Introduction
Dengue is currently the most important
mosquito-borne viral pathogen that affects humans. Estimates
indicate that 3 billion people live in tropical and subtropical
regions where they are at risk of infection. There are reports of
~96 million symptomatic episodes and ~20,000 cases of mortality
each year (1). Dengue ranges
clinically from asymptomatic or self-limiting non-severe dengue
(with or without warning symptoms) to severe dengue, which is
characterized variously by severe plasma leakage, severe bleeding
or severe organ involvement (2).
The clinical presentation is difficult to predict, although the
presence of warning signs in the first day warrant strict
observation and medical intervention. The triage of patients who
are likely to require intensive care during hospitalization would
reduce the risk of mortality. The factors proposed for increased
risk of severe clinical manifestations remain poorly characterized.
Age, sex, social status, genetic background, sickle cell anemia,
uremia, bronchial asthma, allergies, hypertension, chronic renal
failure, concurrent bacterial infections, and diabetes mellitus may
adversely influence the clinical presentation of an infection
(3). The identification of
patients for intensive monitoring in a well-resourced
high-dependency unit with medical and nursing staff trained in
dengue and critical care management may improve clinical outcomes
(2).
Host factors and the genetic variability of the
virus are other crucial factors. Dengue virus (DENV) is a single
positive RNA filament, belonging to the genus Flavivirus (FV) and
constitutes four large groups or serotypes that infect humans.
These are classified as DENV-1, -2, -3 and -4. Despite the
remarkable genetic similarity between the four serotypes, these
viruses have epidemiological variability and specific immunological
response (3).
Like all FVs, DENV has a high rate of genetic
diversity, due to recombination and mutations at selective
pressures in its genome (2). For
this reason, each serotype has at least five genetic variants that
are sufficiently different to explain changing clinical and
epidemiological behavior (4).
Traditionally, severe forms of dengue, including dengue hemorrhagic
fever, are due to the phenomenon known as antibody dependent
enhancement, in which the presence of antibodies against a
determined serotype enhances viral replication within infected
cells in a second infection with a different serotype (5). However, it has been demonstrated
that certain strains of DENV are naturally aggressive and are able
to generate severe dengue from the first infection (6).
The prognosis of DENV infection is determined by a
balance between the rate of viral replication and the efficiency of
the immune system for viremia clearance (7). Innate immunity is crucial at the
beginning of infection and includes interferon (IFN)-γ action,
cytokine-mediated signaling, chemotaxis, and complement activity,
among others. The processes triggered by these molecules are
activated at days 0–1 following DENV infection and decline 3–4 days
thereafter. The upregulation of this early innate immune response
generates a reduction in DENV replication during days 0–3 (6,7).
Deficiencies in specific and non-specific immune-driven antiviral
responses result in unrestricted DENV replication and dissemination
in the host, with the possible appearance of severe clinical
manifestations (7). Comorbidities
that may lead to immunologic deficiencies in the host, such as
diabetes, are risk factors for severe dengue (3). However, viral genetic variations may
also effectively evade the immune response with severe clinical
consequences. As such, the genetic variations of dengue and its
association with clinical manifestations are an interesting topic
of study with scarce evidence.
A recent study has suggested that a rapid pattern of
mutations in DENV-1 is possible within an epidemic (evolutionary
process in situ) (8),
although it is not known whether this may induce a change in the
virus transmission pattern and its pathogenicity. In order to
examine the genetic variability of DENV during an epidemic, the NS5
protein gene was examined. This gene, with low variability, was
sequenced to establish whether during an epidemic there were
genetic variations of the virus that may be differentiated
phylogenetically and whether any variant was associated (through a
case-control design) with severe clinical behavior. In addition,
in silico, the possible consequences of these genetic
variations generated in the NS5 viral protein and their possible
impact on the innate immune response were evaluated.
Patients and methods
Patients
The present study was conducted from June
2007-December 2008 in patients with acute fever who consulted at
the Public Health Services of the State of Colima (Colima, Mexico;
632,000 habitants). Antecubital venous blood samples were collected
from 462 febrile patients (male:female, 228:234; age range, 6–80
years old) who attended medical units. The samples were collected
under aseptic conditions with a Vacutainer tube with EDTA
maintained at 4°C for a maximum of 6 h. The serum was separated and
stored at −70°C until processing. The inclusion criterion was the
presence of fever with an evolution between 2 and 7 days with a
follow up on diagnosis, and the medical management received.
Exclusion criteria included patients from other locations outside
the State of Colima, age <5 or >80 years, a frank septic
focus (abscesses and pneumonia), immunosuppressive states
(diabetes, HIV/AIDS and chemotherapy) or fever onset during
hospital stay. First-contact physicians evaluated all patients and
the intake was made prior to deciding their hospitalization or
discharge. Sequential convenience sampling was used to obtain a
constant proportion of cases by each participating medical center
throughout the study.
Patient data were registered, including age, sex,
place of residence, time of evolution with fever, initial
presumptive diagnosis and final diagnosis recorded by the medical
staff of each unit. In cases that required hospitalization, their
diagnosis of discharge was recorded. Patients participated
voluntarily in the present study and signed statements of informed
consent to use their blood samples, following the guidelines of the
project as approved by the Institutional Ethics Committee of the
Department of Health (Colima, Mexico).
Sample processing
Total RNA was extracted from blood samples using an
RNeasy mini kit (Qiagen, Inc., Valencia, CA, USA). RNA samples were
treated with DNase I (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) for 10 min at 37°C to remove traces of genomic
DNA, and RNA quality and integrity were assessed by standard
spectrophotometric and electrophoretic methods, respectively. A 10
ng sample of total RNA was subject to reverse
transcription-polymerase chain reaction (RT-PCR) using the
SuperScript® III One-Step RT-PCR System with
Platinum® Taq DNA Polymerase (Invitrogen; Thermo Fisher
Scientific, Inc.). Previously reported consensus primers were
selected to target segments of the NS5 coding region conserved
across several FV species, including DENV-1, -2, -3 and -4
(9). The sequences of the primers
are as follows: Flav100F (forward) 5′-AAY TCI ACI CAI GAR ATG
TAY-3′ and Flav200R (reverse) 5′-CCI ARC CAC ATR WAC CA-3′. RT-PCR
was performed as described previously (9). PCR products were cloned using the
TOPO-XL cloning vector system using the PCR-XL-TOPO 3.5 kb kit
(Invitrogen; Thermo Fisher Scientific, Inc.). The sequencing
reactions of cloning products were performed with universal M13
primers using a Big Dye terminator v3.1 Cycle Sequencing kit
(Applied Biosystems; Thermo Fisher Scientific, Inc.) in a
3100-Avant Genetic Analyzer sequencer (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The sequences of the M13 primers are as
follows: Forward, 5′-CTG GCC GTC GTT TTA C-3′ and reverse 5′-CAG
GAA ACA GCT ATG AC-3′. Novel sequences were deposited in the
GenBank database (accession no. MF073185-95; www.ncbi.nlm.nih.gov/Genbank/).
Bioinformatics analysis
The chromatograms and sequences obtained from three
different clones from each PCR product were analyzed using the
GeneStudio™ Pro v2.2.0.0. software (GeneStudio, Inc., Suwanee, GA,
USA). By eliminating the sequences of the primers, 765 bp
nucleotide sequences were obtained and translated into 255 amino
acids. Peptide sequences were predicted from obtained DNA sequences
using the Transeq online program (www.ebi.ac.uk/emboss/transeq/) and aligned using
ClustalW (www.ebi.ac.uk/Tools/msa/clustalo/) together with
peptide sequences extracted from GenBank via homology search.
Evolutionary history among the DENV-1 sequences were inferred
through the construction of Minimum Spanning Networks utilizing the
program PopArt v1.7 (Allan Wilson Centre, Otago, New Zealand). Once
clades were identified their divergence was estimated by
calculating the mean uncorrected p-distance between them using the
program MEGA v7.0 (10). The
identified chimeras in both clades were modeled in the I-TASSER
server (11) after being aligned
with the NS5 RNA-dependent RNA polymerase domain [Protein Database
(PDB_: 2J7U A-chain)] (12) and
inserted to complete the peptide sequence (chimera). Subsequently,
models with the highest C-score were evaluated in the MolProbity
server (13–15). The conformational changes were
computed in the web-interface elNemo (16) to identify sections susceptible to
interact with other biomolecules.
Docking
The interaction of the chimeras of both clades was
evaluated with the following receptors associated with the
infectious process of DENV-1: Human type I interferon receptor
(PDB:2HYM A) (17), Janus kinase
1 (JAK-1; PDB:3EYH) (18), plasma
platelet activating factor acetylhidrolase (PDB:3D59 A,B) (19), unphosphorylated signal transducer
and activator of transcription factor (STAT)-1 (PDB: 1YVL A)
(20), platelet factor 4 (PDB:
1F9Q A,B,C.D) (21),
interleukin-1β (2I1B) (22) and
interleukin-6 (PDB: 1ALU) (23).
Preliminary docking was carried out with the
receptors proposed by GRAMM-X Protein-Protein Docking Web Server
v.1.2.0 (24,25) in order to identify the most likely
contact surfaces in both clades with these biological targets.
Subsequently, the protein-protein in silico molecular
coupling was performed on the server ClusPro 2.0 (Structural
Bioinformatics Lab, Boston University, Boston, MA, USA) (26–28) masking only the amino acids
corresponding to the base of human type I interferon receptor
(PDB:2HYM A) and there were no further restrictions of attraction
or repulsion on the other couplings.
In the analysis of the results, four aspects were
evaluated to discriminate between the poses obtained: i) All
analyzed complexes are products of balanced algorithms hydrophobic
and electrostatically; ii) couplings were in accordance with the
results of the conformational changes obtained in the web-interface
ElNemo and similar to those obtained in the GRAMM-X Protein-Protein
Docking web server v.1.2.0; iii) they were equivalent in both
clades for comparison purposes; and iv) priority was given to
energy between protein centers (reported in Table II) to the lowest of the total
complex.
 | Table IICoupling energies in the center of
clade 1 and 2 with possible NS5 receptor RNA-dependent RNA
polymerase. |
Table II
Coupling energies in the center of
clade 1 and 2 with possible NS5 receptor RNA-dependent RNA
polymerase.
| Receptor PDB | Complex clade 2
(cal/mol) | Complex clade 1
(kcal/mol) |
|---|
| Janus kinase-1
(3EYH) | −818.7 | −702.2 |
| Type I interferon
receptor (2HYM A) | −663.1 | −650.2 |
| PPAFA (3D59
A,B) | −884.1 | −877.0 |
| Interleukin-6
(1ALU) | −765.8 | −764.1 |
| Unphosphorylated
STAT1 (1YVL A) | −800.7 | −801.2 |
| Platelet FACTOR 4
(1F9Q A,B,C.D) | −710.1 | −714.3 |
| Interleukin-1β
(2I1B) | −650.6 | −654.6 |
Statistical analysis
Student's t-test was used to compare normally
distributed mean values of the measurement variables. Qualitative
data were compared using the chi-square test. The association
between genetic variant (clades) and the risk for sever dengue was
estimated by odds ratios (ORs) and 95% confidence intervals (CIs;
crosstabs procedure). All statistical analyzes were performed using
the SPSS statistical software, version 20 (IBM Corp., Armonk, NY,
USA).
Results
Identification of patients with DENV
A total of 462 febrile patients were included in the
present study, whose characteristics and initial clinical diagnosis
are presented in Table I. Of the
patients included, 31 were positive for FV. The obtained sequences
in the present study corresponded to DENV-1, without finding
another serotype of dengue, nor another FV. The sensitivity and
specificity of the clinical diagnosis regarding the diagnosis of
dengue fever were 58.06% (95% CI, 39.08–77.05%) and 58% (95% CI,
53.23–62.78%), respectively. The positive predictive value was
9.05% (95% CI, 4.81–13.28%).
| Table IDescription of febrile population
analyzed. |
Table I
Description of febrile population
analyzed.
| Variable | Patients (n) |
|---|
| Sex | |
| Female | 234 |
| Male | 228 |
| Age (years) | |
| 6–20 | 232 |
| 21–40 | 144 |
| 41–60 | 65 |
| 60–80 | 21 |
| Community | |
| Urban | 411 |
| Rural | 51 |
| Zone | |
| Coastal | 307 |
| Non-coastal | 155 |
| Initial
diagnosis | |
| Unspecific
fever | 180 |
| Dengue | 201 |
| Respiratory
Infection | 14 |
| Influenza | 12 |
| Tonsilitis | 9 |
|
Rhinopharyngitis | 7 |
| Meningitis | 7 |
|
Pharyngotonsillitis | 6 |
| Urosepsis | 4 |
| Urinary
infection | 4 |
| Hepatitis | 3 |
| Pneumonia | 2 |
| Typhoid | 2 |
| Dysentery
syndrome | 1 |
| Ricketsiosis | 1 |
| Acute
pyelonephritis | 1 |
| Rheumatic
fever | 1 |
| Typhoid | 1 |
| Encephalitis | 2 |
| Parasitosis | 1 |
| Anemia | 1 |
| Tonsillitis | 2 |
Analysis of NS5 sequences
A number of the 31 DENV sequences were identified,
with 11 different patterns remaining. The evolutionary analysis
revealed the presence of two groups with differences of 1.3%
(standard error ± 0.04%) in their nucleotide sequences as indicated
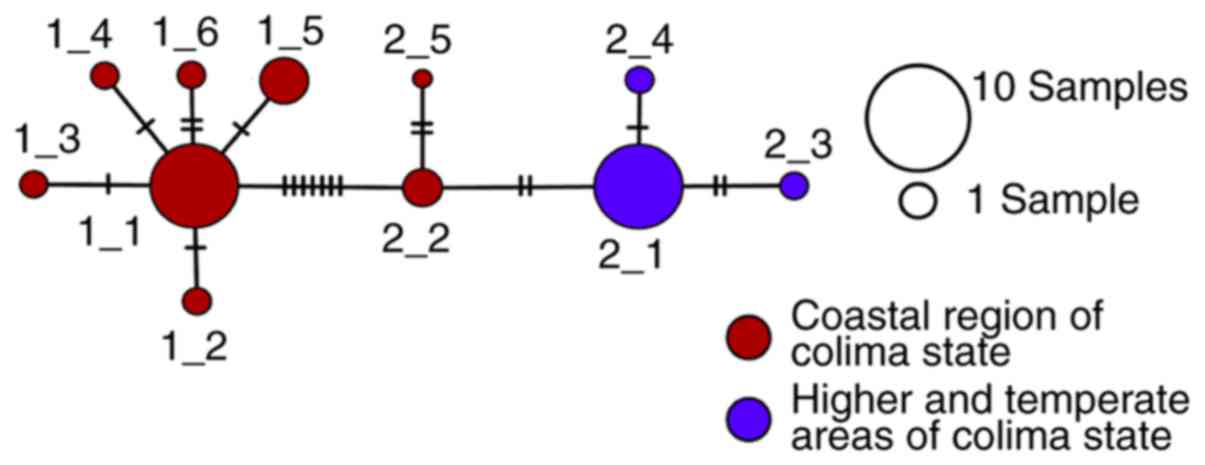
in the minimum spanning network of Fig. 1.
Of the nucleotide differences between the clades,
only two generated amino acid changes in the NS5 protein.
Considering the nucleotide sequence of GenBank KJ651911.1 of the
NS5 gene, these variations are 372A/G and 496G/A. Referring to the
GenBank protein sequence AJL35015.1, the variations are at position
124Ile/Met and 166Gly/Ser, in clade 1/2, respectively. These
positions are equivalent to 8593/8717 of the viral genome and
positions 2833/2875 of the viral polyprotein.
Dividing the patients with dengue according to the
virus clade, there were 16 patients with clade 1 and 15 with clade
2. Epidemiologically, it was found that clade 1 (Fig. 1) was the first to appear during
2007 and was located almost exclusively on the warm coastal region
of Colima (Municipalities of Manzanillo and Tecoman). Clade 2
viruses (Fig. 1) were observed at
the end of 2008, predominantly in the higher and temperate areas of
the state (Municipalities of Colima, Villa de Álvarez and
Cuahutemoc). Regarding the clinical behavior of the 16 patients
with clade 1 virus, only 1 progressed to severe dengue that
required hospitalization for 3 days, with a clinical diagnosis of
hemorrhagic dengue. Of the 15 patients corresponding to clade 2, 5
had severe dengue with the following clinical diagnoses: 2 patients
with hemorrhagic dengue, 1 with hepatitis/pneumonitis, 1 with viral
hepatitis and 1 with probable rickettsiosis. The patient with
probable rickettsiosis succumbed to mortality. These patients had a
mean hospital stay of 5.3 days. Infection with clade 2 virus
significantly increased the risk of having severe dengue 10-fold
(OR, 10; 95% CI, 1.03–97.04; P=0.03), determined by a crude
analysis without considering other host factors.
Given this association of a clade with the severity
of the disease, an in silico analysis was performed to
determine the effects on the NS5 protein of the two amino acid
variations that mark the difference between the two clades. For
this, a modeling of the NS5 protein was initially performed and a
docking methodology was subsequently used to determine its
interaction with other proteins previously implicated in the
pathophysiology of similar infections.
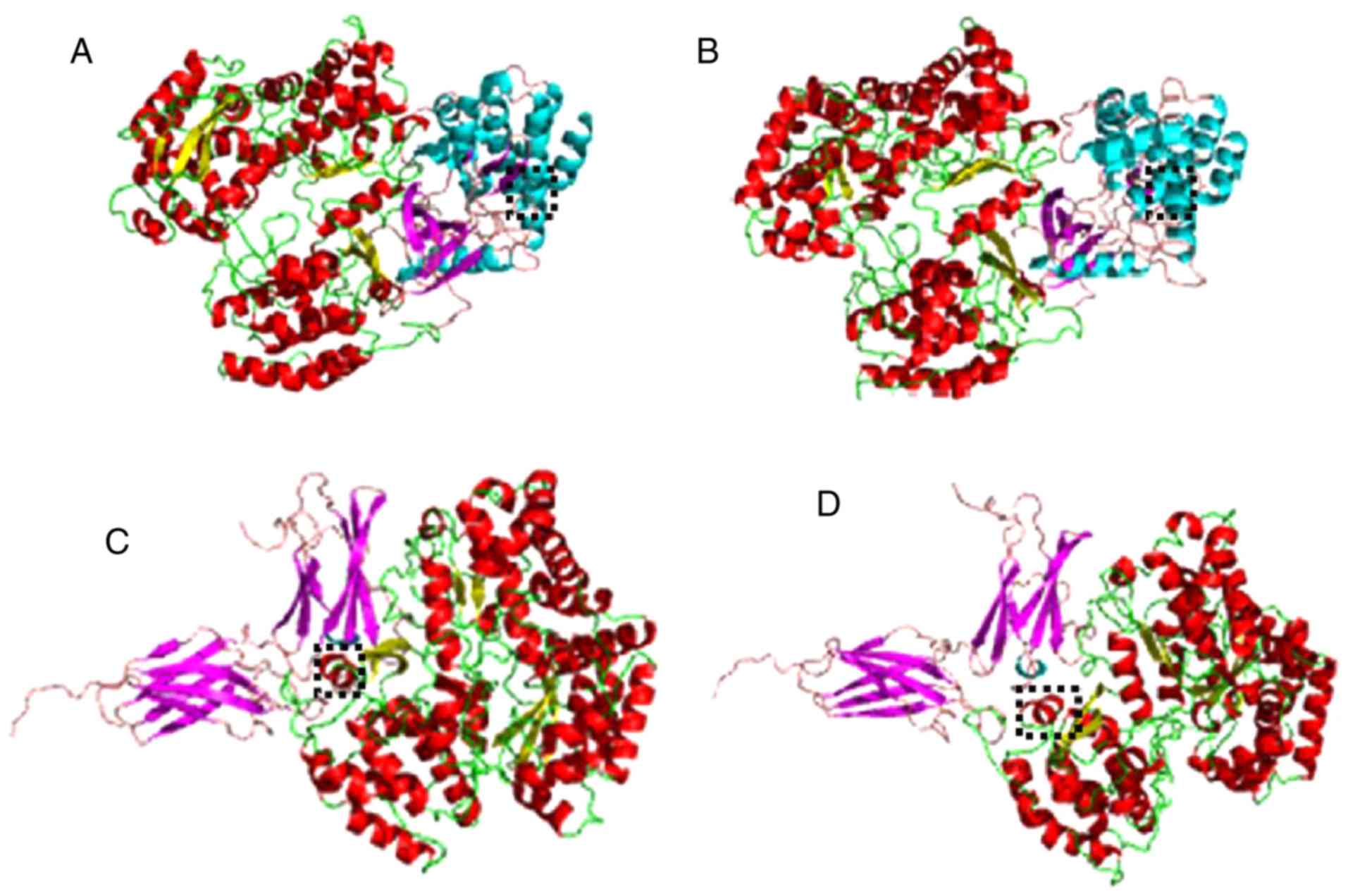
Modeling of NS5 clade 1 and 2
The chimeras corresponding to clade 1 and 2 were
analyzed. It was demonstrated that residues that are different
between clade 1 and 2 are located in critical regions, as amino
acid 166 and neighbors form part of an alpha helix that serves an
important role in the interaction between NS5 and other
proteins.
Docking
The present results (Table II) suggest that clade 2 had a
greater attraction with human type I interferon receptor and JAK-1,
which was also demonstrated by the three-dimensional
representations of Fig. 2.
The greatest difference in affinities between the
two NS5s with the other proteins was observed in the coupling with
JAK-1, with a difference of −101 kcal/mol in favor of clade 2. In
order to identify which of the two amino acids changes of this
clade had more relevance in these couplings, new chimeric NS5s were
generated where a single amino acid was changed with respect to
clade 1. These new chimeras had the following variations: 124Ile
and 166Ser; or 124Met and 166Gly. The Ile/Ser variant had energies
of −690.4 and −660.4 kcal/mol, whereas the variant Met/Gly had
energies of −781.2 and −656.6 kcal/mol, for the JAK-1 and
interferon receptor 1, respectively. Taking as reference the
composition of clade 1 (Ile/Gly), which exhibited energies of
coupling of JAK-1 and interferon receptor 1 of −702.2 and −650.2
kcal/mol, respectively, it may be inferred that the greater
affinity of clade 2 with JAK-1 depends mostly on the amino acid
124Met, whereas its greater affinity with the interferon receptor
does not depend mainly on some of the analyzed amino acids.
Discussion
The findings of the present study suggest that
during an epidemic caused by DENV-1, the clinical behavior may be
associated with genetic variants of the virus, independently to the
presence of other risk factors. This result was relevant, as it had
recently been estimated that in some regions of the world, among
the 4 DENV serotypes, DENV-1 exhibits the highest risk of severe
disease (29). A viral variant
reported in the present study, called clade 2, generated a 10-fold
increased risk of severe infection. The difference associated with
severe disease was given only by two amino acid changes in NS5,
whereas residues Met124 and Ser166 in clade 2 replaced the Ile and
Gly of clade 1, respectively. Previously it had been demonstrated
that subtle sequence changes in viral proteins may generate
alterations in their biological behavior. For example, in studies
performed by Zulkarnain et al (30) and Ishak et al (31) based on the comparison of the amino
acid and nucleotide sequences in dengue type 1 protein E, NS3, NS4A
and NS5. The change of few amino acid residues may modify the
replication characteristics of the virus in in vitro
experiments. In particular, in the NS5 RNA-dependent RNA
polymerase, there are amino acids that are critical for some of
their functions. Studies of this protein from other flaviviruses,
such as Langat (32) and West
Nile Virus NY99 (33), have
demonstrated that some residues (623, 635, 641, 643 and 653) are
important for the protein to serve a role as an antagonist of type
I interferon (α/β), and suppressing the JAK-STAT signaling
pathway.
In the present study, it was demonstrated that
changes in NS5 may generate changes in the protein structure with
possible implications in their interactions, though in
silico tests. The amino acid at position 166 and neighboring
residues are part of an α helix that serves an important role in
the interaction between NS5 and other host proteins according to
in silico couplings. This α helix is preferably located in
the center of the most probable pose of coupling, which infers that
it is a critical region in the NS5. When comparing the coupling
energies, no difference >7 kcal/mol was observed between clades
in the complexes with interleukins, STAT1 and with the plasma
platelet activating factor, thus it was assumed that the clinical
differences associated with the viral variants are not associated
with variations in the interaction with these proteins. However,
the interaction difference of the two NS5 variant (clade 1 vs. 2)
with the interferon type 1 receptor was appreciable (13.1 kcal).
The closest approach in clade 2 of the critical area of residue 166
of the NS5 with the center of the receiver was visible. This may
mean that NS5 clade 2 is a more efficient antagonist of type 1
interferon by interfering with its receptor.
However, the most important difference was in the
coupling with JAK-1, being −101 kcal/mol in favor of clade 2, and
in which a greater number of residues participate and allow a
better penetration. This suggests that genetic variants 372A/G and
496G/A (protein variants 124Ile/Met and 166Gly/Ser) in NS5 of
DENV-1 condition the degree of antagonism with type 1 interferon
due to interruption of JAK-STAT1. It has recently been reported, in
similar viruses, that replication increases through inhibiting the
JAK-STAT signaling pathway (34).
It has also been evidenced that the JAK/STAT3
pathway is critical in chemokine production from DV-infected
hepatocytes. The response of interferon or cytokines to dengue
infection was part of the innate immunity initiated at day 0–1
following DENV infection, and declines 3–4 days thereafter. The
upregulation of these early innate immune responses coincides, as
it should from an immune surveillance perspective, with a drop in
DENV viral replication during days 0–3 of the fever (35). A variation in the impairment of
the function of type 1 interferon, either at the level of its
receptor or by interfering with its signaling pathway (JAK-STAT)
may cause variations in the clinical course of the disease. A
better coupling of the NS5 variant of clade 2 with the interferon 1
receptor and with JAK-1 may cause the association found in the
present study, between this genetic variant of dengue with a more
severe clinical course. Interestingly, none of the patients
presented a simultaneous infection among the identified DENV1
strains; therefore, dengue disease progression remains unknown when
both NS5 genetic variations are present in those patients. Thus,
the biological interactions of DENV-1 NS5 need to be tested
experimentally in future studies.
It was observed that for coupling with JAK-1, the
most relevant amino acid was 124Met. This variation in DENV-1 has
only previously been reported in two samples from Singapore and
Cambodia (ABW82089.1 and ACL99206.1, respectively) recollected in
2005 and 2007 (28). This was
relevant, as a report by Yung et al (29), which analyzed the cases of dengue
in Singapore from 2005–2011, demonstrated that among the four
serotypes of the virus, DENV-1 had the highest risk of severe
disease. This may be due to the presence of this viral variant in
the analyzed epidemic outbreaks of Singapore. In other regions of
the world, DENV-2 and -3 were the most associated with severe
disease (36,37). The amino acid 166Ser is not the
most common residue in DENV-1 and has been previously reported in
only three sequences from Argentina, Puerto Rico and Thailand in
samples from 2009, 2010 and 2011, respectively (GenBank:
AHF50483.1, AHI43747.1 and AKL88404.1) (36). However, they have not previously
reported a sequence with the two changes that were identified in
the present study in clade 2 (124Met and 166GSer). It is notable
that the residue 124Met was commonly reported in homologous
sequences of DENV-2 (example GenBank: AOE23003.1) and DENV-3
(example GenBank: ANS59201.1). Therefore, it may be interesting to
know if the amino acid equivalent to 124Met of the NS5 in DENV-2
and -3 has an important effect in the interaction with JAK-1.
A limitation of the present study is the fact that
an analysis to corroborate the present docking observations at the
molecular level was not performed. Ongoing experimental studies at
our laboratory are being performed to explore the key role of
interferon-α/β in the JAK-STAT signaling pathway.
An important epidemiological aspect was that during
a similar epidemic outbreak caused by a same serotype of dengue in
our region, there were genetic, geographic and clinical variations
over time (data not shown). Clade 2 (associated with severity)
occurred one year after the onset of the outbreak in higher
altitudes, cooler temperatures, and more severe forms of the
disease, including a mortality due to multiple organ failure, which
was clinically confused with a rickettsiosis. It is difficult to
establish whether a genetic variant of DENV-1 was introduced from
another region at the end of the epidemic outbreak and replaced the
previous variant; or if it was a de novo mutation in the
course of the epidemic outbreak that generated the new variant. It
has recently been suggested that a rapid pattern of mutations in
DENV-1 is possible within an epidemic (evolutionary process in
situ). Although an epidemic outbreak of dengue is maintained
over time with the same virus serotype, it does not imply that it
is necessarily the same virus from the genetic and pathogenic point
of view.
In conclusion, the genetic variants 372G and 496A of
NS5 from DENV-1 were associated with a severe clinical course of
the disease in an epidemic outbreak in a region of Mexico. The
variations generate changes in the protein that probably most
effectively block the innate immune response, by affecting the
function of type 1 interferon, either at the level of its receptor
or by interfering with its JAK-STAT signaling pathway. Biological
testing and studies in other populations are necessary.
Acknowledgments
The present study was completed using equipment
resources obtained through grants from the INFRAESTRUCTURA-CONACYT
(grant no. 270485), FOSISS-CONACYT (grant no. 272792) and
FORDECyT-CONACYT (grant no. 2009/1-000000117535).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Gubler DJ: Dengue and dengue hemorrhagic
fever. Clin Microbiol Rev. 11:480–496. 1998.PubMed/NCBI
|
|
2
|
Kuno G, Chang GJ, Tsuchiya KR, Karabatsos
N and Cropp CB: Phylogeny of the genus Flavivirus. J Virol.
72:73–83. 1998.PubMed/NCBI
|
|
3
|
Leitmeyer KC, Vaughn DW, Watts DM, Salas
R, Villalobos I, Ramos C and Rico-Hesse R: Dengue virus structural
differences that correlate with pathogenesis. J Virol.
73:4738–4747. 1999.PubMed/NCBI
|
|
4
|
Descloux E, Cao-Lormeau VM, Roche C and De
Lamballerie X: Dengue 1 diversity and microevolution, French
Polynesia 2001–2006: Connection with epidemiology and clinics. PLoS
Negl Trop Dis. 3:e4932009. View Article : Google Scholar
|
|
5
|
Halstead SB: Dengue in the Americas and
Southeast Asia: Do they differ? Rev Panam Salud Publica.
20:407–415. 2006. View Article : Google Scholar
|
|
6
|
Rico-Hesse R, Harrison LM, Salas RA, Tovar
D, Nisalak A, Ramos C, Boshell J, de Mesa MT, Nogueira RM and da
Rosa AT: Origins of dengue type 2 viruses associated with increased
pathogenicity in the Americas. Virology. 230:244–251. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Diamond MS: Evasion of innate and adaptive
immunity by flaviviruses. Immunol Cell Biol. 81:196–206. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hapuarachchi HC, Koo C, Kek R, Xu H, Lai
YL, Liu L, Kok SY, Shi Y, Chuen RL, Lee KS, et al: Intra-epidemic
evolutionary dynamics of a Dengue virus type 1 population reveal
mutant spectra that correlate with disease transmission. Sci Rep.
6:225922016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maher-Sturgess SL, Forrester NL, Wayper
PJ, Gould EA, Hall RA, Barnard RT and Gibbs MJ: Universal primers
that amplify RNA from all three flavivirus subgroups. Virol J.
5:162008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar S, Stecher G and Tamura K: MEGA7:
Molecular evolutionary genetics analysis version 7.0 for bigger
datasets. Mol Biol Evol. 33:1870–1874. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roy A, Kucukural A and Zhang Y: I-TASSER:
A unified platform for automated protein structure and function
prediction. Nat Protoc. 5:725–738. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yap TL, Xu T, Chen YL, Malet H, Egloff MP,
Canard B, Vasudevan SG and Lescar J: Crystal structure of the
dengue virus RNA-dependent RNA polymerase catalytic domain at
1.85-angstrom resolution. J Virol. 81:4753–4765. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Davis IW, Leaver-Fay A, Chen VB, Block JN,
Kapral GJ, Wang X, Murray LW, Arendall WB III, Snoeyink J,
Richardson JS and Richardson DC: MolProbity: All-atom contacts and
structure validation for proteins and nucleic acids. Nucleic Acids
Res. 35:W375–W383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen VB, Arendall WB III, Headd JJ, Keedy
DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS and
Richardson DC: MolProbity: All-atom structure validation for
macromolecular crystallography. Acta Crystallogr D Biol
Crystallogr. 66:12–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen VB, Davis IW and Richardson DC: KING
(Kinemage, Next Generation): A versatile interactive molecular and
scientific visualization program. Protein Sci. 18:2403–2409. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suhre K and Sanejouand YH: On the
potential of normal-mode analysis for solving difficult
molecular-replacement problems. Acta Crystallogr D Biol
Crystallogr. 60:796–799. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Quadt-Akabayov SR, Chill JH, Levy R,
Kessler N and Anglister J: Determination of the human type I
interferon receptor binding site on human interferon-alpha2 by
cross saturation and an NMR-based model of the complex. Protein
Sci. 15:2656–2668. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Williams NK, Bamert RS, Patel O, Wang C,
Walden PM, Wilks AF, Fantino E, Rossjohn J and Lucet IS: Dissecting
specificity in the Janus kinases: The structures of JAK-specific
inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase
domains. J Mol Biol. 387:219–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Samanta U and Bahnson BJ: Crystal
structure of human plasma platelet-activating factor
acetylhydrolase: Structural implication to lipoprotein binding and
catalysis. J Biol Chem. 283:31617–31624. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mao X, Ren Z, Parker GN, Sondermann H,
Pastorello MA, Wang W, McMurray JS, Demeler B, Darnell JE Jr and
Chen X: Structural bases of unphosphorylated STAT1 association and
receptor binding. Mol Cell. 17:761–771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang J, Doyle M, Faulk T, Visentin G,
Aster R and Edwards B: Crystal structure of platelet factor 4
mutant 2: Full wwPDB X-ray Structure Validation Report. Sept
27–2017, http://files.rcsb.org/pub/pdb/validation_reports/f9/1f9q/1f9q_full_validation.pdf.
|
|
22
|
Priestle JP, Schär HP and Grütter MG:
Crystallographic refinement of interleukin 1 beta at 2.0
Aresolution. Proc Natl Acad Sci USA. 86:9667–9671. 1989. View Article : Google Scholar
|
|
23
|
Somers W, Stahl M and Seehra JS: 1.9
Acrystal structure of interleukin 6: Implications for a novel mode
of receptor dimerization and signaling. EMBO J. 16:989–997. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tovchigrechko A and Vakser IA: GRAMM-X
public web server for protein-protein docking. Nucleic Acids Res.
34:W310–W314. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tovchigrechko A and Vakser IA: Development
and testing of an automated approach to protein docking. Proteins.
60:296–301. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kozakov D, Hall DR, Beglov D, Brenke R,
Comeau SR, Shen Y, Li K, Zheng J, Vakili P, Paschalidis ICh and
Vajda S: Achieving reliability and high accuracy in automated
protein docking: Cluspro, PIPER, SDU, and stability analysis in
CAPRI rounds 13-19. Proteins. 78:3124–3130. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kozakov D, Brenke R, Comeau SR and Vajda
S: PIPER: An FFT-based protein docking program with pairwise
potentials. Proteins. 65:392–406. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Comeau SR, Gatchell DW, Vajda S and
Camacho CJ: ClusPro: An automated docking and discrimination method
for the prediction of protein complexes. Bioinformatics. 20:45–50.
2004. View Article : Google Scholar
|
|
29
|
Yung CF, Lee KS, Thein TL, Tan LK, Gan VC,
Wong JG, Lye DC, Ng LC and Leo YS: Dengue serotype-specific
differences in clinical manifestation, laboratory parameters and
risk of severe disease in adults, Singapore. Am J Trop Med Hyg.
92:999–1005. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zulkarnain E, Hotta S and Takegami T:
Molecular comparison of dengue type 1 Mochizuki strain virus and
other selected viruses concerning nucleotide and amino acid
sequences of genomic RNA: A consideration of viral epidemiology and
variation. Microbiol Immunol. 38:581–585. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ishak H, Takegami T, Kamimura K and Funada
H: Comparative sequences of two type 1 dengue virus strains
possessing different growth characteristics in vitro. Microbiol
Immunol. 45:327–331. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park GS, Morris KL, Hallett RG, Bloom ME
and Best SM: Identification of residues critical for the interferon
antagonist function of Langat virus NS5 reveals a role for the
RNA-dependent RNA polymerase domain. J Virol. 81:6936–6946. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Laurent-Rolle M, Boer EF, Lubick KJ,
Wolfinbarger JB, Carmody AB, Rockx B, Ashour J, Shupert WL,
Holbrook MR, Barrett AD, et al: The NS5 protein of the virulent
West Nile virus NY99 strain is a potent antagonist of type I
interferon-mediated JAK-STAT signaling. J Virol. 84:3503–3515.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang J, Chen S, Liao Y, Zhang E, Feng S,
Yu S, Li LF, He WR, Li Y, Luo Y, et al: Mitogen-Activated Protein
Kinase Kinase 2, a Novel E2-interacting protein, promotes the
growth of classical swine fever virus via attenuation of the
JAK-STAT signaling pathway. J Virol. 90:10271–10283. 2016.
View Article : Google Scholar :
|
|
35
|
Chiappelli F, Santos SM, Caldeira Brant
XM, Bakhordarian A, Thames AD, Maida CA, Du AM, Jan AL, Nahcivan M,
Nguyen MT and Sama N: Viral immune evasion in dengue: Toward
evidence-based revisions of clinical practice guidelines.
Bioinformation. 10:726–733. 2014. View Article : Google Scholar
|
|
36
|
Díaz FJ, Black WC IV, Farfán-Ale JA,
Loroño-Pino MA, Olson KE and Beaty BJ: Dengue virus circulation and
evolution in Mexico: A phylogenetic perspective. Arch Med Res.
37:760–773. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Espinoza-Gómez F, López-Lemus AU,
Rodriguez-Sanchez IP, Martinez-Fierro ML, Newton-Sánchez OA,
Chávez-Flores E and Delgado-Enciso I: Detection of sequences from a
potentially novel strain of cell fusing agent virus in Mexican
Stegomyia (Aedes) aegypti mosquitoes. Arch Virol. 156:1263–1267.
2011. View Article : Google Scholar : PubMed/NCBI
|