Introduction
Cadmium (Cd) is an environmentally abundant toxic
metal, which is considered a serious threat to human health
(1). Cd has a long biological
half-life, and exhibits organ accumulation. One of the primary
target organs of Cd is the kidney (2). In addition to accumulation in
proximal tubular cells, Cd exposure directly damages the
glomerulus, thus resulting in proteinuria (3). However, little is currently known
regarding the molecular mechanisms underlying its cytotoxic
effects.
The glomerular filtration barrier (GFB) consists of
fenestrated endothelial cells, the glomerular basement membrane and
podocyte slit diaphragms (4). It
achieves size selectivity of the glomerular filter, permitting
filtration of water and small-sized solutes in the plasma (5). Surrounding the glomerular
capillaries, podocytes are highly specialized epithelial cells that
maintain the structural and functional integrity of the GFB. As
interdigitating foot processes separated by a slit diaphragm,
podocytes regulate the passage of proteins from the capillary lumen
to Bowman's space (6). Podocyte
dysfunction impairs size selectivity of the glomerular filter, thus
leading to proteinuria, hypoalbuminuria and edema (5). In addition, podocyte phenotype is
determined by the expression of CD2-associated protein (CD2AP) and
synaptopodin (7). Podocyte
dedifferentiation over the course of collapsing focal segmental
glomerulosclerosis results in the loss of synaptopodin and
cytoskeleton markers (8,9). Oxidized low-density lipoprotein
stimulation re-organizes F-actin filaments and promotes podocyte
migration in a focal adhesion kinase (FAK)-dependent manner
(10).
Mitogen-activated protein kinases (MAPKs) are a
family of serine/threonine protein kinases, which regulate numerous
biological cellular processes, including cell proliferation,
apoptosis and F-actin cytoskeletal formation (11). The three major MAPK pathways in
mammalian cells are extracellular signal-regulated kinases, c-Jun
N-terminal kinase (JNK) and p38 MAPK (11). JNK, also termed stress-activated
protein kinase (SAPK), is activated by stress signals, including
heat shock, ultraviolet light, osmotic stress and metabolic toxins
(12–14). Activated JNK translocates to the
nucleus and activates c-Jun and c-Fos, thus resulting in the
formation of activating protein-1 (AP-1) (15). AP-1 recognizes either
12-0-tetradecanoylphorbol-13-acetate response elements or cAMP
response elements, and activates the expression of a series of
stress-response genes (16).
The present study aimed to examine the effects of
low-dose Cd (4 μM) on human renal podocytes (HRPs). The
results indicated that Cd activates the JNK pathway, thus
increasing c-Jun and c-Fos expression. However, Cd had no effect on
proliferation, viability, apoptosis and alignment of the F-actin
cytoskeleton of HRPs. In addition, treatment with SP600125, a JNK
inhibitor, alongside Cd did not alter cell proliferation,
viability, apoptosis and alignment of the F-actin cytoskeleton of
HRPs compared with SP600125 treatment alone.
Material and methods
Cell culture
HRPs were originally provided by Dr MA Saleem
(University of Bristol, Bristol, UK), and were cultured in
RPMI-1640 medium (Corning, Inc., Corning, NY, USA) supplemented
with 10% fetal bovine serum (Gibco™; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), 100 U/ml penicillin and 100 μg/ml
streptomycin. The cells were grown in a humidified atmosphere
containing 5% CO2 at 37°C. CdCl2 was
purchased from Sigma-Aldrich; Merck KgaA (Darmstadt, Germany) and
was dissolved in PBS, with a stock concentration of 1 mM. In the
present study, 4 μM CdCl2 was considered low-dose
treatment. SP600125 was purchased from Cell Signaling Technology,
Inc. (Danver, MA, USA) and was dissolved in dimethyl sulfoxide.
Cells were pretreated with 10 μM SP600125 for 1 h prior to
treatment with Cd. Control cells were only treated with 10
μM SP600125 for 1 h. Experimental cells were pretreated with
10 μM SP600125 for 1 h, and then were treated with Cd for 1,
2, 6, 12 and 24 h. All treatments were at 37°C.
Western blotting
Protein samples were extracted from HRPs using
radioimmunoprecipitation assay buffer [20 mM Tris (pH 7.5), 150 mM
NaCl, 50 mM NaF, 1% NP-40, 0.1% deoxycholate, 0.1% sodium dodecyl
sulfate, 1 mM EDTA] supplemented with the protease inhibitors
aprotonin (1 μg/ml), leupeptin (10 μg/ml) and
phenylmethylsulfonyl fluoride (1 mM). Equal amounts of protein [40
μg; protein concentration was determined using BCA assay
(Bio-Rad, Hercules, CA, USA)] were separated by 8% SDS-PAGE
(Beyotime Institute of Biotechnology, Haimen, China) and were then
transferred onto a polyvinylidene fluoride membrane. The membrane
was blocked with 5% non-fat milk in Tris-buffered saline containing
1% Tween-20 (TBST; Cell Signaling Technology, Inc.) at room
temperature for 2 h prior to incubation with primary antibodies
overnight at 4°C. After washing three times with TBST, the membrane
was incubated with a peroxidase-conjugated secondary antibody at
room temperature for 2 h. The primary antibodies used in the
present study were as follows: Rabbit anti-SAPK/JNK (9258), rabbit
anti-phosphorylated (p)-SAPK/JNK (4668), rabbit anti-c-Fos (2250),
rabbit anti-c-Jun (9165) and rabbit anti-GAPDH (2118) (Cell
Signaling Technology, Inc.). The secondary antibody used was goat
anti-rabbit immunoglobulin G (7074; Cell Signaling Technology,
Inc.). The immunoblots were developed using enhanced
chemiluminescence reagents (EMD Millipore, Billerica, MA, USA), and
relative blot intensities were semi-quantified using ImageJ
software (version 1.4.3.67; National Institutes of Health,
Bethesda, MA, USA).
Cell proliferation and viability
assay
HRP proliferation was evaluated using the MTT assay
kit (Cayman Chemical Company, Ann Arbor, MI, USA) according to the
manufacturer's protocol. Briefly, HRPs were plated at a density of
9×103 cells/well in a 96-well plate and were cultured
overnight. Subsequently, the cells were treated with Cd, SP600125
or a combination of Cd and SP600125 for 24 h, after which 10
μl MTT solution was added to each well and incubated for 4 h
at 37°C. The combination of SP600125 and Cd treatment was as
follows: cells were pretreated with SP600125 for 1 h, and then
co-treated with Cd and SP600125 for 24 h. The crystals were
solubilized by the addition of 110 μl DMSO, and colorimetric
intensity was analyzed using a 96-well plate reader (Molecular
Devices, LLC, Sunnyvale, CA, USA) at a wavelength of 490 nm. HRP
viability was assessed using a trypan blue exclusion assay.
Following treatment, HRPs were washed and incubated in 0.05%
trypsin for 2 min at 37°C. Subsequently, the cells were
disaggregated into a single cell suspension and diluted 9:1 with
0.4% trypan blue (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China). The percentage of unstained cells was
determined under an OLYMPUS CKX41 microscope (Olympus Corporation,
Tokyo, Japan).
Immunofluorescence
HRPs were grown until confluent (80%) on
fibronectin-coated glass chamber slides and were then treated with
4 μM Cd for 24 h. The medium was then aspirated, and
monolayers were washed with PBS, fixed with 4% paraformaldehyde at
room temperature for 20 min and washed three times with PBS for 15
min. Subsequently, the cells were permeabilized with 0.3% Triton
X-100 for 10 min, washed three times with PBS for 15 min and
incubated with a rabbit polyclonal antibody against CD2AP (1:50;
#2135; Cell Signaling Technology, Inc.) or synaptopodin (1:50;
ab224491; Abcam, Cambridge, MA, USA) overnight at 4°C. The cells
were then incubated with an Alexa Fluor 546-labeled donkey anti
rabbit secondary antibody (1:200; A10040; Thermo Fisher Scientific,
Inc.) for 2.5 h at room temperature. Images of the slides were
captured using an Olympus FSX100 Imaging system (Olympus
Corporation) with an excitation wavelength of 546 nm.
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) analysis
Apoptosis of the HRPs was determined by Annexin
V-FITC and PI staining using an assay kit (Neobioscience, Shenzhen,
China) according to the manufacturer's protocol. Briefly, following
treatment with Cd, SP600125 or a combination of Cd and SP600125 for
24 h, cells were trypsinized and resuspended into a single cell
suspension. Subsequently, 1×106 cells were stained with
Annexin V-FITC (0.025%) for 3 min and PI (20 μg/ml) for 10
min at room temperature in the dark. Positive staining of the cells
was detected using a FACSAria II flow cytometer, and data were
analyzed using the FACSDiva acquisition and analysis software
(v6.1.3) (both from BD Biosciences, San Jose, CA, USA).
Phalloidin-labeling
HRPs were grown until confluent (80%) on
fibronectin-coated glass chamber slides. Following exposure to 4
μM Cd, SP600125 or a combination of 4 μM Cd and
SP600125 for 24 h, the medium was aspirated and the monolayer was
fixed for 5 min in 3.7% formaldehyde solution in PBS. The cells
were then permeabilized with 0.3% Triton X-100 in PBS, and stained
with 5 μg/ml phalloidin-tetramethylrhodamine B
isothiocyanate (Sigma-Aldrich; Merck KGaA) in PBS for 1 h at room
temperature. DAPI staining was used to visualize the nuclei. Images
were captured using a confocal microscope (LSM 880; Carl Zeiss AG,
Oberkochen, Germany).
Statistical analyses
Data are presented as means ± SD. Statistical
significance was assessed using one-way analysis of variance
followed by Tukey's post hoc test or Student's t-test. Statistical
analyses were performed using SPSS 17.0 statistical software
package (SPSS Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Low-dose Cd activates the JNK pathway in
HRPs
Cd activates JNK signaling and stimulates downstream
effectors of JNK in HepG2 cells (17). In the present study, the effects
of 4 μM Cd exposure on the JNK pathway in HRPs were
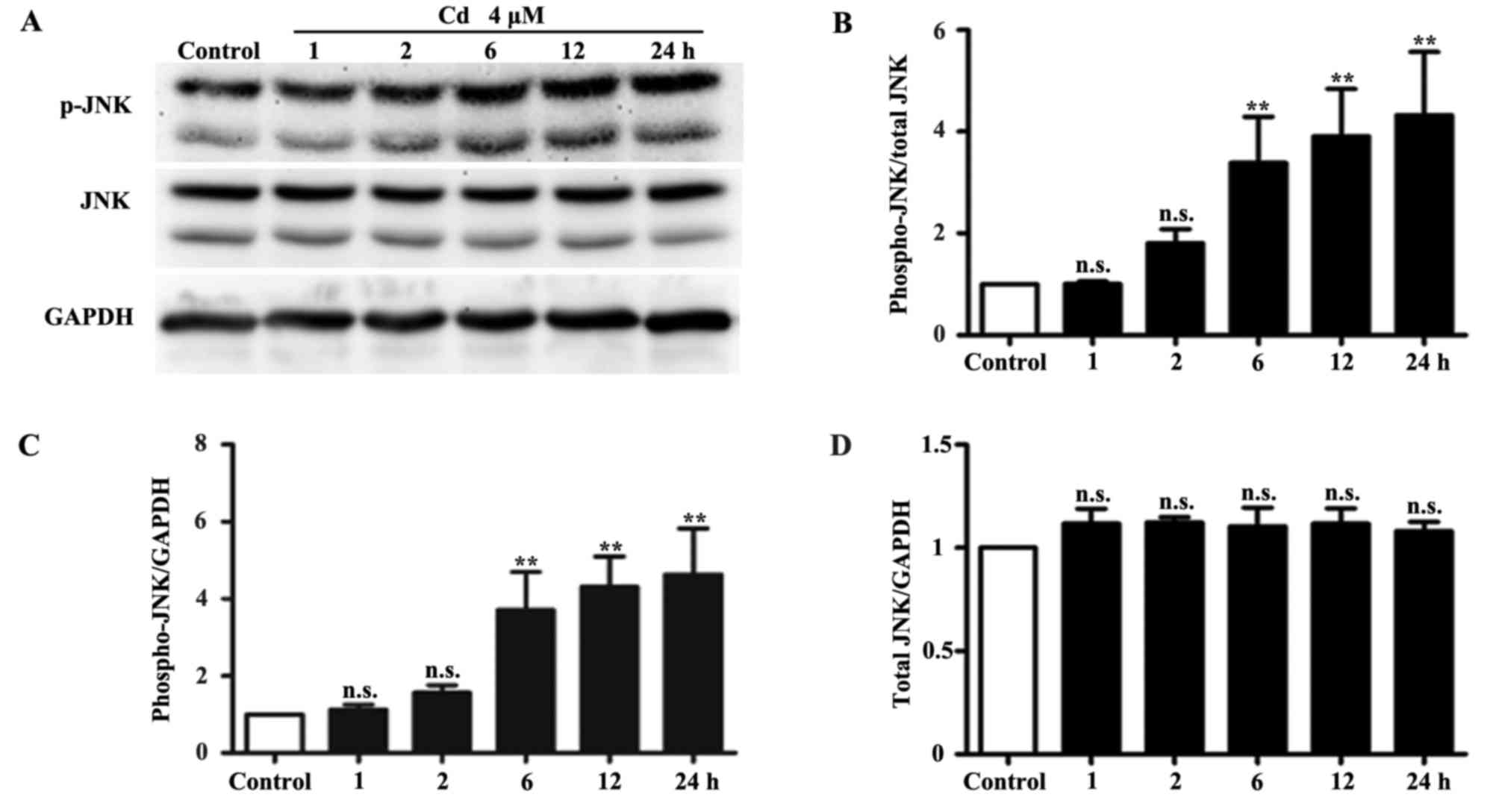
determined by western blotting. The results indicated that the
expression levels of p-JNK were significantly increased following
treatment of HRPs with 4 μM Cd for 6, 12 and 24 h.
Conversely, the protein expression levels of total JNK and the
internal control GAPDH remained unchanged (Fig. 1). In addition, the protein
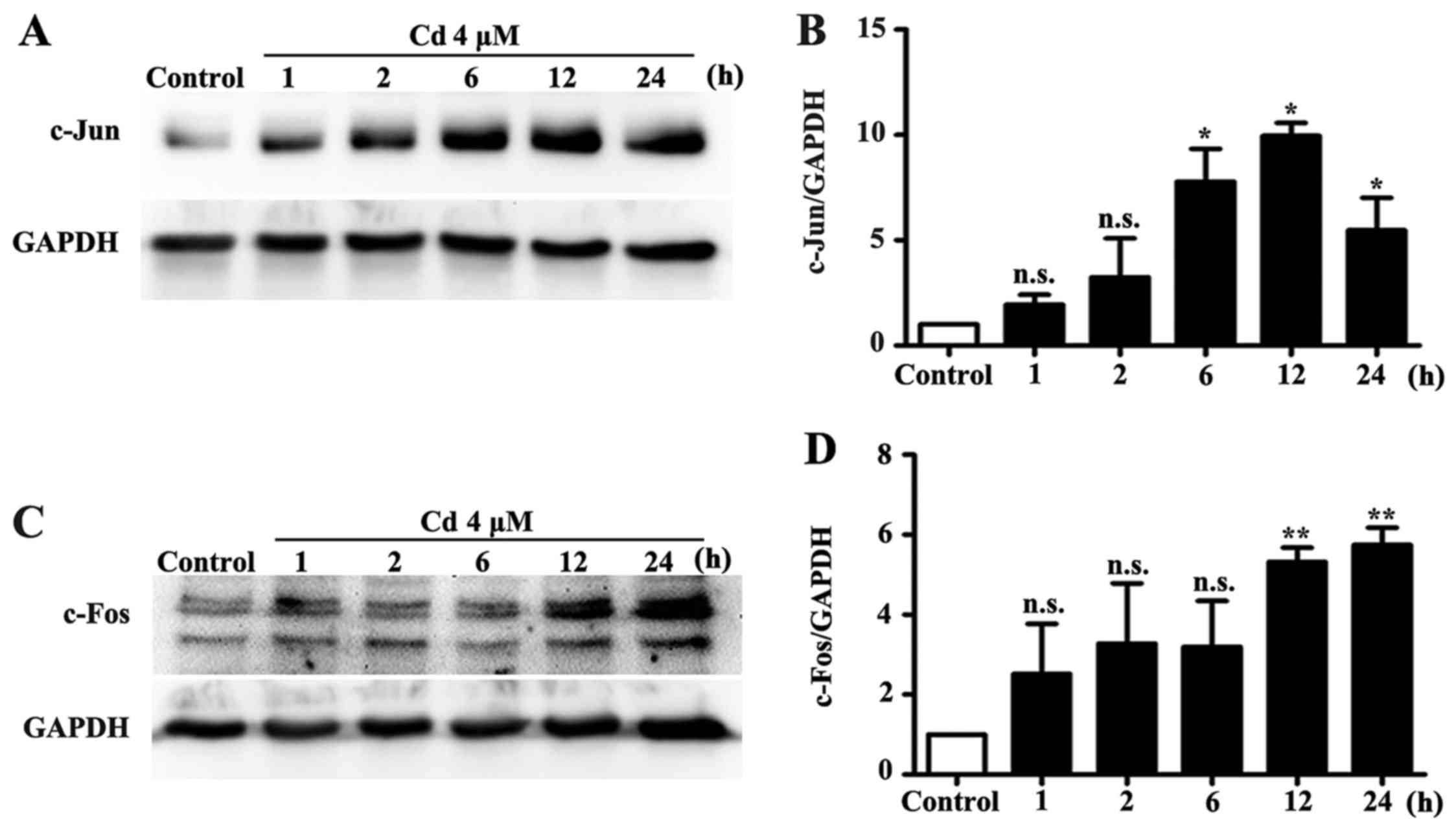
expression levels of c-Jun and c-Fos were significantly increased
by Cd treatment (Fig. 2).
Low-dose Cd exposure does not affect
proliferation and the expression of cell type-specific HRP
markers
Cd inhibits the proliferation of numerous cell types
(18,19). In the present study, the MTT assay
was performed to examine the effects of Cd on HRP proliferation.
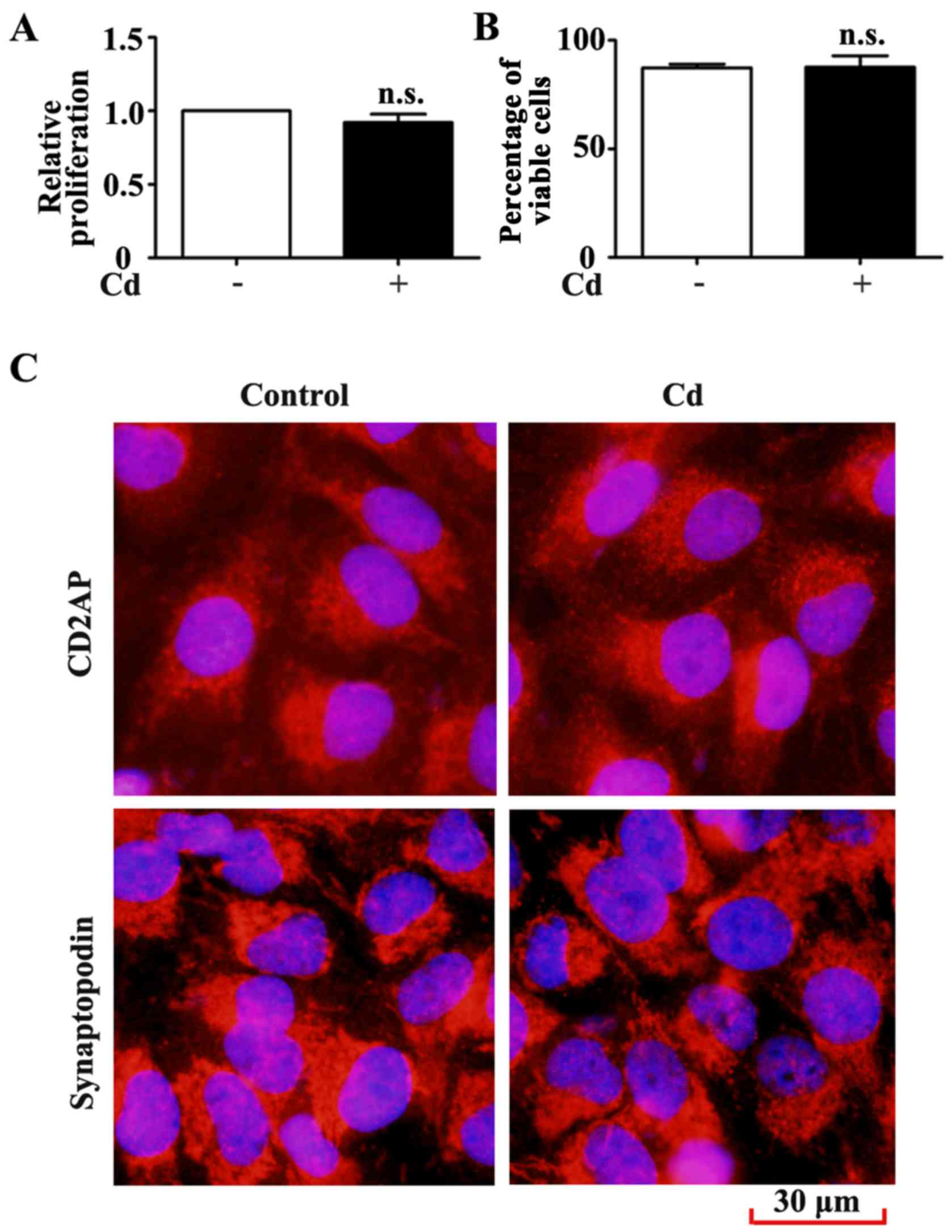
The results indicated that 4 μM Cd did not significantly
inhibit HRP proliferation after 24 h (Fig. 3A). The results of the trypan blue
exclusion assay also demonstrated that HRP viability remained
unchanged following Cd treatment (Fig. 3B). Specific markers of podocytes
include CD2AP and synaptopodin (7). In response to extracellular stimuli,
podocytes may undergo a phenotypic alteration, thus resulting in
the loss of terminal differentiation markers (20,21). HRP characteristics were examined
by immunofluorescence staining for CD2AP and synaptopodin. CD2AP
and synaptopodin expression was unchanged in HRPs following 24 h
exposure to 4 μM Cd (Fig.
3C). These results suggested that low-dose Cd exposure may not
alter the proliferation, viability and phenotype of HRPs.
Effects of low-dose Cd combined with
SP600125 on HRP proliferation and viability
SP600125 is a specific inhibitor of the JNK pathway
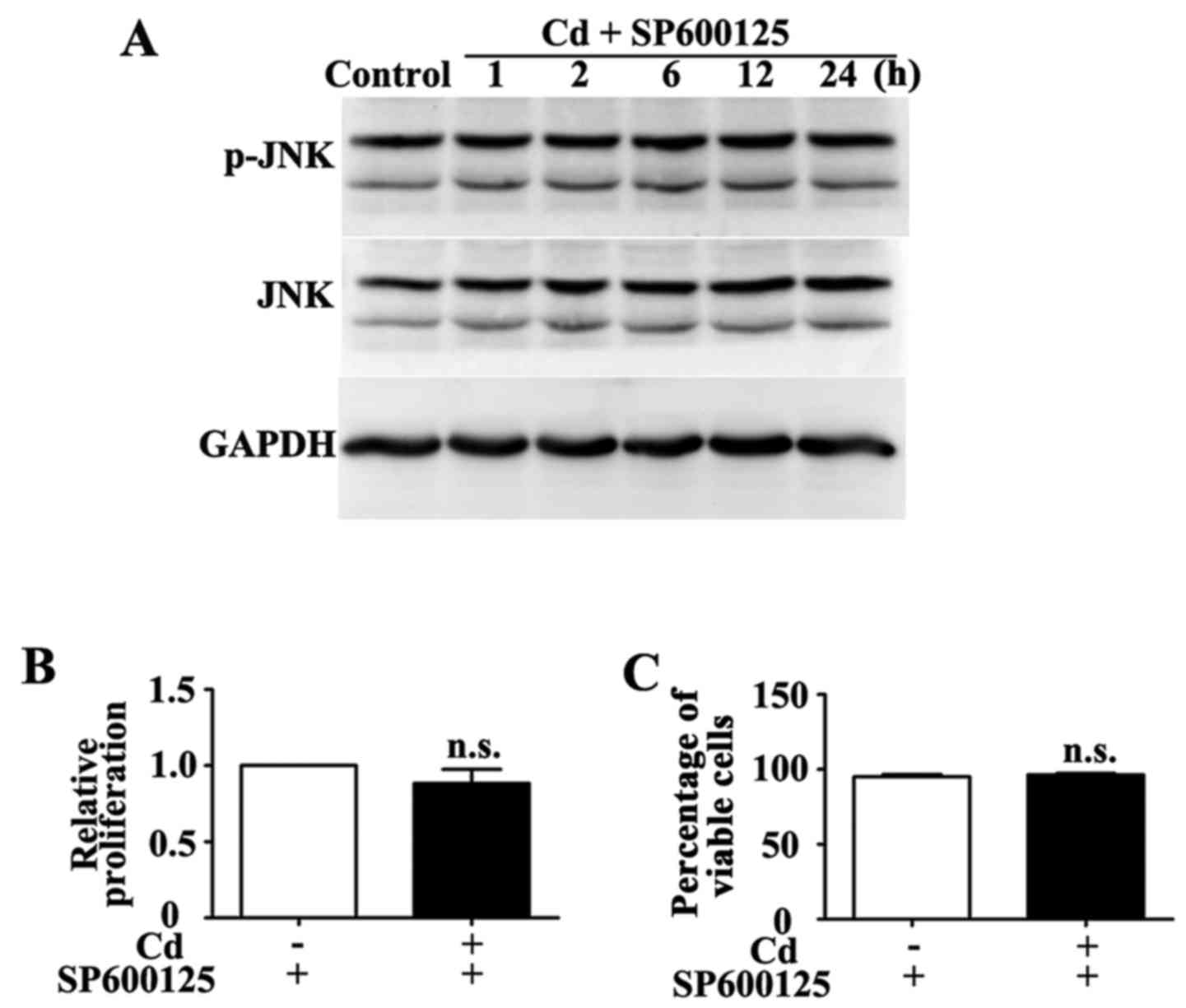
(22). Co-treatment with 10
μM SP600125 for 1 h inhibited Cd-induced phosphorylation of
JNK (Fig. 4A). Conversely, HRP
proliferation and viability were similar in the group treated with
a combination of SP600125 and Cd compared with in the group treated
with SP600125 alone (Fig. 4B and
C).
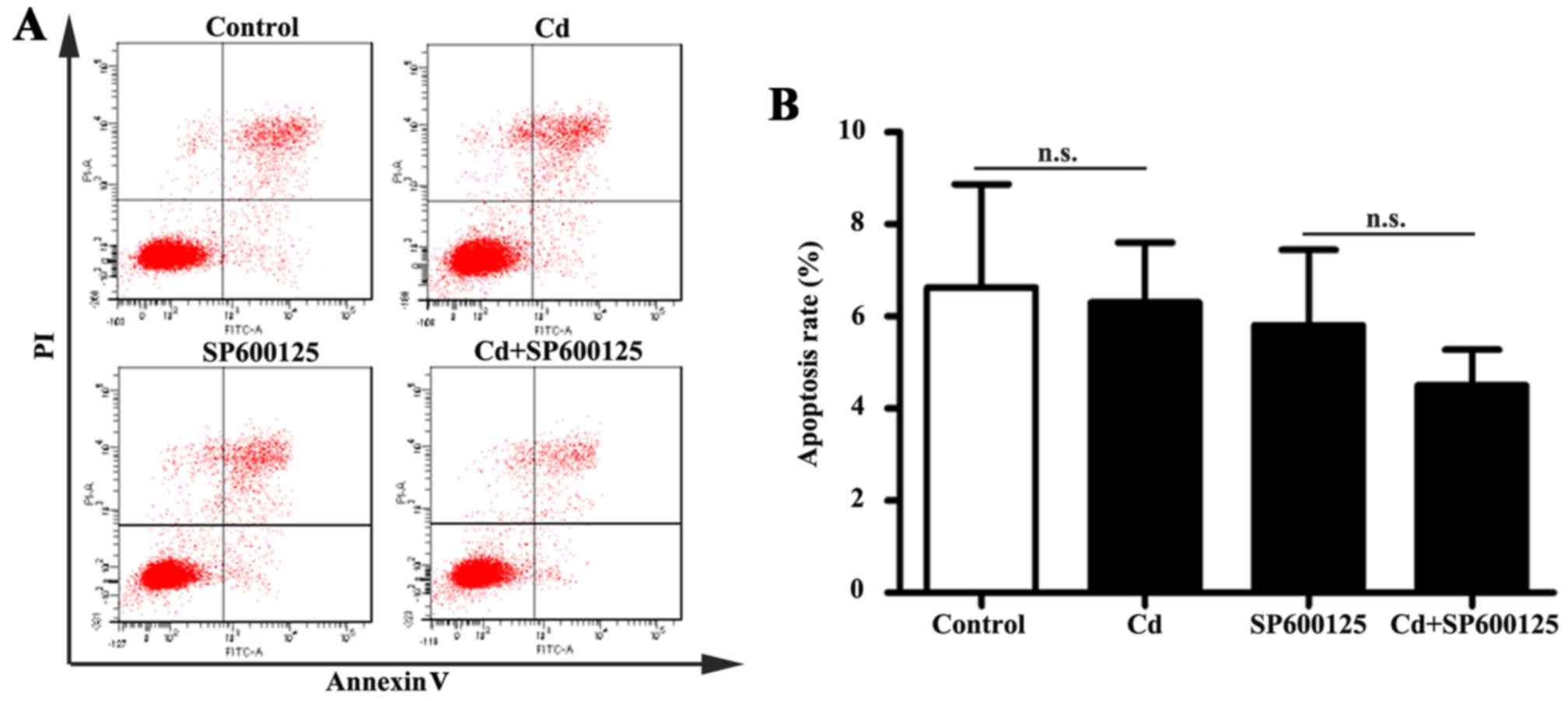
Effects of low-dose Cd exposure on HRP
apoptosis
The JNK pathway mediates apoptotic responses in
numerous cell types (23).
Following exposure to 4 μM Cd for 24 h, apoptosis of HRPs
was examined by Annexin V-FITC/PI double-labeled flow cytometry. No
significant alterations in the apoptotic rate were detected in
Cd-treated HRPs (Fig. 5). In
addition, the apoptotic rate of HRPs treated with a combination of
SP600125 and Cd was similar to that of HRPs treated with SP600125
alone. These results indicated that 4 μM Cd does not affect
apoptosis of HRPs.
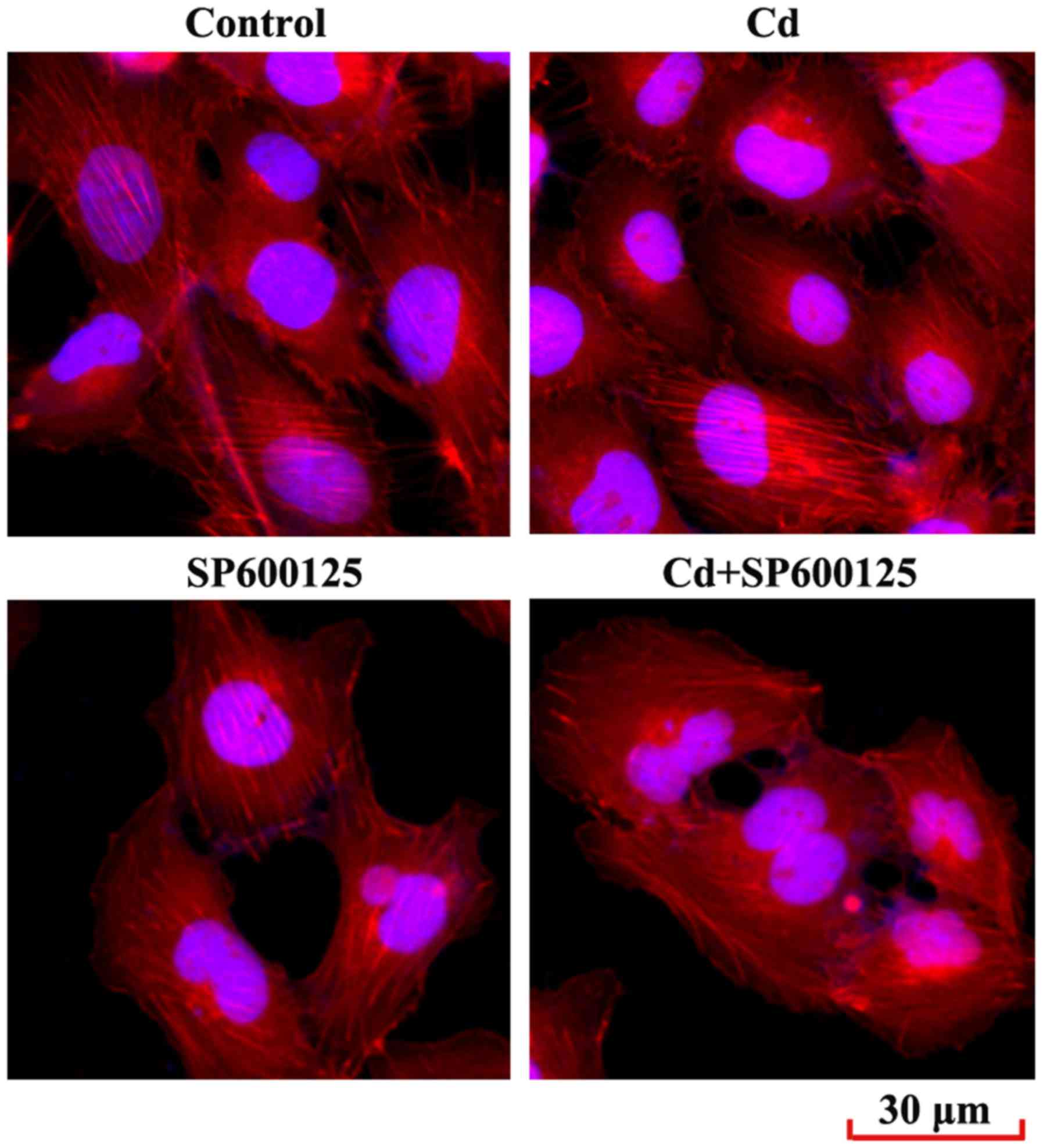
Effects of low-dose Cd exposure on the
F-actin cytoskeleton of HRPs
Reorganization of the cytoskeleton is a hallmark of
podocyte injury (24). The
present study examined the podocyte cytoskeleton by immunostaining
the F-actin cytoskeleton-interacting molecule phalloidin. The
results demonstrated that F-actin arrangement in podocytes treated
with Cd was not disrupted compared with in the control group
(Fig. 6). In addition, SP600125,
or a combined exposure to SP600125 and Cd, did not significantly
alter F-actin arrangement in podocytes.
Discussion
Cd has been reported to induce toxicological effects
in the human kidney (25). The
present study aimed to explore the effects of low-dose Cd on HRPs,
which are the major component of the GFB. The results demonstrated
that treatment with 4 Cd increased phosphorylation of JNK, and the
expression levels of c-Jun and c-Fos. However, Cd treatment did not
affect the proliferation, viability, apoptosis and cytoskeleton of
HRPs. In addition, inhibition of the JNK pathway did not
significantly affect Cd-treated HRPs. These findings indicated that
Cd activates the JNK pathway in podocytes but does not induce
cytotoxic effects.
Depending on the experimental settings, Cd may
differentially affect specific cell types in the glomerulus
(12,26). As previously reported, although
the vascular system is the primary target of Cd toxicity, a low
concentration of Cd does not affect the proliferation and apoptosis
of human umbilical vein endothelial cells or human renal glomerular
endothelial cells (HRGECs) (26–28). However, Cd may increase apoptosis
of human renal mesangial cells (HRMCs) and decrease their
proliferation (12). Podocytes
are a terminally differentiated and highly specialized cell type in
the glomerulus. Previous studies have suggested that podocyte
proliferation may not be affected in response to injury (29,30). In addition, Cd affects cellular
functions other than proliferation and apoptosis. A previous study
demonstrated that low-dose Cd induces vascular hyperpermeability
and disruption of endothelial barrier integrity via the Cd-induced
membrane dissociation of vascular endothelial cadherin and
β-catenin in HRGECs (26).
Therefore, the effects of Cd on podocyte permeability require
further investigation.
MAPKs are activated by numerous cellular stressors
(11). Oxidative stress is one of
the major mechanisms underlying Cd toxicity. The production of
reactive oxygen species (ROS) induced by Cd causes severe toxic
effects in numerous types of tissues and organs (31). JNK is one of the proteins
activated in response to elevated ROS levels, which serves a
critical role in the apoptotic process (32). At a concentration between 5 and 40
μM, Cd may induce apoptosis of BJAB cells by increasing DNA
fragmentation and caspase-3 activity (33). In addition, at a concentration of
4 μM, Cd inhibits the proliferation of HRMCs via activation
the JNK pathway (12). In the
present study, treatment with 4 μM Cd increased
phosphorylation of JNK, and c-Jun and c-Fos expression. However,
activation of the JNK pathway did not induce apoptosis of HRPs. A
previous study also indicated that Cd treatment increased p-JNK
expression, but did not affect apoptosis of endothelial cells
(26). Therefore, JNK-regulated
cell functions may vary depending on cell type, stimulus, and the
duration and strength of kinase activities (31,34). In addition to MAPKs, the
phosphatidylinositol 3-kinase/Akt pathway, hypoxia inducible
factor-1α and nuclear factor-κB are involved in Cd-induced signal
transduction (35–37). Activation of other signaling
pathways may compensate the effects mediated by the JNK
pathway.
An intact actin cytoskeleton is crucial for the
maintenance of podocyte membrane tension, shape and glomerular
function, and is associated with signaling events at the slit
diaphragm (38). F-actin is one
of the major components of the cytoskeleton (39). Synaptopodin is a proline-rich
protein that is expressed in differentiated podocytes and
orchestrates actin organization through its interaction with
F-actin (40,41). JNK is associated with cytoskeletal
integrity, and is involved in 4-hydroxy-2-non- enal-mediated actin
remodeling in endothelial cells (42). In the present study, Cd activated
the JNK pathway, and increased the expression levels of c-Jun and
c-Fos, but had no significant effect on synaptopodin distribution.
A previous study also reported that the F-actin cytoskeleton was
unchanged in HRMCs by Cd treatment, despite activation of the JNK
pathway (12). Numerous signaling
cascades may be involved in reorganization of the actin
cytoskeleton (43). For example,
high glucose-induced F-actin rearrangement in mouse podocytes was
mediated by activation of the Fyn/Rho-associated coiled-coil
forming protein kinase signaling pathway (44). Furthermore, the FAK/p38 signaling
pathway regulates cytoskeletal arrangement in mouse podocytes
(10). Therefore, activation of
the JNK pathway may not be sufficient for rearrangement of the
F-actin cytoskeleton in podocytes.
In conclusion, although low-dose Cd increased the
phosphorylation of JNK, and the expression levels of c-Jun and
c-Fos, it had no significant effect on proliferation, apoptosis and
F-actin cytoskeletal rearrangement of HRPs. Further studies are
required to address the molecular mechanisms underlying Cd-induced
loss of glomerular barrier integrity.
Acknowledgments
Not applicable.
Notes
[1]
Funding
The present study was supported by grants from the
Science and Technology Development Plan of Shandong Province (grant
no. 2013GSF11805), the National Natural Science Foundation of China
(grant no. 81370269) and the Shandong Taishan Scholarship (JL).
[2] Availability
of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
JL conceived and designed the experiments; XC, YX,
ZC, HS, XL performed the experiments; JL, XC, DX and CMK analyzed
the data; JL, XC and CMK wrote the paper.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Eichler TE, Ransom RF and Smoyer WE:
Differential induction of podocyte heat shock proteins by prolonged
single and combination toxic metal exposure. Toxicol Sci.
84:120–128. 2005. View Article : Google Scholar
|
|
2
|
Liu J, Liu Y and Klaassen CD:
Nephrotoxicity of CdCl2 and Cd-metallothionein in
cultured rat kidney proximal tubules and LLC-PK1 cells. Toxicol
Appl Pharmacol. 128:264–270. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prozialeck WC, Edwards JR and Woods JM:
The vascular endothelium as a target of cadmium toxicity. Life Sci.
79:1493–1506. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pavenstädt H, Kriz W and Kretzler M: Cell
biology of the glomerular podocyte. Physiol Rev. 83:253–307. 2003.
View Article : Google Scholar
|
|
5
|
Sugano Y, Lindenmeyer MT, Auberger I,
Ziegler U, Segerer S, Cohen CD, Neuhauss SC and Loffing J: The
Rho-GTPase binding protein IQGAP2 is required for the glomerular
filtration barrier. Kidney Int. 88:1047–1056. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inoue K and Ishibe S: Podocyte endocytosis
in the regulation of the glomerular filtration barrier. Am J
Physiol Renal Physiol. 309:F398–F405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Testagrossa L, Azevedo Neto R, Resende A,
Woronik V and Malheiros D: Immunohistochemical expression of
podocyte markers in the variants of focal segmental
glomerulosclerosis. Nephrol Dial Transplant. 28:91–98. 2013.
View Article : Google Scholar
|
|
8
|
Barisoni L, Schnaper HW and Kopp JB:
Advances in the biology and genetics of the podocytopathies:
Implications for diagnosis and therapy. Arch Pathol Lab Med.
133:201–216. 2009.PubMed/NCBI
|
|
9
|
Shankland SJ, Eitner F, Hudkins KL,
Goodpaster T, D'Agati V and Alpers CE: Differential expression of
cyclin-dependent kinase inhibitors in human glomerular disease:
Role in podocyte proliferation and maturation. Kidney Int.
58:674–683. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu M, Fan M, Zhen J, Lin J, Wang Q, Lv Z
and Wang R: FAK contributes to proteinuria in hypercholesterolaemic
rats and modulates podocyte F-actin re-organization via activating
p38 in response to ox-LDL. J Cell Mol Med. 21:552–567. 2017.
View Article : Google Scholar
|
|
11
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen X, Li J, Cheng Z, Xu Y, Wang X, Li X,
Xu D, Kapron CM and Liu J: Low dose cadmium inhibits proliferation
of human renal mesangial cells via activation of the JNK pathway.
Int J Environ Res Public Health. Oct 7–2016.(Epub ahead of print).
View Article : Google Scholar
|
|
13
|
Subramanian D, Bunjobpol W and Sabapathy
K: Interplay between TAp73 protein and selected activator protein-1
(AP-1) family members promotes AP-1 target gene activation and
cellular growth. J Biol Chem. 290:18636–18649. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hess J, Angel P and Schorpp-Kistner M:
AP-1 subunits: Quarrel and harmony among siblings. J Cell Sci.
117:5965–5973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Woodgett JR, Avruch J and Kyriakis JM:
Regulation of nuclear transcription factors by stress signals. Clin
Exp Pharmacol Physiol. 22:281–283. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsai JS, Chao CH and Lin LY: Cadmium
activates multiple signaling pathways that coordinately stimulate
Akt activity to enhance c-Myc mRNA stability. PLoS one.
11:e01470112016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu C, Zhang R, Sun C, Zhang H, Xu C, Liu
W, Gao W, Huang S and Chen L: Resveratrol prevents cadmium
activation of Erk1/2 and JNK pathways from neuronal cell death via
protein phosphatases 2A and 5. J Neurochem. 135:466–478. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao X, Yu L, Moore AB, Kissling GE,
Waalkes MP and Dixon D: Cadmium and proliferation in human uterine
leiomyoma cells: Evidence of a role for EGFR/MAPK pathways but not
classical estrogen receptor pathways. Environ Health Perspect.
123:331–336. 2015.
|
|
20
|
Barisoni L: Podocyte biology in segmental
sclerosis and progressive glomerular injury. Adv Chronic Kidney
Dis. 19:76–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meyrier A: Mechanisms of disease: Focal
segmental glomerulosclerosis. Nat Clin Pract Nephrol. 1:44–54.
2005. View Article : Google Scholar
|
|
22
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eichler T, Ma Q, Kelly C, Mishra J, Parikh
S, Ransom RF, Devarajan P and Smoyer WE: Single and combination
toxic metal exposures induce apoptosis in cultured murine podocytes
exclusively via the extrinsic caspase 8 pathway. Toxicol Sci.
90:392–399. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shen X, Jiang H, Ying M, Xie Z, Li X, Wang
H, Zhao J, Lin C, Wang Y, Feng S, et al: Calcineurin inhibitors
cyclosporin A and tacrolimus protect against podocyte injury
induced by puromycin aminonucleoside in rodent models. Sci Rep.
6:320872016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Provias JP, Ackerley CA, Smith C and
Becker LE: Cadmium encephalopathy: A report with elemental analysis
and pathological findings. Acta Neuropathol. 88:583–586. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li L, Dong F, Xu D, Du L, Yan S, Hu H,
Lobe CG, Yi F, Kapron CM and Liu J: Short-term, low-dose cadmium
exposure induces hyperpermeability in human renal glomerular
endothelial cells. J Appl Toxicol. 36:257–265. 2016. View Article : Google Scholar
|
|
27
|
Dong F, Guo F, Li L, Guo L, Hou Y, Hao E,
Yan S, Allen TD and Liu J: Cadmium induces vascular permeability
via activation of the p38 MAPK pathway. Biochem Biophys Res Commun.
450:447–452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Prozialeck WC, Edwards JR, Nebert DW,
Woods JM, Barchowsky A and Atchison WD: The vascular system as a
target of metal toxicity. Toxicol Sci. 102:207–218. 2008.
View Article : Google Scholar
|
|
29
|
Kriz W: Progressive renal
failure–inability of podocytes to replicate and the consequences
for development of glomerulosclerosis. Nephrol Dial Transplant.
11:1738–1742. 1996.PubMed/NCBI
|
|
30
|
Rennke HG: How does glomerular epithelial
cell injury contribute to progressive glomerular damage? Kidney Int
Suppl. 45:S58–S63. 1994.PubMed/NCBI
|
|
31
|
Nemmiche S: Oxidative signaling response
to cadmium exposure. Toxicol Sci. 156:4–10. 2017.
|
|
32
|
Pineda-Molina E, Klatt P, Vázquez J,
Marina A, García de Lacoba M, Pérez-Sala D and Lamas S:
Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for
redox-induced inhibition of DNA binding. Biochemistry.
40:14134–14142. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nemmiche S, Chabane-Sari D, Kadri M and
Guiraud P: Cadmium-induced apoptosis in the BJAB human B cell line:
Involvement of PKC/ERK1/2/JNK signaling pathways in HO-1
expression. Toxicology. 300:103–111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J and Kapron CM: Differential
induction of MAP kinase signalling pathways by cadmium in primary
cultures of mouse embryo limb bud cells. Reprod Toxicol.
29:286–291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee JC, Son YO, Pratheeshkumar P and Shi
X: Oxidative stress and metal carcinogenesis. Free Radic Biol Med.
53:742–757. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang H, Li L, Wang Y, Dong F, Chen X, Liu
F, Xu D, Yi F, Kapron CM and Liu J: NF-k B signaling
maintains the survival of cadmium-exposed human renal glomerular
endothelial cells. Int J Mol Med. 38:417–422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu F, Wang B, Li L, Dong F, Chen X, Li Y,
Dong X, Wada Y, Kapron CM and Liu J: Low-dose cadmium upregulates
VEGF expression in lung adenocarcinoma cells. Int J Environ Res
Public Health. 12:10508–10521. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Faul C, Asanuma K, Yanagida-Asanuma E, Kim
K and Mundel P: Actin up: Regulation of podocyte structure and
function by components of the actin cytoskeleton. Trends Cell Biol.
17:428–437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Heinonen MT, Kanduri K, Lähdesmäki HJ,
Lahesmaa R and Henttinen TA: Tubulin- and actin-associating GIMAP4
is required for IFN-γ secretion during Th cell differentiation.
Immunol Cell Biol. 93:158–166. 2015. View Article : Google Scholar
|
|
40
|
Müller-Krebs S, Kihm LP, Madhusudhan T,
Isermann B, Reiser J, Zeier M and Schwenger V: Human RAGE antibody
protects against AGE-mediated podocyte dysfunction. Nephrol Dial
Transplant. 27:3129–3136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim EY, Suh JM, Chiu YH and Dryer SE:
Regulation of podocyte BK(Ca) channels by synaptopodin, Rho, and
actin microfilaments. Am J Physiol Renal Physiol. 299:F594–F604.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Usatyuk PV and Natarajan V: Role of
mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced
actin remodeling and barrier function in endothelial cells. J Biol
Chem. 279:11789–11797. 2004. View Article : Google Scholar
|
|
43
|
Ispanovic E and Haas TL: JNK and PI3K
differentially regulate MMP-2 and MT1-MMP mRNA and protein in
response to actin cytoskeleton reorganization in endothelial cells.
Am J Physiol Cell Physiol. 291:C579–C588. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lv Z, Hu M, Ren X, Fan M, Zhen J, Chen L,
Lin J, Ding N, Wang Q and Wang R: Fyn mediates high glucose-induced
actin cytoskeleton reorganization of podocytes via promoting ROCK
activation in vitro. J Diabetes Res 2016. 5671803:2016. View Article : Google Scholar
|