Introduction
Traumatic brain injury (TBI) accounts for 15% of
body trauma worldwide and usually leads to neurological dysfunction
or mortality (1). Although great
efforts have been made to investigate the damage mechanism of TBI,
this highly complex disorder is not completely understood. Neuronal
injury following TBI can be classified into primary and secondary
damage; the latter is induced following the primary injury and may
persist for a prolonged time due to adverse biochemical changes
(2). Several signaling cascades
mediate secondary neuronal injury, including oxidative stress,
overload of intracellular calcium, cytoskeletal and mitochondrial
dysfunction, and inflammatory cell infiltration (3). In particular, a sterile inflammatory
response induced by various inflammatory mediators has been
suggested to serve a key role in secondary brain injury (4).
The nuclear factor-κB (NF-κB)/Rel proteins are a
family of transcription factors including five members, namely p50,
p52, p65/RelA, RelB and c-Rel, which are able to form homo- or
heterodimers with the ability to transmit receptor signals to the
nucleus (5). As a key factor in
the inflammatory response, NF-κB regulates a number of genes,
including apoptotic mediators, growth factors, inflammatory
cytokines, adhesion molecules and enzymes, all of which are
involved in crucial cellular pathophysiology processes (6–9).
Previous studies have shown that NF-κB contributes to neuronal
injury by inducing proinflammatory cytokines, but conversely also
promotes cell survival through upregulating the expression of
anti-apoptotic mediators such as B-cell lymphoma 2 (Bcl-2) and
Bcl-extra large (10). This
reciprocal interaction between pro- and anti-apoptotic pathways
indicates that NF-κB serves a dual role in the regulation of
neuronal survival in pathological conditions, with positive and
negative effects on brain damage and neuroprotection (11–15).
Previous studies have demonstrated that double peaks
of cerebral NF-κB activity occur following experimental neonatal
hypoxia-ischemia and subarachnoid hemorrhage (16,17). However, the full details of NF-κB
activation following TBI remain obscure. Thus, the present in
vitro study was designed with the aim of investigating the
activation time course of NF-κB and the expression of p65 and c-Rel
subunits in primary cultured cortical neurons following transection
injury.
Materials and methods
Primary culture of cortical neurons
A primary cortical neuron culture was prepared using
an established technique with certain modifications (18). Embryos from wild-type female mice
of the strain BALB/c were used in the study. In brief, cerebral
cortices were removed from the embryos at 15–17 days, stripped of
meninges and blood vessels and minced with
Ca2+/Mg2+-free Hank's balanced saline
solution (cat. no. 14170112; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) with the aid of a dissection microscope. The
cortex was dissected free and treated with 0.125% trypsin for 5 min
at 37°C. The trypsin-containing supernatant was then discarded.
Subsequently, the tissues were washed three times with precooled
phosphate-buffered saline (PBS), and then triturated in PBS with
fire-polished glass pipettes. The neuron suspension was filtered
through a 22-µm filter into a 15-ml polypropylene conical
tube, and centrifuged at 1,500 × g for 5 min at 4°C. The sediment
was resuspended in neurobasal medium (cat. no. 21103-049) with B27
(cat. no. 10889-038) (both from Thermo Fisher Scientific, Inc.),
100 U/ml penicillin and 0.1 mg/ml streptomycin. Finally, the
neurons were plated onto poly-D-lysine-coated 6-well plates at a
density of 1×106 cells/well. The cells were maintained
at 37°C in a humidified 5% CO2 atmosphere. Half of the
culture medium was replaced with fresh medium every 3 days. All
experiments were performed following 10–12 days in vitro.
Ethical approval for use of the embryonic tissue was given by the
Medical Ethics Committee of Jinling Hospital (Nanjing, China) with
the following reference number: SYXK 2012-0047. All surgical
procedures were performed in accordance with guidelines of the
Jinling Animal Care and Use Committee.
Traumatic neuronal injury model
A transection model was performed as described
previously (19). This
transection model used a slender plastic needle to scrape adherent
cells from a culture dish, which tore the soma while leaving a
large proportion of cells intact (20). Briefly, each well of a 6-well
plate was manually scratched with a sterile plastic needle in the
pattern of a 9×9-square grid with 4-mm spacing between the lines.
Cell cultures were then placed in an incubator at 37°C until a
designated post-trauma time point was reached. After 1, 6, 12, 24,
36 and 48 h, the neurons were collected for analysis using an
electrophoretic mobility shift assay (EMSA), western blotting,
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR), typan blue staining and lactate dehydrogenase (LDH)
release assay. Since a scratch injury initially activates neurons
at the wound edge and later expands to the entire neuron monolayer,
the entire culture on each dish was used for all experiments.
Cell nuclear protein extraction
Proteins from the nucleus and cytoplasm of the cells
were extracted according to the methods described in previous
studies (21,22). The primary cultured neurons were
washed twice with PBS and scraped in cold PBS. The clustered
neurons were then resuspended with 200 µl ice-cold buffer A,
which is composed of 10 mM HEPES (pH 7.9), 2 mM MgCl2,
10 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol (DTT) and 0.5 mM
phenylmethylsulfonyl fluoride (PMSF; all from Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany). The homogenate was subsequently
incubated on ice for 20 min, and 20 µl 10% Nonidet P40
solution was added (Sigma-Aldrich; Merck KGaA). The mixture was
stirred vortically for 30 sec and spun by centrifugation for 10 min
at 5,000 × g, 4°C. The supernatant comprising cytoplasmic protein
was discarded. The precipitated nuclear pellet was resuspended in
40 µl buffer B, which comprised 20 mM HEPES (pH 7.9), 420 mM
NaCl, 1.5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF
and 25% (v/v) glycerol. The mixture was incubated on ice for 60 min
with intermittent mixing, and then centrifuged at 13,000 × g at 4°C
for 15 min. The supernatant, which contained nuclear proteins, was
collected and stored at −80°C for EMSA and western blotting.
EMSA
EMSA was performed using a commercial kit (Gel Shift
assay system; Promega Biotech Co., Ltd., Beijing, China) to detect
NF-κB DNA binding activity. Double-stranded consensus
oligonucleotide probe (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) was
end-labeled with [γ-32P] ATP (Free Biotech, Beijing,
China) and T4 polynucleotide kinase. Neuron nuclear protein (2
µg in 7 µl) was incubated at room temperature for 10
min with 2 µl gel shift binding 5X buffer. The mixture was
then incubated for 20 min with 1 µl 32P-labeled
oligonucleotide probe. Subsequently, the reaction was stopped by
adding 1 µl gel loading buffer and the mixture was resolved
on a native 4% polyacrylamide gel in 0.5X Tris-borate-EDTA buffer.
Following electrophoresis at 250 V for 90 min, when the bromophenol
blue dye had migrated down three-quarters of the gel, the gel was
dried with plastic wrap on a gel dryer prior to exposure to X-ray
film (Fuji Hyperfilm; Fujifilms Holdings Corporation, Tokyo, Japan)
at −70°C. Autoradiography and quantification of the
autoradiographic signal were performed by analysis of the X-ray
film using Un-Scan-It software (version 7.1; Silk Scientific, Inc.,
Orem, UT, USA).
Western blotting
The nuclear protein previously stored at −80°C was
boiled in loading buffer [containing 1.0 M TrisHCl (pH 8.5), 8%
(w/v) lithium dodecyl sulfate, 40% (v/w) glycerol, 2 mM EDTA, 0.5 M
DTT and tracking dye in distilled/deionized water; Beyotime
Institute of Biotechnology, Nantong, China] for 5 min. BCA Protein
Quantification kit (Beyotime Institute of Biotechnology) was used
to measure the protein levels. Then, 45 µg protein samples
were loaded on a gel for 12% SDS-PAGE, separated
electrophoretically, and then transferred to a polyvinylidene
fluoride membrane. Before incubation with antibodies, the membrane
was blocked with blocking buffer (cat. no. P0023B; Beyotime
Institute of Biotechnology) at 4°C overnight. The membrane was
incubated in the primary antibody at 4°C overnight. The primary
antibodies were rabbit anti-rat p65 (sc-372) and c-Rel (sc-272)
(1:200 dilution), and rabbit anti-rat histone 3 antibody (sc-8654;
1:200 dilution; each from Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) was used as a loading control. After washing three times
in TBS containing 0.1% Tween-20, the horseradish peroxidase-labeled
secondary antibody (cat. no. A0208; Beyotime Institute of
Biotechnology) was applied at a 1:5,000 dilution for 1 h at room
temperature. Afterwards, the blot was washed three times, and the
blotted protein bands were visualized by enhanced chemiluminescence
(ECL; Amersham; GE Healthcare Life Sciences, Little Chalfont, UK).
Relative protein levels were estimated from the mean pixel density
using ImageJ software (version 1.51j8; National Institutes of
Health, Bethesda, MD, USA), normalized to histone 3, and calculated
as target protein expression/histone 3 ratios.
RT-qPCR
Total RNA was extracted from fresh neurons with
TRIzol reagent (Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol and immediately reverse transcribed to cDNA
with a PrimeScript RT reagent kit (Takara Biotechnology Co., Ltd.,
Dalian, China). The primers were synthesized by Invitrogen (Thermo
Fisher Scientific, Inc.). The primer sequences were as follows:
Interleukin-1β (IL-1β) forward, GACAGGATGCAGAAGGAGATTACT and
reverse, TGATCCACATCTGCTGGAAGGT; tumor necrosis factor-α (TNF-α)
forward, TCTCATTCCTGCTTGTGGC and reverse, CACTTGGTGGTTGCTTACG;
caspase-3 forward, GACTGGAAAGCCGAAACTC and reverse,
GGCAAGCCATCTCCTCATC; Bcl-2 forward, TGGGATGCTGGAGATGCG and reverse,
AGGCTGGAAGGAGAAGATGC; β-actin forward, AGGCACCAGGGCGTGAT and
reverse, CTCAGGCTGGAAGGAGAAGAT. The reactions were conducted in 96
well optical PCR plates using Applied Biosystems StepOnePlus
Real-Time PCR system (Thermo Fisher Scientific, Inc.) following the
kit protocol. Reactions were performed in a 20-µl volume of
reaction mix with SYBR Premix Ex Taq (2X) 10 µl, forward
primer (10 µM) 0.4 µl, reverse primer (10 µM)
0.4 µl, ROX reference dye (50X) 0.4 µl, DNA template
2.0 µl and dH2O 6.8 µl (Takara
Biotechnology Co., Ltd.). The thermal cycler protocol comprised:
Stage 1, 95°C, 30 sec for denaturation; and stage 2, 95°C, 5 sec;
60°C, 30 sec. Stages 1 and 2 were repeated 40 times, alternatingly.
The 2−ΔΔCq method (23) was used for analyzing the data.
Immunofluorescent labeling
For double-immunostaining, cells in 6-well plates
were fixed with 4% formaldehyde for 10 min at room temperature and
then washed three times with PBS. The fixed cells were blocked by
treatment with immunostaining blocking buffer (Beyotime Institute
of Biotechnology) for 1 h at room temperature. Subsequently, the
cells were incubated overnight at 4°C with the primary antibodies:
Mouse anti-neuronal nuclei (anti-NeuN; MAB377; 1:200 dilution; EMD
Millipore, Billerica, MA, USA), rabbit anti-p65 (sc-372) or
anti-c-Rel (sc-272) (1:200; Santa Cruz Biotechnology, Inc.). Goat
anti-rabbit IgG (A23220) and goat anti-mouse IgG (A23410) (1:200
dilution; both from Abbkine, Inc., Redlands, CA, USA) were used as
secondary antibodies (incubation for 1 h at room temperature).
After washing, neurons were incubated with
4′,6-diamidino-2-phenylindole (DAPI) for 5 min at room temperature
to stain the cell nuclei. Fluorescence microscopy imaging was
performed using a ZEISS HB050 inverted microscope system (Zeiss AG,
Oberkochen, Germany).
Cell viability analysis
Primary cultured neuron viability was quantified by
measuring the release of the cytosolic enzyme, LDH, as previous
described (24). The enzyme
activity was determined using an assay kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Briefly,
the cells were treated with LDH release agent (which served as the
maximum viability), and the medium containing the detached cells
was collected and centrifuged. The supernatant was used for the
assay of LDH activity. A spectrophotometer was used to measure the
optical density (OD) value at 490 nm. The percentage of damaged
cells was calculated according to the following equation:
(OD490sample−OD490media) /
(OD490maximum−OD490media) ×100, where
OD490media was the OD of media without cells, and
OD490maximum was the OD of cells treated with LDH
release agent.
Furthermore, a trypan blue staining assay was
performed to confirm the results of the LDH assay. Following each
treatment, cells were stained with 0.4% trypan blue (Beyotime
Institute of Biotechnology) for 5 min at room temperature.
Unstained cells were regarded as viable, and stained cells were
regarded as dead. The total cell number and the number of trypan
blue-positive cells were counted using a light microscope in a
blinded manner. The percentage of surviving cells was calculated
using the formula: Number of stained cells / number of total cells
×100.
Statistical analysis
SPSS 15.0 software (SPSS, Inc., Chicago, IL, USA)
was used to conduct the statistical analysis. Values are presented
as the mean ± standard error of the mean. Statistical comparisons
between groups were performed using one-way analysis of variance
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
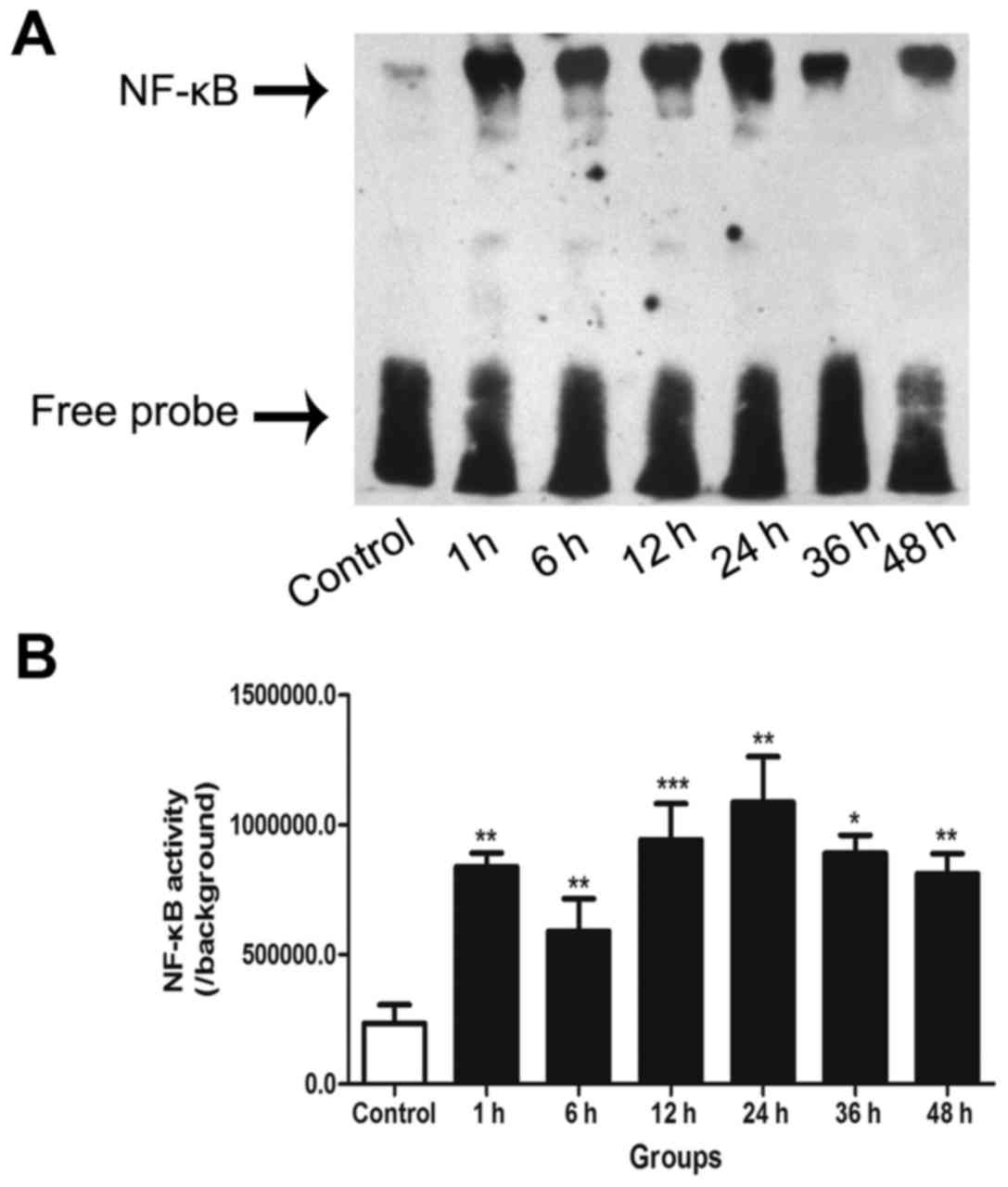
EMSA for NF-κB activity
NF-κB DNA-binding activity in the cultured neurons
was detected by EMSA. The results demonstrated that NF-κB activity
was significantly higher at all post-trauma time points compared
with that in the control group. Furthermore, following an initial
peak at 1 h, a distinct second peak of NF-κB activation was
observed at 24 h in the neurons following the insult induced by
transection (Fig. 1).
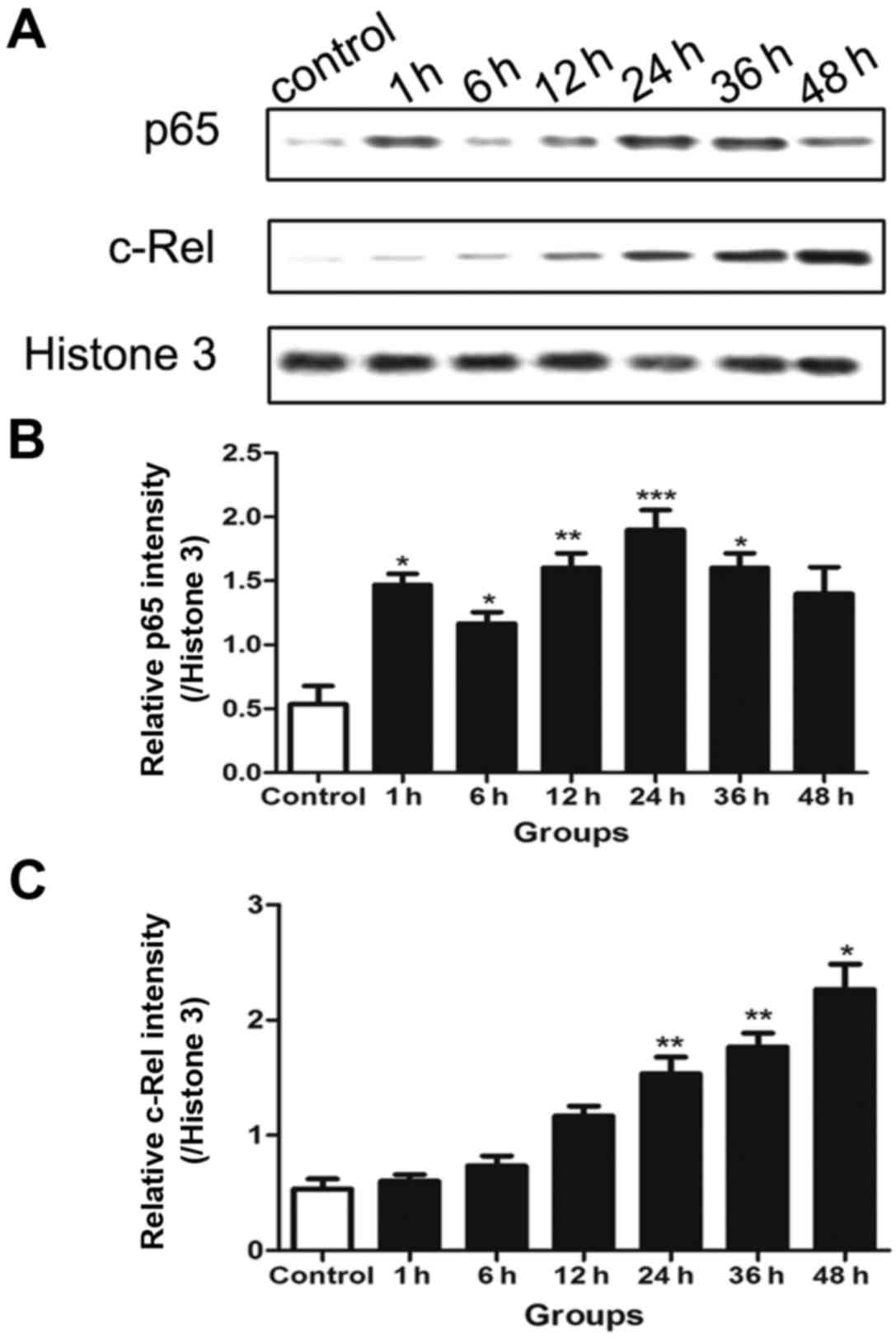
Protein levels of p65 and c-Rel in the
nucleus
Western blotting was performed to detect the protein
levels of p65 and c-Rel. The results reveal that the pattern of p65
expression in the nucleus was consistent with the activation of
NF-κB, and also presented double peaks. The first peak was at 1 h
and the second was at 24 h (Fig.
2B). However, the c-Rel expression increased over time and was
significantly increased compared with that in the control in the
later stage (24–48 h) following traumatic neuronal injury, while no
significant differences were detected from the control during the
earlier period following transection injury (Fig. 2C).
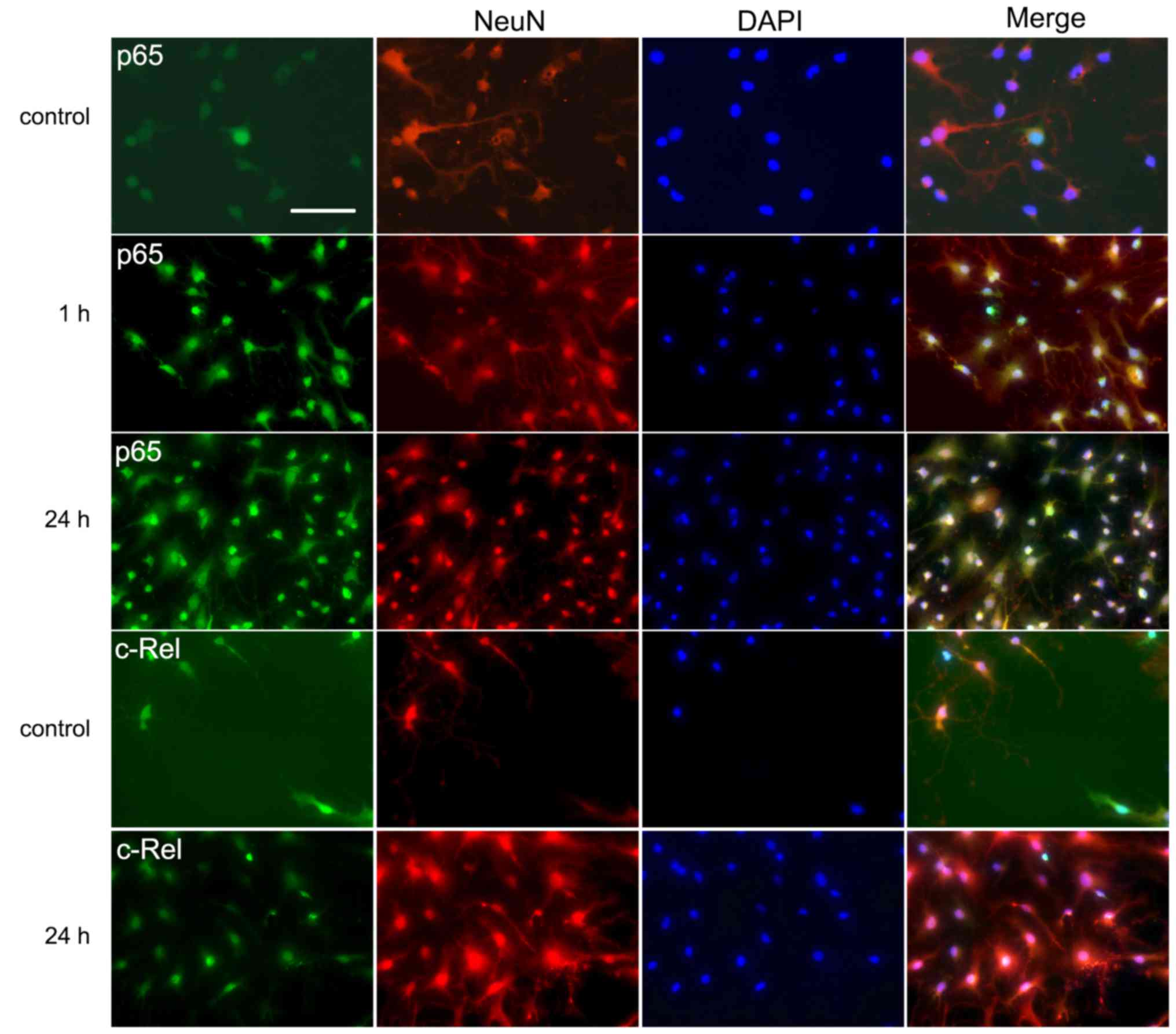
Immunofluorescent labeling of p65 and
c-Rel
Immunofluorescence analysis was performed to
investigate the expression and distribution of the two main
subunits of NF-κB, namely p65 and c-Rel. This type of single cell
analysis is advantageous when studying signal transduction as it
helps to identify the cell types involved. Double labeling with
antibodies against the neuron-specific NeuN protein and either p65
or c-Rel revealed colocalization in the neuronal cells. In the
control groups, the neurons displayed staining of the two NF-κB
subunits in the cytoplasm but weak or no staining in the nuclei
(Fig. 3). However, in the 1 and
24 h groups, a marked increase of immunoreactivity for p65 in the
nuclei was detected (Fig. 3).
Increased nuclear staining for c-Rel was also observed in the 24 h
groups (Fig. 3).
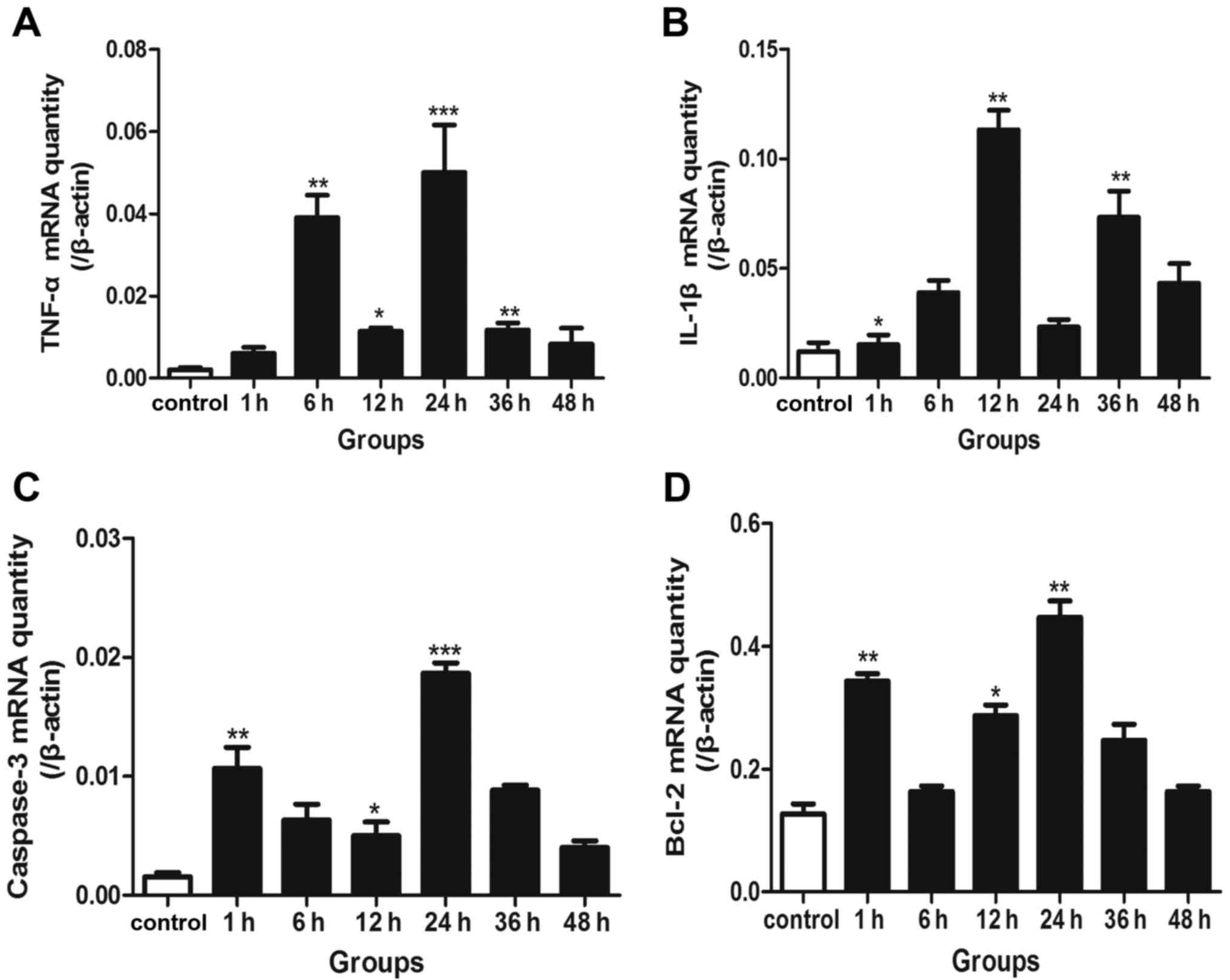
RT-qPCR for the detection of IL-1β,
TNF-α, caspase-3 and Bcl-2 mRNA
In the neurons following transection injury, the
expression of TNF-α mRNA increased and exhibited two peak phases at
6 and 24 h (Fig. 4A). The mRNA
expression of another inflammatory mediator, IL-1β was
significantly increased at 12 and 36 h (Fig. 4B). Furthermore, caspase-3 mRNA
expression reached peak levels in the 1 h and 24 groups (Fig. 4C), accordant with the NF-κB
activation pattern. The mRNA levels of Bcl-2 were also elevated at
1 and 24 h, respectively (Fig.
4D).
LDH release detection and trypan blue
staining
LDH quantification was used to evaluate the neuronal
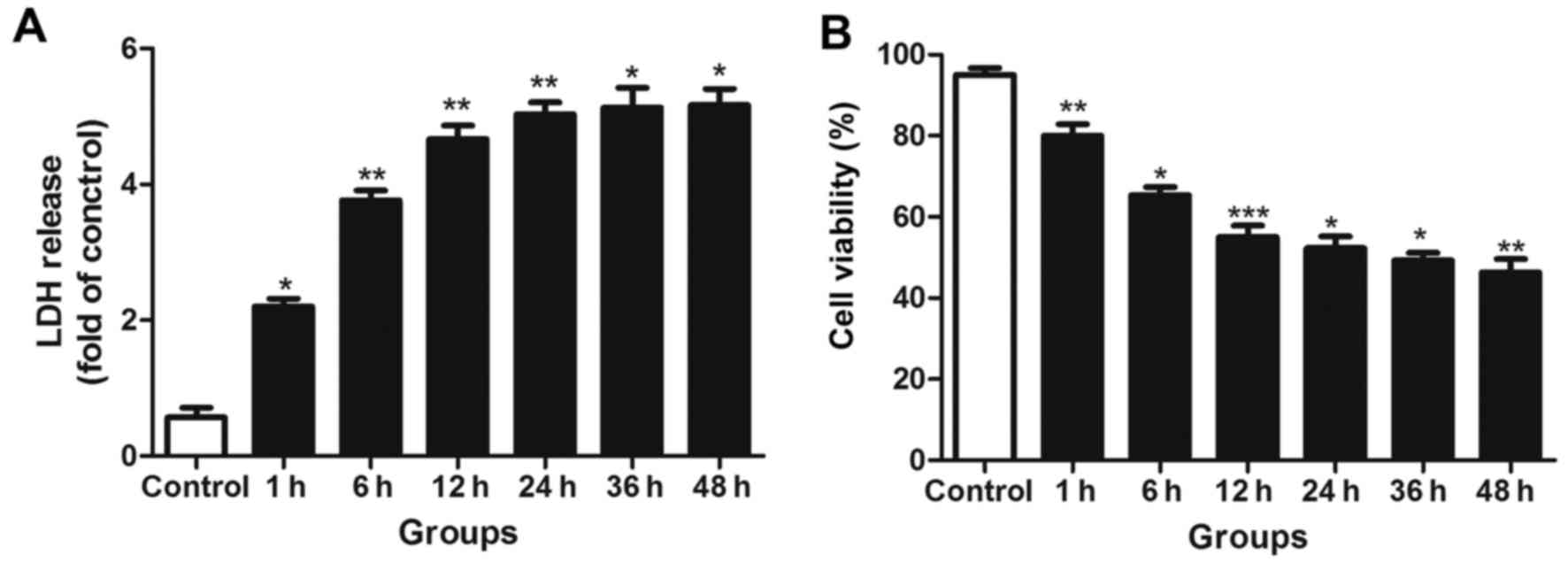
injury at different time-points following transection injury. LDH
leakage was significantly increased in the scratched groups
compared with the control groups (Fig. 5A). However, there was no
significant further increase in LDH leakage during the late NF-κB
activity peak, indicating that no sustained injury of the neurons
occurred.
The trypan blue staining results indicated that the
number of surviving neurons was significantly decreased during the
early activation phase of NF-κB, but no further marked reductions
in cell survival were observed in the later phase (Fig. 5B).
Discussion
In the present study, it was demonstrated that
biphasic activation of NF-κB is induced in primary cultured neurons
following transection injury, with an early peak of NF-κB activity
at 1 h followed by a second late-phase peak at 24 h. Furthermore,
the two main subunits of NF-κB, p65 and c-Rel exhibited different
changes. The alterations in the protein levels of p65 in the
nucleus corresponded with the changes in NF-κB activities, while
the protein levels of the c-Rel subunit were particularly elevated
during the late period; these observations were confirmed by
immunofluorescence. The downstream gene transcriptions of NF-κB
presented similar biphasic changes following the NF-κB activation.
These findings suggest for the first time, to the best of our
knowledge, that biphasic activation of NF-κB may be induced
following traumatic neuronal injury.
In the central nervous system, NF-κB is widely
distributed in neurons, microglia, astrocytes and oligodendrocytes.
In response to external stimuli, NF-κB is released from binding to
its inhibitor, IκB, when the latter is degraded and the free NF-κB
translocates to the nucleus where the transcription of downstream
genes is activated (25).
Numerous studies have demonstrated that the NF-κB pathway serves an
essential role in the pathophysiological processes of various
central system diseases, including TBI and subarachnoid hemorrhage.
Among these studies, some have suggested a neuroprotective role of
NF-κB (26,27), while others reported a
neurodestructive role of NF-κB (28–30). Nijboer et al (17) described the biphasic activation of
NF-κB in a hypoxic-ischemic brain damage model and suggested that
NF-κB may serve different roles in neurons at different
time-points, that is, during early brain injury and later brain
repair. In addition, another study observed the biphasic expression
of NF-κB in experimental models of subarachnoid hemorrhage
(16). However, whether NF-κB
activities present similar changes following TBI remains poorly
understood. Previous studies by the present research team indicated
that the biphasic activation of NF-κB occurs following TBI in rats
(31). In the present study, a
transection model of primary cultured neurons was employed in which
to detect NF-κB activities following neuronal damage in
vitro. Notably, NF-κB exhibited two peaks of significantly
increased activity at 1 and 24 h following transection. The
expression of different subunits of NF-κB, namely p65 and c-Rel was
further investigated in the present study. Previous studies have
demonstrated that the activation of a distinct combination of NF-κB
subunits may result in the differential regulation of target genes
and the induction of diverse genetic programs that dictate the cell
fate (32–34). Importantly, Pizzi et al
(15) demonstrated opposing roles
for the NF-κB/Rel factors p65 and c-Rel in the modulation of neuron
survival elicited by glutamate and IL-1β. Previous studies have
also suggested that numerous cerebral system diseases have
different etiologies yet similar mechanisms (35). Based on these observations, the
expression and distribution of p65 and c-Rel were detected in the
present study to investigate their involvement in the physiological
processes in neurons following transection injury. The results
demonstrated that p65 and c-Rel exhibited different expression
phases following neuronal damage, suggesting that these two
different subunits may participate in different pathophysiological
processes.
LDH leakage is considered to be associated with the
disruption of cell membrane integrity, which may occur following
the apoptotic or necrotic death of mature neurons (36). In the current study, LDH leakage
exhibited a significant increase during the early peak in injured
neurons but did not markedly increase further during the later
phase. Similar results were observed with trypan blue staining.
Thus, it may be inferred that the early peak in NF-κB activity
promoted neuron death, while the late one did not significantly
aggravate neuron damage, which may be beneficial for neuron
survival.
The mRNA levels of target genes of NF-κB were
further investigated in the present study. Previous studies have
demonstrated that NF-κB is able to provide cell protection through
upregulating the expression of anti-apoptotic factors such as Bcl-2
(37). Bcl-2 binds to Bax and
Bcl-2 antagonist (Bak) thereby preventing Bax/Bak pore formation in
the mitochondrial membrane. In the current study, Bcl-2 mRNA
expression altered in a similar manner to NF-κB activity. However,
caspase-3, a crucial mediator of apoptosis for neurons, was also
elevated following the peak NF-κB activation. The pro- and
anti-apoptotic genes presented the same tendency to increase as
NF-κB activity increased, which is confusing with regard to the
neuronal fate. NF-κB activation has been described to promote brain
damage via the induction of pro-inflammatory cytokines, including
TNF-α and IL-1β (38). However,
inflammatory cytokines may also provide a neuroprotection-like
effect in certain conditions by promoting growth, repair, and
ultimately, enhanced functional recovery (39,40). In the present study, TNF-α and
IL-1β displayed a biphasic expression pattern, leading to the
hypothesis that TNF-α and IL-1β may provide protective effects in
the later phase of traumatic neuronal injury. The reciprocal
interaction between the pro- and anti-apoptotic signals regulated
by NF-κB and its role in inflammatory cytokine production therefore
complicate the prediction of the effect of NF-κB on neuronal
injury. The exact mechanism requires further investigation in
future studies. The present research team are carrying out
additional investigations to discover the complex association
between them.
In conclusion, the present study observed for the
first time that biphasic activation of NF-κB occurred following
traumatic neuronal injury in primary cultured neurons. In addition,
the p65 and c-Rel subunits of NF-κB were elevated during different
phases post-injury. These preliminary data indicate that NF-κB may
serve dual roles in the determination of neuronal fate. It is
possible that the final decision of a neuron to live or die is made
relatively late following injury However, the present study is a
pilot study and has certain limitations, such as a lack of
interventions and limited exploration of subunits. Future research
aimed at NF-κB-based interventions and therapeutic approaches to
combat TBI are likely be valuable.
Acknowledgments
Not applicable.
Abbreviations:
|
NF-κB
|
nuclear factor-κB
|
|
TBI
|
traumatic brain injury
|
|
EMSA
|
electrophoresis mobility shift
assay
|
Notes
[1]
Funding
The present study was supported by the National
Natural Science Foundation, China (grant nos. 81171170 and
81371294) and the Nature Science Foundation of Jiangsu Province,
China (grant no. BK2010459).
[2] Availability
of data and materials
The data and materials used or analyzed during the
current study are available from the corresponding author on
reasonable request.
[3] Authors'
contributions
HZ and CH designed the experiments. HZ, DZ, HY and
HL carried out the experiments. HZ, ZZ and CZ analyzed the
experimental results. HZ, QC and ZY wrote the manuscript and
critically revised it for important intellectual content. All
authors read and approved the manuscript.
[4] Ethics
approval and consent to participate
Ethical approval was provided by the Medical Ethics
Committee of Jinling Hospital. All surgical procedures were
performed in accordance with guidelines of the Jinling Animal Care
and Use Committee.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Lescot T, Abdennour L, Degos V, Boch AL
and Puybasset L: Management of severe traumatic brain injury.
Presse Med. 36:1117–1126. 2007.In French. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen T, Liu W, Chao X, Zhang L, Qu Y, Huo
J and Fei Z: Salvianolic acid B attenuates brain damage and
inflammation after traumatic brain injury in mice. Brain Res Bull.
84:163–168. 2011. View Article : Google Scholar
|
|
3
|
Greve MW and Zink BJ: Pathophysiology of
traumatic brain injury. Mt Sinai J Med. 76:97–104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu J, Goh SJ, Tng PY, Deng YY, Ling EA and
Moochhala S: Systemic inflammatory response following acute
traumatic brain injury. Front Biosci (Landmark Ed). 14:3795–3813.
2009. View Article : Google Scholar
|
|
5
|
Bhakar AL, Tannis LL, Zeindler C, Russo
MP, Jobin C, Park DS, MacPherson S and Barker PA: Constitutive
nuclear factor-kappa B activity is required for central neuron
survival. J Neurosci. 22:8466–8475. 2002.PubMed/NCBI
|
|
6
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baldwin AS Jr: The NF-kappa B and I kappa
B proteins: New discoveries and insights. Annu Rev Immunol.
14:649–683. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mattson MP and Meffert MK: Roles for
NF-kappaB in nerve cell survival, plasticity, and disease. Cell
Death Differ. 13:852–860. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shimada M, Satoh N and Yokosawa H:
Involvement of Rel/NF-kappaB in regulation of ascidian notochord
formation. Dev Growth Differ. 43:145–154. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang R, Xue YY, Lu SD, Wang Y, Zhang LM,
Huang YL, Signore AP, Chen J and Sun FY: Bcl-2 enhances
neurogenesis and inhibits apoptosis of newborn neurons in adult rat
brain following a transient middle cerebral artery occlusion.
Neurobiol Dis. 24:345–356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Liu J, Yao S, Li F, Xin L, Lai M,
Bracchi-Ricard V, Xu H, Yen W, Meng W, et al: Nuclear factor kappa
B signaling initiates early differentiation of neural stem cells.
Stem Cells. 30:510–524. 2012. View Article : Google Scholar
|
|
12
|
Biscetti F, Ghirlanda G and Flex A:
Therapeutic potential of high mobility group box-1 in ischemic
injury and tissue regeneration. Curr Vasc Pharmacol. 9:677–681.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Basu S, Rajakaruna S and Menko AS:
Insulin-like growth factor receptor-1 and nuclear factor κB are
crucial survival signals that regulate caspase-3-mediated lens
epithelial cell differentiation initiation. J Biol Chem.
287:8384–8397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Youssef S and Steinman L: At once harmful
and beneficial: The dual properties of NF-kappaB. Nat Immunol.
7:901–902. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pizzi M, Goffi F, Boroni F, Benarese M,
Perkins SE, Liou HC and Spano P: Opposing roles for NF-kappa B/Rel
factors p65 and c-Rel in the modulation of neuron survival elicited
by glutamate and interleukin-1beta. J Biol Chem. 277:20717–20723.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
You WC, Li W, Zhuang Z, Tang Y, Lu HC, Ji
XJ, Shen W, Shi JX and Zhou ML: Biphasic activation of nuclear
factor-kappa B in experimental models of subarachnoid hemorrhage in
vivo and in vitro. Mediators Inflamm. 2012:7862422012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nijboer CH, Heijnen CJ, Groenendaal F, May
MJ, van Bel F and Kavelaars A: A dual role of the NF-kappaB pathway
in neonatal hypoxic-ischemic brain damage. Stroke. 39:2578–2586.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen T, Fei F, Jiang XF, Zhang L, Qu Y,
Huo K and Fei Z: Down-regulation of Homer1b/c attenuates
glutamate-mediated excitotoxicity through endoplasmic reticulum and
mitochondria pathways in rat cortical neurons. Free Radic Biol Med.
52:208–217. 2012. View Article : Google Scholar
|
|
19
|
Zhao Y, Luo P, Guo Q, Li S, Zhang L, Zhao
M, Xu H, Yang Y, Poon W and Fei Z: Interactions between SIRT1 and
MAPK/ERK regulate neuronal apoptosis induced by traumatic brain
injury in vitro and in vivo. Exp Neurol. 237:489–498. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tecoma ES, Monyer H, Goldberg MP and Choi
DW: Traumatic neuronal injury in vitro is attenuated by NMDA
antagonists. Neuron. 2:1541–1545. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou ML, Shi JX, Hang CH, Cheng HL, Qi XP,
Mao L, Chen KF and Yin HX: Potential contribution of nuclear
factor-kappaB to cerebral vasospasm after experimental subarachnoid
hemorrhage in rabbits. J Cereb Blood Flow Metab. 27:1583–1592.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rogers B, Yakopson V, Teng ZP, Guo Y and
Regan RF: Heme oxygenase-2 knockout neurons are less vulnerable to
hemoglobin toxicity. Free Radic Biol Med. 35:872–881. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Hatic H, Kane MJ, Saykally JN and Citron
BA: Modulation of transcription factor Nrf2 in an in vitro model of
traumatic brain injury. J Neurotrauma. 29:1188–1196. 2012.
View Article : Google Scholar
|
|
25
|
Baeuerle PA and Baltimore D: I kappa B: A
specific inhibitor of the NF-kappa B transcription factor. Science.
242:540–546. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lezoualc'h F, Sagara Y, Holsboer F and
Behl C: High constitutive NF-kappaB activity mediates resistance to
oxidative stress in neuronal cells. J Neurosci. 18:3224–3232.
1998.PubMed/NCBI
|
|
27
|
Mattson MP, Goodman Y, Luo H, Fu W and
Furukawa K: Activation of NF-kappaB protects hippocampal neurons
against oxidative stress-induced apoptosis: Evidence for induction
of manganese superoxide dismutase and suppression of peroxynitrite
production and protein tyrosine nitration. J Neurosci Res.
49:681–697. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grilli M, Pizzi M, Memo M and Spano P:
Neuroprotection by aspirin and sodium salicylate through blockade
of NF-kappaB activation. Science. 274:1383–1385. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Clemens JA, Stephenson DT, Dixon EP,
Smalstig EB, Mincy RE, Rash KS and Little SP: Global cerebral
ischemia activates nuclear factor-kappa B prior to evidence of DNA
fragmentation. Brain Res Mol Brain Res. 48:187–196. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qin ZH, Wang Y, Nakai M and Chase TN:
Nuclear factor-kappaB contributes to excitotoxin-induced apoptosis
in rat striatum. Mol Pharmacol. 53:33–42. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hu YC, Sun Q, Li W, Zhang DD, Ma B, Li S,
Li WD, Zhou ML and Hang CH: Biphasic activation of nuclear factor
kappa B and expression of p65 and c-Rel after traumatic brain
injury in rats. Inflamm Res. 63:109–115. 2014. View Article : Google Scholar
|
|
32
|
Lin SC, Wortis HH and Stavnezer J: The
ability of CD40L, but not lipopolysaccharide, to initiate
immunoglobulin switching to immunoglobulin G1 is explained by
differential induction of NF-kappaB/Rel proteins. Mol Cell Biol.
18:5523–5532. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Perkins ND: Achieving transcriptional
specificity with NF-kappaB. Int J Biochem Cell Biol. 29:1433–1448.
1997. View Article : Google Scholar
|
|
34
|
Roshak AK, Jackson JR, McGough K,
Chabot-Fletcher M, Mochan E and Marshall LA: Manipulation of
distinct NFkappaB proteins alters interleukin-1beta-induced human
rheumatoid synovial fibroblast prostaglandin E2 formation. J Biol
Chem. 271:31496–31501. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Leker RR and Shohami E: Cerebral ischemia
and trauma-different etiologies yet similar mechanisms:
Neuroprotective opportunities. Brain Res Brain Res Rev. 39:55–73.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu J, Wang H, Ding K, Zhang L, Wang C, Li
T, Wei W and Lu X: Luteolin provides neuroprotection in models of
traumatic brain injury via the Nrf2-ARE pathway. Free Radic Biol
Med. 71:186–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kuwana T and Newmeyer DD: Bcl-2-family
proteins and the role of mitochondria in apoptosis. Curr Opin Cell
Biol. 15:691–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mattson MP and Camandola S: NF-kappaB in
neuronal plasticity and neurodegenerative disorders. J Clin Invest.
107:247–254. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cederberg D and Siesjö P: What has
inflammation to do with traumatic brain injury? Childs Nerv Syst.
26:221–226. 2010. View Article : Google Scholar
|
|
40
|
Barone FC and Feuerstein GZ: Inflammatory
mediators and stroke: New opportunities for novel therapeutics. J
Cereb Blood Flow Metab. 19:819–834. 1999. View Article : Google Scholar : PubMed/NCBI
|