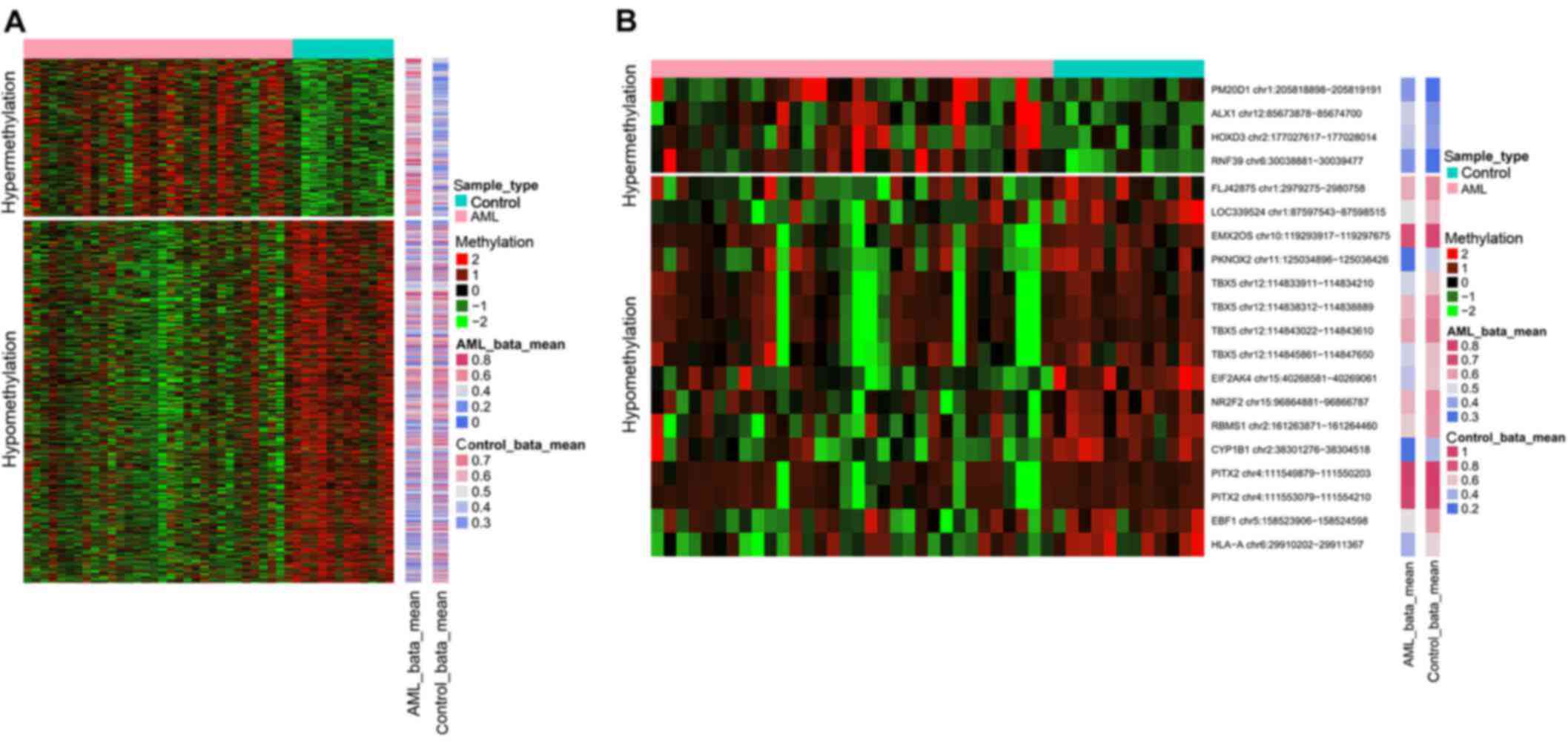
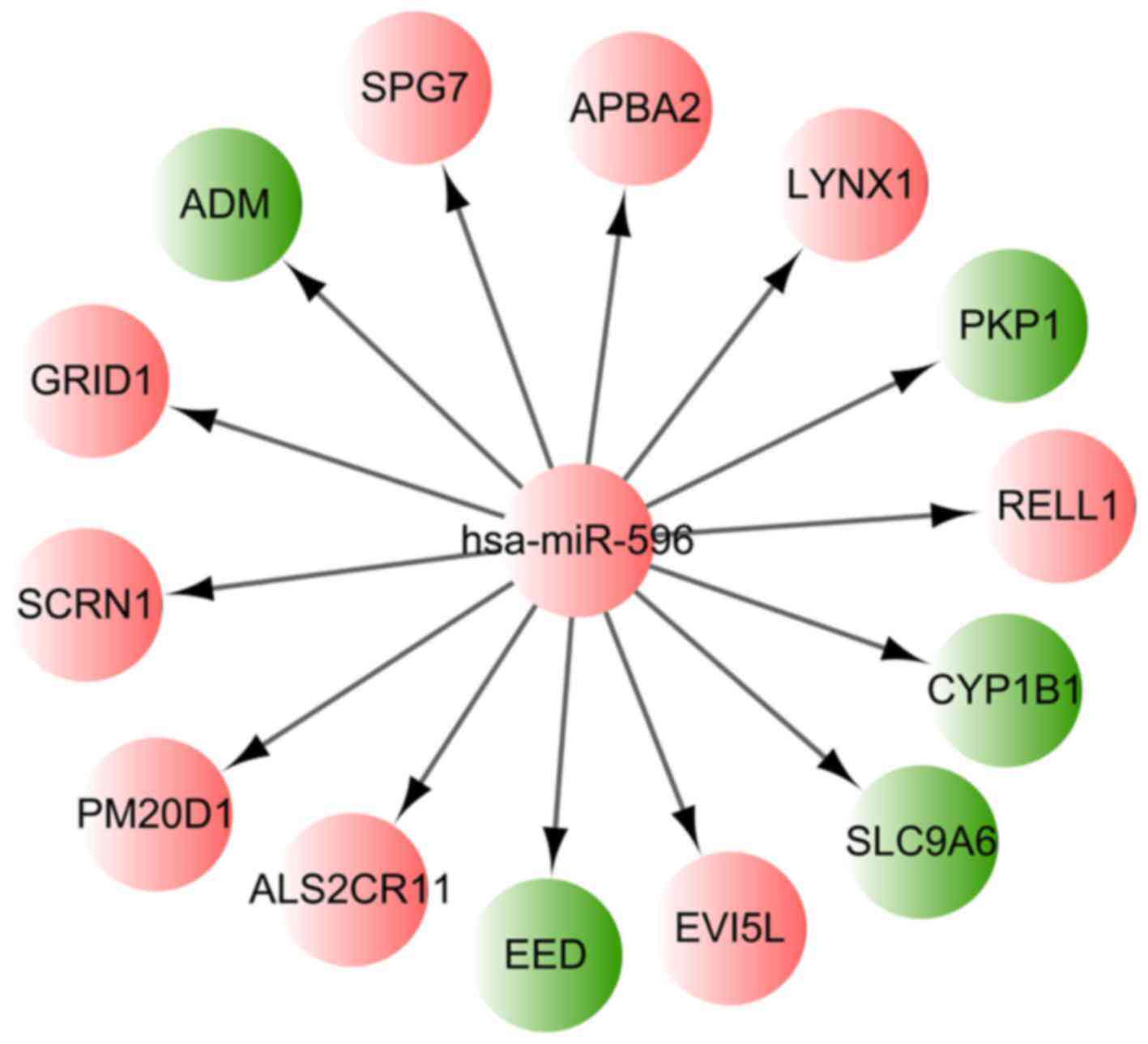
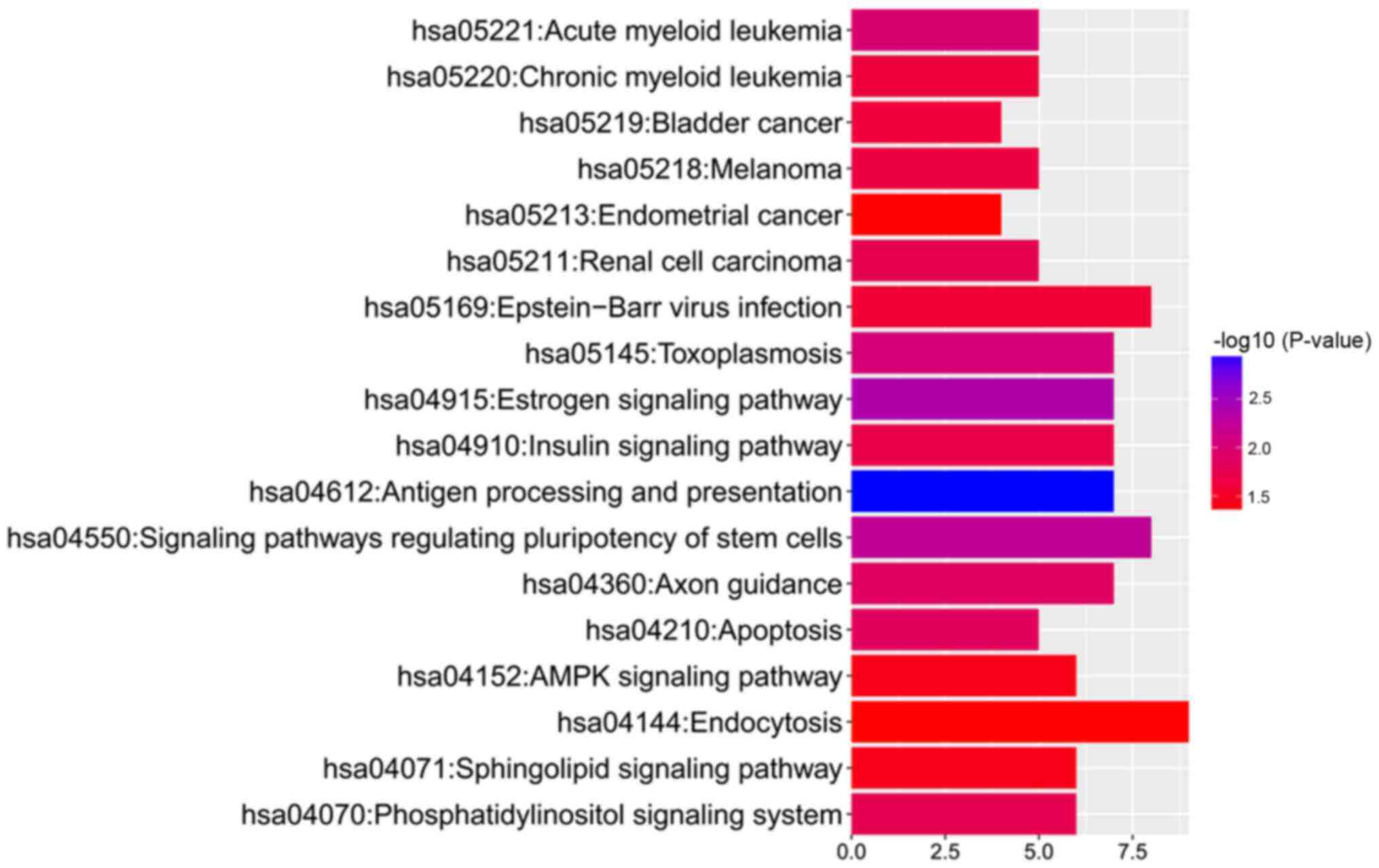
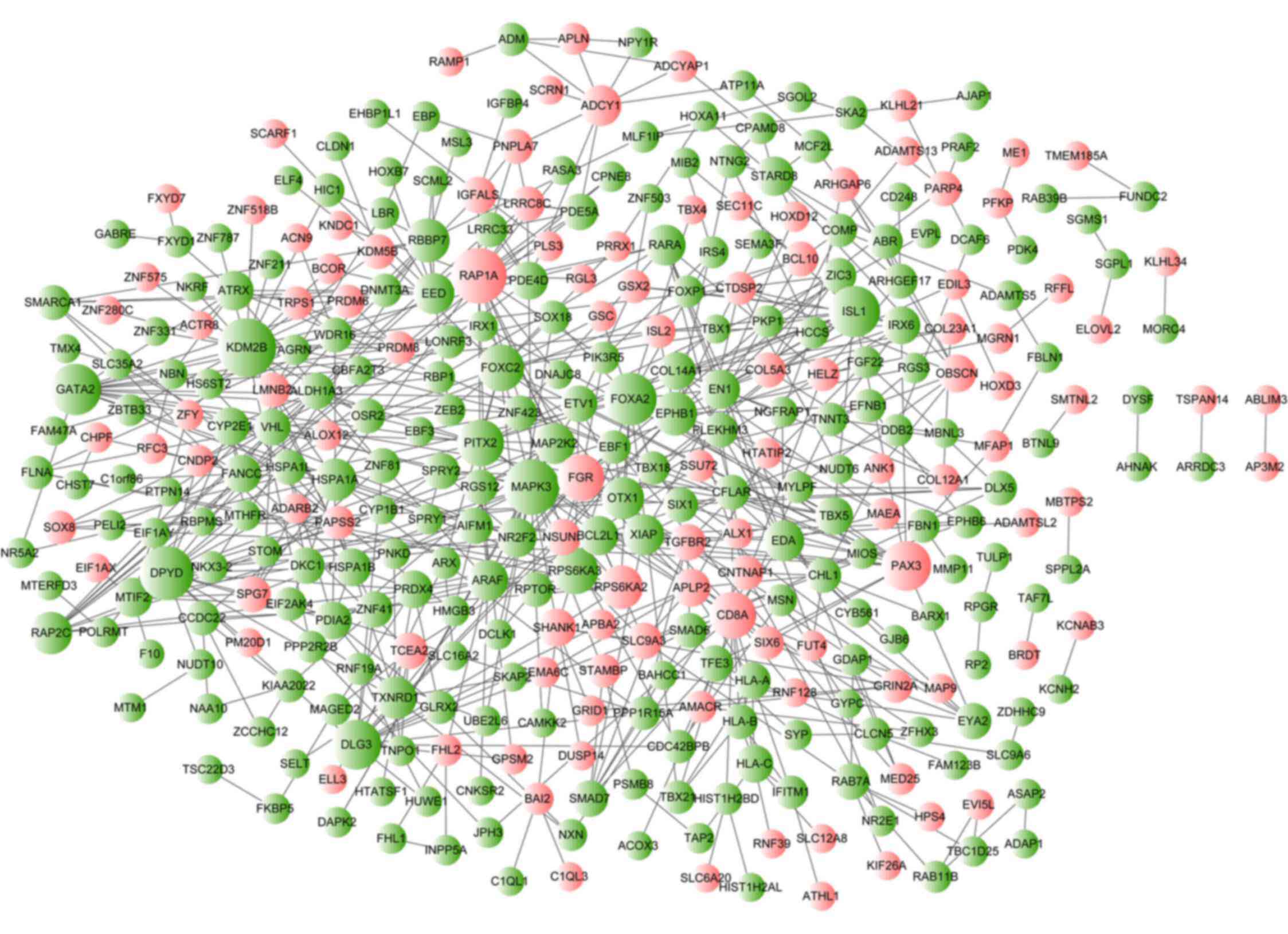
|
1
|
Grove CS and Vassiliou GS: Acute myeloid
leukaemia: A paradigm for the clonal evolution of cancer. Dis Model
Mech. 7:941–951. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Estey EH and Ayalew T: Acute myeloid
leukemia: 2012 update on diagnosis, risk stratification, and
management. Am J Hematol. 87:89–99. 2012. View Article : Google Scholar
|
|
3
|
Ferrara F and Schiffer CA: Acute myeloid
leukaemia in adults. Lancet. 381:484–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou M and Tong X: Downregulated Poly-C
binding protein-1 is a novel predictor associated with poor
prognosis in Acute Myeloid Leukemia. Diagn Pathol. 10:1472015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kwon HY, Bajaj J, Ito T, Blevins A, Konuma
T, Weeks J, Lytle NK, Koechlein CS, Rizzieri D, Chuah C, et al:
Tetraspanin 3 is required for the development and propagation of
acute myelogenous leukemia. Cell Stem Cell. 17:152–164. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Creutzig U, van den Heuvel-Eibrink MM,
Gibson B, Dworzak MN, Adachi S, de Bont E, Harbott J, Hasle H,
Johnston D, Kinoshita A, et al: AML Committee of the International
BFM Study Group: Diagnosis and management of acute myeloid leukemia
in children and adolescents: Recommendations from an international
expert panel. Blood. 120:3187–3205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li S, Garrett-Bakelman FE, Chung SS,
Sanders MA, Hricik T, Rapaport F, Patel J, Dillon R, Vijay P, Brown
AL, et al: Distinct evolution and dynamics of epigenetic and
genetic heterogeneity in acute myeloid leukemia. Nat Med.
22:792–799. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen C, Liu Y, Lu C, Cross JR, Morris JP
IV, Shroff AS, Ward PS, Bradner JE, Thompson C and Lowe SW:
Cancer-associated IDH2 mutants drive an acute myeloid leukemia that
is susceptible to Brd4 inhibition. Genes Dev. 27:1974–1985. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Figueroa ME, Lugthart S, Li Y,
Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J,
van Putten W, Skrabanek L, et al: DNA methylation signatures
identify biologically distinct subtypes in acute myeloid leukemia.
Cancer Cell. 17:13–27. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Akalin A, Garrett-Bakelman FE, Kormaksson
M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D,
Hess JL, et al: Base-pair resolution DNA methylation sequencing
reveals profoundly divergent epigenetic landscapes in acute myeloid
leukemia. PLoS Gene. 8:e10027812012. View Article : Google Scholar
|
|
11
|
Tao YF, Ni J, Lu J, Wang N, Xiao PF, Zhao
WL, Wu D, Pang L, Wang J, Feng X and Pan J: The promoter of miR-663
is hypermethylated in Chinese pediatric acute myeloid leukemia
(AML). BMC Med Genet. 14:742013. View Article : Google Scholar
|
|
12
|
Rodríguez-Pardo VM, Aristizabal JA, Jaimes
D, Quijano SM, de los Reyes I, Herrera MV, Solano J and Vernot JP:
Mesenchymal stem cells promote leukaemic cells aberrant phenotype
from B-cell acute lymphoblastic leukaemia. Hematol Oncol Stem Cell
Ther. 6:89–100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blau O, Hofmann WK, Baldus CD, Thiel G,
Serbent V, Schümann E, Thiel E and Blau IW: Chromosomal aberrations
in bone marrow mesenchymal stroma cells from patients with
myelodysplastic syndrome and acute myeloblastic leukemia. Exp
Hematol. 35:221–229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Roela RA, Carraro DM, Brentani HP, Kaiano
JHL, Simão DF, Guarnieiro R, Lopes LF, Borojevic R and Brentani MM:
Gene stage-specific expression in the microenvironment of pediatric
myelodysplastic syndromes. Leuk Res. 31:579–589. 2007. View Article : Google Scholar
|
|
15
|
Barrett T, Troup DB, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, et al: NCBI GEO: Archive for functional genomics data
sets - 10 years on. Nucleic Acids Res. 39:D1005–D1010. 2011.
View Article : Google Scholar
|
|
16
|
von der Heide EK, Neumann M, Vosberg S,
James AR, Schroeder MP, Ortiz-Tanchez J, Isaakidis K, Schlee C,
Luther M, Jöhrens K, et al: Molecular alterations in bone marrow
mesenchymal stromal cells derived from acute myeloid leukemia
patients. Leukemia. 31:1069–1078. 2017. View Article : Google Scholar
|
|
17
|
Pérez C, Martínez-Calle N, Martín-Subero
JI, Segura V, Delabesse E, Fernandez-Mercado M, Garate L, Alvarez
S, Rifon J, Varea S, et al: TET2 mutations are associated with
specific 5-meth-ylcytosine and 5-hydroxymethylcytosine profiles in
patients with chronic myelomonocytic leukemia. PLoS One.
7:e316052012. View Article : Google Scholar
|
|
18
|
Bibikova M, Lin Z, Zhou L, Chudin E,
Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, Vollmer E, et al:
High-throughput DNA methylation profiling using universal bead
arrays. Genome Res. 16:383–393. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Warden CD, Lee H, Tompkins JD, Li X, Wang
C, Riggs AD, Yu H, Jove R and Yuan YC: COHCAP: An integrative
genomic pipeline for single-nucleotide resolution DNA methylation
analysis. Nucleic Acids Re. 41:e117. 2013. View Article : Google Scholar
|
|
20
|
Robinson MD, Kahraman A, Law CW, Lindsay
H, Nowicka M, Weber LM and Zhou X: Statistical methods for
detecting differentially methylated loci and regions. Front Genet.
5:3242014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dweep H and Gretz N: miRWalk2.0: A
comprehensive atlas of microRNA-target interactions. Nat Methods.
12:6972015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 28: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar
|
|
23
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
24
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:D447–D452. 2015. View Article : Google Scholar
|
|
25
|
Lopes CT, Franz M, Kazi F, Donaldson SL,
Morris Q and Bader GD: Cytoscape Web: An interactive web-based
network browser. Bioinformatics. 26:2347–2348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davis AP, Murphy CG, Johnson R, Lay JM,
Lennon-Hopkins K, Saraceni-Richards C, Sciaky D, King BL,
Rosenstein MC, et al: The comparative toxicogenomics database:
Update 2013. Nucleic Acids Res. 39:1067–1072. 2011. View Article : Google Scholar
|
|
27
|
Bauer-Mehren A, Bundschus M, Rautschka M,
Mayer MA, Sanz F and Furlong LI: Gene-disease network analysis
reveals functional modules in mendelian, complex and environmental
diseases. PLoS One. 6:e202842011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schübeler D: Function and information
content of DNA methylation. Nature. 517:321–326. 2015. View Article : Google Scholar
|
|
29
|
Wen L, Li J, Guo H, Liu X, Zheng S, Zhang
D, Zhu W, Qu J, Guo L, Du D, et al: Genome-scale detection of
hypermethylated CpG islands in circulating cell-free DNA of
hepatocellular carcinoma patients. Cell Res. 1376:252015.
|
|
30
|
Jung M, Kadam S, Xiong W, Rauch TA, Jin SG
and Pfeifer GP: MIRA-seq for DNA methylation analysis of CpG
islands. Epigenomics. 7:695–706. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Larrick JW, Larrick JW and Mendelsohn AR:
Uncoupling Mitochondrial Respiration for Diabesity. Rejuvenation
Res. 19:337–340. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Long JZ, Svensson KJ, Bateman LA, Lin H,
Kamenecka T, Lokurkar IA, Lou J, Rao RR, Chang MR, Jedrychowski MP,
et al: The Secreted Enzyme M20D1 Regulates Lipidated Amino Acid
Uncouplers of Mitochondria. Cell. 166:424–435. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matsuda J, Hosoda K, Itoh H, Son C, Doi K,
Tanaka T, Fukunaga Y, Inoue G, Nishimura H, Yoshimasa Y, et al:
Cloning of rat uncoupling protein-3 and uncoupling protein-2 cDNAs:
Their gene expression in rats fed high-fat diet. FEBS Lett.
418:200–204. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Warner JT, Evans WD, Webb DKH and Gregory
JW: Body composition of long-term survivors of acute lymphoblastic
leukaemia. Med Pediatr Oncol. 38:165–172. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Heller G, Weinzierl M, Noll C, Babinsky V,
Ziegler B, Altenberger C, Minichsdorfer C, Lang G, Döme B,
End-Pfützenreuter A, et al: Genome-wide miRNA expression profiling
identifies miR-9-3 and miR-193a as targets for DNA methylation in
non-small cell lung cancers. Clin Cancer Res. 18:1619–1629. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Endo H, Muramatsu T, Furuta M, Uzawa N,
Pimkhaokham A, Amagasa T, Inazawa J and Kozaki K: Potential of
tumor-suppressive miR-596 targeting LGALS3BP as a therapeutic agent
in oral cancer. Carcinogenesis. 34:560–569. 2013. View Article : Google Scholar
|