Introduction
Platelets are anucleate blood cells which serve
crucial functions in thrombosis under physiological and
pathological conditions. They are critical for maintaining the
integrity of the vascular system and are the first-line defense
against hemorrhage. On encountering subendothelial matrix exposed
by an injury to a vessel, platelets adhere to the matrix, are
activated and become adhesive to other platelets, leading to
further aggregation (1).
Platelets are involved in the pathogenesis of
atherosclerosis-associated diseases, including coronary artery
diseases and stroke. During platelet activation, the release of
several mediators, including adenosine trisphosphate (ATP) and
thromboxane A2, occurs in conjunction with relative
intracellular Ca+2 ([Ca+2]i) mobilization,
attracting additional platelets towards the injured endothelium and
resulting in the thickening of the initial platelet monolayer.
Finally, fibrinogen binds to its specific platelet receptor,
completing the final common pathway for platelet aggregation.
Platelet activation has also been associated with
the key steps of cancer progression, and platelets have been
proposed to influence the development of malignancies via regulated
events that may trigger the pathobiology of cancer growth (2). Platelets also interact with cancer
cells and contribute to the critical steps of cancer metastasis,
including tumor cell migration, invasion, and arresting of the
tumor cell within the vasculature (2,3).
During platelet activation, their contents may be released into the
peritumoral space and subsequently enhance tumor cell extravasation
and metastasis (4).
The chemically similar platinum (Pt) group elements
(PGEs), including Pt, palladium, rhodium, ruthenium, osmium, and
iridium (Ir), have exceptional catalytic qualities. These metals
are resistant to chemical corrosion over a wide range of
temperatures, having a high melting point, high mechanical strength
and remarkable ductility (5). Ir
is rich in the Earth's crust, with an average mass fraction of 1
µg/kg in crustal rock. In nature, Ir is found in alluvium
deposits accompanied by Pt and other PGEs. This metal is obtained
from Pt ores, and is also derived as a by-product in nickel mining
and industry (6). Various metal
complexes have been identified for use in anticancer therapy,
leading to an increasing amount of associated research. In the drug
development industry, metal complexes represent a highly
resourceful platform. Apart from deviations in the metal and
oxidation state, metal ions have variable geometries and
coordination numbers that allow the modification of their chemical
reactivity in terms of kinetics (ligand exchange rates) and
thermodynamics (including metal-ligand bond strength and redox
potentials). Metals, as well as their ligands, are involved in
numerous biological activities, ranging from outer-sphere
recognition of target sites to the activity of any released ligands
and ligand-centered redox processes (7).
Organometallic Ir (III) complexes are particularly
promising in terms of anticancer treatments. Previous research has
focused on Ir (III) compounds due to their potential anti-tumor
activity and low toxicity towards normal tissues (8,9).
Furthermore, Ir complexes exert potent antiangiogenic effects by
activating distinct antiangiogenic signaling pathways (8), and antiangiogenic therapy is
considered a promising cancer treatment strategy. On the basis of
these observations, our group obtained novel biologically active Ir
(III) derivatives by developing a new Ir (III) compound, also
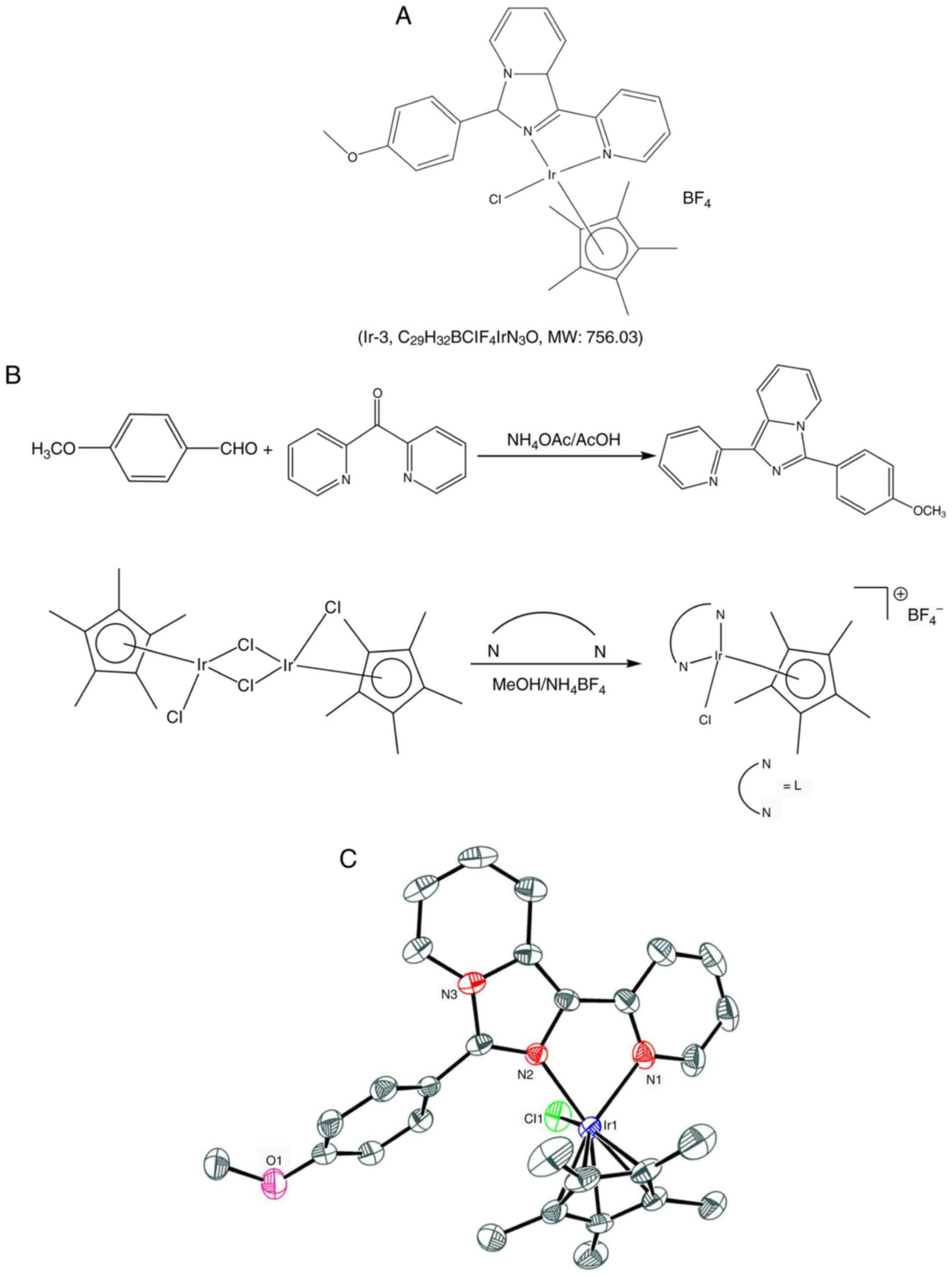
referred to as Ir-3 (Fig. 1).
Although certain experimental and animal studies have demonstrated
that Ir-based compounds have potent anticancer activity, to date,
no study has investigated their effects on platelet aggregation.
The preliminary results of the present study revealed that Ir-3
exhibits potent antiplatelet activity in humans, encouraging
further examination of the characteristics and functional activity
of Ir-3 in platelet activation. The results of the present study
provide valuable evidence for the development of a novel class of
Ir-3-based antiplatelet agents.
Materials and methods
Chemicals and reagents
Thrombin, collagen, arachi-donic acid (AA),
luciferin-luciferase, U46619, phorbol 12,13-dibutyrate (PDBu),
nitroglycerin (NTG), heparin, prostaglandin E1
(PGE1), 5,5-dimethyl-1-pyrroline N-oxide (DMPO),
SQ22536, 1H-[1,2,4]oxadiazolo[4,3-a]quinox-alin-1-one (ODQ), and
bovine serum albumin (BSA) were purchased from Sigma-Aldrich; Merck
KGaA (Darmstadt, Germany). Fura-2AM was obtained from Molecular
Probes; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Anti-phosphorylated (p)-p38 mitogen-activated protein kinase (MAPK)
Thr180/Tyr182 monoclonal antibodies (mAbs; Cat. no. 9211) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Anti-p38 MAPK (Cat. no. 9217), anti-p-c-Jun N-terminal kinase (JNK;
Thr183/Tyr185; Cat. no. 9251), and anti-p44/42 extracellular
signal-regulated kinase (ERK) mAbs (Cat. no. 9107), as well as
anti-phospholipase Cγ2 (PLCγ2; Cat. no. 3872), anti-p-(Tyr759)
PLCγ2 (Cat. no. 3874), anti-p-(Ser) protein kinase C (PKC)
substrate (pleckstrin; p-p47; Cat. no. 2261), anti-JNK (Cat. no.
9252), and anti-p-p44/p42 ERK (Thr202/Tyr204) polyclonal antibodies
(pAbs; Cat. no. 9101) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Anti-p-protein kinase B (Akt)
(Ser473; Cat. no. 9271) and anti-Akt mAbs (Cat. no. 2920) were
purchased from Biovision (Mountain View, CA, USA). An
anti-pleckstrin (p47) pAb (Cat. no. GTX17020) was purchased from
Gene Tex (Irvine, CA, USA). A Hybond-P polyvinylidene fluoride
(PVDF) membrane, an enhanced chemiluminescence western blotting
detection reagent, horseradish peroxidase (HRP)-conjugated donkey
anti-rabbit immunoglobulin G (IgG; Cat. no. RPN4301) and sheep
anti-mouse IgG (Cat. no. RPN4201) were purchased from GE Healthcare
Life Sciences (Little Chalfont, UK). A fluorescein isothiocyanate
(FITC)-conjugated anti-human CD42P (P-selectin) mAb (Cat. no.
304904) was obtained from Bio Legend, Inc. (San Diego, CA,
USA).
Synthesis of
1-(2-pyridyl)-3-(3-methoxyphenyl)imidazo[1,5-a] pyridine (L)
Ammonium acetate (1.93 g, 25 mM), 4-methoxy
benzaldehyde (1.02 g, 7.5 mM) and glacial acetic acid (25 ml) were
added to a degassed mixture of di-pyridin-2-yl-metha-none (0.9 g, 5
mM), and the mixture was placed in an oil bath maintained at 120°C
under a nitrogen atmosphere for 18 h. The reaction mixture was then
cooled to room temperature, poured into ice cold water, and
extracted with chloroform (3×70 ml). The organic layer was dried
over anhydrous sodium sulfate, and then filtered. The residue was
chromatographed through silica gel (ethylacetate/hexane = 1:3) to
yield a yellow solid. The characteristics of this solid were as
follows: Melting point: 105–110°C; 1H nuclear magnetic
resonance (NMR; 400 MHz, CDCl3): δ 8.72–8.70 (d, 1H,
J=8 Hz), 8.65–8.64 (d, 1H, J=4 Hz), 8.27–8.25 (d, 1H,
J=8 Hz), 8.2–8.19 (d, 1H, J=8 Hz), 7.78–7.73 (t, 3H,
J=10 Hz), 7.13–7.07 (m, 3H, J=8 Hz), 6.96–6.92 (t,
1H, J=8 Hz), 6.67–6.64 (t, 1H, J=6 Hz), 3.9 (s, 3H);
13C NMR (400 MHz, CDCl3) 160.0, 155.1, 148.9,
138.0, 136.1, 130.1, 129.9, 129.7, 122.5, 121.7, 121.5, 120.8,
120.3, 119.8, 114.4, 113.6, 55.3; ultraviolet-visible
spectrophotometry (UV-Vis; λabs, nm): 380, 321, 300,
238; electrospray ionization-mass spectrometry (ESI-MS; m/z): 301
(M+).
Synthesis of [Ir(Cp*)(L)Cl]BF4
(Ir-3)
[Ir(Cp*)(Cl)2]2 (0.16 g, 0.2
mM) in methanol (10 ml) solution was added dropwise to 10 ml
methanolic solution of
1-(2-pyridyl)-3-(3-methoxy-phenyl)imidazo[1,5-a]pyridine (L)
(0.12 g, 0.4 mM), and the solution was stirred at room temperature
for 3 h. Subsequently, NH4BF4 (200 mg, 0.60
mM) was added to the initial pale yellow solution, changing the
color to orange. After 24 h, the solution was evaporated and the
solid obtained was filtered. The residue was washed with diethyl
ether (40 ml) and dried under a vacuum. The desired product was
recrystallized from the dichloromethane/hexane mixture, yielding
orange micro-crystals. The characteristics of this solid were as
follows: 1H NMR (400 MHz, dimethyl sulfoxide [DMSO]-D6):
δ 8.82–8.81 (d, 1H, J=4 Hz), 8.52–8.39 (m, 3H), 8.17–8.10
(m, 3H), 7.54–7.47 (m, 2H), 7.31–7.29 (d, 2H, J=8 Hz),
7.15–7.11 (t, 1H, J=8 Hz), 3.90 (s, 3H), 1.29 (s, 15H);
UV-Vis (λabs, nm) (ε, M−1 cm−1):
406 (1649), 385 (2372), 367 (1854), 282 (2871), 242 (2881); ESI-MS
(m/z): 664.08 [M-BF4]+ (Fig. 1).
Platelet aggregation
The present study was approved by the Institutional
Review Board of Taipei Medical University (Taipei, Taiwan; approval
no. TMU-JIRB-N201612050) and conformed to the directives of the
Declaration of Helsinki. All human volunteers involved in the
present study provided written informed consent. Human platelet
suspensions were prepared as described previously (10). Human blood was collected from 20
healthy individuals (aged between 20 and 30 years old, 12 females
and 8 males) of Taipei Medical University who had taken no drugs or
other substances that would interfere with the experiment for at
least 14 days prior to collection between July 2017 and August
2017. The collected blood was mixed with an acid-citrate-dextrose
solution (9:1, v/v) and centrifuged at 120 × g for 10 min at 37°C
to separate the platelet-rich plasma (PRP). Following
centrifugation, the PRP was supplemented with 0.5 µM
PGE1 and 6.4 IU/ml heparin, incubated for 10 min at 37°C
and centrifuged at 500 × g for 10 min at room temperature. The
collected pellets were mixed with 5 ml Tyrode's solution, pH 7.3
[containing (mM) NaCl 11.9, KCl 2.7, MgCl2 2.1,
NaH2PO4 0.4, NaHCO3 11.9, and
glucose 11.1]. Then, apyrase (1.0 U/ml), PGE1 (0.5
µM) and heparin (6.4 IU/ml) were added, and the mixture was
incubated for 10 min at 37°C. Following centrifugation of the
suspensions at 500 × g for 10 min at room temperature, the washing
procedure was repeated. The washed platelets were finally suspended
in Tyrode's solution containing BSA (3.5 mg/ml). The final
Ca2+ concentration in the Tyrode solution was 1 mM. A
platelet aggregation test was performed using a Lumi-Aggregometer
(Payton Associates, Scarborough, ON, Canada) as described
previously (10). Various
concentrations of Ir-3 or a solvent control (0.1% DMSO) were
preincubated with platelet suspensions (3.6×108
cells/ml) for 3 min prior to the addition of various concentrations
of agonists (1 µg/ml collagen, 0.01 U/ml thrombin, 1
µM U46619 or 120 µM AA). The extent of platelet
aggregation was calculated as the percentage with respect to the
control (absence of Ir-3) in light transmission units when the
reaction had proceeded for 6 min. For an ATP release assay, 20
µl luciferin-luciferase was added 1 min prior to the
addition of the agonist, and the amount of ATP released was
compared with that released by the control using a
Lumi-Aggregometer.
Measurement of [Ca2+]i
mobilization using Fura-2AM fluorescence
The [Ca2+] concentration was measured
using the calcium sensitive dye Fura-2AM, as described previously
(10). In brief, citrated whole
blood was centrifuged at 120 × g for 10 min at room temperature,
and the supernatant was collected and incubated with 5 µM
Fura-2AM for 1 h at 37°C. The Fura-2AM-preincubated platelets were
washed with Tyrode's solution and treated with Ir-3 in the presence
of 1 mM CaCl2, and then stimulated with collagen (1
µg/ml). Fura-2 fluorescence was measured using excitation
wavelengths of 340 and 380 nm and emission at 510 nm with a
spectrofluo-rometer (Hitachi FL Spectrophotometer F-4500; Hitachi,
Ltd., Tokyo, Japan).
Detection of lactate dehydrogenase
Washed platelets (3.6×108 cells/ml) were
preincubated with 20–100 µM Ir-3 or the solvent control
(0.1% DMSO) for 20 min at 37°C. Following incubation, supernatant
(10 µl) was added to a Fuji Dri-Chem slide lactate
dehydrogenase (LDH)-PIII (Fujifilm Holdings Corporation, Tokyo,
Japan). The absorbance of the supernatant was measured at 540 nm
using a UV-Vis spectrophotometer (UV-160; Shimadzu Corporation,
Kyoto, Japan). A maximal value of LDH was recorded in platelets
lysed with Triton.
Flow cytometric analysis
Platelet surface P-selectin expression was examined
using flow cytometric analysis. Washed platelets were prepared as
aforementioned. Aliquots of platelet suspensions
(3.6×108 cells/ml) were treated with either the solvent
control (0.1% DMSO) or Ir-3 (10 and 20 µM), and
FITC-conjugated P-selectin (2 µg/ml) mAbs were then added
and incubated for 3 min at 37°C. This was followed by the addition
of collagen (1 µg/ml) to trigger platelet activation, for 1
min at 37°C. The suspensions were then assayed for
fluorescein-labeled platelets using a flow cytometer (FACScan
System; BD Biosciences, San Jose, CA, USA) with FACSuite™ software
(version 1.0.5.3841; BD Biosciences). Data were collected from
50,000 platelets per experimental group, and the platelets were
identified on the basis of their characteristic forward and
orthogonal light-scattering profiles. All experiments were repeated
at least three times to ensure reproducibility.
Western blotting
Washed platelets (1.2×109 cells/ml) were
preincubated with Ir-3 (10 and 20 µM) or the solvent control
(0.1% DMSO) for 3 min at 37°C, and 1 µg/ml collagen was
added to trigger platelet activation for 5 min at 37°C. The
reaction was then stopped, and the platelets were immediately
resuspended in 200 µl lysis buffer [containing (mM) 50
HEPES, 5 EDTA, 50 NaCl and 1% Triton X-100]. Proteins were
quantified using an ELISA reader at 570 nm. Samples containing 80
µg of protein were separated through 12% sodium dodecyl
sulfate gel electrophoresis, and the proteins were
electrotrans-ferred to PVDF membranes using a Bio-Rad semi-dry
transfer unit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
blots were then blocked with Tris-buffered saline in Tween-20
(TBST; 10 mM Tris-base, 100 mM NaCl, and 0.01% Tween-20) containing
5% BSA for 1 h at room temperature, and probed with the
aforementioned primary antibodies (diluted 1:1,000 in TBST) for 2 h
at 4°C. The membranes were incubated with HRP-conjugated anti-mouse
IgG for anti-ERK, anti-p38 and anti-Akt or anti-rabbit IgG for
anti-phosphorylated MAPKs, anti-p-Akt, anti-p-PLCγ2, anti-PLCγ2,
anti-p-PKC substrate and anti-JNK (diluted 1:3,000 in TBST) for 1 h
at 4°C. An enhanced chemiluminescence system was used to detect
immunoreactive bands, and their optical density was quantified
using Bio-profil Biolight software (version V2000.01; Vilber
Lourmat, Marne-la-Vallée, France).
Measurement of OH radical formation in
platelet suspensions and Fenton reaction solution through electron
spin resonance (ESR) spectrometry
Electron spin resonance spectrometry was performed
on a Bruker EMX ESR spectrometer (Bruker Corporation, Billerica,
MA, USA) as described previously (11). Platelet suspensions
(3.6×108 cells/ml) or Fenton reaction solution (50
µM FeSO4+2 mM H2O2) were
preincu-bated with 0.1% DMSO or Ir-3 (10 and 20 µM) for 3
min at room temperature, with or without the addition of 1
µg/ml collagen. Following incubation of the suspensions for
5 min, 100 µM DMPO was added prior to the execution of ESR
spectrometry. The ESR spectra were recorded using a quartz flat
cell designed for aqueous solutions. The spectrometer was operated
at a power of 20 mW, frequency of 9.78 GHz, scan range of 100 G and
receiver gain of 5×104. The modulation amplitude was 1
G, the time constant was 164 ms, and scanning was performed for 42
sec, with the spectra being the sum of three scans.
Statistical analysis
The experimental results are expressed as the mean ±
standard error of the mean, and are accompanied by the number of
observations (n). The n values refer to the number of experiments,
and each experiment was conducted using different blood donors. The
between-group differences in the experiments were assessed through
one-way analysis of variance (ANOVA). When the ANOVA indicated
significant differences among the group means, the groups were
compared using the Student-Newman-Keuls method. Statistical
analyses were performed using SAS (version 9.2; SAS Institute,
Inc., Cary, NC, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Inhibitory effects of Ir-3 on platelet
aggregation in washed human platelets
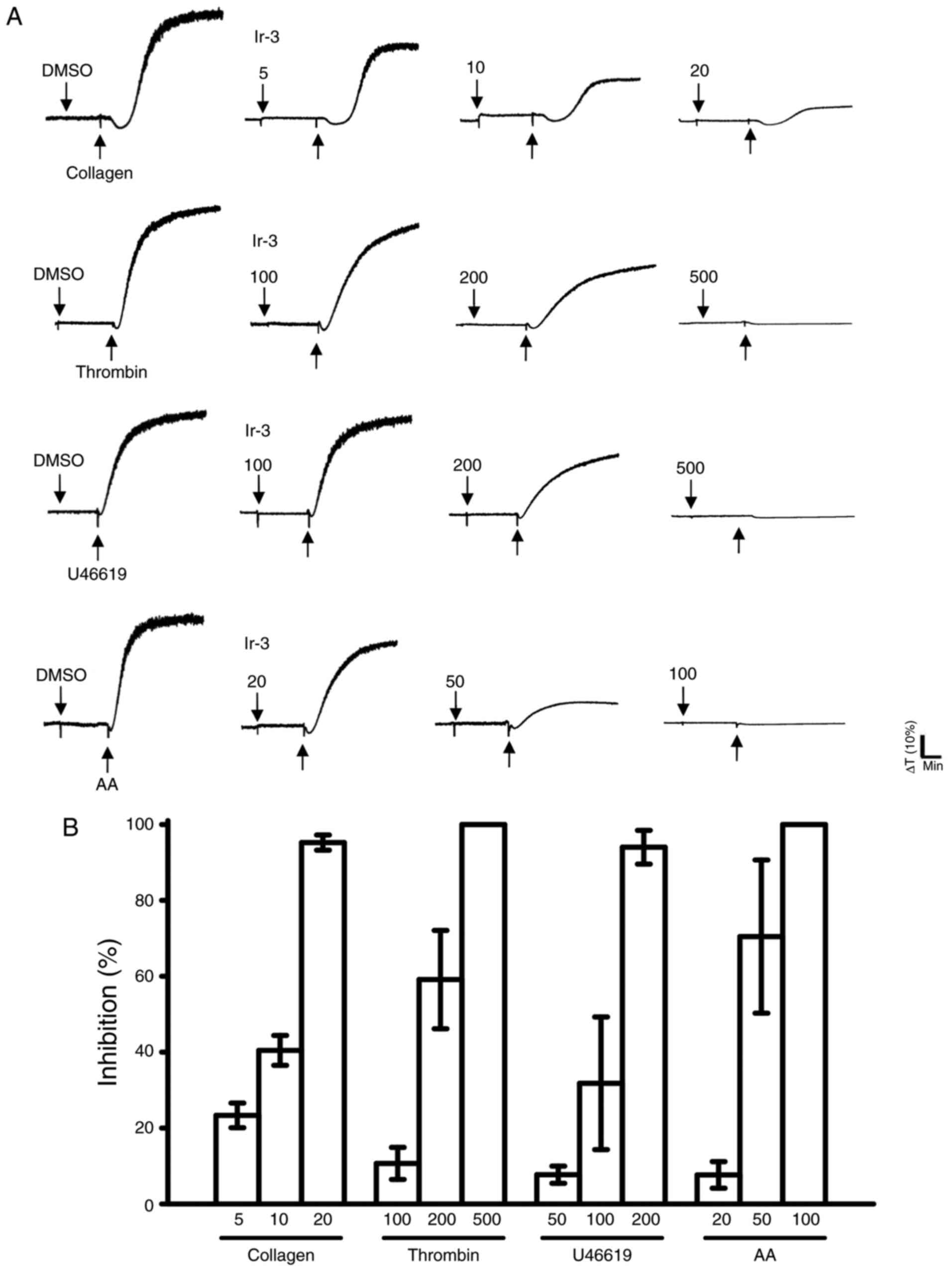
Ir-3 (5–20 µM; Fig. 2) inhibited platelet aggregation in
collagen (1 µg/ml)-stimulated human platelets in a
concentration-dependent manner. Ir-3 (20–100 µM)
demonstrated moderate activity against platelet aggregation in
platelets stimulated by 120 µM AA. Furthermore, Ir-3
(100–500 µM) had relatively weak activity against 0.01 U/ml
thrombin or 1 µM U46619, a prostaglandin endoperoxide
stimulant, indicating that Ir-3 had more potent inhibitory activity
against collagen stimulation compared with other agonists (Fig. 2B). The solvent control, 0.1% DMSO,
did not affect platelet aggregation (Fig. 2A). Furthermore, aspirin (20, 50
and 100 µM) inhibited platelet aggregation stimulated by 1
µg/ml collagen in a concentration-dependent manner, with a
half maximal inhibitory concentration of ~50 µM (n=3; data
not shown). Therefore, Ir-3 is ~5 times more potent than aspirin at
inhibiting collagen-stimulated platelet aggregation. In the
following experiments, 1 µg/ml collagen was used as an
agonist for investigating potential inhibitory mechanisms of Ir-3
in human platelets.
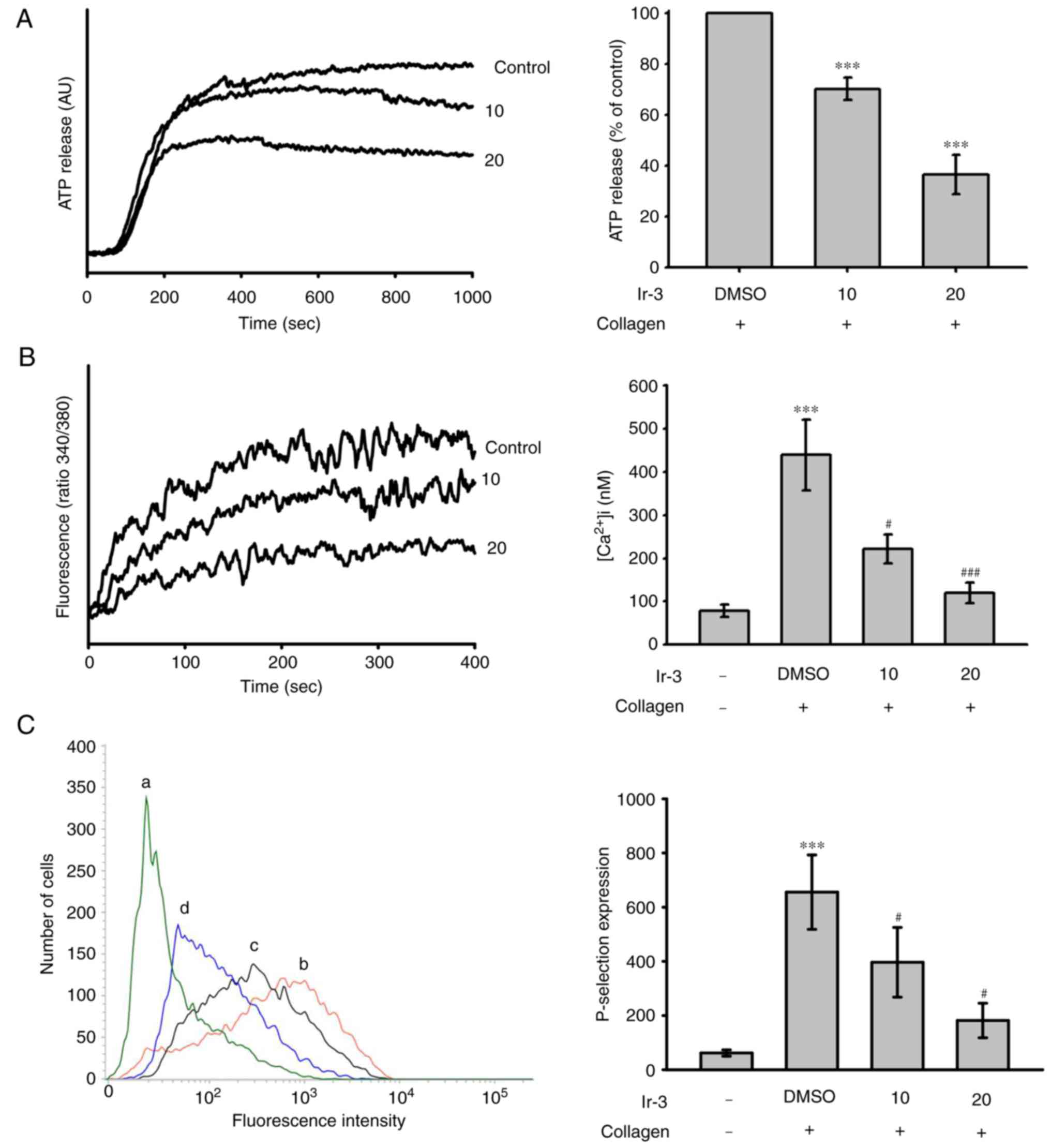
Effects of Ir-3 on ATP release reaction,
relative [Ca+2] mobilization and surface P-selectin
expression
The activation of platelets releases the granular
contents, including adenosine diphosphate (ADP)/ATP and
Ca+2 from the dense granules, and surface P-selectin
from the α-granules, to the external environment where they are
involved in substantial platelet aggregation. In the present study,
Ir-3 (10 and 20 µM) inhibited the ATP release reaction (10
µM, 33.2±1.1%; 20 µM, 67.8±3.7%; Fig. 3A) and relative [Ca2+]i
mobilization (resting control, 78.4±14.3 nM; collagen-stimulated,
438.9±82.6 nM; 10 µM, 221.9±33.3 nM; 20 µM,
119.1±23.8 nM; n=3, Fig. 3B) in
platelets stimulated by 1 µg/ml collagen. The corresponding
statistical data are presented in the right panels of Fig. 3. In quiescent platelets,
P-selectin is located on the inner wall of the α-granules. Platelet
activation exposes the inner walls of the granules to the outside
of the cell (12). Ir-3 treatment
significantly reduced collagen-induced surface P-selectin
expression, as demonstrated by the corresponding statistical data
in the right panel (resting control, 61.7±11.5; collagen-activated,
655.7±137.3; 10 µM, 396.7±128.8; 20 µM, 181.7±63.8;
n=3; Fig. 3C).
Influence of Ir-3 on LDH release and
cyclic nucleotide formation in washed human platelets
The aggregation curves of platelets pre-incubated
with 100 µM Ir-3 for 10 min and washed two times with Tyrode
solution were not significantly different from those of platelets
pre-incubated with the solvent control (0.1% DMSO) under equivalent
conditions (Fig. 4A),
preliminarily indicating that the effects of Ir-3 on platelet
aggregation are reversible and non-cytotoxic. Furthermore, the LDH
results revealed that Ir-3 (20, 50, and 100 µM) incubated
with platelets for 20 min did not significantly increase LDH
activity or exert cytotoxic effects on platelets (Fig. 4B), demonstrating that Ir-3 does
not affect platelet permeability or induce platelet cytolysis.
Furthermore, 100 µM SQ22536, an adenylate cyclase inhibitor,
and 10 µM ODQ, a guanylate cyclase inhibitor, significantly
reversed the inhibition of collagen-induced platelet aggregation
mediated by 1 µM PGE1 or 10 µM NTG
(Fig. 4C and D). Neither SQ22536
nor ODQ significantly reversed the inhibition of collagen-induced
platelet aggregation mediated by 20 µM Ir-3 (Fig. 4C and D), indicating that the
mechanisms of Ir-3-mediated inhibition of platelet aggregation are
independent of increasing cyclic nucleotide formation.
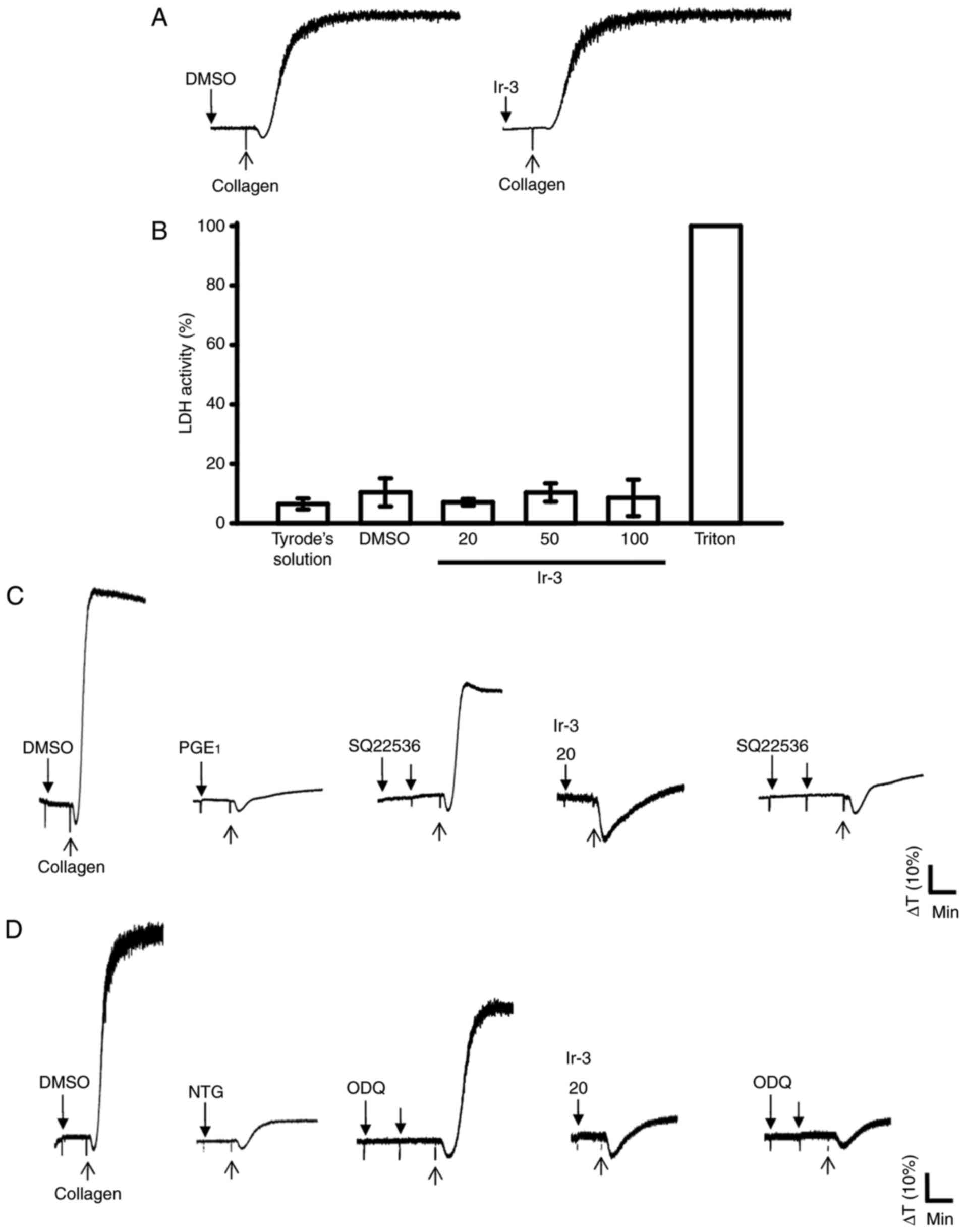 | Figure 4Influence of Ir-3 on cytotoxicity,
LDH release and cyclic nucleotide formation in human platelets. (A)
Washed platelets were pre-incubated with the solvent control (0.1%
DMSO) or Ir-3 (100 µM) for 10 min and subsequently washed
two times with Tyrode's solution. Collagen (1 µg/ml) was
then added to trigger platelet aggregation. (B) Washed platelets
(3.6×108/ml) were pre-incubated with the solvent control
(0.1% DMSO) or Ir-3 (20, 50, and 100 µM) for 20 min, and a
10-µl aliquot of the supernatant was deposited on a Fuji
Dri-Chem slide LDH-PIII. For other experiments, washed platelets
(3.6×108 cells/ml) were pre-incubated with (C) 1
µM PGE1, (D) 10 µM NTG, or Ir-3 (20
µM) in the absence or presence of 100 µM SQ22536 or
10 µM ODQ, and were subsequently treated with 1 µg/ml
collagen to induce platelet aggregation. Data are presented as the
mean ± standard error of the mean (n=3). Profiles in sections A, C
and D represent the four independent experiments. LDH, lactate
dehydrogenase; PGE1, prostaglandin E1; NTG,
nitroglycerin; ODQ, 1H-[1,2,4]
oxadiazolo[4,3-a]quinoxalin-1-one. |
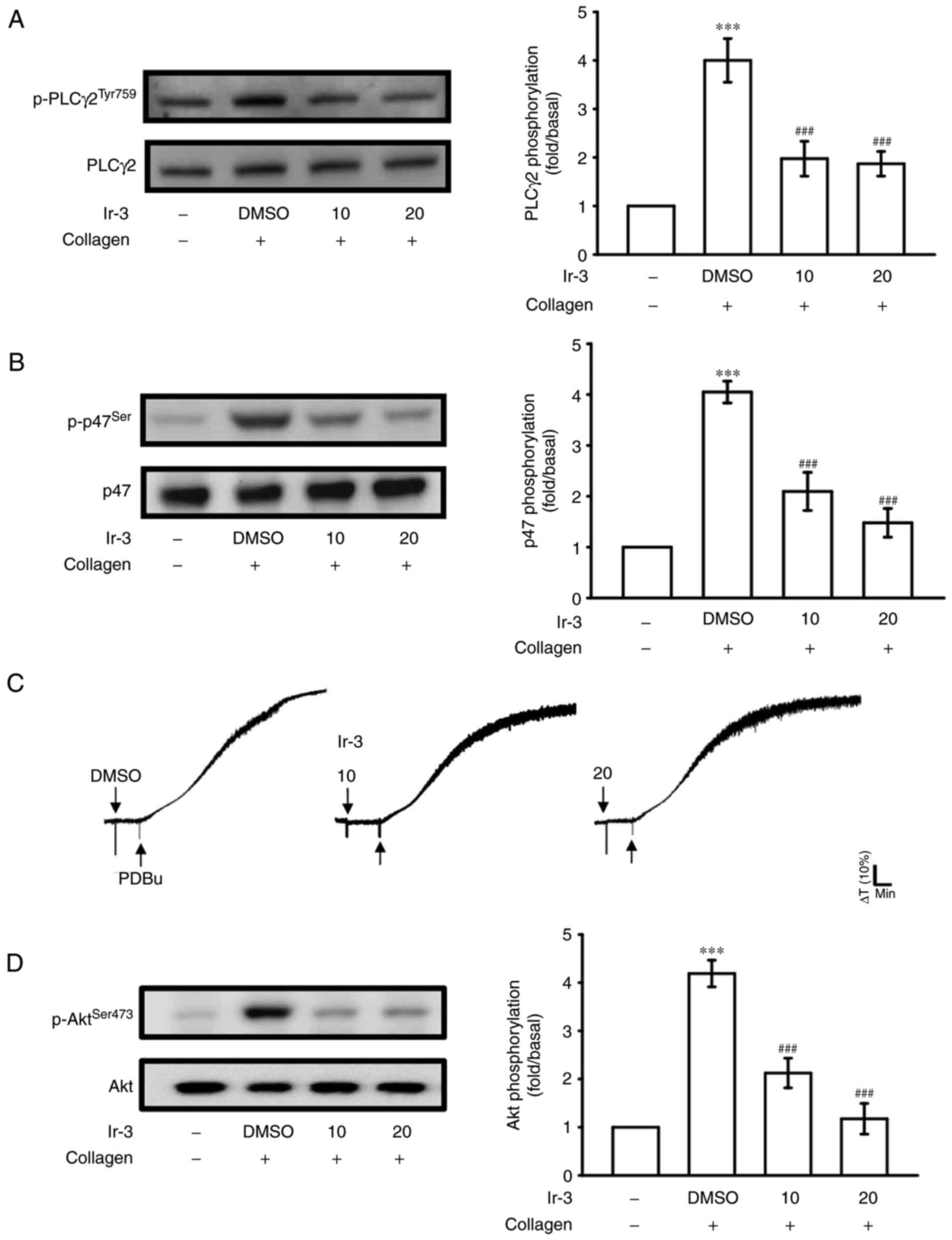
Regulatory character of Ir-3 on the
PLCγ2/PKC cascade and Akt activation
PLCs hydrolyze phosphatidylinositol 4,5-bisphosphate
to generate the secondary messengers inositol 1,4,5-trisphosphate
(IP3) and diacylglycerol (DAG). IP3 triggers
relative [Ca2+] mobilization and DAG activates PKC,
yielding a ~47-kDa protein that is predominantly phosphorylated
(p47 protein; pleckstrin) and leads to the ATP release reaction
(13). Fig. 3A and B illustrate the inhibitory
effects of Ir-3 against ATP release and relative [Ca2+]
mobilization. The influence of Ir-3 on the phosphorylation of the
PLCγ2-PKC signaling cascade was then investigated further. At 10
and 20 µM, Ir-3 significantly reduced PLCγ2 phosphorylation
and PKC activation (pleckstrin phosphorylation) in
collagen-stimulated platelets compared with the DMSO control
(Fig. 5A and B). However, Ir-3
had no significant effects on platelet aggregation stimulated by
150 nM PDBu, a PKC activator (Fig.
5C), indicating that Ir-3 does not directly disturb PKC
activation but may interfere with upstream regulators of PKC,
including PLCγ2. Akt is a serine/threonine-specific protein kinase
that serves a key function in multiple cellular processes,
including platelet activation, cell proliferation, apoptosis and
cell migration (14). Ir-3 (10
and 20 µM) significantly inhibited collagen-induced Akt
phosphorylation (Fig. 5D)
compared with the DMSO control, demonstrating the involvement of
Akt signaling pathway inhibition in Ir-3-mediated inhibition of
platelet activation.
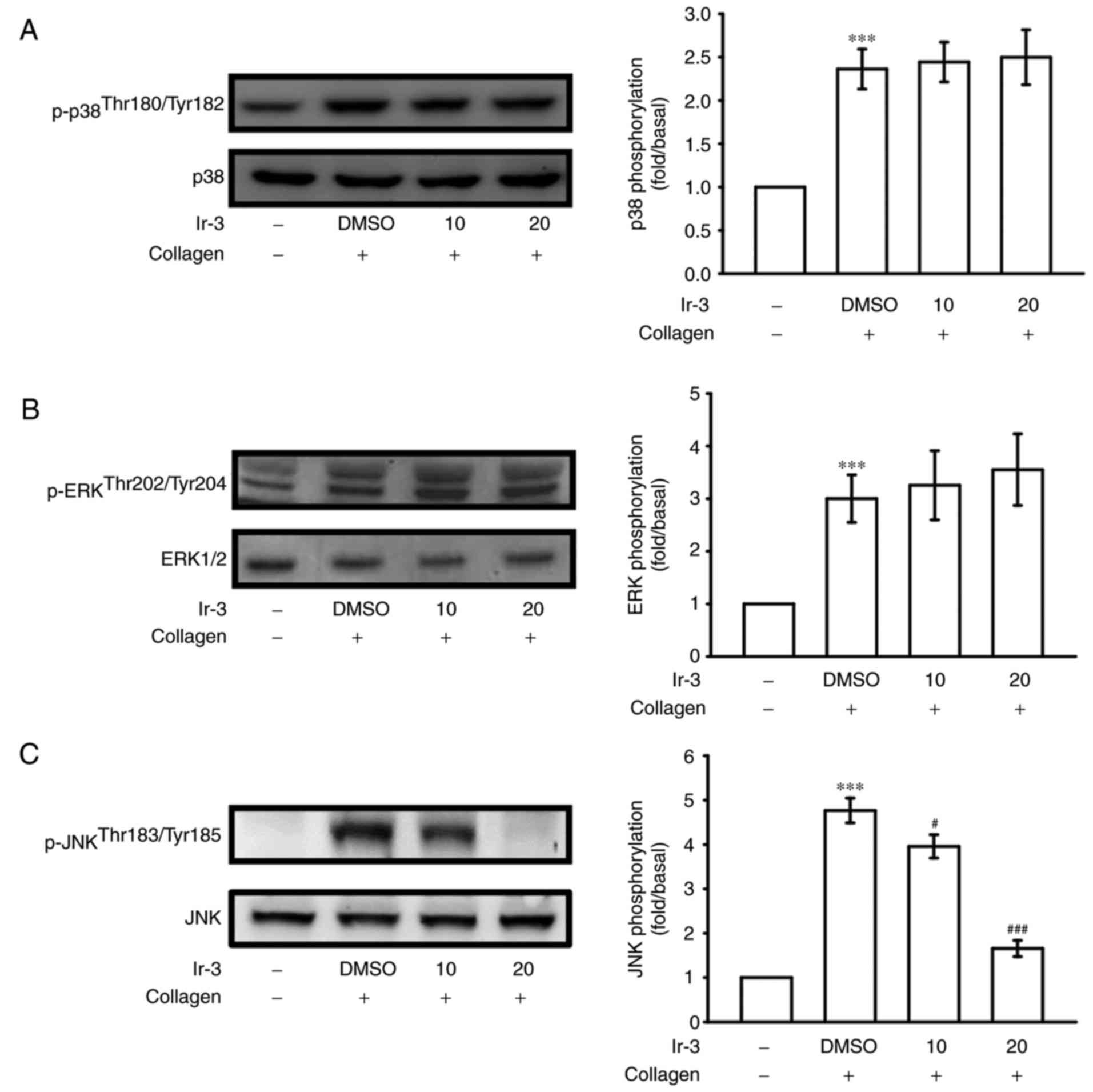
Inhibitory effects of Ir-3 on p38 MAPK,
ERKs and JNK1 phosphorylation
To investigate the inhibitory mechanisms of Ir-3 in
platelet activation, several signaling molecules of the MAPK
phosphorylation pathway were evaluated. MAPKs, including p38 MAPK,
ERKs, and JNKs, regulate major cellular responses in eukaryotic
organisms and contribute to cell proliferation, migration,
differentiation, and apoptosis (15). ERKs, JNK1, and p38 MAPK have been
identified in platelets (16).
Although collagen-induced p38 MAPK and ERK phosphorylation was
unaffected by Ir-3, JNK1 phosphorylation was suppressed by 10 and
20 µM Ir-3 in a concentration-dependent manner (Fig. 6). These results suggest that only
JNK1 may be involved in the antiplatelet activity of Ir-3.
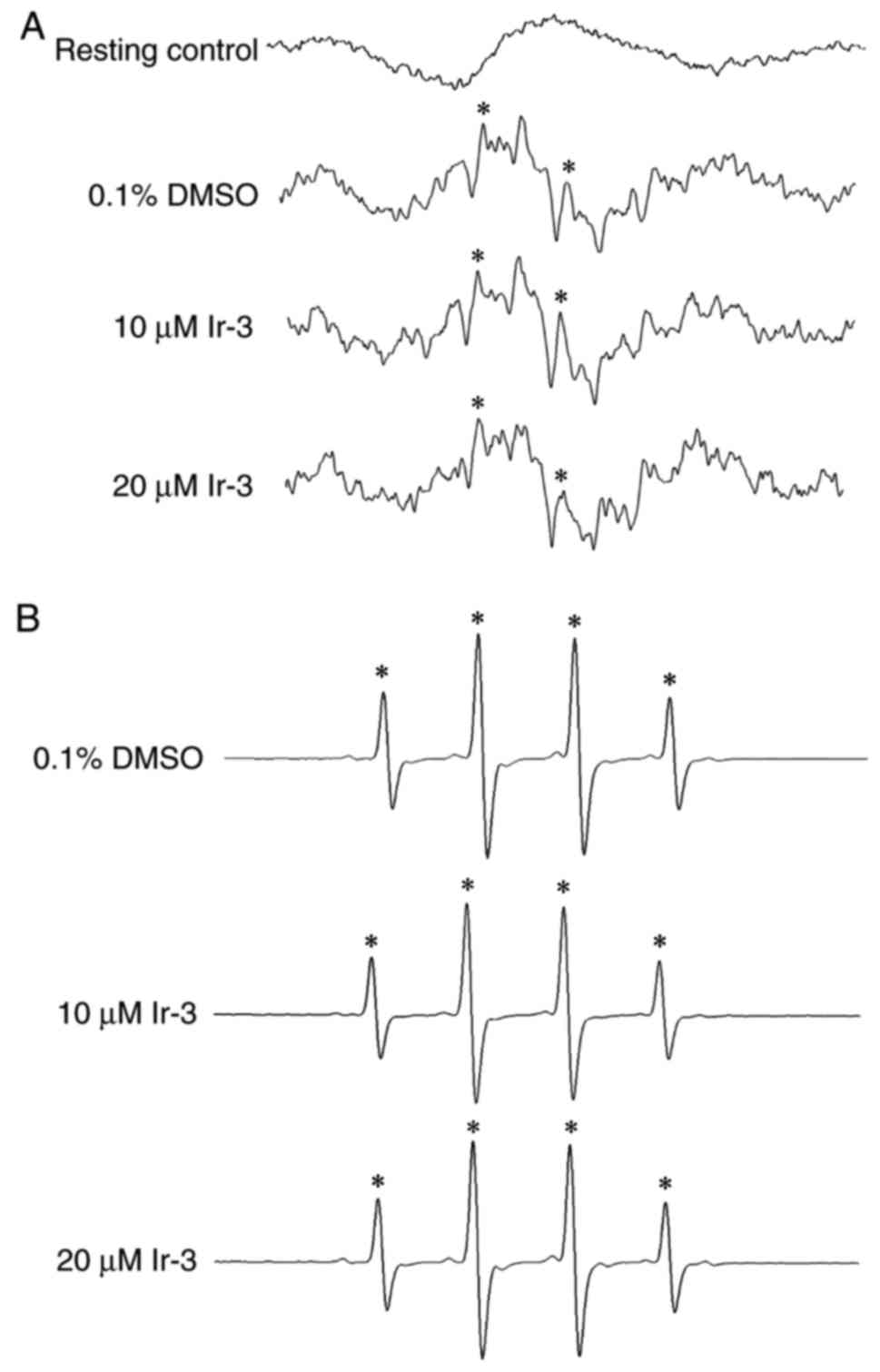
Determination of the function of the OH
radical (OH·) in the Ir-3-mediated inhibition of platelet
aggregation through ESR spectrometry
An ESR signal indicative of OH· formation was
observed in collagen-stimulated platelet suspensions and Fenton
reaction solution (cell-free system; Fig. 7A and B). A typical OH· signal
(aN =aH = 14.8 G) and a long-lived g=2.005
radical detectable using the spin trap DMPO were observed in
collagen-stimulated platelets, whereas this signal was not detected
in resting platelets (Fig. 7A and
B). Treatment with 10 and 20 µM Ir-3 did not notably
diminish the OH· signals in collagen-stimulated platelet
suspensions or Fenton reaction solution (Fig. 7), suggesting that the
Ir-3-mediated inhibition of platelet activation may not be mediated
through the reduction of free radical formation.
Discussion
Platelet activation is associated with thrombotic
events in patients with cancer (17). Chemotherapeutics may amplify this
effect and stimulate vascular thromboembolic events (VTEs) by
aggravating endothelial damage, increasing platelet aggregation and
increasing oxidative damage, leading to vascular toxicity (18). Among Pt-based chemotherapeutics,
cisplatin has a high prevalence of treatment-associated VTEs
(19). Gemcitabine in combination
with a Pt-based agent is associated with increased thrombotic and
vascular side effects (20,21). Therefore, research has focused on
the development of novel metal-based agents for the inhibition of
platelet activation to treat vascular disease, reduce toxic side
effects and overcome Pt resistance. To the best of our knowledge,
the present study is the first to demonstrate that Ir-3, an unique
synthetic Ir (III)-derived compound, displays effective
antiplatelet activity in human platelets in addition to its
antitumor activity.
Platelets adhere to subendothelial matrix proteins,
including collagen, thus altering their shape and releasing their
granular contents, which include ATP, Ca+2 and
P-selectin. Intracellular [Ca2+] mobilize as a result of
several agonists, including collagen, thrombin and AA, to
phosphorylate the Ca2+/calmodulin-dependent myosin light
chain (20 kDa), which is involved in the release of serotonin and
ATP (22), and platelet
aggregation. Therefore, the inhibition of [Ca2+]
mobilization and ATP production are crucial for assessing the
antiplatelet effects of a compound. In the present study, Ir-3
inhibited platelet aggregation to different degrees, depending on
the agonist used (either collagen, U46619, AA or thrombin),
indicating that Ir-3 did not act at the specific individual
receptors of these agonists. Therefore, Ir-3 may exert its
inhibitory effects on stimulated platelets through a common
signaling cascade.
Furthermore, platelet activation by collagen,
substantially alters PLC expression, and results in IP3
and DAG production, which activates PKC and consequently induces
p47 phosphorylation (13). PKC
activation triggers specific responses, assisting in the
transmission of activating signals in different cellular
compartments. The PLCγ family is comprised of the isozymes PLCγ1
and PLCγ2, with PLCγ2 being involved in collagen-dependent
signaling in platelets (23).
Ir-3 diminished collagen-induced PLCγ2-PKC activation; however,
Ir-3 was not effective on PKC activation as it did not inhibit
PDBu-induced platelet aggregation, suggesting that the
Ir-3-mediated inhibition of platelet activation involves PLCγ2
downstream signaling. This result also explains how Ir-3 was more
efficacious at inhibiting platelet aggregation induced by collagen
than that induced by thrombin, U46619, and AA.
Cyclic nucleotides are important modulators of
platelet activation (24), and
intracellular cyclic AMP- and cyclic GMP are involved in the
inhibition of human platelet activation. At elevated levels, cyclic
nucleotides prevent several platelet responses and reduce
[Ca2+] levels through Ca2+ uptake by the
dense tubular system, thereby suppressing PLC and PKC activation
(24). Therefore, cyclic AMP and
cyclic GMP synergistically inhibit platelet activation.
Furthermore, neither SQ22536, an inhibitor of adenylate cyclase,
nor ODQ, an inhibitor of guanylate cyclase, significantly reversed
the Ir-3-mediated inhibition of collagen-induced platelet
aggregation. Therefore, the Ir-3-mediated mechanisms were
independent of increasing cyclic nucleotide formation in
platelets.
Akt is a downstream molecule of phosphoinositide
3-kinase (PI3K). Akt-deficient mice reported defective effects in
agonist-induced platelet activation, suggesting that Akt regulates
platelet activation, and such regulation may have consequences
concerning thrombosis (14,25). Therefore, selective inhibitors of
Akt isoforms or its activating proteins, including individual PI3K
isoforms, may be attractive antithrombotic therapy targets
(14). In addition, MAPKs are
activated by specific MAPK kinases (MEKs). Specifically, MEK1/2,
MEK3/6 and MEK4/7 activate ERKs, p38 MAPK and JNKs, respectively
(26). Cytosolic phospholipase
A2 (cPLA2) is a substrate of p38 MAPK
activity induced by various agonists, including von Willebrand
factor (vWF) and thrombin (27).
Therefore, p38 MAPK is essential for cPLA2 stimulation
and AA release (28). This may
explain how Ir-3 was less able to inhibit p38 MAPK activation and
thrombin- or AA-stimulated platelet aggregation. ERK activation is
involved in platelet aggregation but requires prior ATP release,
which triggers a P2X1-mediated
Ca2+ influx and activates ERKs, thereby increasing the
phosphorylation of myosin light-chain kinase (27). JNK1 is the most recently
identified MAPK in platelets, and therefore its activation status
and function are poorly established. It is activated by several
agonists, including thrombin, vWF, collagen, and ADP (27). In addition, a previous study
demonstrated that an increased bleeding time, decreased integrin
αIIbβ3 activation and severe granule secretion
impairment occur in JNK−/− platelets (29). Therefore, the inhibition of JNK
phosphorylation may serve an important function in platelet
activation. In accordance with these results, the results of the
present study demonstrated that Ir-3 markedly inhibited
collagen-induced JNK1 phosphorylation.
Reactive oxygen species produced through platelet
activation, including hydrogen peroxide and OH·, may affect cells
that they come in contact with, for example endothelial cells,
thereby increasing platelet reactivity during thrombus formation.
Free radical species act as secondary signals that increase
[Ca2+] levels during the initial phase of platelet
activation, and PKC is involved in the receptor-mediated production
of free radicals in platelets (30). In addition, hydrogen peroxide
produced by platelets is converted into OH·, because platelet
aggregation is inhibited by OH· scavengers (30). The ESR spectrometry results from
the present study provide direct evidence that Ir-3 does not
significantly reduce OH· formation in collagen-stimulated platelet
suspensions and Fenton reaction solution. In addition, Ir tissue
distribution and excretion in rats following sub-chronic oral
administration of Ir (III) chloride hydrate (1–1,000 ng/ml) for 90
days was previously determined (31). Concerning the distribution, the
majority of Ir was located in the kidney and spleen, and smaller
amounts were observed in the lungs, liver and brain. Notably, a
slight increase of brain Ir levels was observed with increasing
doses of Ir, indicating the ability of Ir to cross the blood-brain
barrier. Furthermore, the dose-dependent increase of Ir levels in
the serum demonstrated that the Ir was distributed in the blood
compartment. For excretion, the body clearance of Ir primarily took
place by elimination via feces, and this was also dose-associated
(31).
In conclusion, the results of the present study
demonstrate that the novel Ir-3 compound inhibited platelet
activation by inhibiting signaling pathways, including the
PLCγ2-PKC cascade, and subsequently suppressing Akt and JNK1
activation. These alterations reduced granule secretion (including
ATP release, [Ca2+] levels and surface P-selectin
expression) and ultimately inhibited platelet aggregation. However,
additional studies are required to investigate the involvement of
other unidentified mechanisms of the Ir-3-mediated inhibition of
platelet activation. Nevertheless, Ir-3 represents a potential
chemotherapeutic agent for the treatment of cancer. In addition, it
may be used as an antiplatelet agent for treating thromboembolic
disorders or the interplay between platelets and tumor cells which
contributes to tumor cell proliferation and progression.
Acknowledgments
The present study was supported by grants from the
Ministry of Science and Technology of Taiwan (MOST
104-2622-B-038-003, MOST 104-2320-B-038-045-MY2 and MOST
106-2320-B-038-012), Shin Kong Wu Ho-Su Memorial Hospital-Taipei
Medical University (SKH-TMU-103-01), and the University Grants
Commission, India (MRP-MAJOR-CHEM-2013-5144; 69/2014 F. No.
10-11/12UGC).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Jayakumar T, Yang CH, Geraldine P, Yen TL
and Sheu JR: The pharmacodynamics of antiplatelet compounds in
thrombosis treatment. Expert Opin Drug Metab Toxicol. 12:615–632.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Belloc C, Lu H, Soria C, Fridman R,
Legrand Y and Menashi S: The effect of platelets on invasiveness
and protease production of human mammary tumor cells. Int J Cancer.
60:413–417. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Felding-Habermann B, Ooole TE, Smith JW,
Fransvea E, Ruggeri ZM, Ginsberg MH, Hughes PE, Pampori N, Shattil
SJ, Saven A, et al: Integrin activation controls metastasis in
human breast cancer. Proc Natl Acad Sci USA. 98:1853–1858. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boucharaba A, Serre CM, Gres S,
Saulnier-Blache JS, Bordet JC, Guglielmi J, Clezardin P and
Peyruchaud O: Platelet-derived lysophosphatidic acid supports the
progression of osteolytic bone metastases in breast cancer. J Clin
Invest. 114:1714–1725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iavicoli I, Cufino V, Corbi M, Goracci M,
Caredda E, Cittadini A, Bergamaschi A and Sgambato A: Rhodium and
iridium salts inhibit proliferation and induce DNA damage in rat
fibroblasts in vitro. Toxicol In Vitro. 26:963–969. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iavicoli I, Fontana L, Marinaccio A,
Alimonti A, Pino A, Bergamaschi A and Calabrese EJ: The effects of
iridium on the renal function of female wistar rats. Ecotoxicol
Environ Safe. 74:1795–1799. 2011. View Article : Google Scholar
|
|
7
|
Romero-Canelón I and Sadler PJ:
Next-generation metal anticancer complexes: Multitargeting via
redox modulation. Inorg Chem. 52:12276–12291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yellol J, Pérez SA, Buceta A, Yellol G,
Donaire A, Szumlas P, Bednarski PJ, Makhloufi G, Janiak C, Espinosa
A and Ruiz J: Novel C, N-cyclometalated benzimidazole ruthenium(II)
and iridium(III) complexes as antitumor and antiangiogenic agents:
A structure-activity relationship study. J Med Chem. 58:7310–7327.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schmitt F, Donnelly K, Muenzner JK, Rehm
T, Novohradsky V, Brabec V, Kasparkova J, Albrecht M, Schobert R
and Mueller T: Effects of histidin-2-ylidene vs. imidazol-2-ylidene
ligands on the anticancer and antivascular activity of complexes of
ruthenium, iridium, platinum, and gold. J Inorg Biochem.
163:221–228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sheu JR, Lee CR, Lin CH, Hsiao G, Ko WC,
Chen YC and Yen MH: Mechanisms involved in the antiplatelet
activity of staphylococcus aureus lipoteichoic acid in human
platelets. Thromb Haemost. 83:777–784. 2000.PubMed/NCBI
|
|
11
|
Chou DS, Hsiao G, Shen MY, Tsai YJ, Chen
TF and Sheu JR: ESR spin trapping of a carbon-centered free radical
from agonist-stimulated human platelets. Free Radic Biol Med.
39:237–248. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Harrison P and Cramer EM: Platelet
alpha-granules. Blood Rev. 7:52–62. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singer WD, Brown HA and Sternweis PC:
Regulation of eukaryotic phosphatidylinositol-specific
phospholipase C and phospholipase D. Annu Rev Biochem. 66:475–509.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Woulfe DS: Akt signaling in platelet and
thrombosis. Expert Rev Hematol. 3:81–91. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: A family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bugaud F, Nadal-Wollbold F, Levy-Toledano
S, Rosa JP and Bryckaert M: Regulation of c-jun-NH2 terminal kinase
and extracellular-signal regulated kinase in human platelets.
Blood. 94:3800–3805. 1999.PubMed/NCBI
|
|
17
|
Lip GY: Chin BS and Blann A: Cancer and
the prothrombotic state. Lancet Oncol. 3:27–34. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ferroni P, Della-Morte D, Palmirotta R,
McClendon M, Testa G, Abete P, Rengo F, Rundex T, Guadagni F and
Roselli M: Platinum-based compounds and risk for cardiovascular
toxicity in the elderly: Role of the antioxidants in
chemoprevention. Rejuvenation Res. 14:293–308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jafri M and Protheroe A:
Cisplatin-associated thrombosis. Anticancer Drugs. 19:927–929.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Barni S, Labianca R, Agnelli G, Bonizzoni
E, Verso M, Mandalà M, Brighenti M, Petrelli F, Bianchini C,
Perrone T and Gasparini G: Chemotherapy-associated thromboembolic
risk in cancer outpatients and effect of nadroparin
thromboprophylaxis: Results of a retrospective analysis of the
PROTECHT study. J Transl Med. 20(179)2011.
|
|
21
|
Dasanu CA: Gemcitabine: Vascular toxicity
and prothrombotic potential. Expert Opin Drug Safe. 7:703–716.
2008. View Article : Google Scholar
|
|
22
|
Kaibuchi K, Sano K, Hoshijima M, Takai Y
and Nishizuka Y: Phosphatidylinositol turnover in platelet
activation; calcium mobilization and protein phosphorylation. Cell
Calcium. 3:323–335. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ragab A, Séverin S, Gratacap MP, Aguado E,
Malissen M, Jandrot-Perrus M, Malissen B, Ragab-Thomas J and
Payrastre B: Roles of the C-terminal tyrosine residues of LAT in GP
VI-induced platelet activation: Insights into the mechanism of PLC
gamma 2 activation. Blood. 110:2466–2474. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Walter U, Eigenthaler M, Geiger J and
Reinhard M: Role of cyclic nucleotide-dependent protein kinases and
their common substrate VASP in the regulation of human platelets.
Adv Exp Med Biol. 344:237–249. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen J, De S, Damron DS, Chen WS, Hay N
and Byzova TV: Impaired platelet responses to thrombin and collagen
in AKT-1-deficient mice. Blood. 104:1703–1710. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang L and Karin M: Mammalian MAP kinase
signaling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Adam F, Kauskot A, Rosa JP and Bryckaert
M: Mitogen-activated protein kinases in hemostasis and thrombosis.
J Thromb Haemost. 6:2007–2016. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Canobbio I, Reineri S, Sinigaglia F,
Balduini C and Torti M: A role for p38 MAP kinase in platelet
activation by von Willebrand factor. Thromb Haemost. 91:102–110.
2004.
|
|
29
|
Adam F, Kauskot A, Nurden P, Sulpice E,
Hoylaerts MF, Davis RJ, Rosa JP and Bryckaert M: Platelet JNK1 is
involved in secretion and thrombus formation. Blood. 115:4083–4092.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wachowicz B, Olas B, Zbikowska HM and
Buczyński A: Generation of reactive oxygen species in blood
platelets. Platelets. 13:175–182. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Iavicoli I, Fontana L, Bergamaschi A,
Conti ME, Pino A, Mattei D, Bocca B and Alimonti A: Sub-chronic
oral exposure to iridium (III) chloride hydrate in female wistar
rats: Distribution and excretion of the metal. Dose-Response.
10:405–414. 2012. View Article : Google Scholar : PubMed/NCBI
|