Introduction
Ulcerative colitis (UC) is a multifactorial chronic
inflammatory bowel disease (1,2).
Although the pathophysiology of UC remains to be elucidated,
experimental and clinical evidence suggests that chronic intestinal
inflammation may be caused by dysfunction of the immune system
(3). Excessive activation of the
immune system results in persistent overproduction of inflammatory
mediators, further recruitment and activation of immune cells,
which exacerbates inflammatory responses (4). Therefore, regulating the secretion
of inflammatory cytokines is vital to the improvement of intestinal
inflammation.
During the pathogenesis of UC, a variety of
inflammatory cytokines and chemokines are released and activate
associated signaling pathways, such as tumor necrosis factor-α
(TNF-α), interleukin (IL)-1β, IL-6 and IL-17, which activate
inflammatory responses (5). The
production of inflammatory mediators is regulated by a variety of
transcription factors. Among these transcription factors, nuclear
factor (NF)-κB serves a crucial role in regulating gene expression
that encode cytokines, chemokines and other mediators associated
with inflammatory responses (6–8).
The transcriptional activity of the transcription
factor NF-κB can be regulated at multiple levels: The
transcriptional activity of NF-κB is activated by degradation of
IκBs, whereas the nuclear transcription potential of p65 is further
regulated by post-translational modifications, such as
phosphorylation, ubiquitination or acetylation (9). Notable, phosphorylation of p65 at
Ser276, Ser529, and or Ser536 is essential for the promotion of
gene expression (10). It has
been demonstrated that phosphorylation of Ser276 is critical for
transactivation of p65 (11).
Phosphorylation at Ser276 p65 promotes the formation of stable
complexes with coactivator CBP/p300 to enhance promoter function
(10,12,13). Previous reports have demonstrated
that mitogen- and stress-activated protein kinase-1 (MSK1), as a
nuclear kinase, phosphorylated p65 at Ser276 and then promoted
transcription of NF-κB target genes (10,14). Furthermore, the activity of MSK1
is regulated by p38 and extracellular signal-regulated kinase
(ERK); MSK1 is directly activated by mitogen-activated protein
kinase (MAPK) and stress-activated protein kinase/p38, and may
mediate activation of cAMP response element-binding protein
(15). In addition, the
regulation of inducible nitric oxide synthase (iNOS) expression is
also associated with NF-κB, which may be activated by complex
kinase pathways including MSK1 (16,17).
UC can cause the body decreased food intake and
nutrient absorption, and accelerated nutrient loss, which may
result in malnutrition (18). It
has previously been demonstrated that enteral nutrition can not
only improve nutritional status, but also promotes mucosal
remodeling and regulation of immune function (19). At present, various enteral
nutrition formulae have been used in the treatment of inflammatory
bowel disease (IBD), and the protective mechanism of intestinal
mucosa has been previously studied (20,21). In the present study, the effects
of exclusive enteral nutrition (EEN) on UC were examined and its
possible mechanism was explored.
Materials and methods
Reagents and antibodies
Dextran sulfate sodium (DSS; molecular weight 36–50
kDa) was obtained from MP Biomedicals, LLC (Santa Ana, CA, USA).
Picrylsulfonic acid solution (TNBS) was obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). DAPI was purchased
from Invitrogen; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Paraformaldehyde was purchased from Yonghua Chemical Technology
(Jiangsu) Co., Ltd. (Changshu, China). Triton X-100 was purchased
from Beyotime Institute of Biotechnology (Haimen, China). Bovine
serum albumin (BSA) was purchased from Roche Diagnostics (Basel,
Switzerland). Radioimmunoprecipitation assay lysis buffer was
purchased from Beyotime Institute of Biotechnology.
Myeloperoxidase (MPO) activity assay kit was
purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing,
China). iNOS assay kit was purchased from Beyotime Institute of
Biotechnology. ELISA kits for mouse IL-1β (EK0394), IL-6 (EK0411),
TNF-α (EK0527) and IL-17 (EK0431) were purchased from Boster
Biological Technology (Pleasanton, CA, USA).
Primary antibodies against phosphorylated (p)-p65
(Ser276; sc-101749; 1:1,000), p65 (sc-109, 1:2,000), p-ERK1/2
(Thr177/Thr160; sc-23759-R; 1:1,000), p-c-Jun N-terminal kinase
(JNK; G-7; sc-6254; 1:1,000), JNK (sc-7345; 1:2,000), p-p38
(Thr180; sc-101758; 1:1,000), p38 (sc-81621; 1:2,000) and β-actin
(sc-47778; 1:2,000) were obtained from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA), and antibodies against ERK1/2 (9102s;
1:2,000), p-MSK1 (Thr581; 9595; 1:1,000) and MSK1 (3489; 1:2,000)
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). IRDye 800-conjugated secondary antibodies (anti-mouse,
072-07-18-06, 1:2,000; anti-rabbit, 072-07-15-06, 1:2,000) were
obtained from Rockland Immunochemicals, Inc. (Pottstown, PA, USA).
Fluorescein isothiocyanate (FITC)-anti-cluster of differentiation
(CD)11b antibodies (11-0113-42) were purchased from eBioscience;
Thermo Fisher Scientific, Inc. EEN was composed of 92% Ensure,
which was purchased from Abbott Pharmaceutical Co. Ltd. (Lake
Bluff, IL, USA); 6% dietary fiber, which was obtained from Hebei
Vilof Agritech Co., Ltd. (Beijing, China); and 2% cod liver oil,
which was obtained from Blackmores (Warriewood, Australia).
Animals
A total of 48 female 6–8 week old C57BL/6 mice,
weighing 18–22 g, and 56 female 6–8 week old Sprague-Dawley rats,
weighing 230–240 g, were supplied by Shanghai Laboratory Animal
Center, China Academy of Sciences (Shanghai, China). Experimental
protocols were performed in accordance with National Institutes of
Health regulations and approved by the Institutional Animal Care
and Use Committee (Shanghai Institute of Biochemistry and Cell
Biology, Chinese Academy of Sciences, Shanghai, China). Throughout
the acclimatization and study periods, all animals had access to
food and water ad libitum and were maintained on in 12-h
light/dark cycle at 21±2°C with a relative humidity of 45±10%.
DSS colitis
Colitis was induced by administration of DSS in
drinking water. Mice were randomly assigned to normal, DSS-treated
and EEN (12.5, 25, 50 or 100%)-treated groups (n=8/group; Table I). Mice received either drinking
regular water (control) or 3% (w/v) DSS drinking water (model) for
7 days, and thereafter were provided regular water for 3 days. EEN
was administered intragastrically from day 1–10 (22).
 | Table IComposition of EEN (g/100 g). |
Table I
Composition of EEN (g/100 g).
| Component | EEN
|
|---|
| 12.5% | 25% | 50% | 100% |
|---|
| Protein | 1.8 | 3.6 | 7.3 | 14.5 |
| Fat | 1.8 | 3.6 | 7.3 | 14.5 |
| Linoleic acid | 1.0 | 2.0 | 4.0 | 7.9 |
| Carbohydrate | 7.0 | 14.0 | 28.0 | 56 |
| Dietary fiber | 0.7 | 1.5 | 2.9 | 5.8 |
| Fish oil | 0.3 | 0.5 | 1.0 | 2 |
TNBS colitis
Following fasting for 8–12 h, rats were anesthetized
with sodium pentobarbital (50 mg/kg, intraperitoneal; Beijing Ouhe
Technology Co., Ltd., Beijing, China), and then a flexible catheter
was inserted into the colon (3.5 cm proximal to the anus). To
induce colitis, TNBS (2 mg in 100 µl 40% ethanol solution)
was slowly administered. To assure the distribution of TNBS within
the entire colon, rats were held upside down in a vertical position
for 1 min and returned to their cages. The control group, received
100 µl 40% ethanol alone through the same technique. Rats
were then given ad libitum access to food and water. EEN
(12.5, 25, 50 or 100%) groups were treated via gavage (n=8/group;
Table I) (22). Body weight changes were recorded
once daily.
Macroscopic assessment and histological
analysis of colonic lesions
Following DSS- and TNBS-induced colitis, animals
were sacrificed, colons were removed, opened longitudinally and
washed with PBS. The colon weights and lengths were measured and
the ratio of weight:length was calculated for each group. Colonic
tissue samples were fixed in 10% neutral-buffered formalin at room
temperature for 24 h, and routinely embedded in paraffin and
processed. Hematoxylin and eosin (H&E) staining was performed
to clarify whether there was a difference in erosion of the lamina
propria mucosa, disappearance of glandular epithelium and increased
inflammatory cell infiltration compared among groups. The
experiment was performed as follows: i) Samples were dewaxed and
rehydrated in a descending alcohol series and washed in PBS; ii)
hematoxylin staining at room temperature for 10 min; iii) washed
with PBS for ~10 min; iv) washed with distilled water; v) 95%
ethanol dehydration for 5 sec; vi) eosin staining at room
temperature for 30 sec; vii) 95% ethanol dehydration for 2 min;
viii) repeat step vii; ix) xylene soak at room temperature for 5
min x) repeat step ix; xi) slides were mounted and evaluated
(original magnification, ×400) with fluorescent microscopy.
Histological analysis was performed as previously described
(23).
Immunofluorescence and
immunohistochemistry of colon tissues
CD11b-positive cell infiltration analysis was
performed on paraffin-embedded colon tissue sections. Briefly, the
sections were deparaffinized at 60°C for 40 min, placed in xylene
for dewaxing for 10 min, rehydrated in a descending alcohol series
and washed in PBS. Following treatment with 3% hydrogen peroxide,
blocking with 3% bovine serum albumin (BSA) at room temperature for
20 min, the sections were incubated for 1 h at room temperature
with FITC-anti-CD11b antibodies (1:100). The slides were then
counter-stained with DAPI for 30 min at room temperature. The
reaction was stopped by thorough washing in water for 5 min. Images
(original magnification, ×400) were acquired by confocal
laser-scanning microscope (Olympus Corporation, Tokyo, Japan).
Settings for image acquisition were identical for control and
experimental tissues.
The expressions of IL-1β, IL-6, TNF-α, p-p38
(Thr180) and p-p65 (Ser276) of the colonic tissues was assessed as
described in a previous study (24).
MPO activity and iNOS activity in colon
tissue of rat colitis model
The colon tissues of rats were collected, accurately
weighed, cut with ophthalmic scissors, homogenized using a
homogenizer, transferred to an EP tube and centrifuged at 600 × g
at 4°C for 5 min, and the cells were collected and tested for MPO
activity and iNOS activity. MPO activity and iNOS activity kits
were used according to the manufacturers’ protocols.
Cytokine quantification by enzyme-linked
immunoassay
Colons from mice in each group were homogenated with
radioimmunoprecipitation assay lysis buffer to extract total
protein. The homogenate was centrifuged at 12,000 × g at 4°C for 15
min. The amount of total extracted protein was determined using a
bicinchoninic acid protein assay kit (Thermo Fisher Scientific,
Inc.). Following blood collection, static agglutination at room
temperature for 2 h, samples were centrifuged at 900 × g for 10
min, and the upper serum was stored at −80°C. The amounts of IL-1β,
IL-6, TNF-α and IL-17 in serum and colon homogenate were measured
with ELISA kits according to the manufacturers’ protocols.
Isolation of peritoneal macrophages
Peritoneal macrophages were obtained from the
peritoneal cavity of mice by injection of PBS. Cells were washed
twice in PBS and suspended in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) containing 10% FBS, 10,000 U/ml penicillin
and 10 mg/ml streptomycin. The macrophages suspended in culture
medium were cultured in 24-well microplates for 40 min at 37°C in a
moist atmosphere of 5% CO2. Non-adherent cells were
removed by washing the plate twice with PBS. The adherent
macrophages were used for western blot analysis.
Western blot analysis
After whole cell lysates were prepared. Western blot
analysis was prepared as described previously (23). Protein samples (40 µg) were
separated by 10% SDS-PAGE and transferred onto nitrocellulose
membranes. The membranes were blocked with 1% BSA at 37°C for 1 h
and incubated with primary antibodies overnight at 4°C, followed by
IRDye800-conjugated secondary antibody for 1 h at 37°C.
Immunoreactive protein was detected with an Odyssey Scanning System
(Odyssey Application Software version 3.0.30; LI-COR Biosciences,
Lincoln, NE, USA).
Statistical analysis
Data were obtained from at least three independent
experiments and are presented as the mean ± standard deviation.
Differences between the groups were assessed by one-way analysis of
variance and Dunnett’s post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
EEN attenuates DSS-induced colitis
symptoms
In the present study, DSS-induced colitis was used
in mice, a well-established preclinical model, exhibiting many
phenotypic features of relevance to human UC (25). In general, DSS-induced colitis is
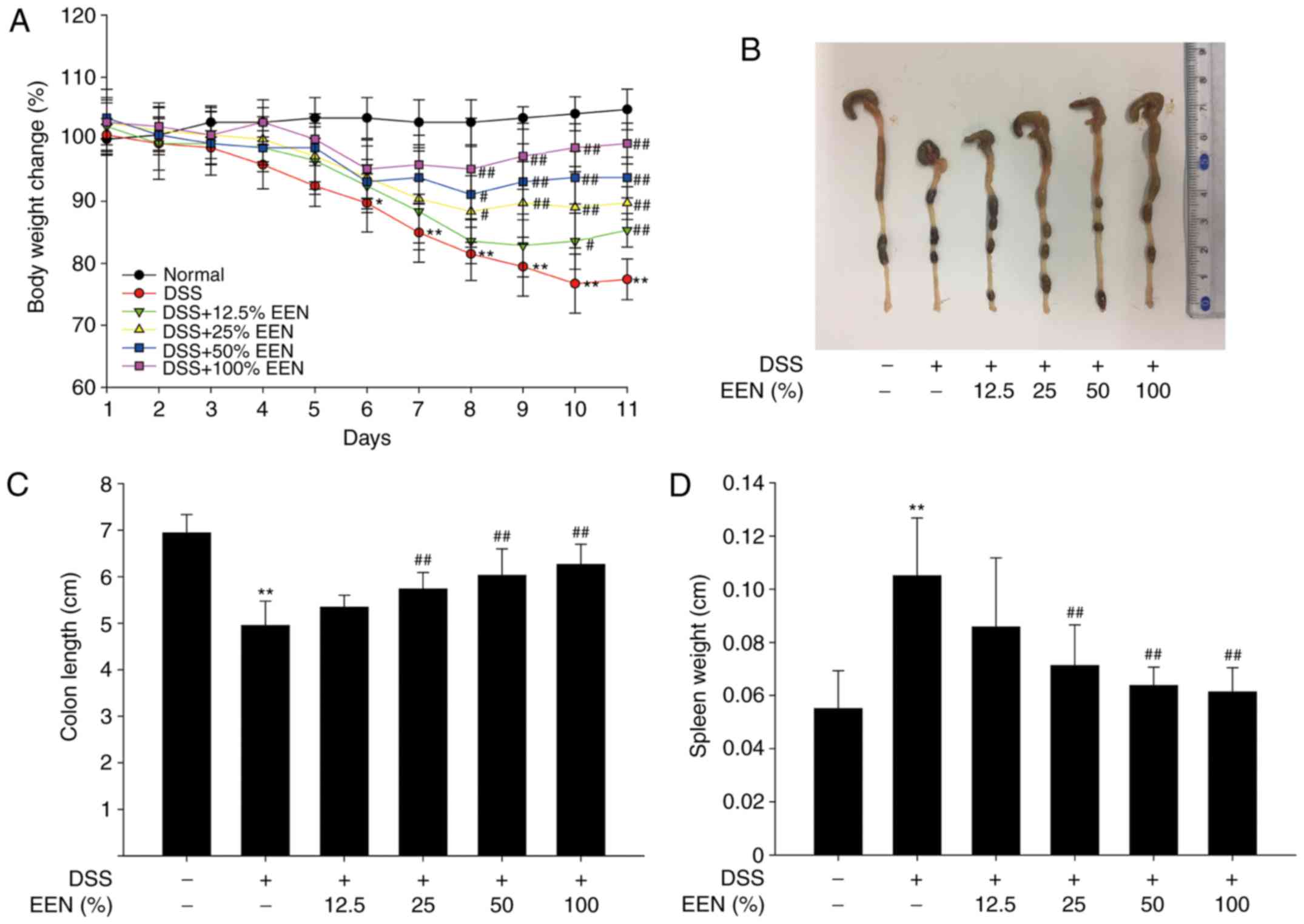
characterized by a marked body weight loss. The present results
demonstrated that DSS-treated mice exhibited marked body weight
loss, whereas EEN groups were significantly different from the DSS
model group (Fig. 1A).
DSS-induced colitis causes colonic shortening in mice (26). Compared with DSS-treated mice,
colonic shortening was significantly attenuated in EEN-treated mice
(Fig. 1B and C). In addition, EEN
ameliorated DSS-induced gain of spleen weight (Fig. 1D). These results suggested that
EEN successfully attenuated DSS-induced colon inflammatory
symptoms.
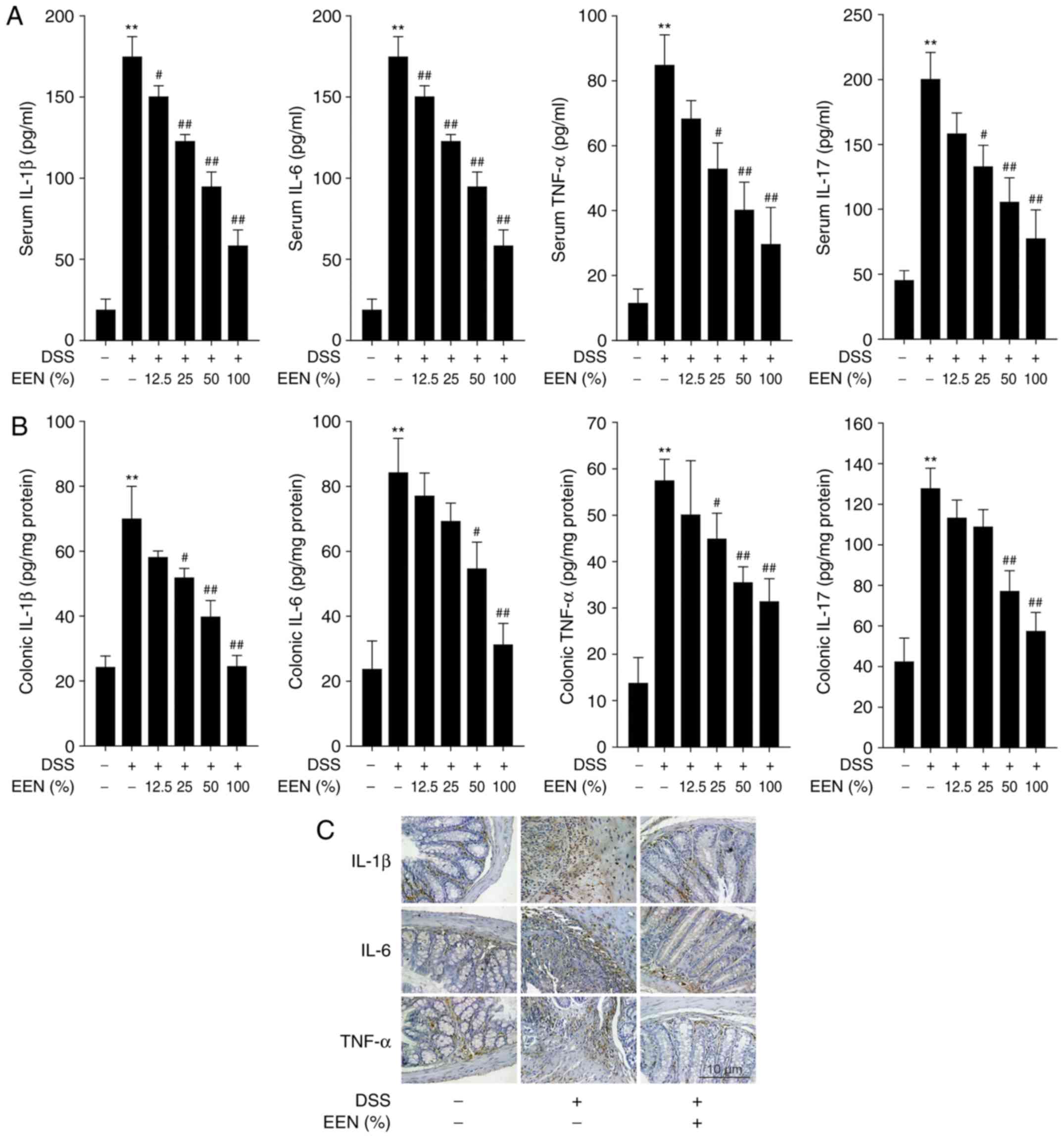
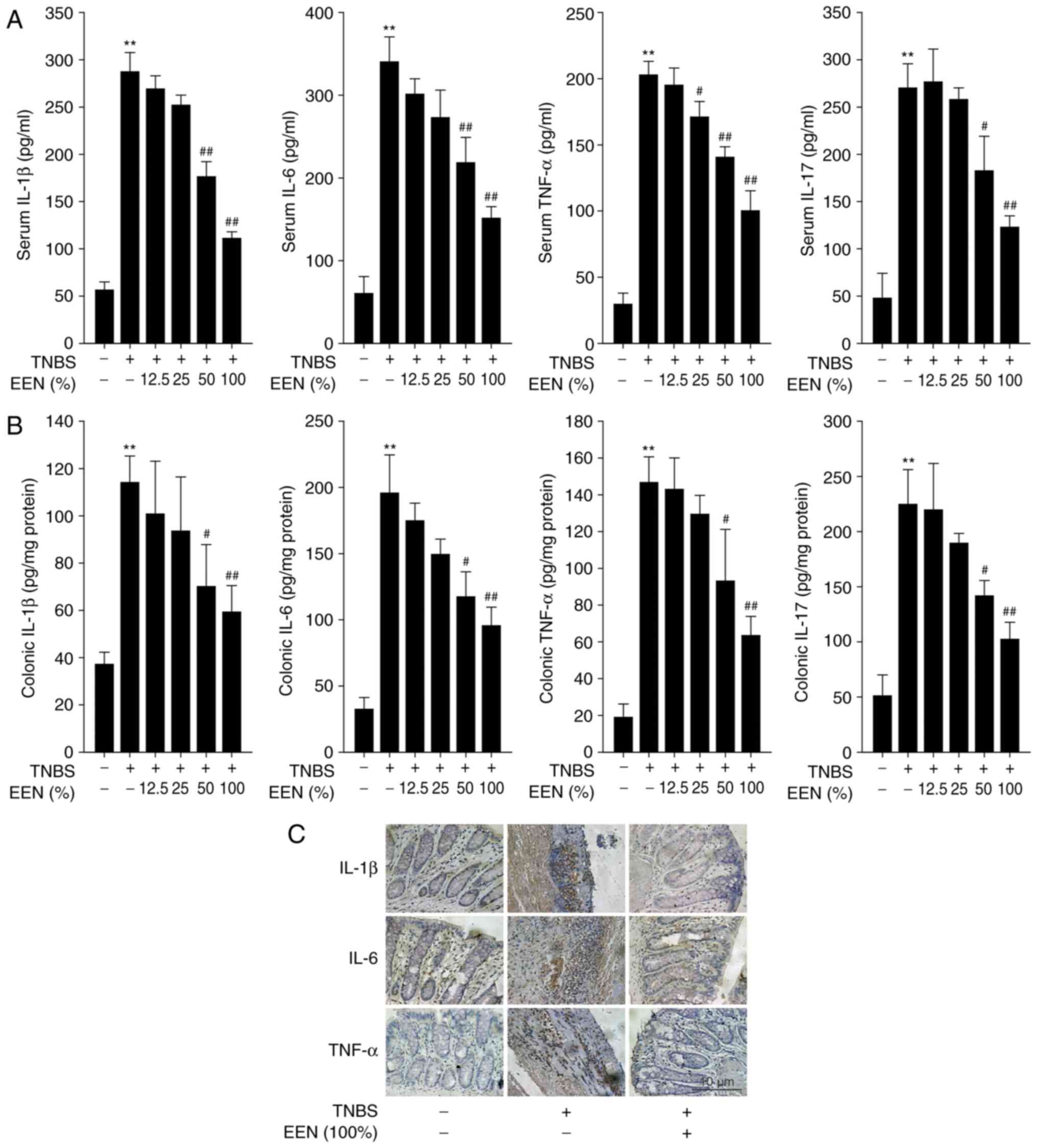
EERN decreases pro-inflammatory cytokines
production in the serum and colon of DSS-induced mice
In general, DSS-induced colitis is characterized by
high levels of cytokine production (27). To assess the effect of EEN on the
inflammatory status of DSS-induced colitis, the levels of
pro-inflammatory cytokines were examined. The expression of IL-1β,
IL-6, TNF-α and IL-17 was increased significantly in the serum of
DSS-induced colitis mice, compared with controls. However, EEN
significantly inhibited the elevated levels of these cytokines
(Fig. 2A). In addition, the
expression of IL-1β, IL-6, TNF-α and IL-17 was also measured in
colon homogenates. EEN significantly inhibited the expression of
IL-1β, IL-6, TNF-α and IL-17 in DSS-induced colitis mice (Fig. 2B). Furthermore, EEN ameliorated
the increased number of IL-1β-, IL-6- and TNF-α-positive cells in
colonic mucosa of DSS-induced mice (Fig. 2C). These results suggested that
EEN inhibited pro-inflammatory cytokines production in the serum
and colon of DSS-induced mice.
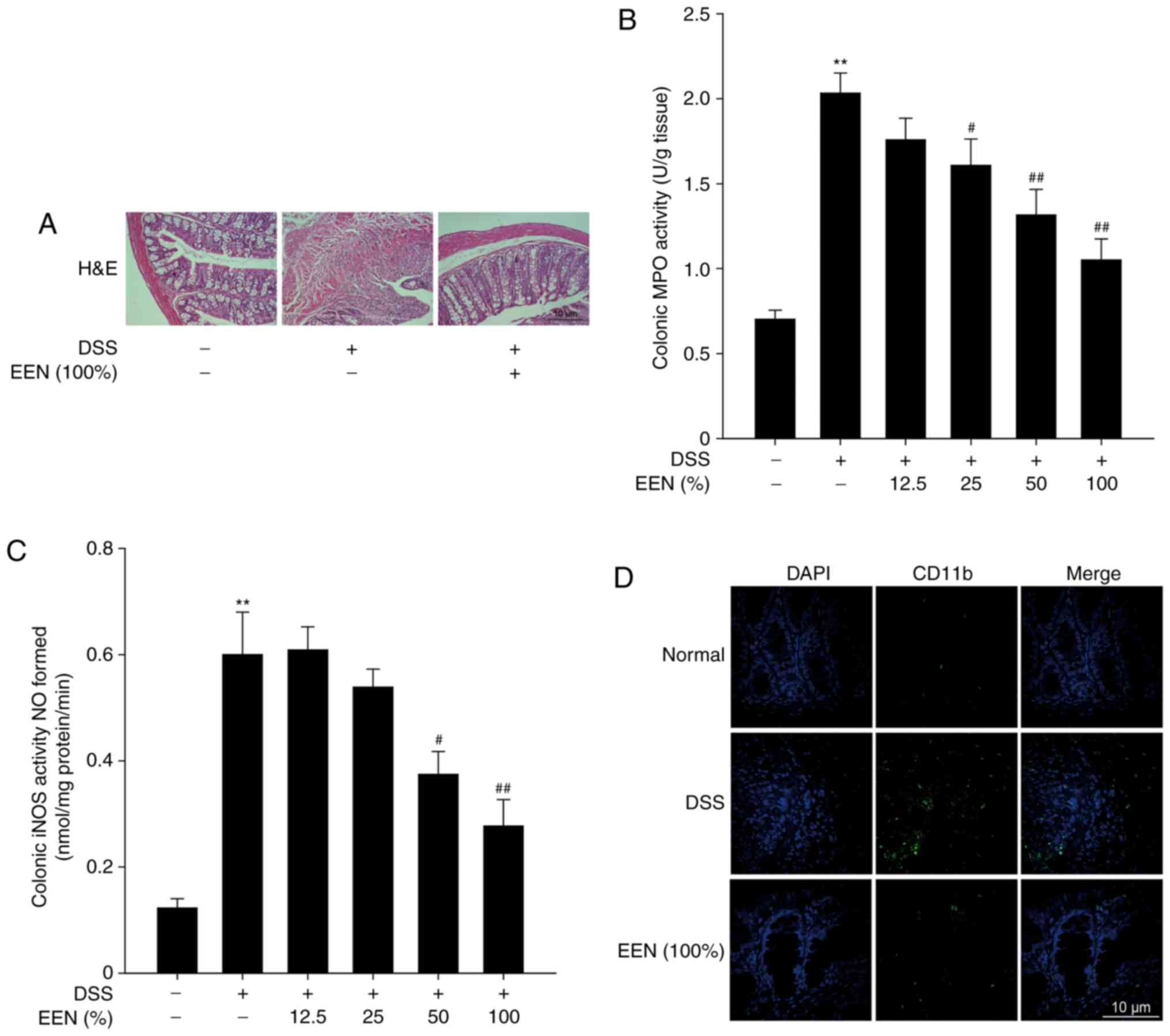
EEN-attenuates colon injury in
DSS-induced colitis mice
Subsequently, a pathological analysis of the colon
tissue was performed to assess the effect of EEN on colon injury.
H&E staining demonstrated that DSS group exhibited marked
erosion of the lamina propria mucosa, disappearance of glandular
epithelium and increased inflammatory cell infiltration compared
with the normal group. Notably, EEN inhibited the infiltration of
inflammatory cells and preserved intact colonic structure, with no
marked ulceration (Fig. 3A). In
addition, EEN significantly inhibited MPO and iNOS activities in
DSS-induced mice (Fig. 3B and C).
It has previously been demonstrated that CD11b is the surface
antigen of many leukocytes, including monocytes, neutrophils,
natural killer cells, granulocytes and macrophages (28). The accumulation of large amounts
of CD11b-positive inflammatory cells was observed in the lesion
site of colonic mucosa of DSS-induced mice. However, EEN reduced
the number of infiltrating CD11b-positive inflammatory cells in
colon tissues (Fig. 3D). These
results suggested that EEN could successfully ameliorate colon
injury in DSS-induced colitis.
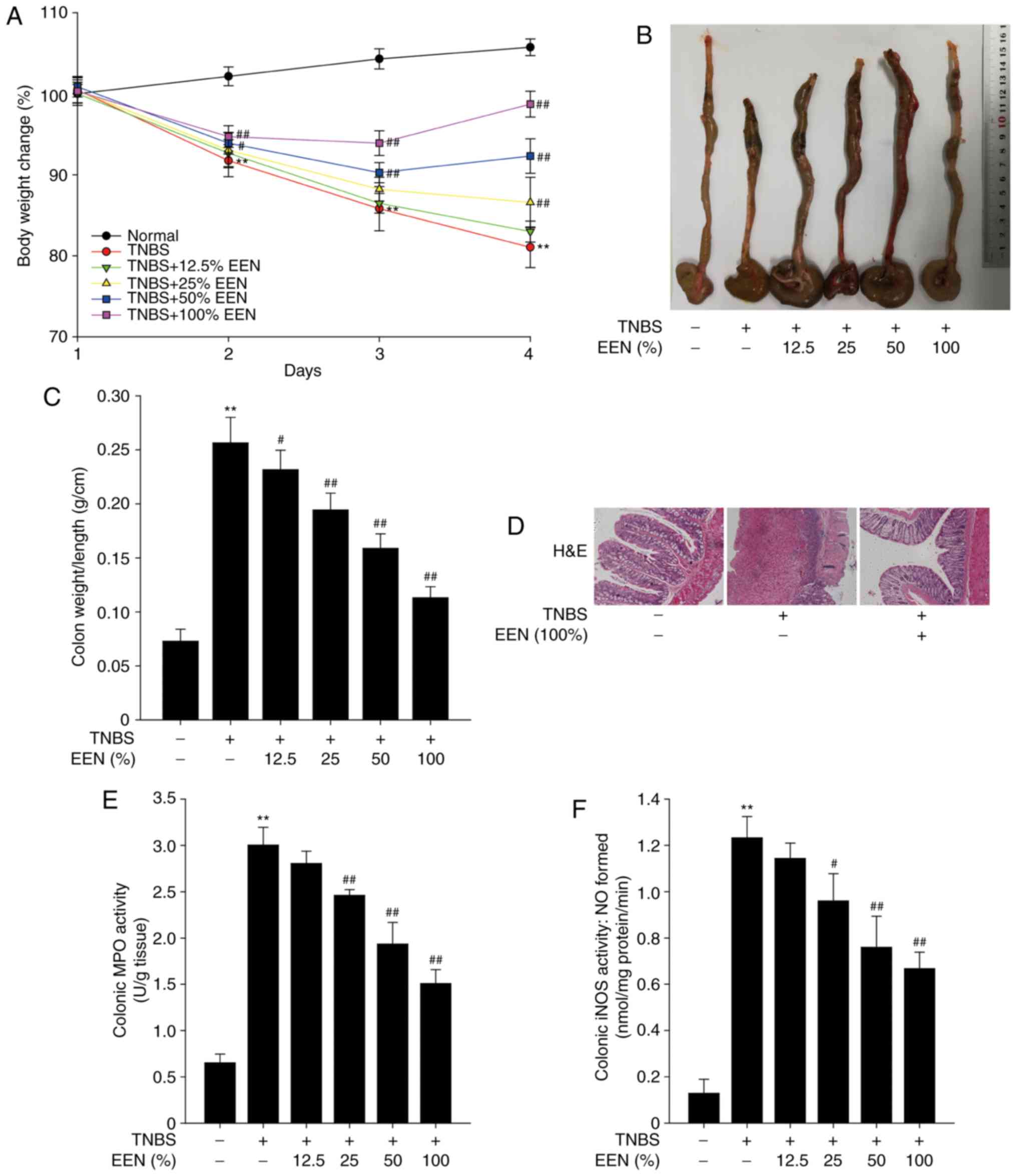
EEN attenuates TNBS-induced colitis
symptoms and colon injury
To further investigate the effect of EEN on colitis,
colitis was induced in rat with TNBS. Similar to the DSS model, the
TNBS-induced mice exhibited weight loss, and EEN groups were
significantly different from the DSS model group (Fig. 4A). TNBS-induced rats exhibited
shortening of the colon, whereas EEN ameliorated the shortening of
the colon in TNBS-induced colitis rat (Fig. 4B). EEN also reduced colonic weight
vs. length ratio in TNBS-induced colitis rats (Fig. 4C). The pathological analysis of
colon tissue demonstrated that the mucosa layer was severely
injured and a large number of inflammatory cells infiltrated in
TNBS-induced colon tissue. However, EEN inhibited the infiltration
of inflammatory cells and maintained the integrity of the colonic
tissue structure (Fig. 4D). In
addition, EEN significantly inhibited MPO an iNOS activities in
TNBS-induced rats (Fig. 4E and
F). These findings indicated that EEN ameliorated colitis
symptoms and colon tissue injury in TNBS-induced colitis rat.
EEN decreases pro-inflammatory cytokine
production in the serum and colon of TNBS-induced rats
As presented in Fig.
5A, the expression of IL-1β, IL-6, TNF-α and IL-17 was
increased significantly in the serum of TNBS-induced colitis rats,
whereas EEN significantly inhibited the elevated levels of these
cytokines. In colon tissue homogenates, EEN also significantly
reduced the production of IL-1β, IL-6, TNF-α and IL-17 in
TNBS-induced colitis rats (Fig.
5B). Furthermore, EEN markedly decreased the number of IL-1β-,
IL-6- and TNF-α-positive cells in colonic mucosa tissues of
TNBS-induced rats (Fig. 5C).
These data demonstrated that EEN suppressed pro-inflammatory
cytokines production in the serum and colon of TNBS-induced
rats.
EEN suppresses phosphorylation of p65 at
Ser276 via inhibition of p38/MSK1 pathway in DSS-colitis mice and
TNBS-colitis rat
The production of pro-inflammatory cytokines is
regulated by a variety of transcription factors, in which NF-κB
serves an important role in regulating inflammatory responses. To
elucidate the anti-inflammation mechanism of EEN, the effect of EEN
on the activation of p65 in peritoneal macrophages of DSS-colitis
mice was examined. Notably, EEN downregulated p-p65 expression in
peritoneal macrophages (Fig. 6A and
B). Furthermore, detection of p-MSK1 expression, a kinase
phosphorylated p65 at Ser276, revealed that EEN was able to
downregulate phosphorylation of MSK1 (Fig. 6A and B). Previous studies have
demonstrated that phosphorylation of MSK1 is regulated by the MAPK
signaling pathway (29–31). Therefore, the effect of EEN on the
MAPK signaling pathway was then examined. As presented in Fig. 6C and D, EEN downregulated
phosphorylation of p38 without significantly affecting
phosphorylation of ERK and JNK in peritoneal macrophages of
DSS-colitis mice.
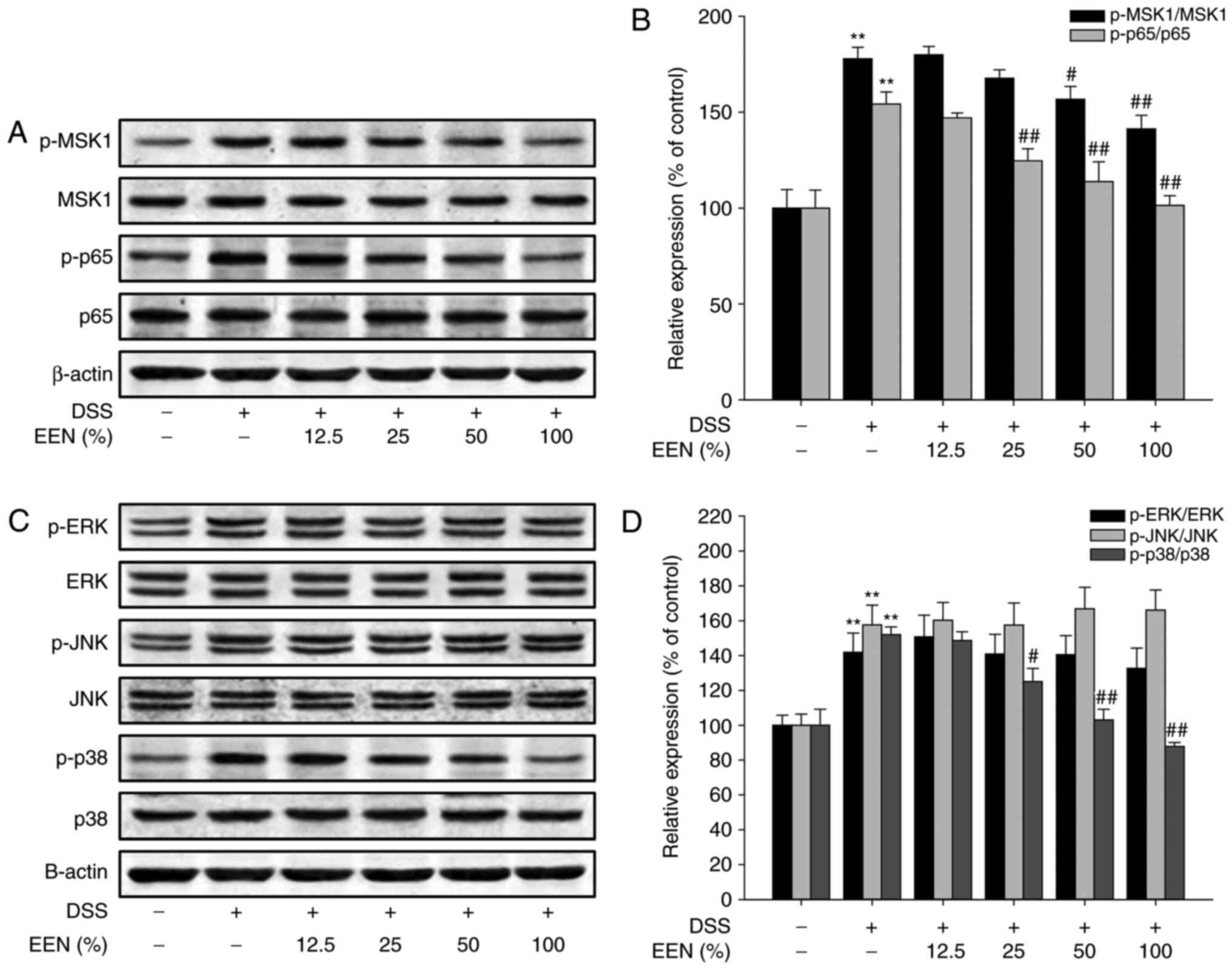 | Figure 6EEN down-regulated p-p65 (Ser276)
expression by inhibiting the p38/MSK1 signaling pathway in
peritoneal macrophages of DSS-colitis mice. (A) Levels of p-MSK1,
MSK1, p-p65, p65 and β-actin were assessed by western blotting in
peritoneal macrophages of DSS-colitis mice. (B) Densitometric
analysis was performed to determine the relative ratios of each
protein. (C) Levels of p-ERK, ERK, p-JNK, JNK, p-p38, p38 and
β-actin were assessed by western blot in peritoneal macrophages of
DSS-colitis mice. (D) Densitometric analysis was performed to
determine the relative ratios of each protein. Data were obtained
from at least three independent experiments and are presented as
the mean ± standard deviation. **P<0.01 vs. normal
group; #P<0.05, ##P<0.01 vs. DSS. EEN,
exclusive enteral nutrition; p, phosphorylated; MSK1, mitogen- and
stress-activated protein kinase-1; DSS, dextran sulfate sodium;
ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal
kinase. |
Then, the expression of related proteins was
examined in colon tissue of DSS- and TNBS-induced colitis models.
EEN downregulated phosphorylation of p38, MSK1 and p65 expression
in colon tissue of DSS-induced colitis mice and TNBS-induced
colitis rats (Fig. 7A–D).
Immunohistochemical results demonstrated that EEN decreased the
number of p-p38- and p-p65-positive cells in colonic tissues of
DSS-induced colitis mice and TNBS-induced colitis rats (Fig. 7E and F). These findings suggest
that EEN exerted the anti-inflammation effect by suppressing
phosphorylation of p65 at Ser276 via inhibition of p38/MSK1 pathway
in DSS-induced colitis mice and TNBS-induced colitis rat.
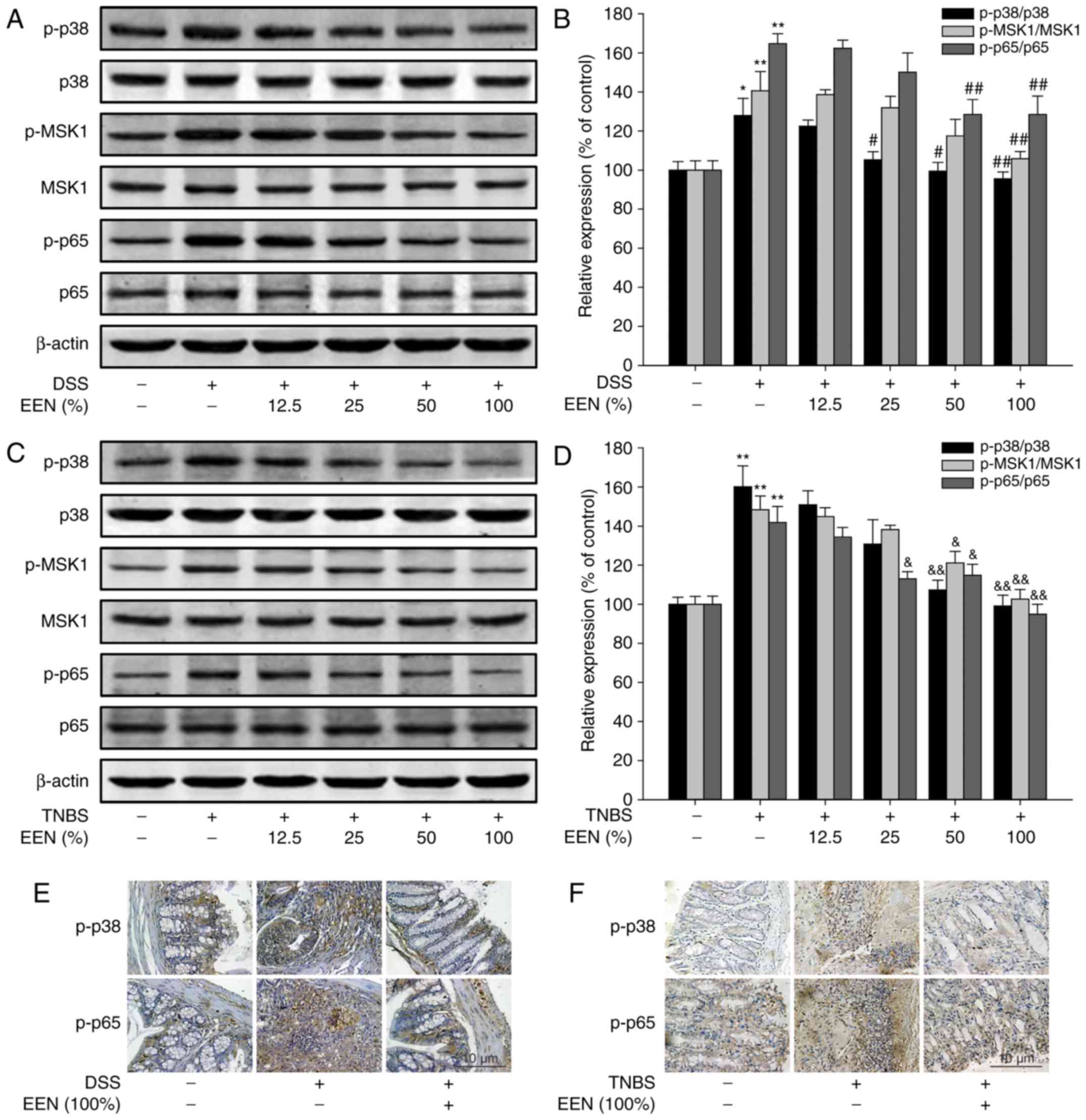 | Figure 7EEN suppressed activation of p65 by
inhibiting the p38/MSK1 signaling pathway in DSS- and TNBS-colitis
mice. (A) Levels of p-p38, p38, p-MSK1, MSK1, p-p65, p65 and
β-actin were assessed by western blotting in colonic tissues of
DSS-colitis mice. (B) Densitometric analysis was performed to
determine the relative ratios of each protein. (C) Levels of p-p38,
p38, p-MSK1, MSK1, p-p65, p65 and β-actin were assessed by western
blotting in colonic tissues of TNBS-colitis rats. (D) Densitometric
analysis was performed to determine the relative ratios of each
protein. The expressions of p-p38 and p-p65 were detected by
immunohistochemistry (original magnification, ×400) in colonic
tissues of (E) DSS- and (F) TNBS-colitis models. Data were obtained
from at least three independent experiments and are presented as
the mean ± standard deviation. *P<0.05,
**P<0.01 vs. normal group; #P<0.05,
##P<0.01 vs. DSS; &P<0.05,
&&P<0.01 vs. TNBS. EEN, exclusive enteral
nutrition; MSK1, mitogen- and stress-activated protein kinase-1;
DSS, dextran sulfate sodium; TNBS, picrylsulfonic acid; p,
phosphorylated. |
Discussion
UC is a prevalent inflammatory bowel disease in
North America and Europe. The annual number of novel occurrences is
1–20 people per 100,000, with a total occurrence of 5–500 per
100,000 affected worldwide (32).
At present, in developing countries, the incidence of UC is
increasing (33,34). There are many clinical treatments
for UC, including corticosteroids, sulfasalazine, classic
immunosuppressive agents, and antibiotics. However, the majority of
these drugs have limitations in wide clinical applications for
their serious side effects (35,36). Therefore, there is an urgent need
to develop novel drugs with minor side effects. The integrity of
the epithelial structure is a key factor in preventing colon
bacteria from migrating to the colon wall. Enteral nutrition from
the circulating blood to colonic epithelial cell membrane is
critical for structural integrity of the colonic epithelium,
because nutrients serve an important role in the differentiation,
proliferation and mucosal healing of colon epithelial cells
(37).
Hundreds of millions of gut microbes parasites in
the intestine (38). Under normal
conditions, intestinal microorganisms and the host exist in a state
of balance (39). Intestinal
microorganisms activate the body’s immune response when the
homeostasis of microorganisms and the host is destroyed, ultimately
contributing to the development of IBD (40). Dietary fiber in EEN can be used by
gut microbes to produce short-chain fatty acids that enhance the
intestinal barrier and thus reduce the damage done to the body by
the inflammatory response (41).
In the present study, the effect of EEN on the body itself was
evaluated.
The DSS-induced colitis model mimics human
inflammatory bowel disease appropriately (42); therefore, this model was used to
investigate the effect of EEN on colitis. In the present study, EEN
attenuated colitis injury and inflammatory symptoms induced by DSS,
such as weight loss, shortening of colon length and colonic tissue
damage. In addition, EEN reduced MPO activity, an index of
inflammation damage, which was a marker of neutrophil infiltration
(43). The release of NO was
modulated by iNOS activity, which damaged the intestinal mucosa and
submucosa cells (44). The
present study demonstrated that EEN also reduced iNOS activity in
colonic tissue. Furthermore, EEN inhibits infiltration of
inflammatory cells by decreasing CD11b-positive cell count. More
importantly, EEN reduced the production of pro-inflammatory
cytokines in serum and colon tissue, including IL-1β, IL-6, TNF-α
and IL-17 (45). Comprehensive
results demonstrated that EEN can alleviate DSS-induced colitis in
mice. The DSS-induced colitis mouse model was established by
letting mice freely drink water containing dissolved DSS.
Therefore, the change of water intake in mice directly affected the
level of DSS-induced colitis. In order to evaluate the protective
effect of EEN on colitis in a comprehensive and systematic way, the
effect of EEN on TNBS-induced colitis was also examined. In
TNBS-induced colitis rats, EEN alleviated colitis injury and
inflammatory symptoms and inhibited production of pro-inflammatory
cytokines. To further investigate the mechanism of EEN, the
activity of NF-κB was examined in peritoneal macrophages of DSS
mice. Western blotting results demonstrated that EEN downregulated
expression of p-p65 at Ser276. Further experiments demonstrated
that EEN suppressed phosphorylation of p65 at Ser276 via inhibition
of the p38/MSK1 pathway. Furthermore, this result was verified in
colonic tissue of DSS- and TNBS-induced colitis models. In
conclusion, EEN alleviated colitis by preventing phosphorylation of
p65 at Ser276 via inhibition of p38/MSK1 pathway.
Previously, great efforts have been made to
understand how NF-κB is activated by a variety of inducers
including bacteria, viruses and cytokines (46). The classic IKKβ-IκBα signaling
pathway is associated with activation of NF-κB via p65 nuclear
translocation-dependent mechanism (47). In addition, the activation of
NF-κB can also be carried out by post-translational modifications
via phosphorylation of p65 at Ser276, Ser529 or Ser536 (48). Among them, Ser276 phosphorylation
of p65 is very important for the activation of NF-κB; whether
inhibiting Ser276 phosphorylation of p65 formation, or mutation of
Ser276 blocking the ability of p65 to transactivate target genes
(49). In addition, Ser276
phosphorylation may inhibit p65 degradation mediated by ubiquitin
ligase SOCS-1 (10). It has
previously been demonstrated that protein kinase A (PKA) catalytic
subunit is associated with Ser276 phosphorylation of p65 formation
(10). Besides PKA, another
report demonstrated that p65 was phosphorylated at Ser276 by MSK1
as a nuclear kinase (10). The
present study demonstrated that phosphorylation of MSK1 expression
was downregulated following EEN treatment. This suggests that EEN
attenuated colitis by regulation of p65 activation via acting on
MSK1. MSK1 is activated by the MAPK/ERK or SAPK2/p38 pathways
(31). In the present study, it
was demonstrated that p38 was inhibited but had no influence on ERK
and JNK following EEN treatment. These data indicated that the
p38/MSK1 pathway is associated with inhibition of p65 activation by
EEN treatment.
These findings suggest that EEN alleviates DSS- and
TNBS-induced colitis and illustrate its anti-inflammatory mechanism
by inhibiting Ser276 phosphorylation of p65 via regulating p38/MSK1
pathway. Therefore, EEN may be an effective therapy for the
treatment UC in humans. However, intestinal flora is complex.
Inflammatory bowel disease is associated with other genetic,
environmental, microbiological and immune factors. The exact
etiology and pathogenesis are still unclear. Furthermore, the
clinical value of EEN requires further investigation with a larger
clinical sample size, collecting more data and conducting an
in-depth study.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81470819).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors’ contributions
TY and CY conceived the idea. TY and QY performed
the literature search. TY wrote the manuscript. TY, QY performed
the majority of the molecular biology experiments; TY, QY, XC
performed the majority of the cellular experiments; TY, LZ, YW and
CY performed the majority of the animal experiments. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Experimental protocols were performed in accordance
with National Institutes of Health regulations and approved by the
Institutional Animal Care and Use Committee (Shanghai Institute of
Biochemistry and Cell Biology, Chinese Academy of Sciences,
Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
D’Arcangelo G and Aloi M: Inflammatory
bowel disease-unclassified in children: Diagnosis and
pharmacological management. Paediatri Drugs. 19:113–120. 2017.
View Article : Google Scholar
|
|
2
|
Mărginean CO, Meliţ LE, Mocanu S and
Mărginean MO: Inflammatory bowel diseases: a burden in pediatrics:
Case series and a review of the literature. Medicine. 96:e63292017.
View Article : Google Scholar
|
|
3
|
Francescone R, Hou V and Grivennikov SI:
Cytokines, IBD, and colitis-associated cancer. Inflamm Bowel Dis.
21:409–418. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Russo RA and Katsicas MM: Autoinflammatory
diseases. Medicina (B Aires). 76:166–172. 2016.In Spanish.
|
|
5
|
Striz I, Brabcova E, Kolesar L and
Sekerkova A: Cytokine networking of innate immunity cells: A
potential target of therapy. Clin Sci (Lond). 126:593–612. 2014.
View Article : Google Scholar
|
|
6
|
Estruch M, Sanchez-Quesada JL,
Ordoñez-Llanos J and Benitez S: Inflammatory intracellular pathways
activated by electronegative LDL in monocytes. Biochim Biophys
Acta. 1861.963–969. 2016.
|
|
7
|
Patel SJ, Jindal R, King KR, Tilles AW and
Yarmush ML: The inflammatory response to double stranded DNA in
endothelial cells is mediated by NFκB and TNFα. PloS One.
6:e199102011. View Article : Google Scholar
|
|
8
|
Kweon SM, Wang B, Rixter D, Lim JH, Koga
T, Ishinaga H, Chen LF, Jono H, Xu H and Li JD: Synergistic
activation of NF-kappaB by nontypeable H. influenzae and S.
pneumoniae is mediated by CK2, IKKbeta-IkappaBalpha, and p38 MAPK.
Biochem Biophys Res Commun. 351:368–375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schmitz ML, Mattioli I, Buss H and Kracht
M: NF-kappaB: A multifaceted transcription factor regulated at
several levels. Chembiochem. 5:1348–1358. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nihira K, Ando Y, Yamaguchi T, Kagami Y,
Miki Y and Yoshida K: Pim-1 controls NF-kappaB signalling by
stabilizing RelA/p65. Cell Death Differ. 17:689–698. 2010.
View Article : Google Scholar
|
|
11
|
Arun P, Brown MS, Ehsanian R, Chen Z and
Van Waes C: Nuclear NF-kappaB p65 phosphorylation at serine 276 by
protein kinase A contributes to the malignant phenotype of head and
neck cancer. Clin Cancer Res. 15:5974–5984. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kawashima T, Murata K, Akira S, Tonozuka
Y, Minoshima Y, Feng S, Kumagai H, Tsuruga H, Ikeda Y, Asano S, et
al: STAT5 induces macrophage differentiation of M1 leukemia cells
through activation of IL-6 production mediated by NF-kappaB p65. J
Immunol. 167:3652–3660. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen YM, Zhao Y, Zeng Y, Yan L, Chen BL,
Leng AM, Mu YB and Zhang GY: Inhibition of Pim-1 kinase ameliorates
dextran sodium sulfate-induced colitis in mice. Dig Dis Sci.
57:1822–1831. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeng S, Zhang QY, Huang J, Vedantham S,
Rosario R, Ananthakrishnan R, Yan SF, Ramasamy R, DeMatteo RP,
Emond JC, et al: Opposing roles of RAGE and Myd88 signaling in
extensive liver resection. FASEB J. 26:882–893. 2012. View Article : Google Scholar :
|
|
15
|
Zhang HM, Li L, Papadopoulou N, Hodgson G,
Evans E, Galbraith M, Dear M, Vougier S, Saxton J and Shaw PE:
Mitogen-induced recruitment of ERK and MSK to SRE promoter
complexes by ternary complex factor Elk-1. Nucleic Acids Res.
36:2594–2607. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Basu S, Pathak S, Pathak SK, Bhattacharyya
A, Banerjee A, Kundu M and Basu J: Mycobacterium avium-induced
matrix metalloproteinase-9 expression occurs in a
cyclooxygenase-2-dependent manner and involves phosphorylation- and
acetylation-dependent chromatin modification. Cell Microbiol.
9:2804–2816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vermeulen L, De Wilde G, Van Damme P,
Vanden Berghe W and Haegeman G: Transcriptional activation of the
NF-kappaB p65 subunit by mitogen- and stress-activated protein
kinase-1 (MSK1). EMBO J. 22:1313–1324. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan Y, Liu Y, Guo H, Jabir MS, Liu X, Cui
W and Li D: Associations between folate and vitamin B12 levels and
inflammatory bowel disease: A meta-analysis. Nutrients. 9:E3822017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang F, Zhao HY, Zhang ST, Gong YZ, Zhang
HF and Zhang C: Effect of enteral nutrition on dextran sulfate
sodium-induced colitis in rats. J Dig Dis. 12:453–458. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park MY, Kwon HJ and Sung MK: Dietary
aloin, aloesin, or aloe-gel exerts anti-inflammatory activity in a
rat colitis model. Life Sci. 88:486–492. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
El-Jamal N, Erdual E, Neunlist M, Koriche
D, Dubuquoy C, Maggiotto F, Chevalier J, Berrebi D, Dubuquoy L,
Boulanger E, et al: Glugacon-like peptide-2: Broad receptor
expression, limited therapeutic effect on intestinal inflammation
and novel role in liver regeneration. Am J Physiol Gastrointest
Liver Physiol. 307:G274–G285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alex P, Zachos NC, Nguyen T, Gonzales L,
Chen TE, Conklin LS, Centola M and Li X: Distinct cytokine patterns
identified from multiplex profiles of murine DSS and TNBS-induced
colitis. Inflamm Bowel Dis. 15:341–352. 2009. View Article : Google Scholar :
|
|
23
|
Yao J, Pan D, Zhao Y, Zhao L, Sun J, Wang
Y, You QD, Xi T, Guo QL and Lu N: Wogonin prevents
lipopolysaccharide-induced acute lung injury and inflammation in
mice via peroxisome proliferator-activated receptor gamma-mediated
attenuation of the nuclear factor-kappaB pathway. Immunology.
143:241–257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao Y, Sun Y, Ding Y, Wang X, Zhou Y, Li
W, Huang S, Li Z, Kong L, Guo Q and Lu N: GL-V9, a new synthetic
flavonoid derivative, ameliorates DSS-induced colitis against
oxidative stress by up-regulating Trx-1 expression via activation
of AMPK/FOXO3a pathway. Oncotarget. 6:26291–26307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cabre E, Mañosa M, Garcia-Sanchez V,
García-Sánchez V, Gutiérrez A, Ricart E, Esteve M, Guardiola J,
Aguas M, Merino O, et al: Phenotypic concordance in familial
inflammatory bowel disease (IBD). Results of a nationwide IBD
Spanish database. J Crohns Colitis. 8:654–661. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fukuda T, Majumder K, Zhang H, Turner PV,
Matsui T and Mine Y: Adenine inhibits TNF-α signaling in intestinal
epithelial cells and reduces mucosal inflammation in a dextran
sodium sulfate-induced colitis mouse model. J Agric Food Chem.
64:4227–4234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Morin C, Blier PU and Fortin S: MAG-EPA
reduces severity of DSS-induced colitis in rats. Am J Physiol
Gastrointest Liver Physiol. 310:G808–G821. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shah KH, Shi P, Giani JF, Janjulia T,
Bernstein EA, Li Y, Zhao T, Harrison DG, Bernstein KE and Shen XZ:
Myeloid Suppressor cells accumulate and regulate blood pressure in
hypertension. Circ Res. 117:858–869. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reber L, Vermeulen L, Haegeman G and
Frossard N: Ser276 phosphorylation of NF-κB p65 by MSK1 controls
SCF expression in inflammation. PloS One. 4:e43932009. View Article : Google Scholar
|
|
30
|
Schiller M, Bohm M, Dennler S, Ehrchen JM
and Mauviel A: Mitogen- and stress-activated protein kinase 1 is
critical for interleukin-1-induced, CREB-mediated, c-fos gene
expression in keratinocytes. Oncogene. 25:4449–4457. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deak M, Clifton AD, Lucocq LM and Alessi
DR: Mitogen- and stress-activated protein kinase-1 (MSK1) is
directly activated by MAPK and SAPK2/p38, and may mediate
activation of CREB. EMBO J. 17:4426–4441. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Burisch J: Crohn’s disease and ulcerative
colitis. Occurrence, course and prognosis during the first year of
disease in a European population-based inception cohort. Dan Med J.
61:B47782014.
|
|
33
|
McDermott F, Hughes ES and Pihl E:
Mortality and morbidity of Crohn’s disease and ulcerative colitis
in Australia. Med J Aust. 1:534–536. 1980.PubMed/NCBI
|
|
34
|
Grimm IS and Friedman LS: Inflammatory
bowel disease in the elderly. Gastroenterol Clin North Am.
19:361–389. 1990.PubMed/NCBI
|
|
35
|
Reich K, Leonardi C, Langley RG, Warren
RB, Bachelez H, Romiti R, Ohtsuki M, Xu W, Acharya N, Solotkin K,
et al: Inflammatory bowel disease among patients with psoriasis
treated with ixekizumab: A presentation of adjudicated data from an
integrated database of 7 randomized controlled and uncontrolled
trials. J Am Acad Dermatol. 76:441–448e2. 2017. View Article : Google Scholar
|
|
36
|
Archer R, Tappenden P, Ren S, Martyn-St
James M, Harvey R, Basarir H, Stevens J, Carroll C, Cantrell A,
Lobo A and Hoque S: Infliximab, adalimumab and golimumab for
treating moderately to severely active ulcerative colitis after the
failure of conventional therapy (including a review of TA140 and
TA262): Clinical effectiveness systematic review and economic
model. Health Technol Assess. 20:1–326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Perdikis DA and Basson MD: Basal nutrition
promotes human intestinal epithelial (Caco-2) proliferation, brush
border enzyme activity, and motility. Crit Care Med. 25:159–165.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zaiss MM, Rapin A, Lebon L, Dubey LK,
Mosconi I, Sarter K, Piersigilli A, Menin L, Walker AW, Rougemont
J, et al: The intestinal microbiota contributes to the ability of
helminths to modulate allergic inflammation. Immunity. 43:998–1010.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vitetta L, Saltzman ET, Nikov T, Ibrahim I
and Hall S: Modulating the gut micro-environment in the treatment
of intestinal parasites. J Clin Med. 5:E1022016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
No authors listed. Inflammatory bowel
disease. Prog Drug Res. 71:117–122. 2016.PubMed/NCBI
|
|
41
|
Jandhyala SM, Talukdar R, Subramanyam C,
Vuyyuru H, Sasikala M and Nageshwar Reddy D: Role of the normal gut
microbiota. World J Gastroenterol. 21:8787–8803. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dae Park D, Yum HW, Zhong X, Kim SH, Kim
SH, Kim DH, Kim SJ, Na HK, Sato A, Miura T and Surh YJ: Perilla
frutescens extracts protects against dextran sulfate sodium-induced
murine colitis: NF-κB, STAT3, and Nrf2 as putative targets. Front
Pharmacol. 8:4822017. View Article : Google Scholar
|
|
43
|
Kono H, Fujii H, Ogiku M, Tsuchiya M,
Ishii K and Hara M: Enteral diets enriched with medium-chain
triglycerides and N-3 fatty acids prevent chemically induced
experimental colitis in rats. Transl Res. 156:282–291. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kawahara R, Yasuda M, Hashimura H, Amagase
K, Kato S and Takeuchi K: Activation of α7 nicotinic acetylcholine
receptors ameliorates indomethacin-induced small intestinal
ulceration in mice. Eur J Pharmacol. 650:411–417. 2011. View Article : Google Scholar
|
|
45
|
Hata K, Andoh A, Shimada M, Fujino S,
Bamba S, Araki Y, Okuno T, Fujiyama Y and Bamba T: IL-17 stimulates
inflammatory responses via NF-kappaB and MAP kinase pathways in
human colonic myofibroblasts. Am J Physiol Gastrointest Liver
Physiol. 282:G1035–G1044. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rivera-Serrano EE and Sherry B: NF-κB
activation is cell type-specific in the heart. Virology.
502:133–143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lim H, Lee H, Noh K and Lee SJ:
IKK/NF-κB-dependent satellite glia activation induces spinal cord
microglia activation and neuropathic pain after nerve injury. Pain.
158:1666–1677. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Durand JK and Baldwin AS: Targeting IKK
and NF-κB for therapy. Adv Protein Chem Struct Biol. 107:77–115.
2017. View Article : Google Scholar
|
|
49
|
Rinkenbaugh AL, Cogswell PC, Calamini B,
Dunn DE, Persson AI, Weiss WA, Lo DC and Baldwin AS: IKK/NF-κB
signaling contributes to glioblastoma stem cell maintenance.
Oncotarget. 7:69173–69187. 2016. View Article : Google Scholar : PubMed/NCBI
|