Introduction
Nonalcoholic fatty liver disease (NAFLD) has
gradually emerged as the most common chronic liver disease in the
majority of the developed world, which is characterized by excess
accumulation of fat in hepatocytes (1). The increasing global prevalence of
NAFLD may be the result of changes in dietary habits and an
increase in sedentary lifestyles (2). It is reported that the prevalence of
NAFLD among adults in China is ~15% (2). Although saturated fatty acid
(SFA)-induced lipotoxicity and endoplasmic reticulum (ER) stress
have relevant roles in the pathogenesis of NAFLD, the exact
molecular mechanisms involved in liver cell injury contributing to
progression from NAS to NASH remains to be elucidated (3,4).
Palmitic acid (PA) is the most abundant SFA in the
circulation and a major lipotoxic inducer. Accumulating evidence
supports that autophagy confers protection against the lipotoxicity
induced by PA (5,6). Autophagy is a catabolic process,
which is crucial in maintaining cellular homeostasis through a
lysosomal degradative pathway, which is involved in various
physiological and pathological processes (7). Established functions for autophagy
in hepatic lipid metabolism, insulin sensitivity and cellular
injury suggest multiple possible mechanisms by which autophagy may
affect the lipid accumulation and hepatocellular injury which
underlie NAFLD (8–10).
In NAFLD, an abnormal increase in intracellular
lipid impairs autophagic degradation (8), in turn, autophagic dysfunction
exacerbates NAFLD by causing ER stress and increasing insulin
resistance (11).
Hyperinsulinemia contributes further to the hepatic defect in
autophagy (12). Although the
exact mechanism remains to be fully elucidated, several mechanisms
underlying NAFLD-associated autophagy dysfunction have been
suggested, including decreased expression of autophagic genes and
reduced levels of degradative lysosomal enzymes (13). However, the most important
contributor is impaired autophagosome maturation, including
vesicular fusion and acidification (14–16). The mechanism is likely to be
multifactorial, however, the important implication is that a
harmful cycle may exist in which hepatic lipid accumulation
promotes insulin resistance, and these events impair autophagy
which worsens steatosis and insulin insensitivity (13). This cycle leads to
self-perpetuating worsening of cellular autophagic function and
steatosis. The numerous potential mechanisms involved in autophagy
in NAFLD suggest that autophagy offers novel approaches for the
treatment of NAFLD.
Previous studies have shown that SREBP-2, a
transcription factor that regulates cholesterol metabolism, can
regulate autophagy under certain circumstances (17). However, whether the regulation of
SREBP-2 enhances autophagic function in hepatocytes remains to be
elucidated, particularly in lipid-overloaded hepatic cells. In the
present study, it was identified that autophagic activity was
impaired in lipid-overloaded human hepatocytes. It was confirmed
that SREBP-2 activated autophagy-associated genes in different
hepatocytes. It was also shown that the overexpression of SREBP-2
partially alleviated the inhibited autophagic activity in
lipid-overloaded hepatocytes. Based on these results, the present
study provided novel insights for potential NAFLD therapies.
Materials and methods
Cell culture and treatment
The HepG2 human cell line was obtained from the
Department of Infectious Diseases of the Union Hospital of Wuhan
(Wuhan, China). The HepG2 cells were cultured in DMEM supplemented
with 10% fetal bovine serum (both from Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 37°C in a humidified
atmosphere containing 5% CO2. HL-7702 cells (Cell Bank
of the Chinese Academy of Sciences, Shanghai, China) were cultured
in RPMI-1640 medium supplemented as above. In vitro models
of hepatic steatosis were induced by treating the cells with PA at
the indicated times (0, 6, 12 and 24 h) and concentrations (0, 100,
200 and 400 µM). Briefly, a 100-mM stock solution of PA
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was prepared in 0.1
N NaOH, and adjusted to 10 mM PA with fatty acid-free BSA solution
(Sigma-Aldrich; Merck KGaA). The PA-BSA complex was then filtered
and added to complete culture medium to achieve a final
concentration of 400 µM. Cells treated with autolysosome
inhibitor CQ (50 µM) were used as a positive control of
autophagosome accumulation. All experimental procedures were
approved by the Ethics Committee of Tongji Medical College,
Huazhong University of Science and Technology (Wuhan, China).
Overexpression and silencing of
SREBP-2
Firstly, a pcDNA3.1+ expression vector containing
the human cDNA of SREBP-2 (pcDNA3-1-14GS0029MU-8) was designed and
synthesized by Invitrogen; Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). For RNA silencing, the recombinant plasmid
pcDNA6.2-GW/EmGFP-miR, expressing a cytomegalovirus promoter-driven
micro155 short hairpin RNA (shRNA) targeting SREBP-2 (SREBP-2
shRNA), was constructed by Invitrogen (Thermo Fisher Scientific,
Inc.). The sequences are shown in Table I.
 | Table IRecombinant plasmid
pcDNA6.2-GW/EmGFP-microRNA sequences. |
Table I
Recombinant plasmid
pcDNA6.2-GW/EmGFP-microRNA sequences.
| Gene | Sequence
(5′-3′) |
|---|
| MR0003-1F |
TGCTGAGGACATTCTGATTAAAGTCCGTTTTGGCCACTGACTGACGGACTTTACAGAATGTCCT |
| MR0003-1R |
CCTGAGGACATTCTGTAAAGTCCGTCAGTCAGTGGCCAAAACGGACTTTAATCAGAATGTCCTC |
| MR0003-2F |
TGCTGTTCCGGTGCCTCCAGAAGGTGGTTTTGGCCACTGACTGACCACCTTCTAGGCACCGGAA |
| MR0003-2R |
CCTGTTCCGGTGCCTAGAAGGTGGTCAGTCAGTGGCCAAAACCACCTTCTGGAGGCACCGGAAC |
| MR0003-3F |
TGCTGTTCAAAGCCTGCCTCAGTGGCGTTTTGGCCACTGACTGACGCCACTGACAGGCTTTGAA |
| MR0003-3R |
CCTGTTCAAAGCCTGTCAGTGGCGTCAGTCAGTGGCCAAAACGCCACTGAGGCAGGCTTTGAAC |
| MR0003-4F |
TGCTGTCACCAAGGACTCTATAGCTCGTTTTGGCCACTGACTGACGAGCTATAGTCCTTGGTGA |
| MR0003-4R |
CCTGTCACCAAGGACTATAGCTCGTCAGTCAGTGGCCAAAACGAGCTATAGAGTCCTTGGTGAC |
Cell transfection
The plasmids were transfected into HepG2 and HL-7702
cells according with Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Briefly, prior to transfection, the cells were seeded at
1×106 per well in a 6-well plate of complete medium with
serum and antibiotics (100 U/ml penicillin G and 100 µg/mg
streptocycin; Gibco; Thermo Fisher Scientific, Inc.). After 24 h,
the cells were transfected with 4 µg of plasmids and 10
µl of Lipofectamine. Following incubation for 4–6 h, the
6-well plates were placed in 2 ml medium containing 10% FBS in a 5%
CO2 incubator overnight. GFP fusion proteins were
observed under a laser scanning microscope system (Olympus
Corporation, Tokyo, Japan).
Western blot analysis
Cells were harvested and lysed in ice-cold RIPA
lysis buffer (Applygen Technologies, Inc., Beijing, China) for 30
min before centrifugation at 12,000 × g for 15 min at 4°C. The
protein concentration of the supernatants was quantified using the
BCA Protein Assay Kit (Applygen Technologies, Inc., Beijing,
China). Whole-cell lysates (30–50 µg) were subjected to
SDS-PAGE (10 or 12%), followed by transfer onto polyvinylidene
difluoride membranes. The membranes were incubated with
anti-microtubule-associated protein 1 light chain 3β (LC3B)
antiserum for human (cat. no. 2775; 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-SREBP-2 (cat. no.
sc-5603; 1:100; Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
anti-P62 (cat. no. 7758; 1:1,000; ABclonal Biotech Co., Ltd.,
Cambridge, MA, USA) and anti-GAPDH (cat. no. 012; 1:1,000; AntGene
Biotech Co., Ltd., Hubei, China) as primary antibodies at 4°C
overnight. Following incubation with horseradish
peroxidase-conjugated secondary antibodies (cat. no. 020; 1:5,000;
AntGene Biotech Co., Ltd.) for 1 h at room temperature. The
immunoreactive bands were visualized by enhanced chemiluminescence
using an ECL western blotting starter kit (Thermo Fisher
Scientific) and imaged on a chemiluminescence gel imaging system
(FluorChem FC3; ProteinSimple, San Jose, CA, USA). Gray-scale
values of various bands were analyzed using the AlphaView SA
software version 3.4.0 (ProteinSimple).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from human hepatic cells with
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) and was reverse
transcribed using PrimeScript™ RT Master mix (Takara Bio, Inc.,
Otsu, Japan). The mRNA for autophagic genes was estimated via
RT-qPCR analysis using a Quantifast SYBR-Green PCR kit (Qiagen,
Inc., Valencia, CA, USA). Briefly, RT-PCR was performed using
SYBR-Green PCR Master mix in a 10 µl volume containing 5
µl SYBR Green, 1 µl cDNA, 0.5 µl forward
primer, 0.5 µl reverse primer and 3 µl
ddH2O. The RT-PCR cycling parameters were 95°C for 10
min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min.
The RT-qPCR analysis was performed on an ABI StepOne™ system. Gene
expression was analyzed in triplicate and normalized to GAPDH. The
relative expression levels were analyzed by the 2−ΔΔCq
method as previously described (18). The primer sequences used for
RT-qPCR analysis are listed in Table
II.
| Table IIPrimers used for autophagic
genes. |
Table II
Primers used for autophagic
genes.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| SREBP-2 |
CCCTTCAGTGCAACGGTCATTCAC |
TGCCATTGGCCGTTTGTGTC |
| LC3B |
AAGGCGCTTACAGCTCAATG |
CTGGGAGGCATAGACCATGT |
| ATG5 |
AAAGATGTGCTTCGAGATGTGT C |
ACTTTGTCAGTTACCAACGTCA |
| ATG7 |
CAGTTTGCCCCTTTTAGTAGTGC CC |
AGCCGATACTCGTTCAGC |
| Beclin1 |
GGTGTCTCTCGCAGATTCATC |
TCAGTCTTCGGCTGAGGTTCT |
| GAPDH C |
TGGGCTACACTGAGCACC |
AAGTGGTCGTTGAGGGCAATG |
Oil Red O staining and triglyceride
assay
Oil Red O staining was performed according to a
previously described procedure (19). Representative photomicrographs
were captured at 400× magnification using a system incorporated
into the microscope. Intracellular triglycerides were assayed using
a triglyceride assay kit (GPO-POD; Applygen Technologies, Inc.,
Beijing, China), according to the manufacturer's recommended
protocol.
Autophagic flux assay using tandem
fluorescent-tagged LC3
Human hepatic cells were transfected with
GFP-mCherry-LC3 plasmid DNA (Changsha Yingrun Biotechnology Co.,
Ltd., Changsha, China) according to the manufacturer's protocol.
After 24 h, the cells were then treated with the different
treatments. The cells were then washed three times with PBS and
fixed with 4% paraformaldehyde for 30 min. Images of the cells were
captured and observed under a confocal laser scanning microscope
(Nikon Corporation, Tokyo, Japan).
Statistical analysis
The results were analyzed using one-way analysis of
variance with the SPSS 19.0 statistical software package (IBM SPSS,
Armonk, NY, USA). All values are expressed as the mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference.
Results
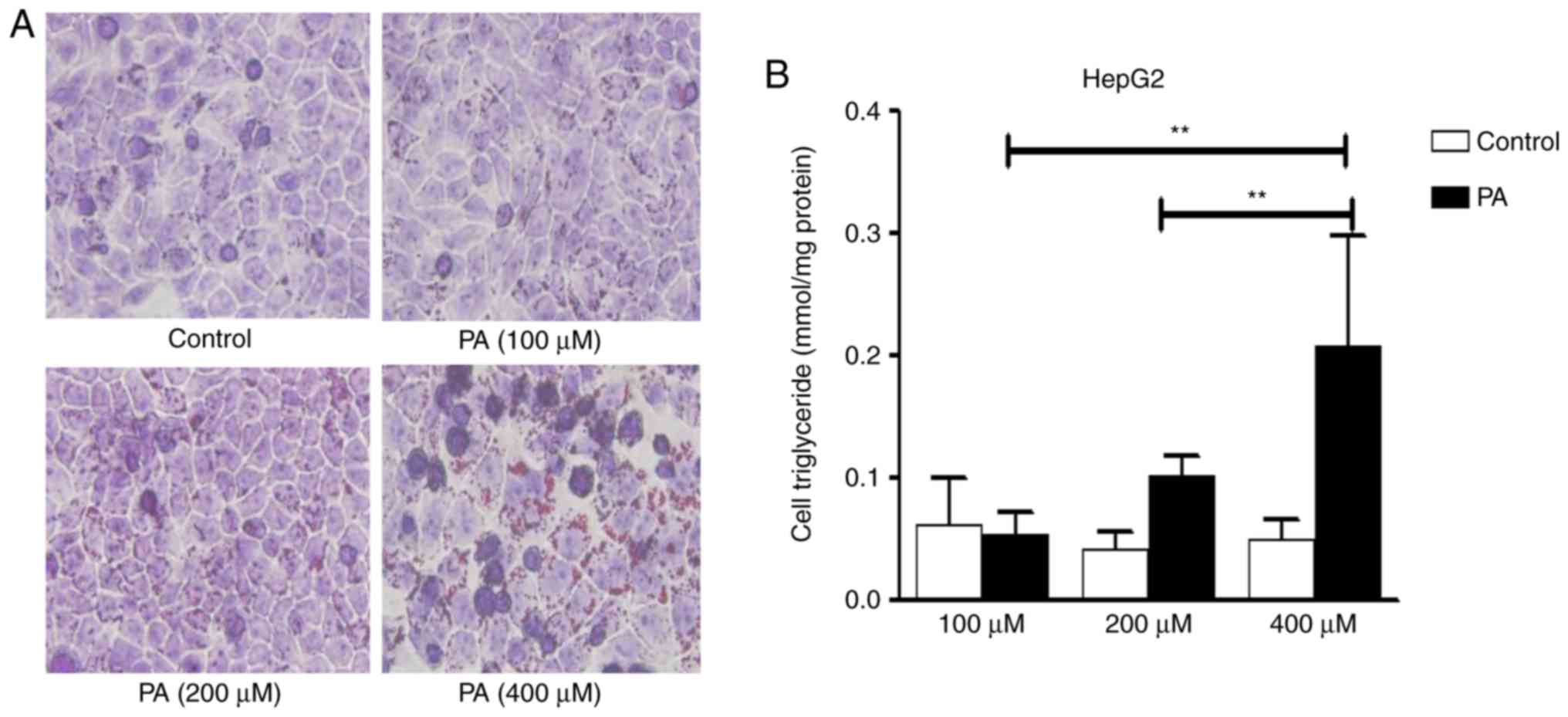
Establishment of a cellular steatosis by
treatment with PA
In order to determine the dysfunction of autophagy
in the pathologic process of NAFLD, a steatosis model was
established with PA treatment at the indicated times and
concentrations. Following PA treatment for 24 h, an increased
number of intracellular lipid droplets were observed in hepacytes,
compared with the control (Fig.
1A). The quantitative analysis of hepatic triglycerides showed
a marked increase in cellular triglyceride levels with PA (400
µM), compared with the control (Fig. 1B). With the increase of PA
concentrations, cell viability gradually decreased. PA (400
µM) led to significant lipid accumulation in hepatocytes
with no marked cell death under the microscope. Therefore, this
concentration PA (400 µM) was used for further
experiments.
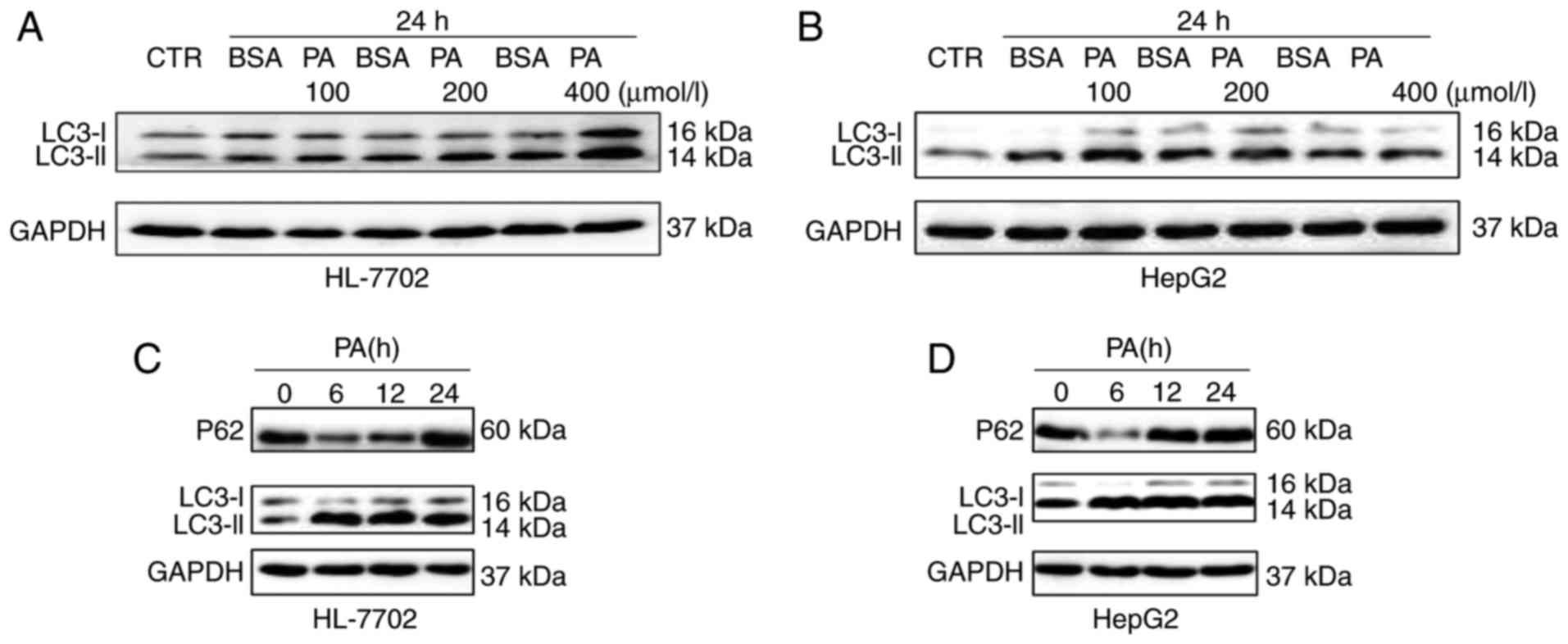
Effects of PA on autophagy in
hepatocytes
To investigate the association between autophagy and
NAFLD at the cellular level, NAFLD cell models were first
established by incubating HL-7702 and human HepG2 hepatoma cells
with PA. In a previous study, PA was shown to induce autophagy in
pancreatic β-cells (20). By
contrast, it is suggested that only oleic acid, not PA, triggers
autophagic responses in hepatocytes (21). However, the effects of PA on
autophagy remain controversial and autophagic dysfunction in
different models of cellular steatosis remains to be elucidated. In
the present study, to elucidate the effects of PA on autophagic
function in different cells (HL-7702 and HepG2), hepatocytes were
treated with PA at different concentrations and different times. As
shown in Fig. 2A, the induction
of PA caused a significant increase in the levels of LC3-II in a
dose-dependent manner. Similar results were observed in HepG2
cells; however, the expression levels of LC3-II were decreased at a
fixed 24 h time point with 400 µM dose (Fig. 2B). The data showed that PA
treatment caused a marked increase in the levels of LC3-II for up
to 24 h in the HL-7702 cells (Fig.
2C). The protein levels of p62, an autophagy substrate known to
be degraded as autophagic flux progresses, were also detected. As
shown in Fig. 2C, a marked
decrease in p62 was detected at 6 h, however, an increase in p62
was detected at 24 h. The expression of LC3-II and P62 were also
examined in HepG2 cells at different time points. These results
showed that PA treatment increased the levels of LC3-II at 6–12 h,
but decreased levels of LC3-II at 24 h. In addition, a marked
decrease in p62 was detected at 6 h, whereas the levels of P62 were
increased at 12–24 h in HepG2 cells (Fig. 2D). These results indicated that PA
may enhance autophagic function at the early stage in HL-7702 and
HepG2, whereas long-term treatment with PA may suppress autophagic
function in the above hepatocytes.
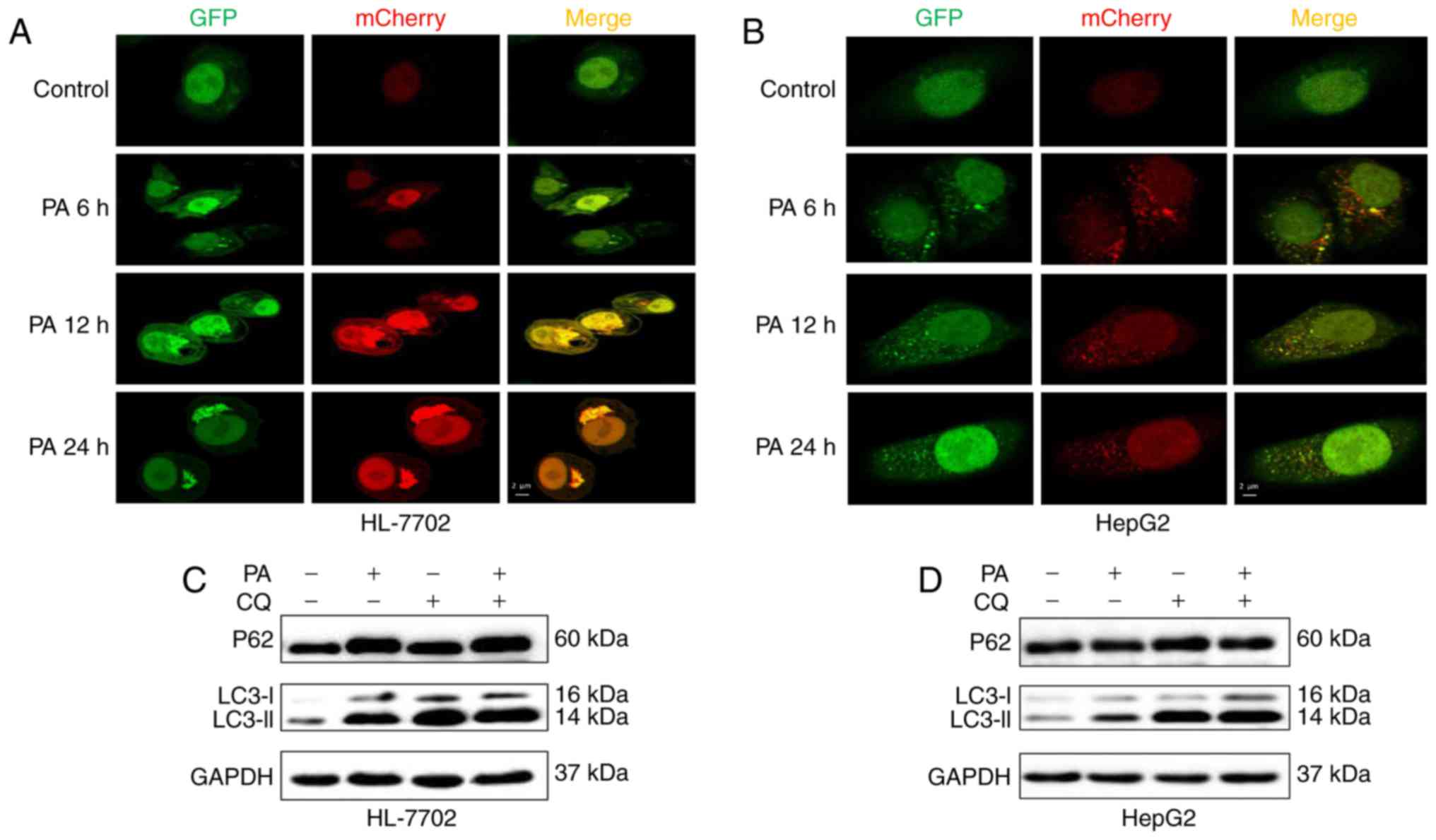
To further monitor autophagic flux induced by PA in
HepG2 and HL-7702 cells, a tandem fluorescent-tagged LC3
(GFP-mCherry-LC3) expression plasmid was used. A low level of basal
autophagy was observed in the hepatocytes (HL-7702 and HepG2 cells)
without PA, as indicated by the low number of yellow and red puncta
(Fig. 3A and B). However, when
the hepatocytes were exposed to PA at an early time-point (6–12 h),
yellow and red punctae were increased, suggesting that autophagic
flux was increased and the accumulation of LC3-II induced by PA
resulted from the induction of autophagosome formation (Fig. 3A and B). However, following
treatment with PA for 24 h, the HL-7702 cells exhibited only
autophagosomal LC3-II (yellow punctae without a concomitant
increase in red punctae), whereas HepG2 cells showed decreased
autophagosomes (yellow punctae) and autolysosomes (red puncta;
Fig. 3A and B), indicating that
the decrease in the numbers of autophagosomes may be due to
decreased autophagosome formation, but not accelerated
turnover.
Autophagosome accumulation may also be due to
lysosomal dysfunction (22). To
determine whether PA impaired autophagosome turnover, the present
study measured autophagic flux via analyzing LC3 turnover using CQ,
which inhibits autophagosome flux at lysosomal degradation, thereby
triggering the accumulation of LC3-II and P62 (23). The combination of PA and CQ
induced an increase in the levels of LC3-II relative to the
control, but no increased expression of p62 or LC3-II, compared
with the PA-treated HL-7702 cells (Fig. 3C), indicating that PA impaired
autophagosome turnover, but did not induce autophagic flux.
Consistent with previous studies, PA inhibited autophagic flux by
impairing lysosomal enzyme activity and reducing autophagosome
clearance (14,24). However, co-treatment with PA and
CQ increased the expression of p62 and induced a higher
accumulation of LC3-II, compared with the PA-treated HepG2 cells
(Fig. 3D), suggesting that
PA-induced autophagic dysfunction was attributed to decreased
autophagosome formation, but not the inhibition of
autophagolysosomal maturation. These effects were coincident with
decreased autophagy in hepatocytes, as shown in fluorescent
microscopy images (Fig. 3A and
B).
Taken together, the above in vitro
experiments suggested that PA induced autophagy in the early stage
of saturated fatty acid loading, whereas the long-term stimulus of
PA impaired autophagic flux or inhibited autophagic activity. The
differential effects of PA on autophagy may be associated with the
duration of PA treatment and the cell type. Of note, it was shown
that different cellular models of NAFLD exhibited different
autophagic dysfunction.
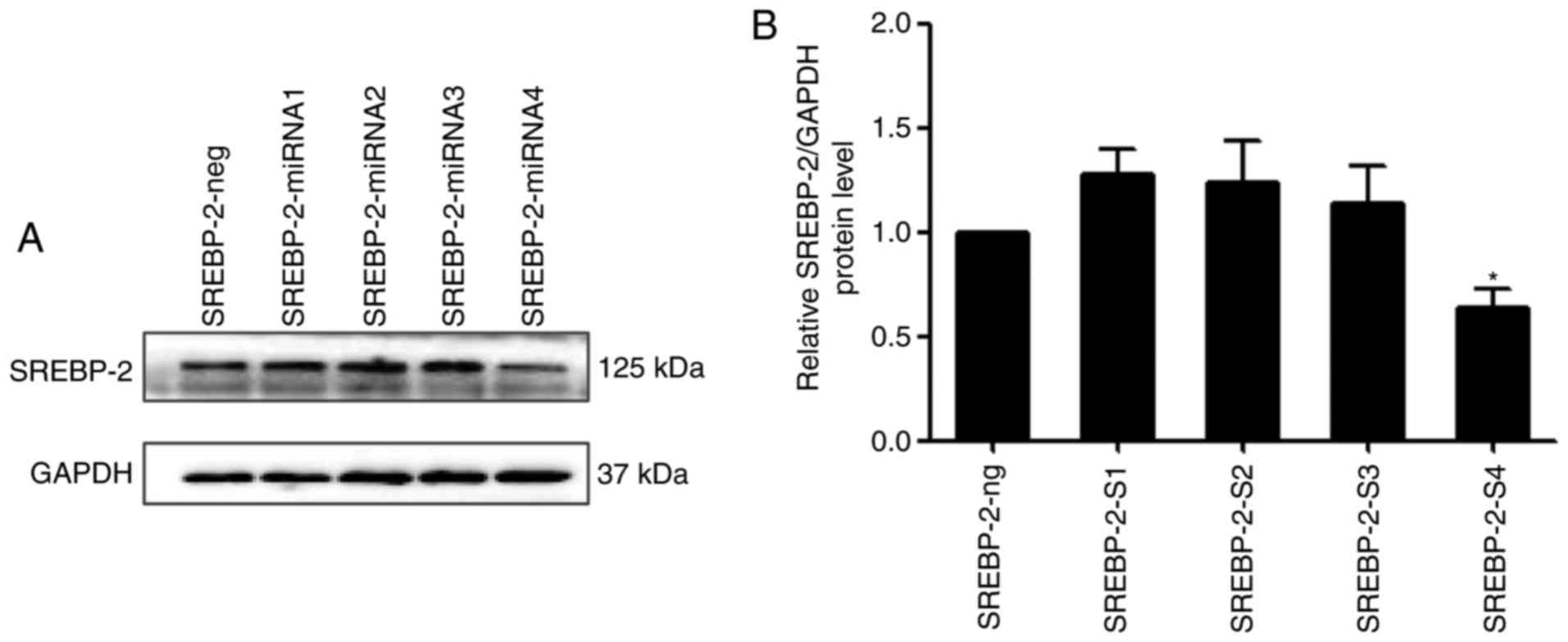
SREBP-2 regulates autophagy-related gene
expression in human liver cells
A previous study demonstrated that SREBP-2 activates
the expression of key autophagic genes during cellular lipid
depletion in 293 cells and HeLa cells (17). However, whether SREBP-2 also
regulates autophagy in hepatocytes remains to be elucidated. To
investigate the role of SREBP-2 in autophagy
pcDNA6.2-GW/EmGFP-miR-SREBP-2 and pcDNA3.1-SREBP-2 expression
vectors were constructed, with four miRNAs targeting the SREBP-2
gene. Immunoblot analysis showed that the inhibitory efficiency of
the fourth pair of miRNAs was more marked, compared with that of
the other three pairs of miRNAs, therefore SREBP-2-4miRNA was used
in the subsequent experiments (Fig.
4A and B).
Loss- and gain-of-function experiments were
performed to ascertain whether SREBP-2 regulated autophagy in
hepatocytes under normal conditions. As shown in Fig. 5A, the knockdown of SREBP-2
efficiently reduced the gene expression of SREBP-2. SREBP-2-miR
reduced the expression of autophagic genes. By contrast, the
overexpression of SREBP-2 increased the expression of these genes
(Fig. 5B). These results
indicated that SREBP-2 activated the expression of autophagic genes
in HL-7702 cells. Similar results were obtained via the transient
overexpression or silencing of SREBP-2 in HepG2 human hepatic cells
(Fig. 5C and D). These gain- and
loss-of function data suggested that the biogenesis of
autophagosomes may be regulated by SREBP-2.
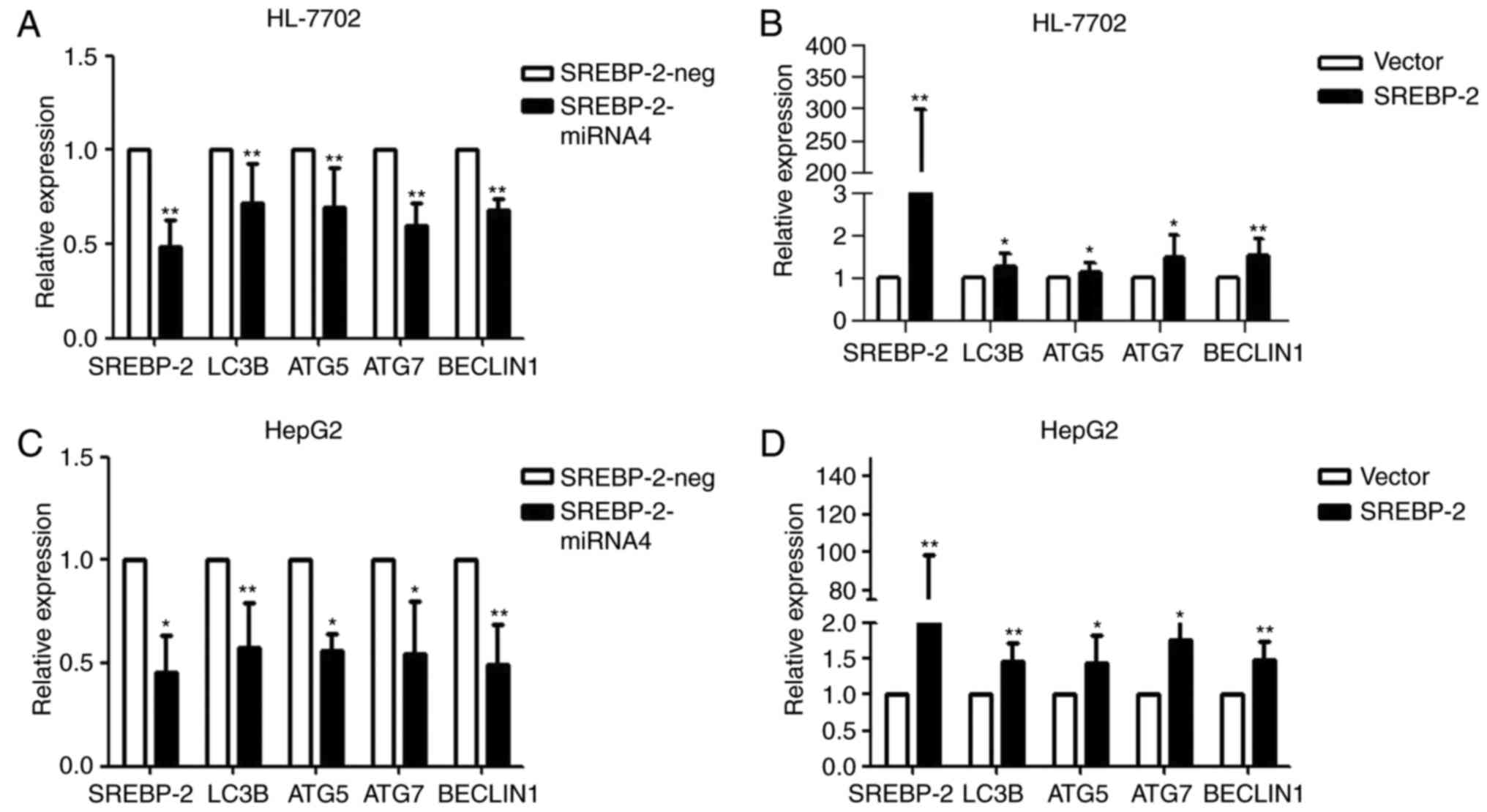 | Figure 5SREBP-2 regulates autophagy-related
gene expression in hepatocytes. HL-7702 cells were transfected with
the (A) modified SREBP-2-miR vector or negative control vector and
(B) SREBP-2-overexpression plasmid vector or empty vector. After 24
h, the cells were harvested for RNA. The mRNA levels of LC3B, ATG5,
ATG7 and Beclin1 were analyzed using RT-qPCR analysis. GAPDH mRNA
was used as an internal control. Relative expression is plotted.
HepG2 cells were harvested for RNA following transfection for 24 h
with (C) modified SREBP-2-miR vector or negative control vector and
(D) SREBP-2-overexpression plasmid vector or empty vector. mRNA
levels of LC3B, ATG5, ATG7 and Beclin1 were analyzed using RT-qPCR
analysis. GAPDH mRNA was used as an internal control.
*P<0.05 and **P<0.01, vs.control.
SREBP-2, sterol regulatory element binding protein-2; miR,
microRNA; ATG, autophagy-related; LC3B, microtubule-associated
protein 1 light chain 3β; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
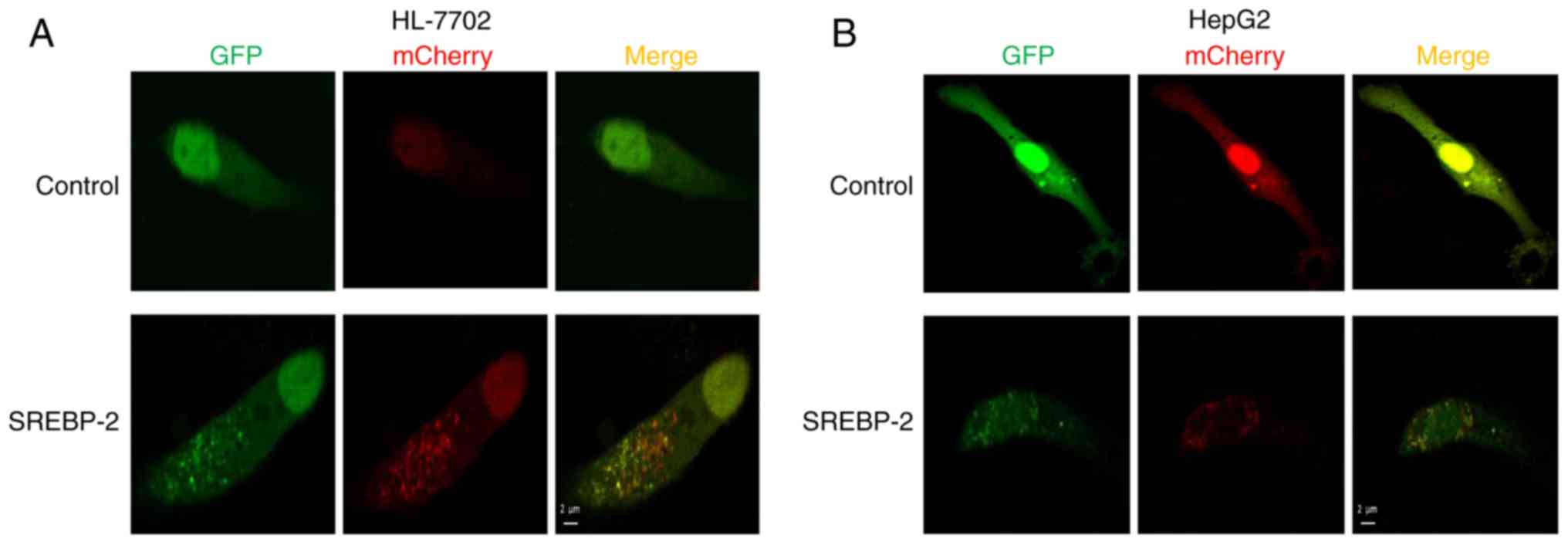
To further investigate the effect of the
overexpression of SREBP-2 on autophagic influx in hepatocytes, the
cells were co-transfected by GFP-mCherry-LC3 and SREBP-2 expression
plasmids or mock plasmids, separately. Confocal fluorescent
microscopy showed that the number of autophagosomal LC3B-II (yellow
puncta on co-localization) and autolysosomal LC3B-II (red) were
increased in hepatocytes transfected with the expressed vector of
SREBP-2, compared with the control (Fig. 6A and B). Collectively, these
results demonstrated that SREBP-2 regulated autophagy in
hepatocytes.
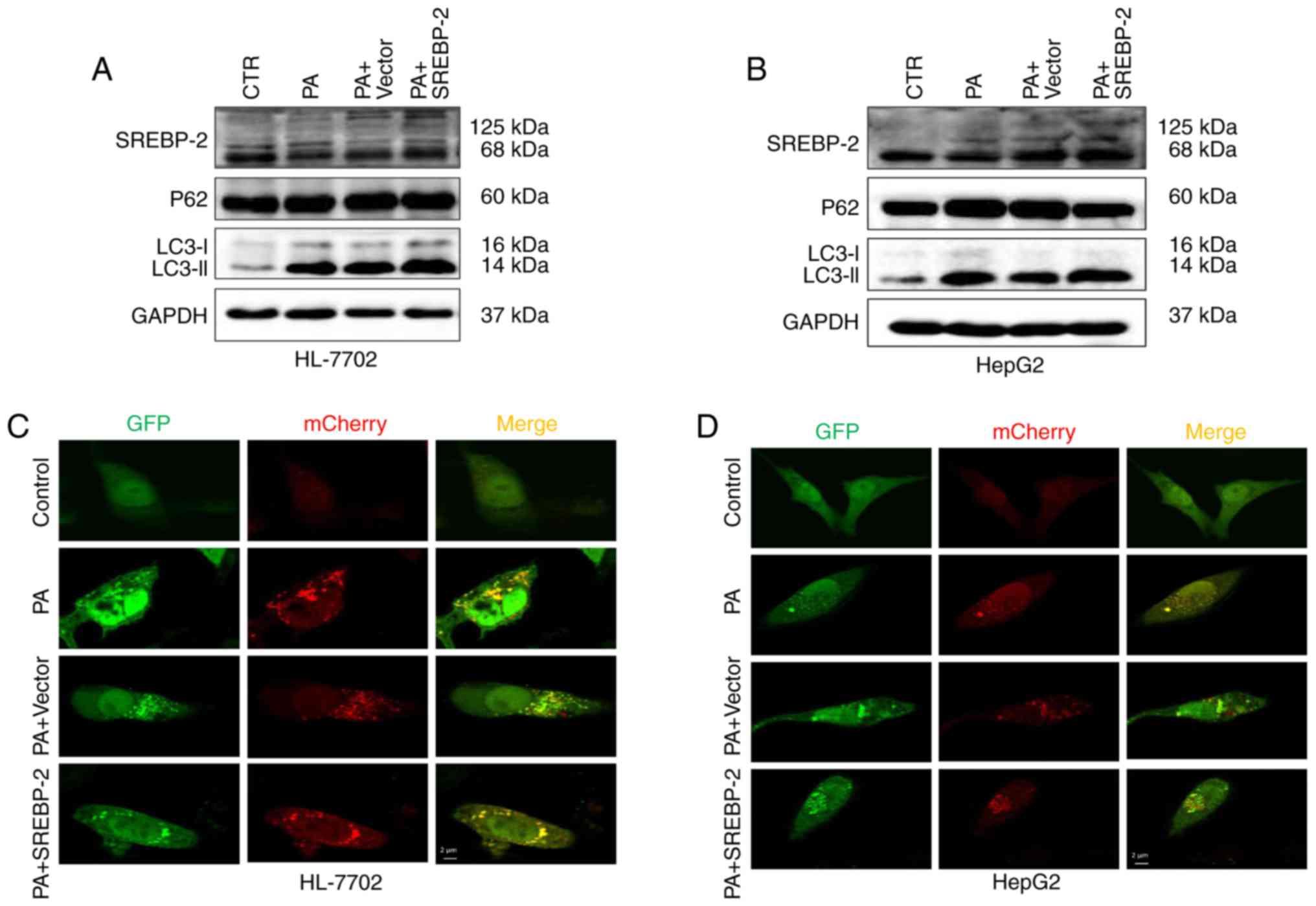
Overexpression of SREBP-2 partly restores
autophagic function in PA-induced steatotic HepG2 cells, but not in
HL-7702 cells
As dysfunctional autophagy in hepatocytes was
observed, the present study the intracellular expression of SREBP-2
was increased via transfection with an SREBP-2-bearing plasmid to
ascertain whether the overexpression of SREBP-2 restored or
alleviated impaired autophagic function in fat-overloaded
hepatocytes. The results showed that the expression of SREBP-2 in
cells transfected with the SREBP-2-bearing vector was higher,
compared with the cells transfected with the control vector
(Fig. 7A). The overexpression of
SREBP-2 marginally increased the levels of LC3-II in HL-7702 cells
treated with PA, compared with the control, whereas no significant
change in the protein expression levels of P62 were observed
(Fig. 7A). Furthermore, using
confocal fluorescence microscopy, it was observed that the HL-7702
cells exhibited only autophagosomal LC3B-II, but no autolysosomal
LC3B-II (Fig. 7C). These data
indicated that the overexpression of SREBP-2 did not restore the
inhibition of autophagic flux, but increased the number of
autophagosomes detected by immunoblotting of the LC3-II protein. By
contrast, the overexpression of SREBP-2 marginally increased the
levels of LC3-II in HepG2 cells with a concomitant decline in P62
(Fig. 7B). Confocal imaging was
also performed to determine the total number of autophagosomes and
autolysosomes. The results revealed that the overexpression of
SREBP-2 induced an increase in the number of autophagosomes and
autolysosomes (Fig. 7D).
Therefore, the overexpression of SREBP-2 partly alleviated the
inhibited autophagic function of HepG2 cells rather than the
inhibition of autophagic flux in HL-7702 cells.
Discussion
SREBPs are basic helix-loop-helix-leucine zipper
transcription factors, which are key in maintaining lipid
homeostasis or lipid metabolism (25). There are three mammalian SREBP
isoforms: SREBP-1a, SREBP-1c and SREBP-2 (25,26). SREBP-1a and SREBP-1c are involved
in fatty acid metabolism, whereas SREBP-2 primarily regulates
cholesterol biosynthesis (27). A
previous study showed that SREBP-2 regulates autophagy in HeLa
cells (17). However, whether
SREBP-2 regulates autophagic function in hepatocytes remains to be
elucidated.
Autophagy, a self-catabolic process, is a conserved
lysosomal degradative pathway, which degrades intracellular
organelles to maintain cellular homeostasis under stress conditions
(28,29). Autophagy is involved in
development, differentiation, survival and homeostasis (30,31). Increasing evidence has indicated
that autophagy regulates lipid metabolism (8). Studies have further demonstrated
that autophagic dysfunction may lead to insulin resistance and ER
stress (11,12,30). In addition, mice with chronic
obesity or insulin resistance, which are prone to NAFLD, have
notably decreased hepatic autophagic activity (12,32). These findings suggest that
alterations in autophagy may have a relevant pathogenic role in
NAFLD. Consequently, therapeutic strategies aimed at the
restoration of autophagic function may provide a novel approach to
preventing NAFLD.
To confirm the derangement of autophagy in the
pathology of NAFLD, the present study used PA to establish an in
vitro model of steatosis. Reports on the effect of PA on
autophagy in different cell models of NAFLD have been
controversial. In a previous study, Mei et al reported that
PA does not induce autophagy in hepatocytes, whereas PA
significantly induced apoptosis (21). By contrast, another study showed
that the levels of autophagy were elevated in hepatoma cells upon
exposure to PA, and PA-induced autophagy was identified to be
independent of the regulation of mammalian target of rapamycin
(mTOR) (33). In the present
study, it was identified that PA induced the activation of
autophagy, but also impaired autophagic function. The data showed
that PA induced autophagy in the early stage of SFA loading,
whereas long-term PA exposure impaired autophagic flux or inhibited
autophagic activity. In the HL-7702 cellular steatosis model, a
longer period of PA treatment inhibited autophagic flux and
autophagosome degradation. By contrast, in the HepG2 cellular
model, a short period of PA treatment activated autophagy, whereas
a prolonged duration of PA inhibited autophagic activity. These
results showed that two cellular steatosis models exhibited
different autophagic dysfunctions: Inhibition of autophagic flux or
subdued autophagic activity.
Although the exact mechanisms affecting this
different outcome remain to be elucidated, several potential
explanations may account for such conflicting results. One
potential mechanistic explanation for the difference is that
elevated ER stress triggers the disruption of autophagic flux.
Accumulating data indicates that ER stress is a potent trigger of
autophagy (10,34,35). A study by González-Rodríguez et
al (10) identified that
short-term treatment with PA triggered the activation of the
unfolding protein response and autophagic flux. By contrast,
prolonged treatment with PA induced ER stress and cell death, in
addition to the inhibition of autophagic flux (10). These results indicated that the
induction of autophagy by PA is dependent on ER stress and the mTOR
signaling pathway, however, sustained or marked ER stress due to
lipid overload may increase the accumulation of unfolded proteins,
triggering liver injury through the disruption of the autophagic
flux and leading to apoptotic cell death (10). Similar results were observed in
another study. Park et al (36) identified that PA promoted mTOR
complex 1-dependent autophagy at an early stage, but prolonged
treatment with PA led to damage of autophagic flux and an
aggravation of apoptosis in H9c2 cells. However, certain studies
have shown that PA induces the activation of autophagy via the
protein kinase C-mediated signaling pathway independent of the ER
stress and mTOR signaling pathways (33,37). The effects of PA on autophagy
remains controversial and may be cell-type dependent (21,33,36); however, accumulating evidence
supports that the activation of autophagy at an early stage confers
a pro-survival effect against lipotoxicity-induced cell death
(38), whereas a longer period of
exposure to PA leads to impaired autophagic function and apoptotic
cell death. It has been reported that the autophagic process is a
cell pro-survival mechanism (29)
and that the evolution of NAFLD may be associated with the dynamic
regulation of autophagy (30).
Understanding the mechanisms of the impairment of hepatic autophagy
has provided key insights into the pathogenesis of NAFLD. The
modulation of autophagy may offer therapeutic potential to prevent
the progression of NAFLD. Previous findings have demonstrated that
SREBP-2 binds to and activates the expression of several autophagic
genes, whereas the knockdown of SREBP-2 suppresses autophagosome
formation and triglyceride mobilization in 293 and HeLa cells
(17). In the present study,
gain- and loss-of-function data demonstrated that SREBP-2 regulated
autophagy-related gene expression in HepG2 cells and HL-7702 cells.
In addition, the overexpression of SREBP-2 increased the number of
autophagosomes. These results indicated that the initiation of
autophagy may be regulated by SREBP-2. However, the mechanism of
SREBP-2 in the regulation of autophagy remains to be elucidated. In
a previous study, a genome-wide binding/ChIP-Seq analysis of
SREBP-2 revealed that SREBP-2 occupied the promoters of several
genes involved in autophagy (17). The effect of SREBP-2 on autophagy
has received less attention, and its underlying mechanisms remain
to be fully elucidated. There has been previous novel insight into
the mechanisms; Seo et al (39) identified that SREBP-2 directly
activated the gene expression of patatin-like phospholipase
domain-containing enzyme 8 (PNPLA8), and PNPLA8 increased autophagy
through inhibiting the phosphorylation of unc-51-like kinase 1
downstream of target of rapamycin complex 1 (TORC1). Therefore, the
SREBP-2/PNPLA8 axis may regulate the initiation of autophagy
through the inhibition of TORC1. These findings suggested that
SREBP-2 may regulate autophagy by involving multiple signaling
pathways. However, whether the modulation of SREBP-2 restored
impaired autophagic activity in the steatotic hepatocytes remains
to be fully elucidated. Subsequently, the present study examined
the effect of the overexpression of SREBP-2 on autophagic function
in fat-overloaded hepatic cells. In the HL-7702 cellular steatosis
model, it was identified that the overexpression of SREBP-2
increased the levels of LC3 II, but not the expression of P62,
suggesting that the modulation of SREBP-2 did not restore the
inhibited autophagic flux. However, the overexpression of SREBP-2
resulted in an increase in the levels of LC3-II and a decrease in
the levels of P62 in PA-treated HepG2 cells, indicating that the
overexpression of SREBP-2 restored the inhibited autophagic
activity. Of note, a previous study reported SREBP-2 did not affect
global autophagic flux (17).
Therefore, it was hypothesized that the overexpression of SREBP-2
may only be involved in the initial stage of autophagy, with no
effect on the late stage of autophagy, including autophagosomes for
fusion with endosomes/lysosomes and metabolite degradation.
The present study provided evidence that, in the
PA-induced cellular steatosis model, PA induced autophagy and
enhanced autophagic flux at the early stage, however, a longer
period of exposure to PA led to impaired autophagic flux. Under the
experimental conditions, two hepatic cell lines presented with
different autophagic dysfunction in response to lipid overload. The
results confirmed that SREBP-2 regulated the expression of hepatic
autophagic genes in normal conditions. Additionally, it was
identified that the overexpression of SREBP-2 partially restored
the inhibited autophagic activity in lipid-overloaded hepatocytes.
Although the role of SREBP-2 in the regulation of hepatocyte
lipogenesis and autophagy requires further investigation, the
present study demonstrated the importance of the role of SREBP-2 in
autophagy in hepatocytes.
In conclusion, the present study demonstrated that
SREBP-2 regulated the expression of autophagy-related genes in
hepatocytes. In addition, the overexpression of SREBP-2 partly
enhanced the inhibited autophagic activity in lipid-overloaded
hepatocytes. Overall, the restoration of aberrant autophagic
function by the modulation of SREBP-2 may be a potential
therapeutic target for the prevention and treatment of NAFLD.
Glossary
Abbreviations
Abbreviations:
|
NAFLD
|
nonalcoholic fatty liver disease
|
|
SREBP-2
|
sterol regulatory element binding
protein-2
|
|
PA
|
palmitic acid
|
|
CQ
|
chloroquine
|
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81370525 and 81170401).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Brunt EM, Wong VW, Nobili V, Day CP,
Sookoian S, Maher JJ, Bugianesi E, Sirlin CB, Neuschwander-Tetri BA
and Rinella ME: Nonalcoholic fatty liver disease. Nat Rev Dis
Primers. 1:150802015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang FS, Fan JG, Zhang Z, Gao B and Wang
HY: The global burden of liver disease: The major impact of China.
Hepatology. 60:2099–2108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheung O and Sanyal AJ: Recent advances in
nonalcoholic fatty liver disease. Curr Opin Gastroenterol.
26:202–208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Neuschwander-Tetri BA: Hepatic
lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis:
The central role of nontriglyceride fatty acid metabolites.
Hepatology. 52:774–788. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li S, Li J, Shen C, Zhang X, Sun S, Cho M,
Sun C and Song Z: Tert-Butylhydroquinone (tBHQ) protects
hepatocytes against lipotoxicity via inducing autophagy
independently of Nrf2 activation. Biochim Biophys Acta. 1841:22–33.
2014. View Article : Google Scholar
|
|
6
|
Wu J, Wu JJ, Yang LJ, Wei LX and Zou DJ:
Rosiglitazone protects against palmitate-induced pancreatic
beta-cell death by activation of autophagy via 5′-AMP-activated
protein kinase modulation. Endocrine. 44:87–98. 2013. View Article : Google Scholar
|
|
7
|
Ryter SW, Cloonan SM and Choi AM:
Autophagy: A critical regulator of cellular metabolism and
homeostasis. Mol Cells. 36:7–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fukuo Y, Yamashina S, Sonoue H, Arakawa A,
Nakadera E, Aoyama T, Uchiyama A, Kon K, Ikejima K and Watanabe S:
Abnormality of autophagic function and cathepsin expression in the
liver from patients with non-alcoholic fatty liver disease. Hepatol
Res. 44:1026–1036. 2014. View Article : Google Scholar
|
|
10
|
González-Rodríguez Á, Mayoral R, Agra N,
Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J,
Lo Iacono O, Corazzari M, Fimia GM, et al: Impaired autophagic flux
is associated with increased endoplasmic reticulum stress during
the development of NAFLD. Cell Death Dis. 5:e11792014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang L, Li P, Fu S, Calay ES and
Hotamisligil GS: Defective hepatic autophagy in obesity promotes ER
stress and causes insulin resistance. Cell Metab. 11:467–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi
J, Liu Z and Cao W: Hepatic autophagy is suppressed in the presence
of insulin resistance and hyperinsulinemia: Inhibition of
FoxO1-dependent expression of key autophagy genes by insulin. J
Biol Chem. 284:31484–31492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Czaja MJ: Functions of autophagy in
hepatic and pancreatic physiology and disease. Gastroenterology.
140:1895–1908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koga H, Kaushik S and Cuervo AM: Altered
lipid content inhibits autophagic vesicular fusion. FASEB J.
24:3052–3065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Inami Y, Yamashina S, Izumi K, Ueno T,
Tanida I, Ikejima K and Watanabe S: Hepatic steatosis inhibits
autophagic proteolysis via impairment of autophagosomal
acidification and cathepsin expression. Biochem Biophys Res Commun.
412:618–625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park HW, Park H, Semple IA, Jang I, Ro SH,
Kim M, Cazares VA, Stuenkel EL, Kim JJ, Kim JS and Lee JH:
Pharmacological correction of obesity-induced autophagy arrest
using calcium channel blockers. Nat Commun. 5:48342014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seo YK, Jeon TI, Chong HK, Biesinger J,
Xie X and Osborne TF: Genome-wide localization of SREBP-2 in
hepatic chromatin predicts a role in autophagy. Cell Metab.
13:367–375. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔC T method. Methods. 25:402–408.
2001. View Article : Google Scholar
|
|
19
|
Pan X, Wang P, Luo J, Wang Z, Song Y, Ye J
and Hou X: Adipogenic changes of hepatocytes in a high-fat
diet-induced fatty liver mice model and non-alcoholic fatty liver
disease patients. Endocrine. 48:834–847. 2015. View Article : Google Scholar
|
|
20
|
Choi SE, Lee SM, Lee YJ, Li LJ, Lee SJ,
Lee JH, Kim Y, Jun HS, Lee KW and Kang Y: Protective role of
autophagy in palmitate-induced INS-1 beta-cell death.
Endocrinology. 150:126–134. 2009. View Article : Google Scholar
|
|
21
|
Mei S, Ni HM, Manley S, Bockus A, Kassel
KM, Luyendyk JP, Copple BL and Ding WX: Differential roles of
unsaturated and saturated fatty acids on autophagy and apoptosis in
hepatocytes. J Pharmacol Exp Ther. 339:487–498. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jaishy B, Zhang Q, Chung HS, Riehle C,
Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE and Abel ED:
Lipid-induced NOX2 activation inhibits autophagic flux by impairing
lysosomal enzyme activity. J Lipid Res. 56:546–561. 2015.
View Article : Google Scholar :
|
|
25
|
Osborne TF and Espenshade PJ: Evolutionary
conservation and adaptation in the mechanism that regulates SREBP
action: What a long, strange tRIP it's been. Genes Dev.
23:2578–2591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shao W and Espenshade PJ: Expanding roles
for SREBP in metabolism. Cell Metab. 16:414–419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wong TY, Lin SM and Leung LK: The flavone
luteolin suppresses SREBP-2 expression and Post-translational
activation in hepatic cells. PLoS One. 10:e01356372015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang K: Autophagy and apoptosis in liver
injury. Cell Cycle. 14:1631–1642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lavallard VJ and Gual P: Autophagy and
non-alcoholic fatty liver disease. Biomed Res Int. 2014:1201792014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Codogno P and Meijer AJ: Autophagy: A
potential link between obesity and insulin resistance. Cell Metab.
11:449–451. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tan SH, Shui G, Zhou J, Li JJ, Bay BH,
Wenk MR and Shen HM: Induction of autophagy by palmitic acid via
protein kinase C-mediated signaling pathway independent of mTOR
(mammalian target of rapamycin). J Biol Chem. 287:14364–14376.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yorimitsu T and Klionsky DJ: Endoplasmic
reticulum stress: A new pathway to induce autophagy. Autophagy.
3:160–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park M, Sabetski A, Kwan Chan Y, Turdi S
and Sweeney G: Palmitate induces ER stress and autophagy in H9c2
cells: Implications for apoptosis and adiponectin resistance. J
Cell Physiol. 230:630–639. 2015. View Article : Google Scholar
|
|
37
|
Cai N, Zhao X, Jing Y, Sun K, Jiao S, Chen
X, Yang H, Zhou Y and Wei L: Autophagy protects against
palmitate-induced apoptosis in hepatocytes. Cell Biosci. 4:282014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu J, Chang F, Li F, Fu H, Wang J, Zhang
S, Zhao J and Yin D: Palmitate promotes autophagy and apoptosis
through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun.
463:262–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim KY, Jang HJ, Yang YR, Park KI, Seo J,
Shin IW, Jeon TI, Ahn SC, Suh PG, Osborne TF and Seo YK:
Corrigendum: SREBP-2/PNPLA8 axis improves non-alcoholic fatty liver
disease through activation of autophagy. Sci Rep. 6:357322016.
View Article : Google Scholar
|