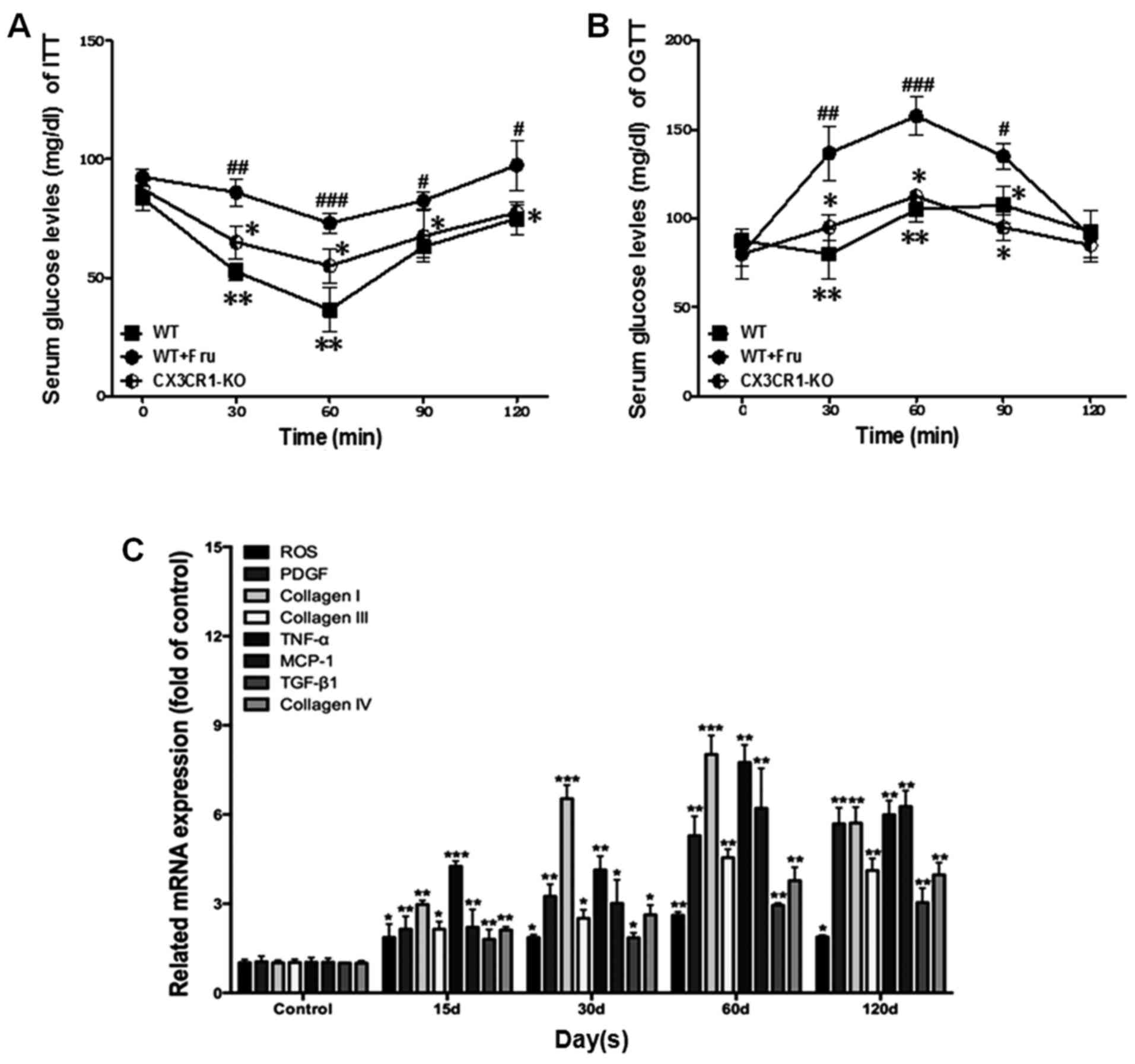
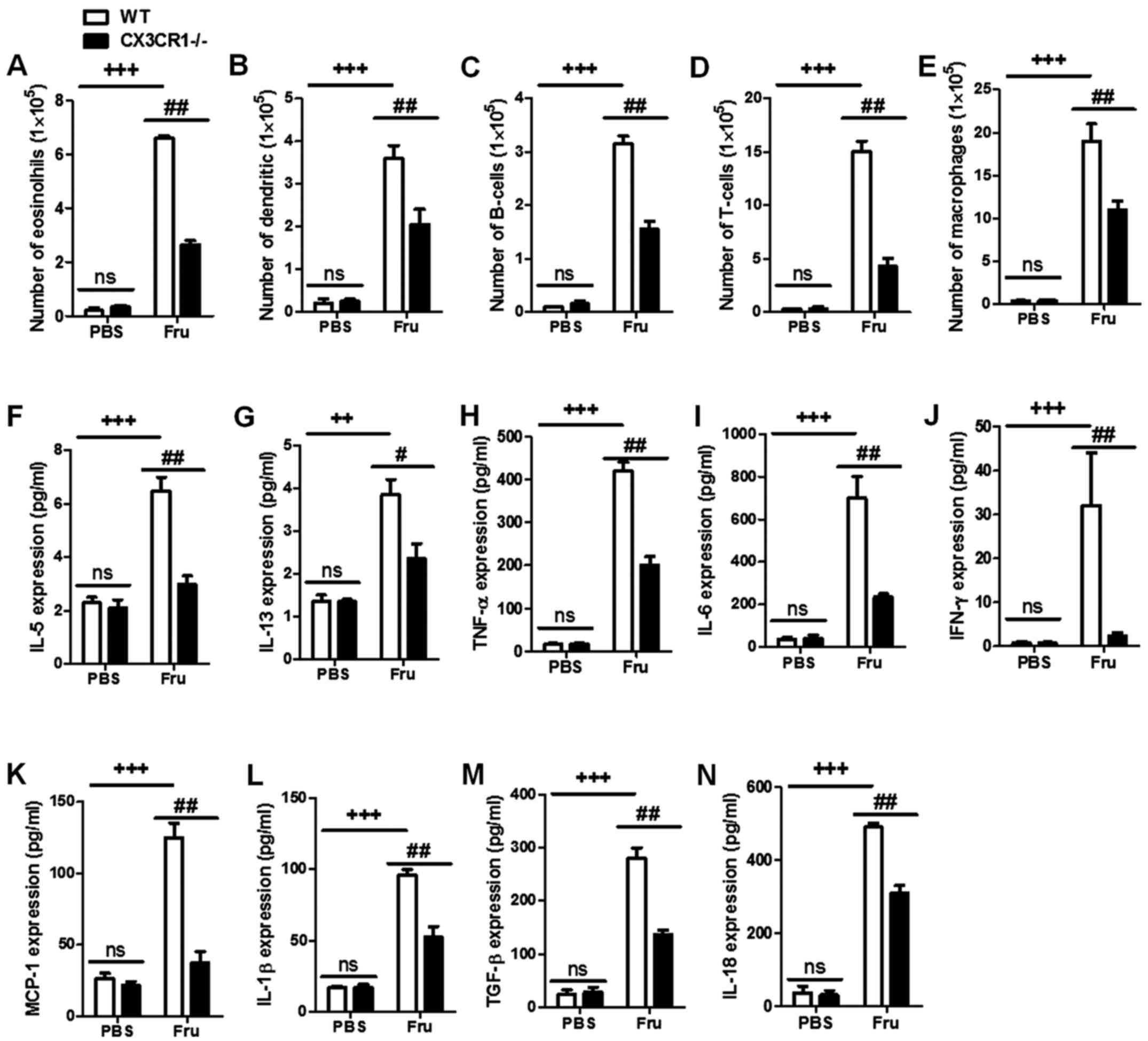
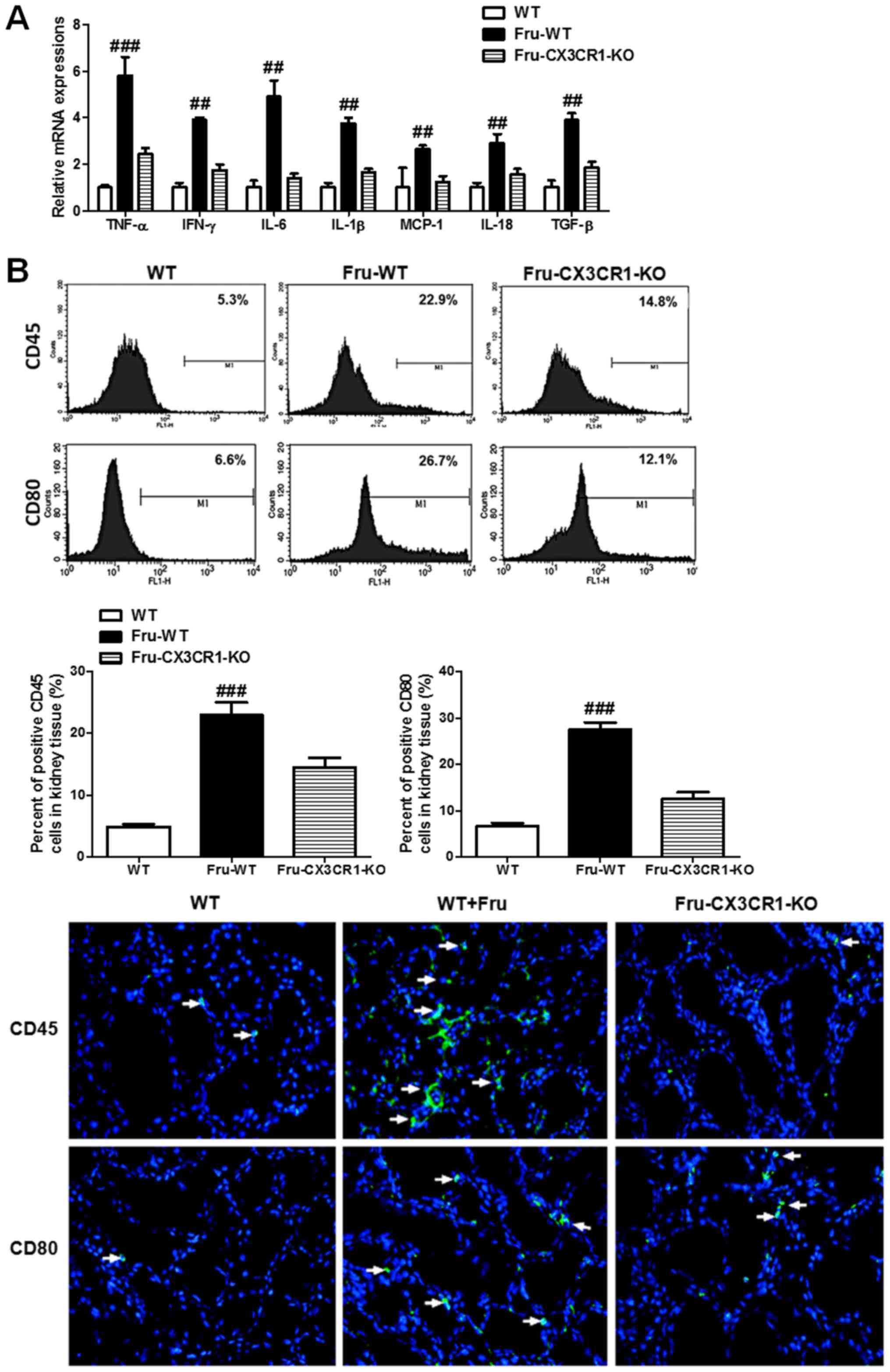
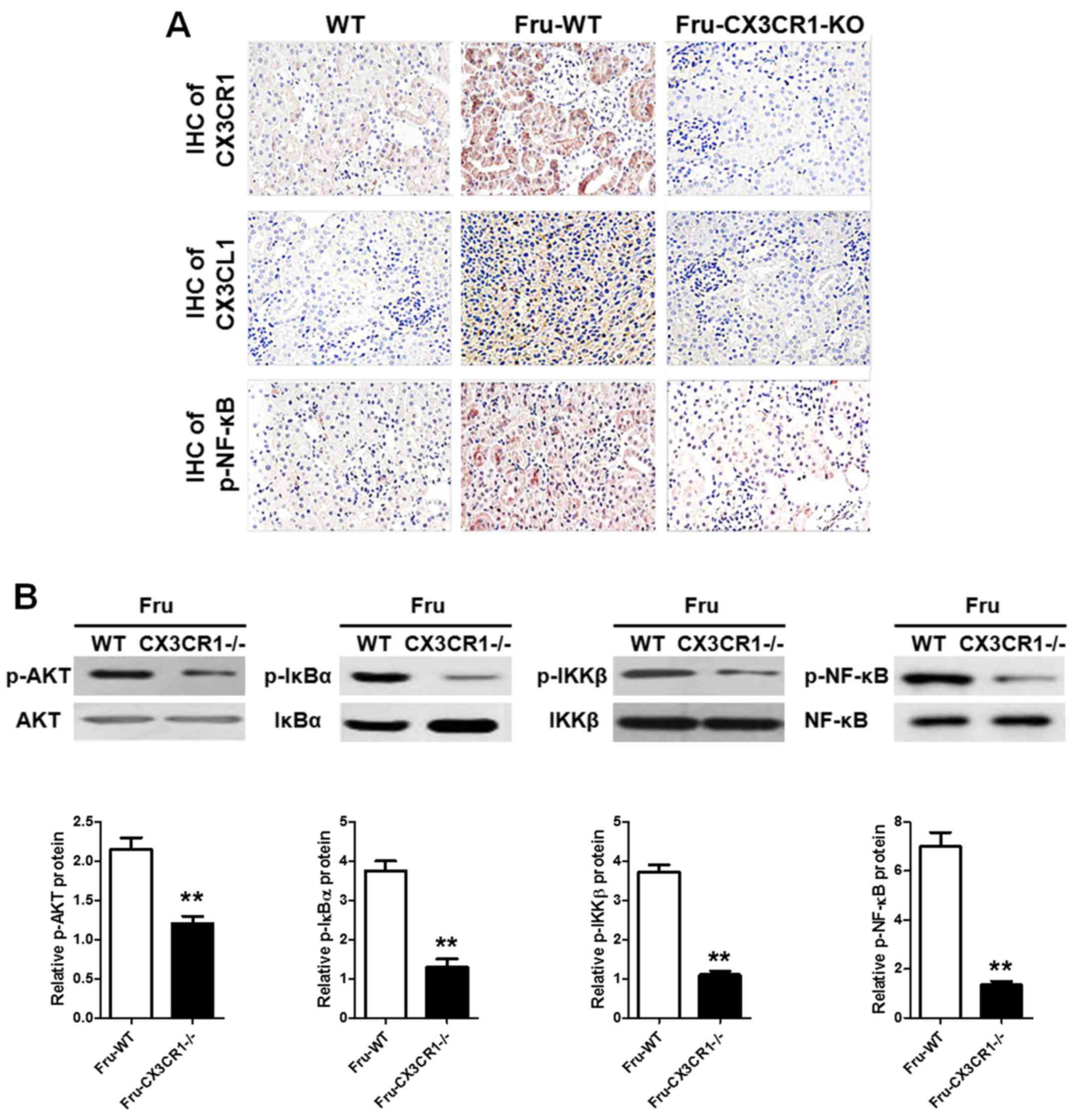
|
1
|
Megyesi J, Andrade L, Vieira JM Jr,
Safirstein RL and Price PM: Coordination of the cell cycle is an
important determinant of the syndrome of acute renal failure. Am J
Physiol Renal Physiol. 283:F810–F816. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou H, Kato A, Yasuda H, Miyaji T,
Fujigaki Y, Yamamoto T, Yonemura K and Hishida A: The induction of
cell cycle regulatory and DNA repair proteins in cisplatin-induced
acute renal failure. Toxicol Appl Pharmacol. 200:111–120. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Price PM, Safirstein RL and Megyesi J:
Protection of renal cells from cisplatin toxicity by cell cycle
inhibitors. Am J Physiol Renal Physiol. 286:F378–F384. 2004.
View Article : Google Scholar
|
|
4
|
Megyesi J, Safirstein RL and Price PM:
Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the
course of cisplatin-induced acute renal failure. J Clin Invest.
101:777–782. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou H, Fujigaki Y, Kato A, Miyaji T,
Yasuda H, Tsuji T, Yamamoto T, Yonemura K and Hishida A: Inhibition
of p21 modifies the response of cortical proximal tubules to
cisplatin in rats. Am J Physiol Renal Physiol. 291:F225–F235. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nowak G, Price PM and Schnellmann RG: Lack
of a functional p21WAF1/CIP1 gene accelerates caspase-independent
apoptosis induced by cisplatin in renal cells. Am J Physiol Renal
Physiol. 285:F440–F450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nath KA: Provenance of the protective
property of p21. Am J Physiol Renal Physiol. 289:F512–F513. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hengst L and Reed SI: Inhibitors of the
Cip/Kip family. Curr Top Microbiol Immunol. 227:25–41.
1998.PubMed/NCBI
|
|
9
|
Sherr CJ and Roberts JM: Inhibitors of
mammalian G1 cyclin-dependent kinases. Genes Dev. 9:1149–1163.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Al-Mohanna MA, Manogaran PS, Al-Mukhalafi
Z, A Al-Hussein K and Aboussekhra A: The tumor suppressor
p16(INK4a) gene is a regulator of apoptosis induced by ultraviolet
light and cisplatin. Oncogene. 23:201–212. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Le HV, Minn AJ and Massagué J:
Cyclin-dependent kinase inhibitors uncouple cell cycle progression
from mitochondrial apoptotic functions in DNA-damaged cancer cells.
J Biol Chem. 280:32018–32025. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mori K: Tripartite management of unfolded
proteins in the endoplasmic reticulum. Cell. 101:451–454. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kleizen B and Braakman I: Protein folding
and quality control in the endoplasmic reticulum. Curr Opin Cell
Biol. 16:343–349. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Price PM, Yu F, Kaldis P, Aleem E, Nowak
G, Safirstein RL and Megyesi J: Dependence of cisplatin-induced
cell death in vitro and in vivo on cyclin-dependent kinase 2. J Am
Soc Nephrol. 17:2434–2442. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu F, Megyesi J and Price PM: Cytoplasmic
initiation of cisplatin cytotoxicity. Am J Physiol Renal Physiol.
295:F44–F52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hirai H, Roussel MF, Kato JY, Ashmun RA
and Sherr CJ: Novel INK4 proteins, p19 and p18 are specific
inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol
Cell Biol. 15:2672–2681. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cordon-Cardo C: Mutations of cell cycle
regulators. Biological and clinical implications for human
neoplasia. Am J Pathol. 147:545–560. 1995.PubMed/NCBI
|
|
19
|
Shankland SJ and Wolf G: Cell cycle
regulatory proteins in renal disease: Role in hypertrophy,
proliferation, and apoptosis. Am J Physiol Renal Physiol.
278:F515–F529. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Franklin DS, Godfrey VL, Lee H, Kovalev
GI, Schoonhoven R, Chen-Kiang S, Su L and Xiong Y: CDK inhibitors
p18(INK4c) and p27(Kip1) mediate two separate pathways to
collaboratively suppress pituitary tumorigenesis. Genes Dev.
12:2899–2911. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nat Rev Drug Discov. 4:307–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|