSCs and ADSCs-V groups (Fig. 1D). However, in the ADSCs-APN
group, the APN concentration began to increase 2 days after
transfection and it reached the peak value on day 7.
ADSCs-APN in the lung
Western blot analysis was used to measure APN
protein expression in the lung of rats after various treatments
(Fig. 1E). It was demonstrated
that expression of APN protein was obviously increased after
ADSCs-APN transplantation (ADSCs-APN group) as compared with levels
in the ADSCs and ADSCs-V groups. The location of GFP in ADSCs was
evaluated by fluorescence microscopy (Fig. 1F). Twenty-four hours after APN
gene transfection into ADSCs, the green fluorescence of GFP was
observed within the cells and GFP was active after several
passages. The frozen section with double fluorescence analysis was
employed to evaluate the location of ADSCs-APN in the lung of rats
(Fig. 1G). Results revealed that
the green fluorescene of APN-GFP and the blue fluorescence of the
cell nuclei were merged, suggesting the engraftment of ADSCs-APN
across lung tissues of rats. The blue nuclei outlined the vascular
wall and the green ADSCs were located around the vessels.
ADSCs-APN attenuate PAH
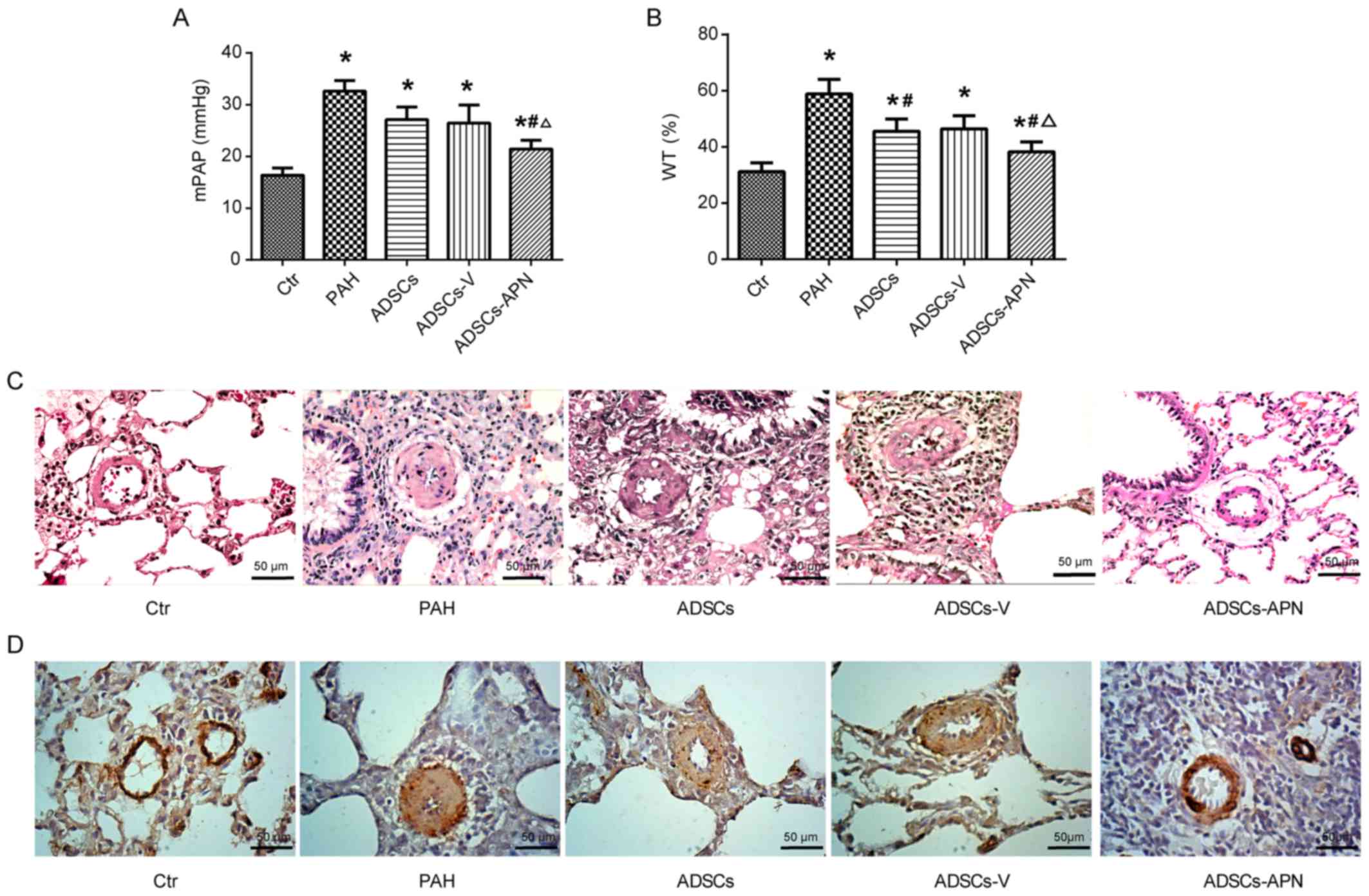
The mPAP results are depicted in Fig. 2A. In the Ctr group, mPAP was
16.38±0.91 mmHg. After MCT administration, PAH was well established
with the mPAP of 32.62±1.77 mmHg. Compared to the PAH group,
moderate decreases in mPAP were noted in the ADSCs and ADSCs-V
groups with 27.11±2.12 and 26.44±2.91 mmHg, respectively. Notably,
although the mPAP in the ADSCs-APN group (21.44±0.89 mmHg) was
still significantly higher than that in the Ctr group, it was
effectively reduced in comparison with the ADSCs and ADSCs-V groups
(P<0.05). These results indicated that ADSCs-APN rather than
ADSCs and ADSCs-V were capable of strongly attenuating PAH.
ADSCs-APN reverse pulmonary vascular
remodeling
To determine the effect of ADSCs-APN on vascular
remodeling, the WT of the pulmonary arteriolae in the rats was
examined (Fig. 2B and C).
Compared to the Ctr group, the WT% was markedly elevated in the PAH
group (P<0.05), suggesting the progression of wall thickening in
the pulmonary arterioles. However, the WT% in both the ADSCs and
ADSCs-V groups was significantly lower than that in the PAH group
(P<0.05). The WT% was further significantly reduced in the
ADSCs-APN group as compared with the ADSCs and ADSCs-V groups
(P<0.05), suggesting that ADSCs-APN transplantation further
prevented arteriolar wall thickening.
α-SMA is a positive marker for smooth muscle cells
(SMCs). The proliferation of pulmonary arterioles was evaluated by
α-SMA expression analysis. Morphometric immunostaining of lung
sections demonstrated that α-SMA expression in the PAH group was
obviously higher than that in the Ctr group (Fig. 2D). After ADSCs and ADSCs-V
treatments, slight suppression of α-SMA expression was found.
Importantly, the α-SMA expression in the ADSCs-APN group was
notably inhibited in comparison with the PAH, ADSCs and ADSCs-V
groups. Thus, the increase in WT of pulmonary arterioles was
positively associated with the enhancement of SMC proliferation.
These results suggested that ADSCs-APN rather than ADSCs and
ADSCs-V was able to powerfully reverse pulmonary vascular
remodeling.
ADSCs-APN improve RV function
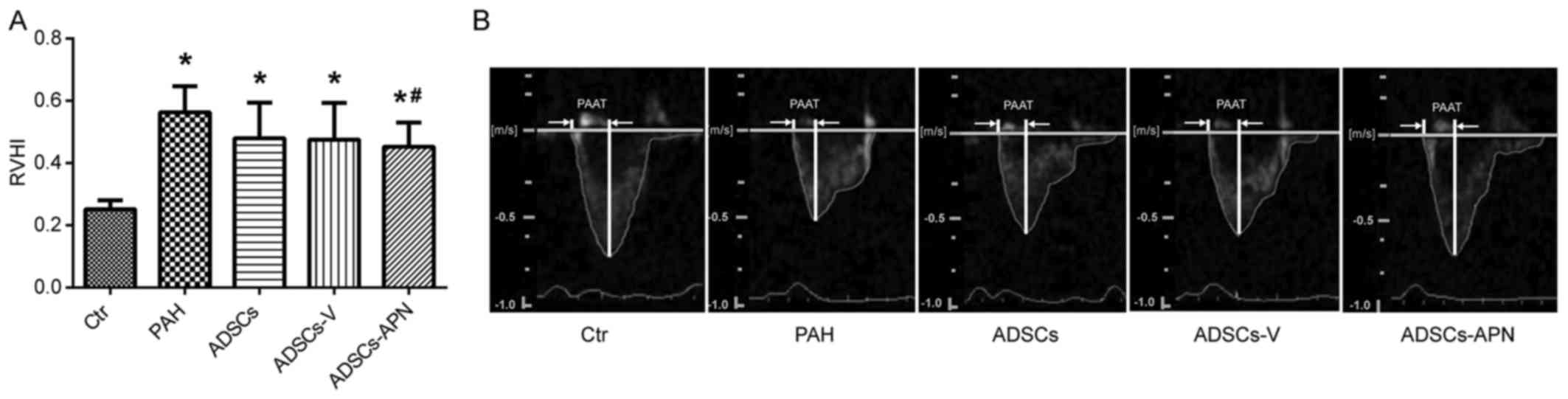
MCT treatment resulted in severe RV hypertrophy with
a sharp increase in RVHI value as compared with the Ctr group
(P<0.05) (Fig. 3A). There was
no significant difference in the RVHI values between the PAH and
ADSCs/ADSCs-V groups (P>0.05). However, compared to the PAH
group, the RVHI value was significantly reduced by the ADSCs-APN
transplantation (P<0.05).
Fig. 3B shows the
pulmonary artery forward flow and the PAAT of rats after diverse
treatments. In the Ctr group, the blood flow through pulmonary
arteries had a typical 'rounded' shape. However, in the PAH group,
the velocity profile of the pulmonary artery was altered to a
'spike and dome' morphology with reduced PAAT. After the ADSC and
ADSCs-V treatments, the echo profiles showed that both PAAT and
ejection time were obviously increased. Furthermore, this profile
in the ADSCs-APN group was further improved and almost similar to
the Ctr group with similar PAAT and morphology. It was suggested
that ADSCs-APN rather than ADSCs and ADSCs-V vigorously attenuated
the high resistance of the pulmonary artery.
The hemodynamic and structural characteristics
including TAPSE, RVEDD, RVESD, RVWT and PAAT/HR measured by
echocardiography are documented in Table I. TAPSE and PAAT/HR in the PAH
group were significantly decreased as compared with the Ctr group
(P<0.05), and this decrease was reversed to a relative high
level in the ADSCs and ADSCs-V groups. ADSCs-APN further
significantly raised the TAPSE and PAAT/HR as compared with the
ADSCs and ADSCs-V groups (P<0.05). As for RVEDD, it was notably
increased in the PAH group, and it was similar among the PAH,
ADSCs, ADSCs-V and ADSCs-APN groups. RVESD and RVWT in the PAH
group were significantly higher than those in the Ctr group
(P<0.05), but after ADSCs, ADSCs-V and ADSCs-APN treatments,
they were non-significantly reduced to relative low levels. There
was no significant difference in all parameters between the ADSCs
and ADSCs-V groups.
 | Table IEchocardiographic parameters of the
PAH rats after various treatments. |
Table I
Echocardiographic parameters of the
PAH rats after various treatments.
| Ctr | PAH | ADSCs | ADSCs-V | APN-ADSCs |
|---|
| TAPSE (mm) | 2.87±0.23 | 1.48±0.10a | 1.97±0.13a,b | 1.87±0.08a,b | 2.43±0.07a,b,c |
| RVEDD (mm) | 2.55±0.18 | 4.23±0.58a | 3.67±0.58a | 3.70±0.32a | 3.71±0.32a |
| RVESD (mm) | 2.36±0.23 | 3.11±0.22a | 2.68±0.53 | 2.72±0.18 | 2.84±0.44 |
| RVWT (mm) | 2.41±0.26 | 2.94±0.23a | 2.68±0.27 | 2.69±0.29 | 2.68±0.27 |
| PAAT/HR | 0.25±0.02 | 0.14±0.02a | 0.18±0.02a,b | 0.18±0.02a,b | 0.22±0.02a,b,c |
APN suppresses the proliferation of
PASMCs via the AMPK/BMP/Smad signaling pathway
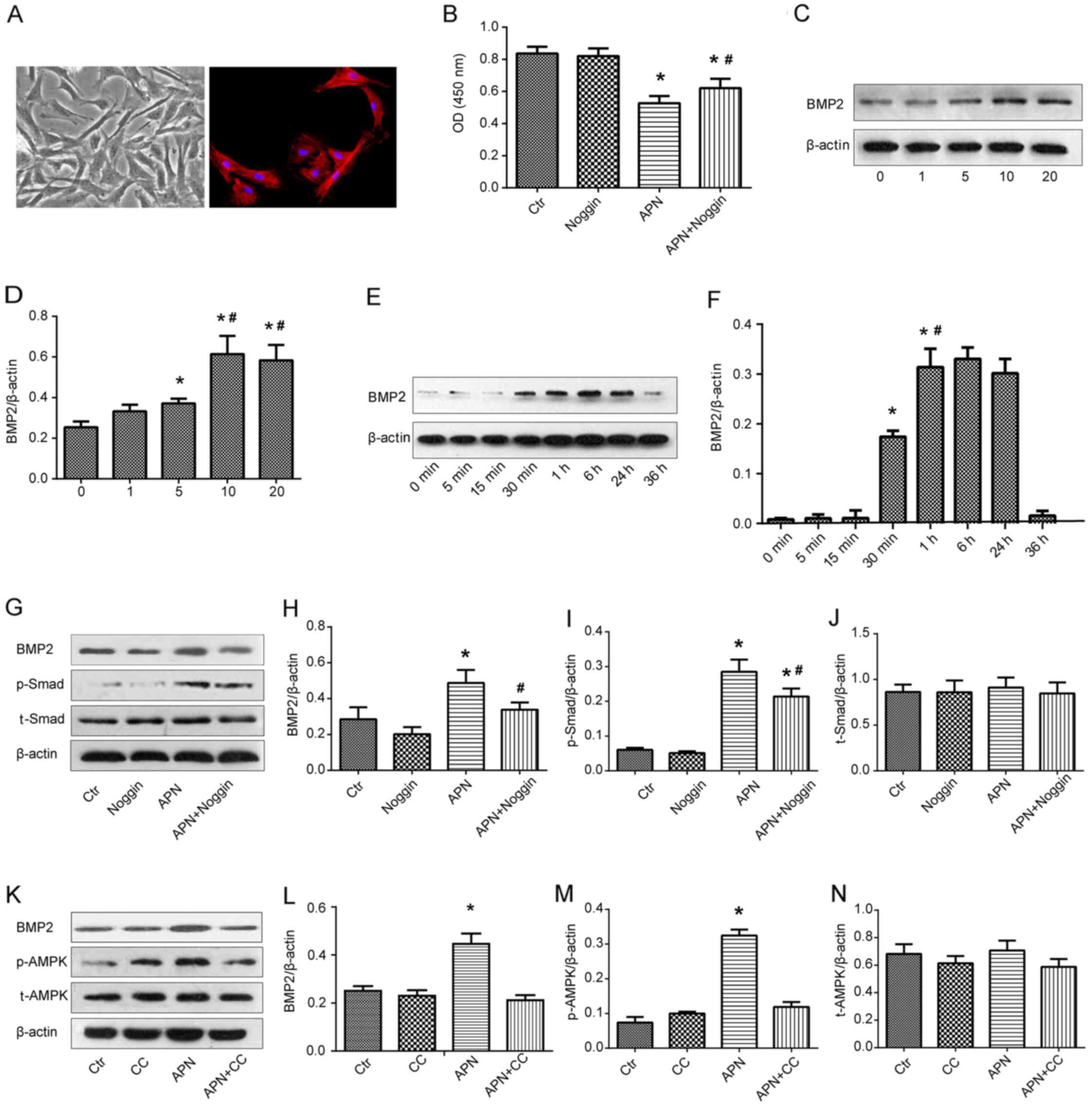
PAMSCs were isolated from the rats bearing PAH and
they were presented as spindle shapes under phase contrast
microscopy (Fig. 4A, left). The
right image in Fig. 4A shows the
immunofluorescence of PASMCs with α-SMA marking.
PASMCs isolated from the rats with PAH were exposed
to APN (10 μg/ml) and/or a specific BMP2 inhibitor noggin
(200 ng/ml) and the viability of PASMCs was analyzed by CCK-8 assay
(Fig. 4B). A similar absorbance
was found between the Ctr and noggin groups. APN markedly reduced
the absorbance as compared with the Ctr and noggin groups
(P<0.05). However, after the combination treatment of APN and
noggin, the absorbance was significantly reversed to a relative
high level in comparison with the APN group (P<0.05). These
results implied that APN inhibited the growth of PASMCs from the
rats bearing PAH and that the inhibitory effect of APN was weakened
by the BMP2 inhibitor.
The effect of APN on BMP2 protein expression was
determined by qualitative and quantitative western blot analysis
(Fig. 4C and D). The expression
of BMP2 was upregulated along with the increase in the APN dosage
(0, 1, 5 and 10 μg/ml), reaching a peak at 10 μg/ml
of APN. There was no significant difference in the BMP2 expression
between treatment with 10 and 20 μg/ml of APN. Therefore,
the optimum dosage of APN was found to be 10 μg/ml. PASMCs
were then incubated with APN (10 μg/ml) for varying time
intervals and BMP2 protein expression was measured by western blot
analysis again (Fig. 4E and F).
APN elevated the expression of BMP2 in a time-dependent manner
between 0 and 6 h and a peak value was found at 6 h. Next, it
gradually decreased and returned to the baseline level at 36 h.
These results indicated that APN upregulated the BMP2 protein
expression in PAMSCs in dosage- and time-dependent manners.
In an attempt to identify the influence of APN on
the BMP/Smad-related signaling pathways, the expression of BMP2,
Smad, p-Smad, AMPK and p-AMPK after treatment with APN, noggin and
CC was evaluated by western blot analysis (Fig. 4G–N). We found that APN treatment
increased the expression of BMP2 and p-Smad proteins, while noggin
downregulated their high expression levels induced by APN (Fig. 4G–I). Similarly, APN treatment
elevated the expression of BMP2 and p-AMPK proteins, while CC
downregulated their strong expression induced by APN (Fig. 4K–M). There was no difference in
the expression of Smad and AMPK proteins among the above groups
(Fig. 4J and N).
Discussion
PAH is a progressive disease characterized by
increased pulmonary arterial pressure, pulmonary vascular
remodeling, and RV dysfunction, which finally leads to right heart
failure and death. The pathogenesis of PAH is still poorly
understood, but it is intensely associated with excessively
proliferative smooth muscle cells that disturb the balance among
vasoactive substances and occlude the pulmonary arterial lumen
(26). How to repair these
damaged cells and restore their function are a major challenge for
tissue regeneration engineering. In recent years, stem cells and
cell-based gene therapy have become the most promising therapeutic
strategies (27). Our results
revealed that ADSCs were successfully isolated from rats and the
APN gene was effectively transfected into the ADSCs. ADSCs were
labeled with GFP to investigate the location of the ADSCs in the
lung and we found that engrafted ADSCs in the lung were located
around vessels. After the effective transplantation of ADSCs-APN
into the rats presenting PAH, mPAP and RVHI were significantly
decreased as compared with the ADSCs and ADSCs-V treatments. The WT
of the small pulmonary arteries, which represents vascular
remodeling, was also significantly reduced after ADSCs-APN
treatment. Hyperplastic smooth muscle cells were evaluated by
H&E and α-SMA immunohistochemical staining of tissue sections,
and PAAT and echocardiographic parameters such as PAAT/HR, RVWT,
RVESD, RVEED and TAPSE were more effectively improved by the
ADSCs-APN treatment rather than ADSCs and ADSCs-V. These results
suggest that antiproliferative activity plays a crucial role in the
APN and ADSC combination therapy for PAH.
Many previous studies have investigated cell therapy
such as EPCs and mesenchymal stromal cells for PAH (28,29). ADSCs are attractive cells for PAH
therapy due to their easy achievement, low immunogenicity,
multilineage differentiation potential, and abundant supply in the
body. We found that ADSCs attenuated PAH and pulmonary arterial
remodeling, which was in agreement with a previous study (30). ADSCs originate from adipose and
have the ability to differentiate into vascular endothelial cells,
vascular smooth muscle cells, and peripheral blood cells, thus
promoting angiogenesis and graft survival (31). Moreover, ADSCs secrete a variety
of cytokines and angiogenic factors to improve the tissue
microenvironment (31,32). ADSCs also recruit host stem cells
to the site of injury and activate the self-repair function
(33).
However, other researchers have reported that ADSCs
have no significant beneficial effect on MCT-induced PAH rats
(6), which may be due to the
differences in MCT dosage and administration method. In this study,
we injected 40 mg/kg MCT to induce the PAH model and mPAP was
elevated to ~33 mmHg, whereas the authors of the previous study
administered 60 mg/kg MCT and mPAP reached up to 55 mmHg. It was
possible that the higher pressure in the pulmonary artery largely
weakened the positive effect of ADSC treatment. Moreover, we
administrated ADSCs via the jugular vein, whereas they used
intratracheal injections. Capillary beds are abundant in the lung
tissues, which are beneficial for ADSC colonization. Therefore, it
is speculated that intratracheal administration would not
effectively improve the effect of ADSCs on PAH.
Increasing knowledge concerning the relationship
between obesity and pulmonary vascular diseases has emerged. An
autopsy study of 76 obese subjects revealed that the obese group
had more extensive pulmonary hypertensive changes as compared with
the control group (34). In
addition, the REVEAL registry, the largest PAH database in the USA,
indicated a higher prevalence of overweight and obese individuals
among those with idiopathic forms of PAH (35). APN is one of the most important
adipocytokines secreted from adipose tissue, which possesses
biological pleiotropic actions in metabolism, immune regulation,
and antiproliferative activities (36). APN may influence the progression
of PAH. The WT of pulmonary vessels and RV hypertrophy were
significantly increased and severe PAH was developed in the
APN-deficient (APN−/−) mice (14,15). APN overexpression was found to
ameliorate pulmonary arterial remodeling and suppress elevated
pulmonary artery pressure (17).
In this study, after treatment with the ADSCs-APN, the hemodynamic
changes and vascular remodeling of PAH were greatly attenuated.
Furthermore, the combination of APN and ADSC therapy was much more
effective than ADSC therapy alone.
To explore the underlying mechanisms in the PAH
therapy here, we evaluated the thickness of the smooth muscle layer
in the lung by examining the immunohistochemical expression of
α-SMA. Compared to the Ctr group, all the treatment groups had a
significantly smaller WT of pulmonary vasculature. The ADSCs-APN
group had a thinner vascular wall than the ADSCs and ADSCs-V
groups. It was suggested that APN attenuated the MCT-induced PAH
and pulmonary arterial remodeling via ameliorating the wall
thickening of the pulmonary arterioles. Moreover, APN was found to
directly suppress the proliferation of PASMCs (25,37). The accumulation of smooth muscle
cells in vessel walls was found to be enhanced and APN suppressed
PAH by directly inhibiting PASMC proliferation in APN-deficient
mice (14,15,17). Therefore, we hypothesized that the
mechanism of reversing vascular remodeling was associated with the
anti-proliferation of PASMCs by APN and we probed the molecular
mechanisms involved in this anti-proliferation process. It is well
known that BMPs interact with receptors and trigger Smad
phosphorylation to propagate the signals into the nucleus and
regulate target gene expression (38). BMPs were found to inhibit the
proliferation of smooth muscle cells through a Smad-dependent
pathway (39,40). A dysfunctional BMP/Smad signaling
pathway was found to be an important risk factor for
hyperproliferative PASMCs (22,41–43). Therefore, we hypothesized that APN
suppresses the proliferation of PASMCs via a BMP/Smad-dependent
pathway.
In the present study, we found that APN partly
reversed the defect of the BMP-stimulated Smad pathway in PASMCs
with PAH. This indicated that the BMP/Smad signaling pathway plays
a crucial part in the anti-proliferation of PASMCs by APN. The
major downstream pathway of APN is the AMPK phosphorylation
pathway. AMPK is not only involved in the regulation of glucose and
lipid metabolism, but also has a variety of other biological
activities, such as inhibition of tumor cell proliferation,
autophagy, stress response and polarity regulation (44). Activation of AMPK was found to
arrest the cell cycle in the G0/G1 phase and restrain tumor growth
by increasing p53 expression (45). Additionally, AMPK activation may
also suppress the proliferation of vascular smooth muscle cells via
an mTOR-dependent pathway (46).
Here, we demonstrated for the first time an involvement of the AMPK
pathway in the BMP/Smad-dependent induction of proliferative
inhibition by APN. Therefore, these results indicate that BMP/Smad
may function as a downstream signaling pathway of AMPK in APN
regulation. Taken together, our findings confirmed that APN
suppressed PASMC proliferation via the AMPK/BMP/Smad pathway.
Besides the canonical BMP/Smad pathway, other
non-Smad pathways such as MAPK kinases and PI3K kinase/Akt may also
be involved in PAH pathogenesis (47–51). Here, we only evaluated the effect
of APN on the Smad signaling pathway. It is yet unclear whether a
non-Smad pathway is also involved in APN-mediated inhibition of
PASMC proliferation and whether there is cross-talk between Smad
and non-Smad pathways. This illustrates the complexity of
understanding the BMP pathway and these issues will be investigated
in our future studies.
We also confirmed that RV function was more
effectively improved by ADSCs-APN transplantation rather than ADSC
therapy alone. Although the observed beneficial effects of APN are
incompletely understood, it is generally believed to exert its
effects by decreasing oxidative stress of myocardial cells
(52). Further studies are
necessary to elucidate these gene-related mechanisms in myocardial
cell lines.
In conclusion, transplantation of ADSCs attenuated
MCT-induced PAH in rats, and transfection of APN into ADSCs further
promoted this therapeutic effect in vivo. The main mechanism
of this effect was that the anti-proliferation of PASMCs by APN
based on the regulation of the AMPK/BMP/Smad pathway suppressed the
hyperplasia of the pulmonary vascular wall. ADSCs are useful and
effective vehicles with which to deliver therapeutic genes
intravenously to the lung to treat PAH. At present, no effective
therapeutic options are available for PAH. This study has important
implications for PAH therapy and provides solid evidence to support
future investigation of regenerative cell-based gene strategies for
the treatment of patients with severe PAH and congenital heart
disease. Therapies restoring APN signaling may present novel
modalities to prevent PAH progression and its complications.
Acknowledgments
The authors are thankful for the financial support
from the National Natural Science Foundation of China (nos.
81400189 and 81270111/H0109) and the Training Project for Young and
Middle-Aged in the Health System of Fujian Province (no.
2013-ZQN-JC-21).
References
|
1
|
Liu K, Liu R, Cao G, Sun H, Wang X and Wu
S: Adipose-derived stromal cell autologous transplantation
ameliorates pulmonary arterial hypertension induced by shunt flow
in rat models. Stem Cells Dev. 20:1001–1010. 2011. View Article : Google Scholar
|
|
2
|
Baber SR, Deng W, Master RG, Bunnell BA,
Taylor BK, Murthy SN, Hyman AL and Kadowitz PJ: Intratracheal
mesenchymal stem cell administration attenuates
monocrotaline-induced pulmonary hypertension and endothelial
dysfunction. Am J Physiol Heart Circ Physiol. 292:H1120–H1128.
2007. View Article : Google Scholar
|
|
3
|
Zhao YD, Courtman DW, Deng Y, Kugathasan
L, Zhang Q and Stewart DJ: Rescue of monocrotaline-induced
pulmonary arterial hypertension using bone marrow-derived
endothelial-like progenitor cells: Efficacy of combined cell and
eNOS gene therapy in established disease. Circ Res. 96:442–450.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luo L, Lin T, Zheng S, Xie Z, Chen M, Lian
G, Xu C, Wang H and Xie L: Adipose-derived stem cells attenuate
pulmonary arterial hypertension and ameliorate pulmonary arterial
remodeling in monocrotaline-induced pulmonary hypertensive rats.
Clin Exp Hypertens. 37:241–248. 2015. View Article : Google Scholar
|
|
5
|
Takemiya K, Kai H, Yasukawa H, Tahara N,
Kato S and Imaizumi T: Mesenchymal stem cell-based prostacyclin
synthase gene therapy for pulmonary hypertension rats. Basic Res
Cardiol. 105:409–417. 2010. View Article : Google Scholar
|
|
6
|
Somanna NK, Wörner PM, Murthy SN, Pankey
EA, Schächtele DJ, St Hilaire RC, Jansen D, Chaffin AE, Nossaman
BD, Alt EU, et al: Intratracheal administration of
cyclooxygenase-1-transduced adipose tissue-derived stem cells
ameliorates monocrotaline-induced pulmonary hypertension in rats.
Am J Physiol Heart Circ Physiol. 307:H1187–H1195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao Q, Liu Z, Wang Z, Yang C, Liu J and
Lu J: Effect of prepro-calcitonin gene-related peptide-expressing
endothelial progenitor cells on pulmonary hypertension. Ann Thorac
Surg. 84:544–552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Iwaguro H, Yamaguchi J, Kalka C, Murasawa
S, Masuda H, Hayashi S, Silver M, Li T, Isner JM and Asahara T:
Endothelial progenitor cell vascular endothelial growth factor gene
transfer for vascular regeneration. Circulation. 105:732–738. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao G, Liu C, Wan Z, Liu K, Sun H, Sun X,
Tang M, Bing W, Wu S, Pang X, et al: Combined hypoxia inducible
factor-1α and homogeneous endothelial progenitor cell therapy
attenuates shunt flow-induced pulmonary arterial hypertension in
rabbits. J Thorac Cardiovasc Surg. 150:621–632. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Granton J, Langleben D, Kutryk MB, Camack
N, Galipeau J, Courtman DW and Stewart DJ: Endothelial NO-synthase
gene-enhanced progenitor cell therapy for pulmonary arterial
hypertension: The PHACeT trial. Circ Res. 117:645–654. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Antonopoulos AS, Margaritis M, Coutinho P,
Shirodaria C, Psarros C, Herdman L, Sanna F, De Silva R, Petrou M,
Sayeed R, et al: Adiponectin as a link between type 2 diabetes and
vascular NADPH oxidase activity in the human arterial wall: The
regulatory role of perivascular adipose tissue. Diabetes.
64:2207–2219. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin Z, Pan X, Wu F, Ye D, Zhang Y, Wang Y,
Jin L, Lian Q, Huang Y, Ding H, et al: Fibroblast growth factor 21
prevents atherosclerosis by suppression of hepatic sterol
regulatory element-binding protein-2 and induction of adiponectin
in mice. Circulation. 131:1861–1871. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Lam KS, Xu JY, Lu G, Xu LY, Cooper
GJ and Xu A: Adiponectin inhibits cell proliferation by interacting
with several growth factors in an oligomerization-dependent manner.
J Biol Chem. 280:18341–18347. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Summer R, Fiack CA, Ikeda Y, Sato K, Dwyer
D, Ouchi N, Fine A, Farber HW and Walsh K: Adiponectin deficiency:
A model of pulmonary hypertension associated with pulmonary
vascular disease. Am J Physiol Lung Cell Mol Physiol.
297:L432–L438. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Medoff BD, Okamoto Y, Leyton P, Weng M,
Sandall BP, Raher MJ, Kihara S, Bloch KD, Libby P and Luster AD:
Adiponectin deficiency increases allergic airway inflammation and
pulmonary vascular remodeling. Am J Respir Cell Mol Biol.
41:397–406. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weng M, Baron DM, Bloch KD, Luster AD, Lee
JJ and Medoff BD: Eosinophils are necessary for pulmonary arterial
remodeling in a mouse model of eosinophilic inflammation-induced
pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol.
301:L927–L936. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakagawa Y, Kishida K, Kihara S, Funahashi
T and Shimomura I: Adiponectin ameliorates hypoxia-induced
pulmonary arterial remodeling. Biochem Biophys Res Commun.
382:183–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Farber HW and Loscalzo J: Pulmonary
arterial hypertension. N Engl J Med. 351:1655–1665. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Momose Y, Aimi Y, Hirayama T, Kataoka M,
Ono M, Yoshino H, Satoh T and Gamou S: De novo mutations in the
BMPR2 gene in patients with heritable pulmonary arterial
hypertension. Ann Hum Genet. 79:85–91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Soubrier F, Chung WK, Machado R, Grünig E,
Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, et
al: Genetics and genomics of pulmonary arterial hypertension. J Am
Coll Cardiol. 62(Suppl 25): D13–D21. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takahashi H, Goto N, Kojima Y, Tsuda Y,
Morio Y, Muramatsu M and Fukuchi Y: Downregulation of type II bone
morphogenetic protein receptor in hypoxic pulmonary hypertension.
Am J Physiol Lung Cell Mol Physiol. 290:L450–L458. 2006. View Article : Google Scholar
|
|
22
|
Upton PD and Morrell NW: The transforming
growth factor-β-bone morphogenetic protein type signalling pathway
in pulmonary vascular homeostasis and disease. Exp Physiol.
98:1262–1266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang S, Fantozzi I, Tigno DD, Yi ES,
Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ and
Yuan JX: Bone morphogenetic proteins induce apoptosis in human
pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol
Physiol. 285:L740–L754. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie L, Lin P, Xie H and Xu C: Effects of
atorvastatin and losartan on monocrotaline-induced pulmonary artery
remodeling in rats. Clin Exp Hypertens. 32:547–554. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weng M, Raher MJ, Leyton P, Combs TP,
Scherer PE, Bloch KD and Medoff BD: Adiponectin decreases pulmonary
arterial remodeling in murine models of pulmonary hypertension. Am
J Respir Cell Mol Biol. 45:340–347. 2011. View Article : Google Scholar :
|
|
26
|
Rabinovitch M: Molecular pathogenesis of
pulmonary arterial hypertension. J Clin Invest. 122:4306–4313.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Farkas L and Kolb M: Vascular repair and
regeneration as a therapeutic target for pulmonary arterial
hypertension. Respiration. 85:355–364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Foster WS, Suen CM and Stewart DJ:
Regenerative cell and tissue-based therapies for pulmonary arterial
hypertension. Can J Cardiol. 30:1350–1360. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suen CM, Mei SH, Kugathasan L and Stewart
DJ: Targeted delivery of genes to endothelial cells and cell- and
gene-based therapy in pulmonary vascular diseases. Compr Physiol.
3:1749–1779. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eguchi M, Ikeda S, Kusumoto S, Sato D,
Koide Y, Kawano H and Maemura K: Adipose-derived regenerative cell
therapy inhibits the progression of monocrotaline-induced pulmonary
hypertension in rats. Life Sci. 118:306–312. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Salgado AJ, Reis RL, Sousa NJ and Gimble
JM: Adipose tissue derived stem cells secretome: Soluble factors
and their roles in regenerative medicine. Curr Stem Cell Res Ther.
5:103–110. 2010. View Article : Google Scholar
|
|
32
|
Rehman J, Traktuev D, Li J, Merfeld-Clauss
S, Temm-Grove CJ, Bovenkerk JE, Pell CL, Johnstone BH, Considine RV
and March KL: Secretion of angiogenic and antiapoptotic factors by
human adipose stromal cells. Circulation. 109:1292–1298. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ii M, Horii M, Yokoyama A, Shoji T, Mifune
Y, Kawamoto A, Asahi M and Asahara T: Synergistic effect of
adipose-derived stem cell therapy and bone marrow progenitor
recruitment in ischemic heart. Lab Invest. 91:539–552. 2011.
View Article : Google Scholar
|
|
34
|
Haque AK, Gadre S, Taylor J, Haque SA,
Freeman D and Duarte A: Pulmonary and cardiovascular complications
of obesity: An autopsy study of 76 obese subjects. Arch Pathol Lab
Med. 132:1397–1404. 2008.PubMed/NCBI
|
|
35
|
Burger CD, Foreman AJ, Miller DP, Safford
RE, McGoon MD and Badesch DB: Comparison of body habitus in
patients with pulmonary arterial hypertension enrolled in the
Registry to Evaluate Early and Long-term PAH Disease Management
with normative values from the National Health and Nutrition
Examination Survey. Mayo Clin Proc. 86:105–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Okamoto Y, Kihara S, Funahashi T,
Matsuzawa Y and Libby P: Adiponectin: A key adipocytokine in
metabolic syndrome. Clin Sci (Lond). 110:267–278. 2006. View Article : Google Scholar
|
|
37
|
Wang ZV and Scherer PE: Adiponectin,
cardiovascular function, and hypertension. Hypertension. 51:8–14.
2008. View Article : Google Scholar
|
|
38
|
García de Vinuesa A, Abdelilah-Seyfried S,
Knaus P, Zwijsen A and Bailly S: BMP signaling in vascular biology
and dysfunction. Cytokine Growth Factor Rev. 27:65–79. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang X, Long L, Southwood M,
Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H,
Trembath RC and Morrell NW: Dysfunctional Smad signaling
contributes to abnormal smooth muscle cell proliferation in
familial pulmonary arterial hypertension. Circ Res. 96:1053–1063.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zeng Y, Liu H, Kang K, Wang Z, Hui G,
Zhang X, Zhong J, Peng W, Ramchandran R, Raj JU, et al: Hypoxia
inducible factor-1 mediates expression of miR-322: Potential role
in proliferation and migration of pulmonary arterial smooth muscle
cells. Sci Rep. 5:120982015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Morty RE, Nejman B, Kwapiszewska G, Hecker
M, Zakrzewicz A, Kouri FM, Peters DM, Dumitrascu R, Seeger W, Knaus
P, et al: Dysregulated bone morphogenetic protein signaling in
monocrotaline-induced pulmonary arterial hypertension. Arterioscler
Thromb Vasc Biol. 27:1072–1078. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang J, Li X, Al-Lamki RS, Wu C, Weiss A,
Berk J, Schermuly RT and Morrell NW: Sildenafil potentiates bone
morphogenetic protein signaling in pulmonary arterial smooth muscle
cells and in experimental pulmonary hypertension. Arterioscler
Thromb Vasc Biol. 33:34–42. 2013. View Article : Google Scholar
|
|
43
|
Yang J, Li X, Al-Lamki RS, Southwood M,
Zhao J, Lever AM, Grimminger F, Schermuly RT and Morrell NW:
Smad-dependent and smad-independent induction of id1 by
prostacyclin analogues inhibits proliferation of pulmonary artery
smooth muscle cells in vitro and in vivo. Circ Res. 107:252–262.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vakana E, Altman JK and Platanias LC:
Targeting AMPK in the treatment of malignancies. J Cell Biochem.
113:404–409. 2012. View Article : Google Scholar
|
|
45
|
Lévy J, Cacheux W, Bara MA, L'Hermitte A,
Lepage P, Fraudeau M, Trentesaux C, Lemarchand J, Durand A, Crain
AM, et al: Intestinal inhibition of Atg7 prevents tumour initiation
through a microbiome-influenced immune response and suppresses
tumour growth. Nat Cell Biol. 17:1062–1073. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee KY, Lee DH and Choi HC: Mesoglycan
attenuates VSMC proliferation through activation of AMP-activated
protein kinase and mTOR. Clin Hypertens. 22:22016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shimpo Lu J, Shimamoto H, Chong A, Hampton
AJ, Spring CR, Yada DJ, Takao M, Onoda M, Yada KI, et al: Specific
inhibition of 38 mitogen-activated protein kinase with FR167653
attenuates vascular proliferation in monocrotaline-induced
pulmonary hypertension in rats. J Thorac Cardiovasc Surg.
128:850–859. 2004. View Article : Google Scholar
|
|
48
|
Ma J, Zhang L, Han W, Shen T, Ma C, Liu Y,
Nie X, Liu M, Ran Y and Zhu D: Activation of JNK/c-Jun is required
for the proliferation, survival, and angiogenesis induced by EET in
pulmonary artery endothelial cells. J Lipid Res. 53:1093–1105.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yu MQ, Liu XS, Wu HX, Xiang M and Xu YJ:
ERK1/2 promotes cigarette smoke-induced rat pulmonary artery smooth
muscle cells proliferation and pulmonary vascular remodeling via
up-regulating cycline1 expression. J Huazhong Univ Sci Technolog
Med Sci. 33:315–322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen XY, Dun JN, Miao QF and Zhang YJ:
Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses
5-hydroxytryptamine-induced pulmonary artery smooth muscle cell
proliferation via JNK and ERK1/2 pathway. Pharmacology. 83:67–79.
2009. View Article : Google Scholar
|
|
51
|
Havrda MC, Johnson MJ, O'Neill CF and Liaw
L: A novel mechanism of transcriptional repression of 27kip1
through Notch/HRT2 signaling in vascular smooth muscle cells.
Thromb Haemost. 96:361–370. 2006.PubMed/NCBI
|
|
52
|
Guo Z, Qi W, Yu Y, Du S, Wu J and Liu J:
Effect of exenatide on the cardiac expression of adiponectin
receptor 1 and NADPH oxidase subunits and heart function in
streptozotocin-induced diabetic rats. Diabetol Metab Syndr.
6:292014. View Article : Google Scholar : PubMed/NCBI
|