Introduction
Abdominal aortic aneurysm (AAA) is a common,
potentially life-threatening, chronic vascular degenerative disease
with a high mortality rate following rupture (1). AAA is commonly found in adult
patients, particularly elderly men and may have severe
complications, including acute kidney injury, myocardial infarction
and stroke (2,3). Although there has been substantial
effort to clarify the mechanism underlying the development of AAA,
there are no effective therapeutic strategies to reduce AAA
development and rupture (4,5).
The apoptosis of vascular smooth muscle cells (VSMCs) is one of the
pathological features of AAA, and is considered to be important in
the development of AAA, possibly through promoting plaque
calcification and medial degeneration, preventing expansive
remodeling (1,6). Therefore, therapeutic strategies
that inhibit or delay VSMC apoptosis are urgently required.
Oxidative stress is a pathological feature, which is
important in the formation and progression of AAA in humans
(7,8). Oxidative damage to VSMCs sharply
decreases the capability to synthesize elastin and collagens,
resulting in the degeneration of aortic walls and eventual rupture
(7-10). Hydrogen peroxide
(H2O2), a highly reactive oxygen species
(ROS), leads to widespread intracellular damage and apoptosis, and
has been widely used to mimic oxidative stress in vitro
(11). Extensive studies using a
H2O2 induced-VSMC injury model have shown
that VSMC apoptosis is a major cellular component in the onset of a
variety of vascular diseases (12,13). This model has attracted
considerable interest for its potential relevance to human AAA, as
it can activate various pathways of apoptosis (14). Therefore, the
H2O2-induced VSMC injury model of AAA was
used in the present study for further investigations.
MicroRNAs (miRNAs) are small conserved,
single-stranded, non-coding RNA molecules (~18-25 nucleotides in
length), which regulate target gene expression through either
inducing transcript degradation or inhibiting translation (15). Increasing evidence has shown that
miRNAs are crucial in the formation of AAA. For example, Wu et
al showed that the upregulation of miRNA (miR)-145 prevented
the formation of AAA in ApoE−/− mice induced by
Angiotensin (Ang) II through modulating the expression of matrix
metalloproteinase (MMP)2 (16).
Maegdefessel et al reported that the inhibition of miR-29b
with a locked nucleic-acid (LNA)-anti-miR-29b led to reduced AAA
expansion and a significant decrease in the aortic rupture rate
with Ang II-treatment (17).
Notably, several studies have shown that various functions in VSMCs
are finely regulated by miRNAs. Iaconetti et al reported
that miR-23b regulated the VSMC phenotypic switch in vitro
and following vascular injury in vivo (18). Lai et al found that
miR-574-5p promoted the cell growth of VSMCs in the progression of
coronary artery disease (19).
However, few studies have been reported on whether miRNAs are
involved in the regulation of VSMC apoptosis in AAA disease.
In the present study, the miRNA expression profile
was examined in peripheral blood from patients with AAA.
Furthermore, using an H2O2-induced VSMC
injury model to mimic the pathological characteristics of AAA, the
role of miR-26a in preventing the apoptosis of VSMCs was examined,
and the role of the phosphatase and tensin homolog
(PTEN)/AKT/mammalian target of rapamycin (mTOR) pathway in the
protective activity of miR-26a against VSMC injury was confirmed.
The results are likely to have important implications for further
elucidating the molecular mechanisms and identifying novel
therapeutic targets in AAA.
Materials and methods
Tissue samples
Peripheral blood samples were obtained from 30
patients with AAA undergoing ascending aorta replacement procedures
at the Department of Vascular Surgery, The First Hospital of Hebei
Medical University (Shijiazhuang, China) from January 2015 to June
2016. Control peripheral blood tissues were obtained from 30 donors
without vascular diseases. The clinicopathological data of the
patients is reported in Table I.
All experimental protocols were approved by the Ethics Committee of
the First Hospital of Hebei Medical University. All experiments
were performed in accordance with the ethical guidelines of the
Ethics Committee of the First Hospital of Hebei Medical University.
Informed consent was obtained from all patients.
 | Table ICharacteristics of AAAs and control
groups. |
Table I
Characteristics of AAAs and control
groups.
|
Characteristics | AAA (n=30) | Control (n=30) |
|---|
| Male/female | 18/12 | 16/14 |
| Age (years) | 65.2±4.5 | 38.3±9.5 |
| Aneurysm diameter,
cm | 7.56±1.21 | – |
| Hypertension
(%) | 5 (16.7) | – |
| Smoking habit
(%) | 12 (40.0) | – |
MicroRNA expression profiling
Total RNA was extracted from the venous peripheral
blood of patients with AAA and controls using a PAXgene Blood RNA
kit (Qiagen GmbH, Hilden, Germany). The RNA quantity and quality
were assessed by NanoDrop ND-1000 spectrophotometry (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and an Agilent 2100 bioanalyzer
(Agilent Technologies, Inc., Santa Clara, CA, USA), respectively.
Total RNA (200 ng) was labeled with fluorescence dye hy3 or hy5
using the miRCURY Hy3/Hy5 Power Labeling kit and hybridized on the
miRCURY™ LNA array (v.16.0; Exiqon A/S, Copenhagen, Denmark)
according to the manufacturer’s protocol. Data were analyzed using
GeneSpring software version 7.3 (Agilent Technologies, Inc.).
Observations with adjusted P-values of P≥0.05 were removed, and
thus excluded from further analysis. The heat map of the 50
microRNAs with the most marked differences was created using a
method of hierarchical clustering by GeneSpring GX, version 7.3
(Agilent Technologies, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
miRNA was prepared using the miRNeasy Mini kit
(Qiagen, Inc., Valencia, CA, USA) and total RNA was prepared using
TRIzol reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer’s protocol. For miRNA reverse transcription, cDNA was
synthesized using the miRNA reverse transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). For mRNA reverse
transcription, cDNA was synthesized using the High Capacity
RNA-to-cDNA kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The RT-qPCR reaction system (30 µl) contained 5
µl cDNA, 15 µl 2X qPCR mix, 1 µl upstream
primer, 1 µl downstream primer and 8 µl double
distilled H2O. The PCR protocol was as follows: 95°C for
15 min, followed by 40 cycles of 94°C for 15 sec, 55°C for 30 sec
and 70°C for 30 sec and a final extension step at 72°C for 5 min
using an Applied Biosystems Prism 7900HT Fast Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Relative
quantification was determined by normalization to U6 or GAPDH. The
primers for RT-qPCR analysis were as follows: Reverse transcription
primer for precursor miR-26a,
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGCCTA-3′; miR-26a,
forward 5′-CGTCCTTCAAGTAATCCAGGA-3′ and reverse
5′-GCAGGGTCCGAGGTATTC-3′; U6 forward 5′-TGCGGGTGCTCGCTTCGCAGC-3′
and reverse 5′-CCAGTGCAGGGTCCGAGGT-3′; PTEN, forward
5′-TGGAAAGGGACGAACTGGTG-3′, and reverse 5′-CATAGCGCCTCTGACTGGGA-3′;
GAPDH, forward 5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse
5′-TGTAGACCATGTAGTTGAGGTCA-3′. The PCR amplification protocol was
as follows: Initial 95°C for 5 min, and 40 cycles of 94°C for 15
sec, 55°C for 30 sec, and 70°C for 30 sec. The RT-qPCR assays were
performed in triplicate and the change in expression level was
calculated using the 2−ΔΔCq method (20).
Cell culture and treatment
Primary vascular smooth muscle cells (VSMCs) were
isolated and obtained using a technique combining explant culture
and enzymatic digestion from the abdominal aorta of five male
Sprague-Dawley rats (age, 3-4 weeks; weight, 80±20 g) obtained from
the Laboratory Animal Center of Hebei Medical University
(Shijiazhuang, China) as described previously (21). All animals were housed in a 12 h
light/dark schedule in a temperature (22±2°C) and humidity
(<40%) controlled room with free access to food and water.
Briefly, the rats were anesthetized with intraperitoneal chloral
hydrate (400 mg/kg; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
and the thoracic aortas were isolated. The adhesive fat and tissues
were removed. The aorta artery was subsequently cut into ~1
mm2 segments after the endothelial cell layer of
endangium was removed. The tissue blocks were cultured in DMEM
supplemented with 10% fetal bovine serum (Hyclone; GE Healthcare
Life Sciences, Logan, UT, USA), 100 U ml−1 penicillin
and streptomycin at 37°C in a humidified atmosphere with 5%
CO2. The VSMCs were treated with different
concentrations of H2O2 (30% w/w solution;
Sigma; EMD Millipore, Billerica, MA, USA) for further measurements.
Cells in the control group were treated with the same medium
without H2O2. All animal procedures were
performed according to approved protocols from the Institutional
Animal Care and Use Committee of the First Hospital of Hebei
Medical University.
Cell transfection
The miR-26a mimics, miR-26a inhibitor, and the
corresponding control vectors were purchased from GeneCopoeia, Inc.
(Guangzhou, China). The PTEN-expressing vector (cat. no. 28298) was
obtained from Addgene, Inc. (Cambridge, MA, USA). The VSMCs
(1.0×106 per well) were seeded and grown overnight in
six-well plates. The following day, transfection was performed
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer’s protocol.
Cell viability
The VSMCs (5×103 per well) were suspended
in DMEM (100 µl) containing 10% fetal bovine serum and
cultured in 96-well plates overnight, following which they were
transfected with miR-26a mimics, miR-26a mimics + pcDNA-PTEN or
negative control oligonucleotides 2 h prior to
H2O2 treatment and incubated for 6 h. The
cell viability was determined using a Cell Counting Kit-8 (CCK-8;
Beyotime Institute of Biotechnology, Jiangsu, China) assay.
Briefly, 10 µl CCK-8 solution was added to each well and
incubated at 37°C in a CO2 cell incubator for 90 min,
and the absorbance rates were then measured at 450 nm using a
microplate reader (Infinite M200; Tecan Austria, GmbH, Grödig,
Austria). All experiments were performed in triplicate.
Cell apoptosis
Cell apoptosis was determined using an Annexin
V-FITC Apoptosis Detection kit (Abcam, Cambridge, UK) according to
the manufacturer’s protocol. Briefly, the VSMCs were seeded in
6-well plates at a density of 1.0×106 cell/well, and
subjected to the various treatments as described above. At the end
of the exposure, the cells were harvested and washed twice with
PBS, and the cells were then stained with Annexin V and propidium
iodide. Following incubation at room temperature in the dark for 15
min, cell apoptosis was analyzed on a FACScan flow cytometer
(Beckman Coulter, Inc., Brea, CA, USA).
Caspase-3 activity assay
The VSMCs were seeded in 6-well plates at a density
of 1×105 cells/well for 24 h. Following treatment for 6
h, caspase-3 activity in cell lysates was determined using a
Caspase-3 Activity Assay kit (Beyotime Institute of Biotechnology),
according to the manufacturer’s protocol.
Detection of ROS generation
The generation of myocardial ROS was assessed using
2′,7′-DCF diacetate (DCF-DA; Sigma-Aldrich; EMD Millipore) as
previously reported (22).
Briefly, the cell culture medium was discarded and the cells were
incubated with 20 µmol/l DCFH for 30 min at 37°C. The cells
were then washed twice with PBS, and were stained at 37°C in the
dark for 20 min, following which they were visualized by
fluorescence microscopy (Leica Microsystems GmbH, Wetzlar,
Germany). The fluorescence levels of the samples were measured
using a fluorescence microplate reader at 488 nm excitation and 525
nm emission wavelengths. Fold-increases in ROS levels were
determined by comparison with the control group.
Bioinformatics
TargetScan 7.0 (targetscan.org/) and miRanda (microrna.org/) target gene prediction software were
used to select PTEN as a target gene of miR-26a. TargetScan target
gene prediction software was used to identify the 2619-2626 site at
the 3′-untranslated region (UTR) of PTEN mRNA as a possible site of
action of miR-26a.
Luciferase assays
The 3′-UTR of PTEN, with wild-type (wt) or mutant
(mut) binding sites for miR-26a, was amplified and cloned into the
pGL3 vector (Promega Corporation, Madison, WI, USA) to generate the
pGL3-wt-PTEN-3′-UTR plasmid or pGL3-Mut-PTEN-3′-UTR plasmid,
respectively. For the luciferase reporter assay, 293 cells
(American Type Culture Collection, Manassas, VA, USA) were
co-transfected with the luciferase reporter vectors and miR-26a
mimics, miR-26a inhibitor or corresponding negative control using
Lipofectamine 2000 reagent. The pRL-TK plasmid (Promega
Corporation) was used as a normalizing control. After 48 h,
luciferase activity was analyzed using the Dual-Luciferase Reporter
Assay system (Promega Corporation) according to the manufacturer’s
protocol.
Western blot assay
Total proteins were extracted from the tissues and
cells using radioimmunoprecipitation assay lysis buffer (Sigma; EMD
Millipore) and quantified with a bicinchoninic acid protein assay
kit (Pierce; Thermo Fisher Scientific, Inc.). The protein samples
(40 µg) were separated by 10% SDS-PAGE (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and transferred onto a
polyvinylidene difluoride membrane (EMD Millipore) and blocked with
5% skimmed milk at room temperature for 1 h. The blots were
incubated with primary antibodies against cleaved caspase-3
(dilution, 1:1,000; cat. no. 9579), total-caspase-3 (dilution,
1:1,000; cat. no. 9665), PTEN (dilution, 1:1,000; cat. no. 9188),
AKT (dilution, 1:1,000; cat. no. 4691), phosphorylated (p-)AKT
(dilution, 1:1,000; cat. no. 5012), mTOR (dilution, 1:1,000; cat.
no. 2893), p-mTOR (dilution, 1:1,000; cat. no. 5536) and β-actin
(dilution, 1:2,000, cat. no. 4970) at 4°C overnight. Then the
membranes were incubated with horseradish peroxidase conjugated
secondary antibodies (1:50,000; cat. no. 7054) for 1 h at room
temperature. All antibodies were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The protein bands were
visualized using ECL detection reagent (GE Healthcare Life
Sciences, Piscataway, NJ, USA). The intensity of protein fragments
was quantified with Quantity One software (4.5.0 basic; Bio-Rad
Laboratories, Inc.).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA). The
results are presented as the mean ± standard deviation. Differences
were analyzed with Student’s t-test between two groups or with
one-way analysis of variance among multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-26a is downregulated in peripheral
blood from patients with AAA
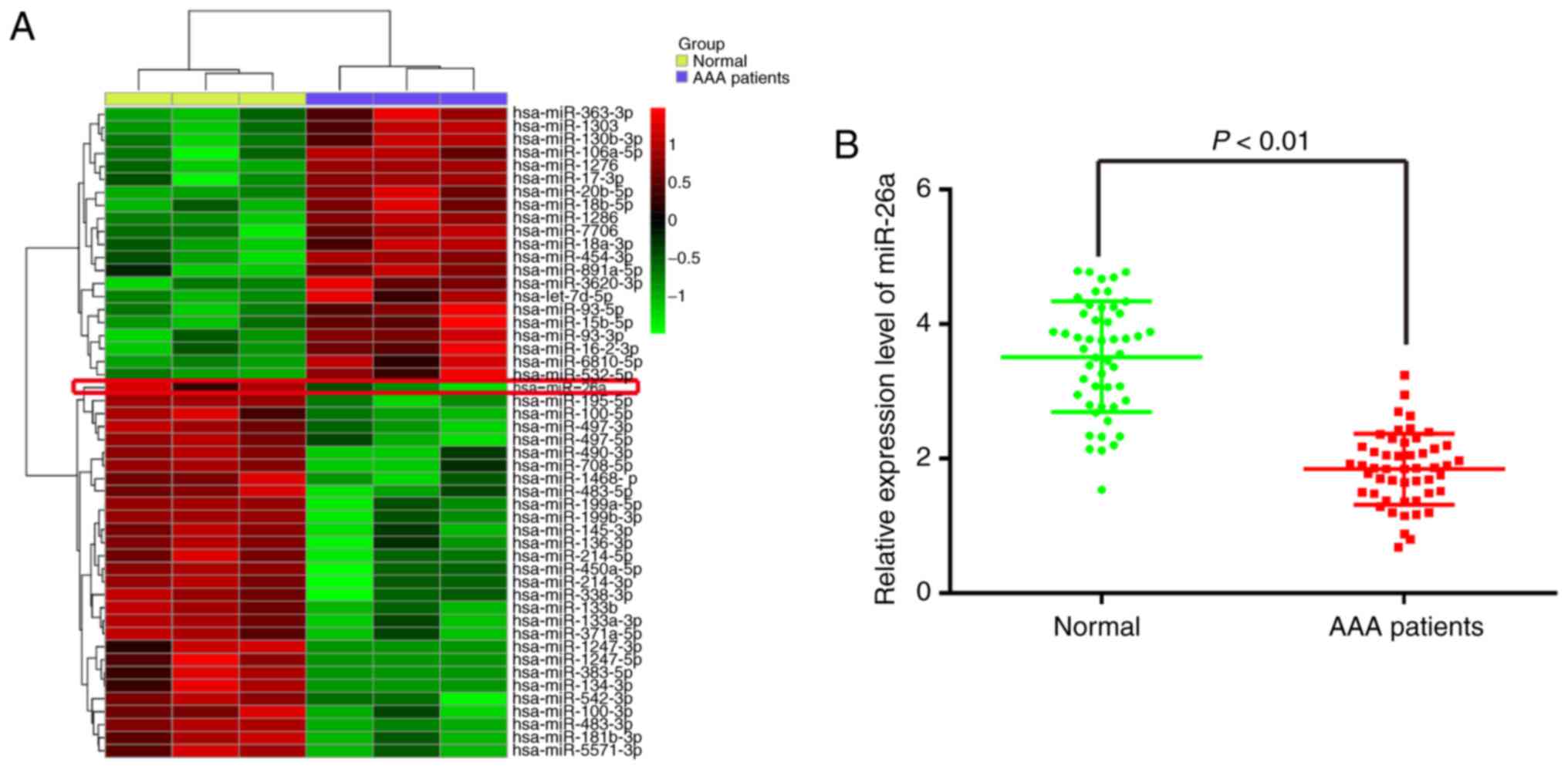
In order to identify miRNAs associated with AAA, a
microRNA array was performed to determine miRNA levels in
peripheral blood from patients with AAA. The data revealed that,
compared with the normal group, 21 miRNAs were upregulated and 29
miRNAs were downregulated in the AAA patient group (Fig. 1A). Among the aberrantly expressed
miRNAs, miR-26a (miR-26a-1-5p) was selected for further
investigation as its expression level was identified as the lowest
in the AAA patient group. Consistent with these results, two
previous studies reported that miR-26a was also found to be
downregulated in AAA, indicating that miR-26a may be involved in
the formation or progression of AAA (23,24). Additionally, previous studies have
reported that miR-26a prevented endothelial cell apoptosis in the
setting of atherosclerosis (25,26). However, whether miR-26a has a
protective effect against the apoptosis of VSMCs in AAA remains to
be elucidated. Therefore, the present study focused on miR-26a for
further investigation.
To validate the expression trend of miR-26a obtained
from the miRNA microarray assay, RT-qPCR analysis was performed to
detect miR-26a in the peripheral blood from 30 patients with AAA
and 30 donors without vascular disease (normal group). As shown in
Fig. 1B, the expression of
miR-26a was significantly downregulated in the peripheral blood
from the patients with AAA, compared with that in the normal group.
These data indicated that miR-26a may be involved in the
pathogenesis of AAA.
miR-26a is downregulated in an
H2O2-induced injury model of VSMCs
As is known, the H2O2-induced
VSMC injury model has been widely used to simulate pathological
conditions of AAA in vitro (12). Following treatment of the VSMCs
with different concentrations of H2O2 for 6
h, cell viability, caspase-3 activity, ROS production, apoptosis,
and the expression of apoptosis-associated proteins were evaluated.
The results showed that H2O2 dose-dependently
inhibited the cell viability of VSMCs, compared with that in the
control group (Fig. 2A).
Treatment with 200 µM H2O2
significantly reduced the cell viability by 50%, compared with that
in the control group. Therefore, 200 µM
H2O2 was selected as the appropriate
concentration in the subsequent experiments. Furthermore, western
blot analysis revealed that the level of cleaved-caspase-3 was
significantly increased in the H2O2-treated
group, compared with the control group, however, the levels of
total cleaved-caspase-3 were not affected (Fig. 2B and C). The percentage of
apoptotic cells in the H2O2-treated group was
significantly increased, compared with that in the control group
(Fig. 2D). These results
indicated that H2O2 treatment markedly
inhibited cell viability and induced cell apoptosis in the VSMCs.
The activity of caspase 3, determined using a commercial assay, was
found to be concomitantly upregulated by H2O2
treatment (Fig. 2E). As ROS and
the resulting oxidative stress in VSMCs is pivotal in the
pathophysiology of AAA, the present study investigated the effect
of H2O2 on the production of ROS using a
DCF-DA assay. Compared with the control group, the production of
intracellular ROS was markedly increased in the
H2O2-treated group (Fig. 2F). The above data indicated that
model establishment was successful. To further examine the role of
miR-26a in VSMC injury, the effect of H2O2 on
the level of miR-26a in VSMCs was determined. As shown in Fig. 2G, it was confirmed that miR-26a
was significantly downregulated in the
H2O2-treated VSMCs, and this effect was
dose-dependent, which was consistent with the results in the
clinical samples. These data provide support for the possible role
of miR-26a in the development of AAA.
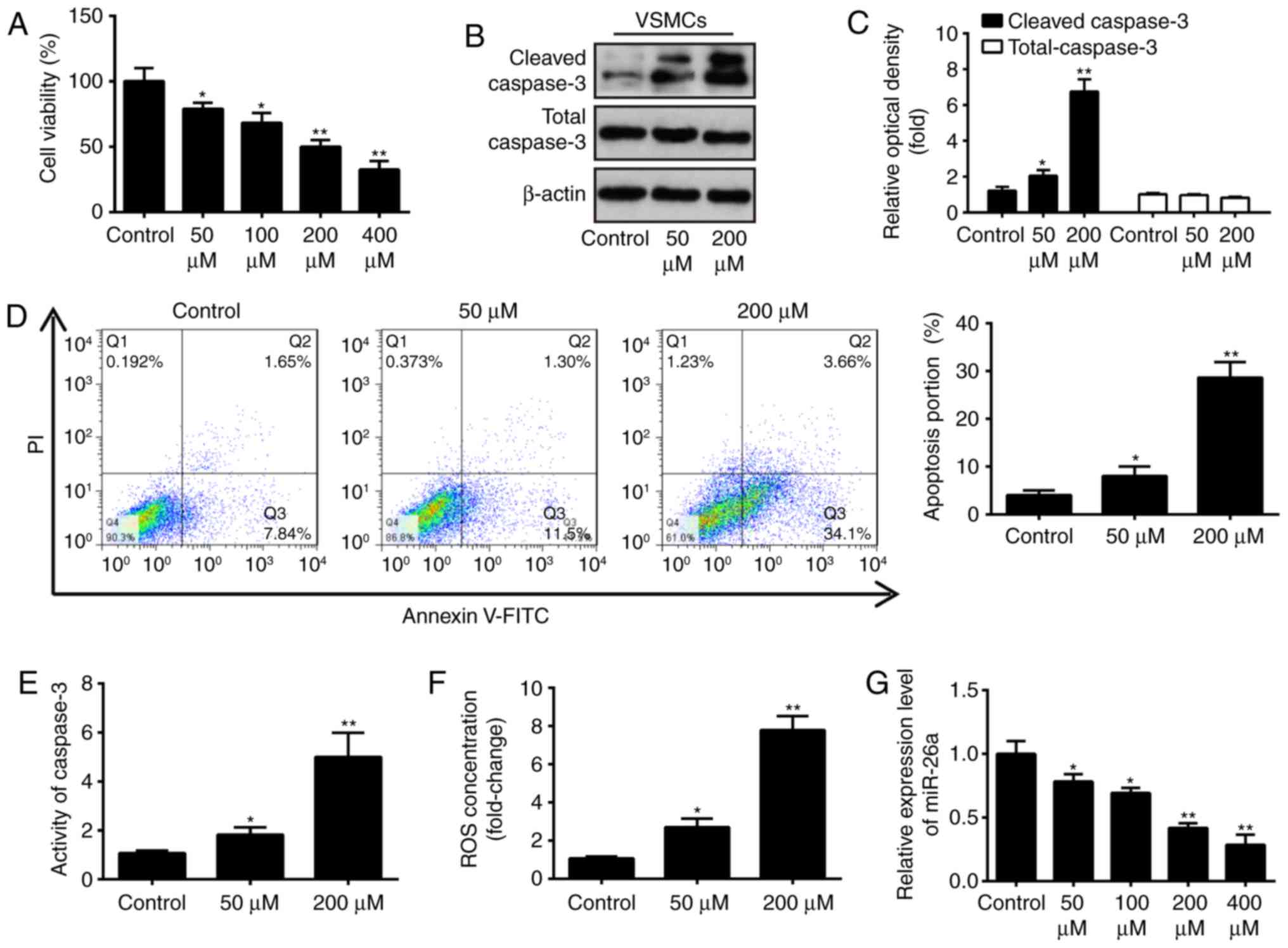 | Figure 2Effect of H2O2
on relative expression of miR-26a in cultured VSMCs. (A) VSMCs were
incubated with 50, 100, 200 or 400 µM
H2O2 for 6 h and cell viability was assessed
using a Cell Counting Kit-8 assay. (B) VSMCs were incubated with 50
and 200 µM H2O2 for 6 h, and the
protein expression levels of cleaved-caspase-3 and total caspase-3
were detected by western blot analysis and (C) statistically
analyzed. (D) VSMCs were incubated with 50 and 200 µM
H2O2 for 6 h, and apoptosis was detected by
flow cytometry. (E) VSMCs were incubated with 50 and 200 µM
H2O2 for 6 h, and the activity of caspase-3
was measured using a commercial kit. (F) VSMCs were incubated with
50 and 200 µM H2O2 for 6 h, and the
production of ROS was detected using 2′,7′-DCF diacetate. (G) VSMCs
were incubated with 50, 100, 200 or 400 µM
H2O2, and the expression of miR-26a was
determined by reverse transcription-quantitative polymerase chain
reaction analysis. Data are presented as the mean ± standard
deviation of three independent experiments. *P<0.05
and **P<0.01, vs. control group. VSMCs, vascular
smooth muscle cells; H2O2, hydrogen peroxide;
miR, microRNA; ROS, reactive oxygen species; PI, propidium
iodide. |
Overexpression of miR-26a attenuates
H2O2-induced VSMC injury
To further examine the role of miR-26a in
H2O2-induced VSMC injury, the miR-26a mimics
was added to the cultured VSMCs 2 h prior to
H2O2 treatment, and incubated for 6 h. The
RT-qPCR analysis showed the successful enhancement of cellular
miR-26a by the miR-26a mimics, compared with that in the negative
control (Fig. 3A). Using a CCK-8
assay, it was demonstrated that the overexpression of miR-26a
restored VSMC proliferation activity following
H2O2 treatment (Fig. 3B). In addition, it was found that
upregulation of miR-26a reversed the promoting effect of
H2O2 on caspase 3 activity and cell apoptosis
in the VSMCs, as determined through a caspase-3 activity assay and
flow cytometry (Fig. 3C and D).
It was also found that the overexpression of miR-26a inhibited the
effect of H2O2 treatment of the production of
ROS (Fig. 3E). Taken together,
these results indicated that the overexpression of miR-26a
protected the VSMCs against H2O2-induced
injury, suggesting that miR-26a may be a key protective factor in
AAA.
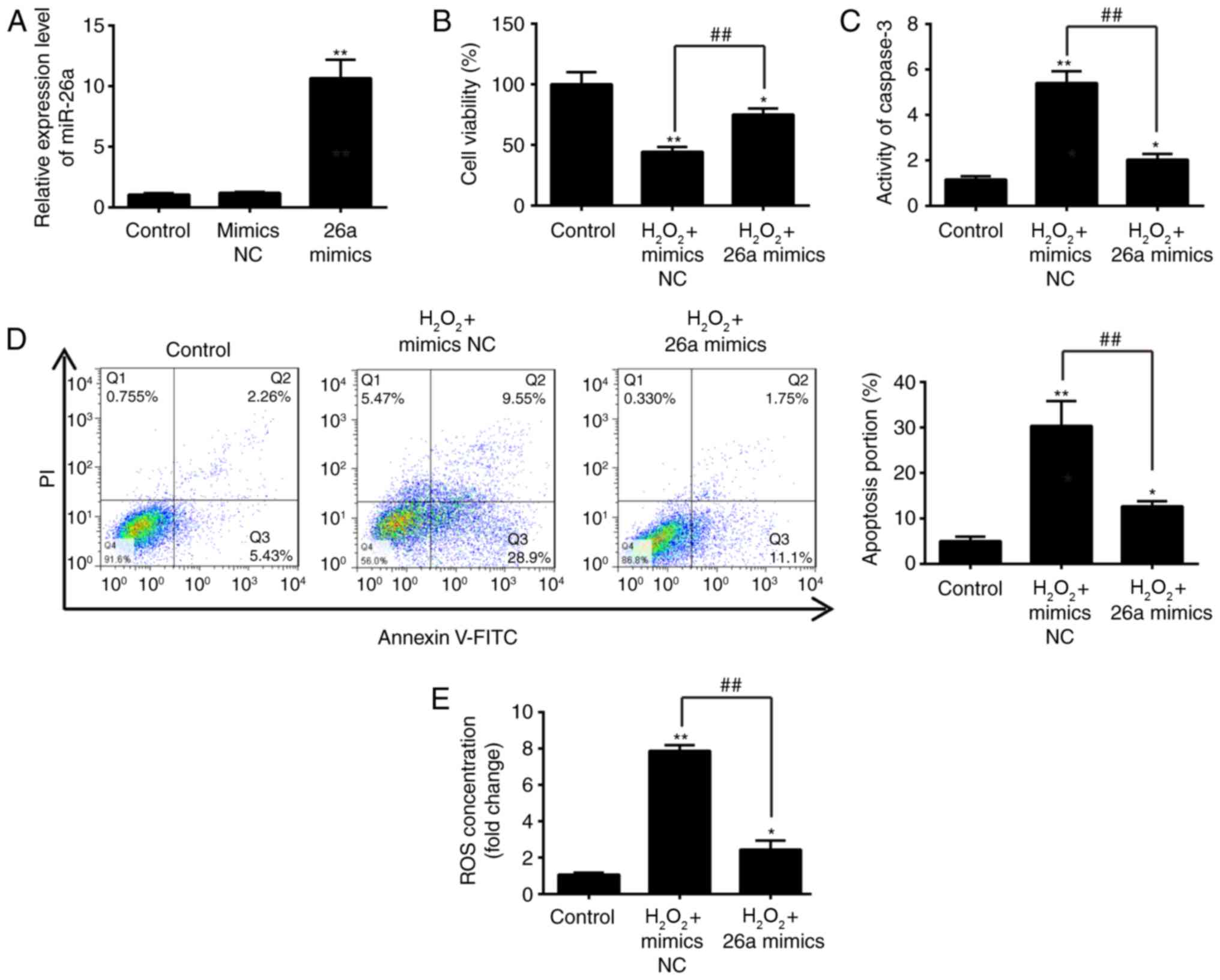 | Figure 3Overexpression of miR-26a inhibits
H2O2-induced VSMC injury. VSMCs were
transfected with miR-26a mimics for 2 h, followed by treatment with
200 µM H2O2 for 6 h, and harvesting of
cells for subsequent experiments. (A) Expression of miR-26a was
determined by reverse transcription-quantitative polymerase chain
reaction analysis. (B) Cell viability was assessed using a Cell
Counting Kit-8 assay. (C) Activity of caspase-3 was measured using
a commercial kit. (D) Apoptosis was detected by flow cytometry. (E)
Production of ROS was detected using 2′,7′-DCF diacetate. Data are
presented as the mean ± standard deviation of three independent
experiments. *P<0.05 and **P<0.01, vs.
control group. ##P<0.01, vs.
H2O2 + mimics NC group. VSMCs, vascular
smooth muscle cells; H2O2, hydrogen peroxide;
miR, microRNA; ROS, reactive oxygen species; PI, propidium iodide;
NC, negative control. |
PTEN is a direct target of miR-26a
To further elucidate the underlying molecular
mechanisms involved in the miR-26a-mediated protective role in the
VSMC injury model, two publicly available databases TargetScan 7.0
(targetscan.org/) and miRanda (microrna.org/) were used to search for the potential
downstream targets of miR-26a. The data in these two public
databases showed that PTEN, an important regulator of the AKT/mTOR
pathway, was potentially a downstream target of miR-26a. As shown
in Fig. 4A and B, the
complementary sequence of miR-26a was found in the 3′-UTR of PTEN
mRNA. Initially, it was found that there are three binding sites of
miR-26a in the 3′-UTR of PTEN mRNA, however, only this site is
valid, the effects of the other two binding sites were weak (data
not shown), suggesting that miR-26a exerts its function through
this binding site. To experimentally validate whether PTEN was a
direct target of miR-26a, a luciferase reporter assay was
performed. The results revealed that the enforced miR-26a
expression significantly attenuated the luciferase activity of the
PTEN-3′UTR wt reporter plasmid, whereas the knockdown of miR-26a
increased luciferase activity. Similarly, the cells co-transfected
with miR-26a mimics, miR-26a inhibitor, and the PTEN-3′UTR mut
reporter plasmid, showed no significant change in luciferase
activity (Fig. 4C). To confirm
that PTEN was regulated by miR-26a, western blot analysis was used.
As shown in Fig. 4D, the
expression of PTEN was markedly downregulated by the overexpression
of miR-26a, but was markedly upregulated by the suppression of
miR-26a, compared with the respective controls. In addition, the
results showed that the mRNA level of PTEN was markedly upregulated
in the H2O2-treated VSMCs in a dose-dependent
manner (Fig. 4E) and the
overexpression of miR-26a attenuated the promoting effect of
H2O2 on the expression of PTEN (Fig. 4F). These results indicated that
miR-26a exerted an inhibitory effect on the expression of PTEN in
the VSMC injury model.
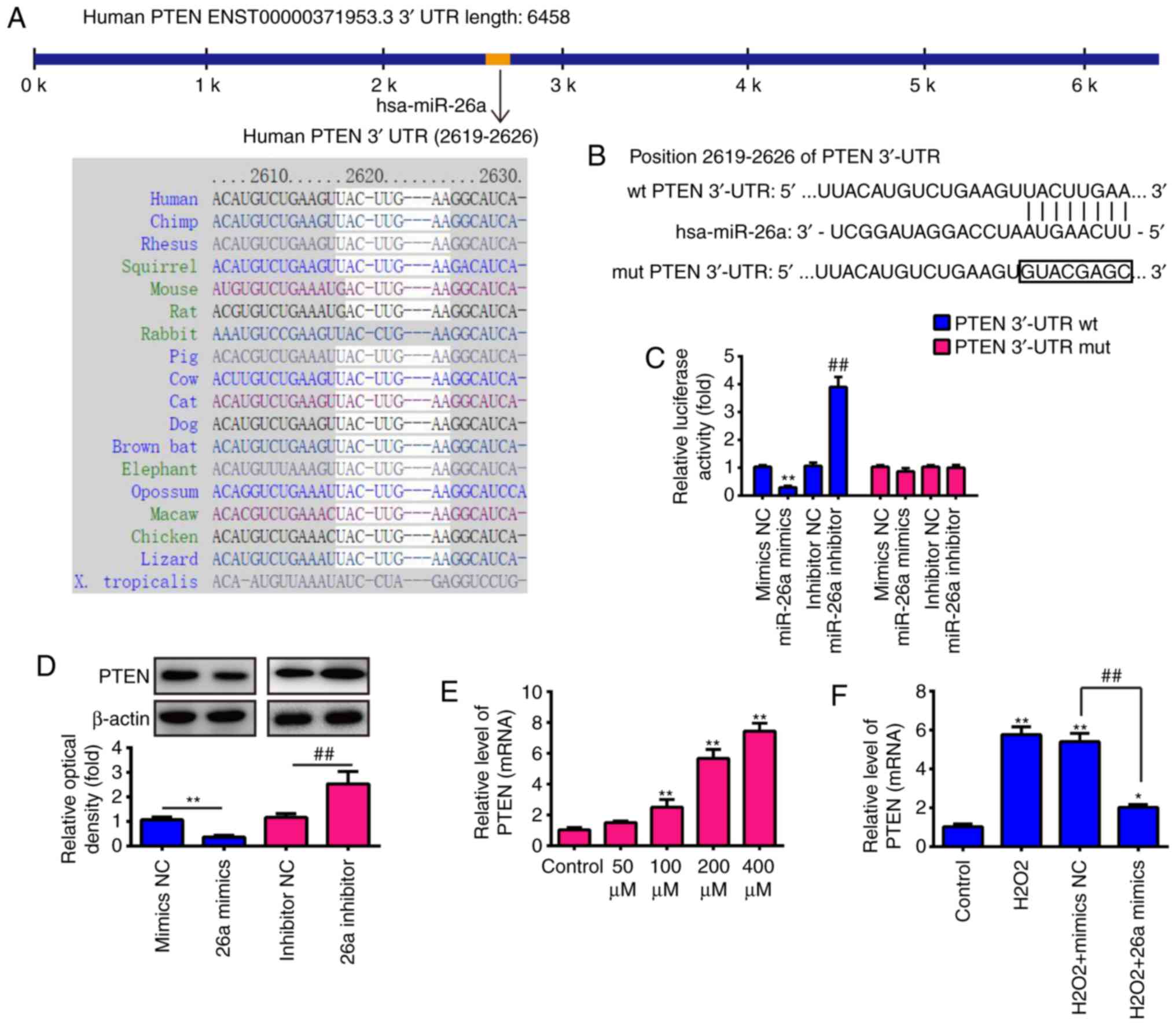 | Figure 4PTEN is a direct target of miR-26a.
(A) Putative binding site of miR-26a and PTEN with (B) mut and wt
3′ UTRs. (C) Luciferase assay of 293 cells co-transfected with
firefly luciferase constructs containing the PTEN wt or mut 3′-UTRs
and miR-26a mimics, mimics NC, miR-26a inhibitor or inhibitor NC,
as indicated (n=3). Data are presented as the mean ± standard
deviation of three independent experiments. **P<0.01,
vs. mimics NC, ##P<0.01, vs. inhibitor NC. (D)
Protein expression of PTEN following transfection with miR-26a
mimic or miR-26a inhibitor was measured by western blot analysis.
(E) VSMCs were incubated with 50, 100, 200 or 400 µM
H2O2 for 6 h, and the mRNA expression of PTEN
was determined by RT-qPCR analysis. (F) VSMCs were transfected with
miR-26a mimics for 2 h, followed by treatment with 200 µM
H2O2 for 6 h, and the mRNA expression of PTEN
was determined by RT-qPCR analysis. Data are presented as the mean
± standard deviation of three independent experiments.
*P<0.05 and **P< 0.01, vs. control
group; ##P<0.01, vs. H2O2 +
mimics NC group. VSMCs, vascular smooth muscle cells;
H2O2, hydrogen peroxide; miR, microRNA; PTEN,
phosphatase and tensin homolog; wt, wild-type; mut, mutant; reverse
transcription-quantitative polymerase chain reaction analysis; NC,
negative control. |
miR-26a protects VSMCs from
H2O2-induced apoptosis by targeting PTEN
As mentioned above, PTEN was a direct target of
miR-26a in VSMCs, therefore, the present study further investigated
whether miR-26a protected VSMCs from
H2O2-induced apoptosis by downregulating
PTEN. The PTEN expression vector, pcDNA-PTEN and miR-26a mimics
were co-transfected into VSMCs 2 h prior to
H2O2 treatment, and incubated for 6 h. It was
found that the expression level of PTEN was significantly
upregulated in the VSMCs following H2O2
treatment, compared with that in the control group, whereas the
H2O2-induced upregulation was decreased by
the overexpression of miR-26a. In addition, the expression level of
PTEN was restored when pcDNA-PTEN was transfected into the
H2O2-treated cells (Fig. 5A). Functionally, these results
revealed that the overexpression of miR-26a attenuated the
reduction in viability of the H2O2-treated
VSMCs, whereas the overexpression of PTEN partly abrogated the
promoting effects of the overexpression of miR-26a on the viability
of H2O2-treated VSMCs (Fig. 5B). It was also found that the
overexpression of miR-26a recovered the injury induced by
H2O2, whereas the overexpression of PTEN
eliminated the protective effect of the overexpression of miR-26a
on the activity of caspase 3, cell apoptosis and production of ROS
(Fig. 5C–E). These data suggested
that miR-26a protected the VSMCs from
H2O2-induced injury by targeting PTEN.
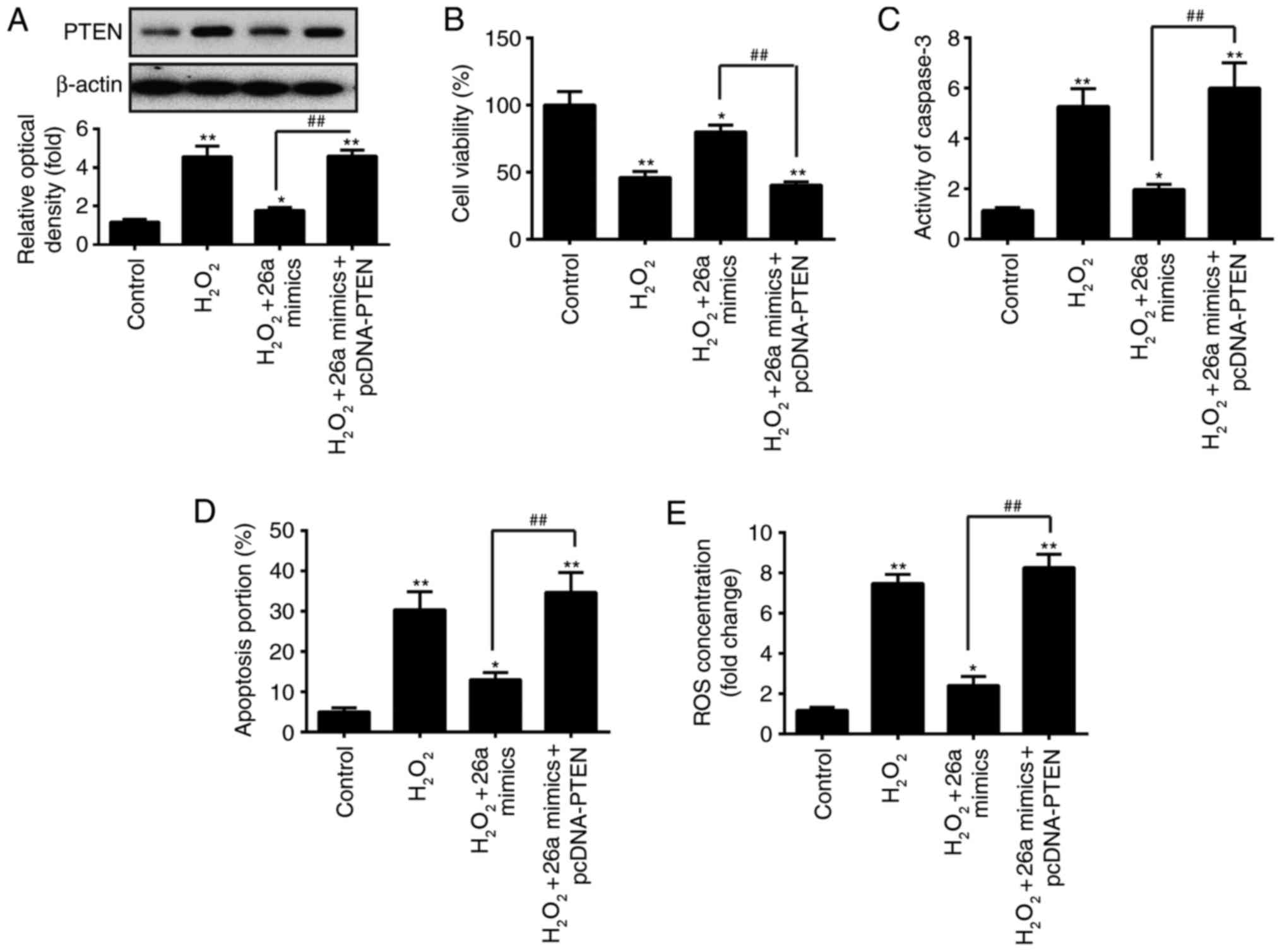 | Figure 5PTEN is involved in the
miR-26a-mediated protective effect on
H2O2-induced VSMC injury. VSMCs were
co-transfected with miR-26a mimics and pcDNA-PTEN for 2 h, followed
by treatment with 200 µM H2O2 for 6 h,
and the cells were harvested for subsequent experiments. (A)
Protein expression of PTEN was measured by western blot analysis.
(B) Cell viability was assessed using a Cell Counting Kit-8 assay.
(C) Activity of caspase-3 was measured using a commercial kit. (D)
Apoptosis was detected by flow cytometry. (E) Production of ROS was
detected using 2′,7′-DCF diacetate. Data are presented as the mean
± standard deviation of three independent experiments.
*P<0.05 and **P<0.01, vs. control group
##P<0.01, vs. H2O2 + miR-26a
mimics group. VSMCs, vascular smooth muscle cells;
H2O2, hydrogen peroxide; miR, microRNA; PTEN,
phosphatase and tensin homolog; ROS, reactive oxygen species. |
miR-26a prevents H2O-induced
VSMC apoptosis through the Akt/mTOR pathway
Subsequently, the molecular pathways responsible for
the protective effects of miR-26a on
H2O2-induced VSMC injury were examined. As
PTEN acts as a major negative regulator of the Akt/mTOR signaling
pathway, which is directly related to the apoptosis of cells
(27,28), further experiments were designed
to investigate the effects of miR-26a on the activation of Akt/mTOR
in H2O2-treated VSMCs. The results of western
blot analysis demonstrated that the protein expression of p-Akt was
reduced in the H2O2-treated VSMCs, compared
with that in the control group, as was the expression of p-mTOR.
The overexpression of miR-26a reversed the inhibitory effect of
H2O2 on the levels of p-Akt and p-mTOR
(Fig. 6A and B). These data
suggested that miR-26a prevented H2O2-induced
VSMC apoptosis through re-activation of the Akt/mTOR pathway.
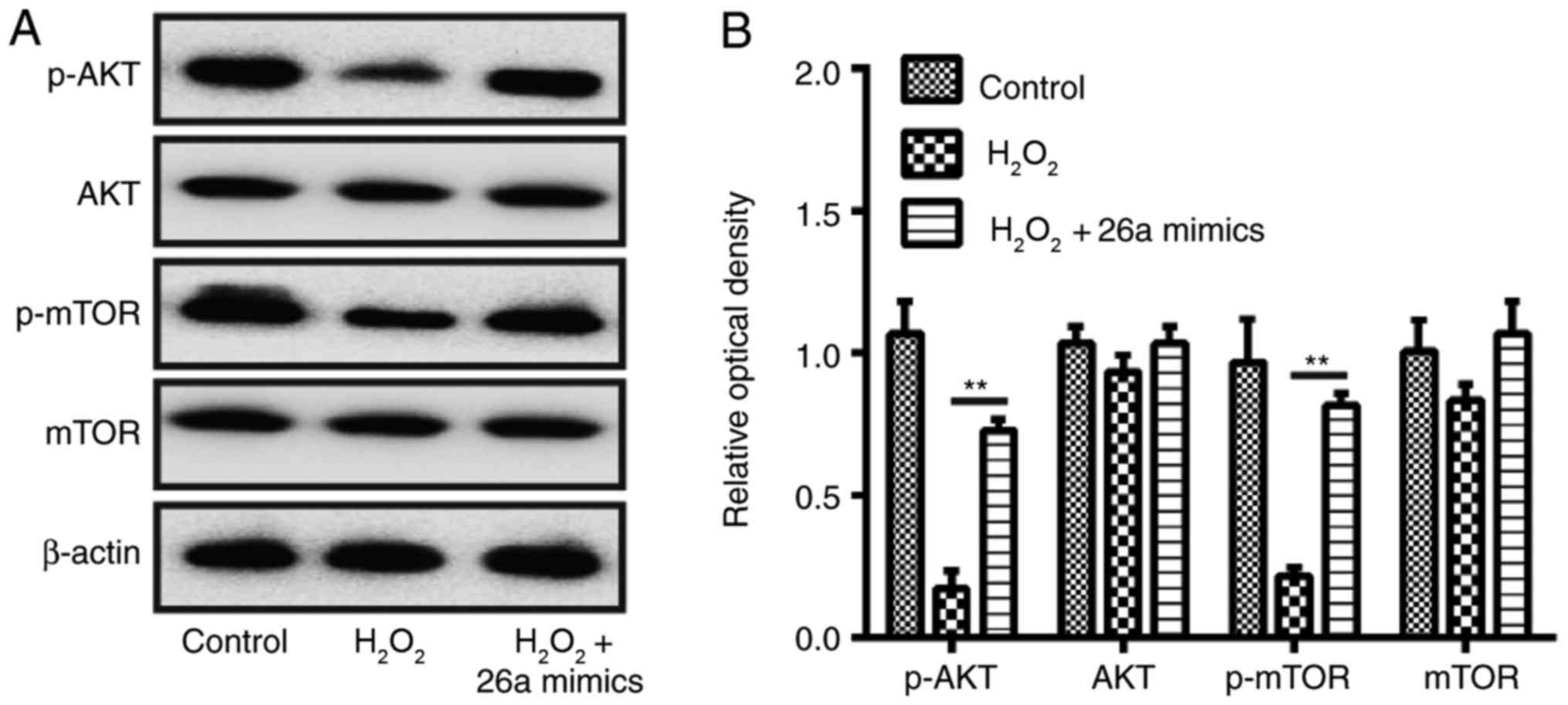 | Figure 6Overexpression of miR-26a restores
activation of the AKT/mTOR pathway in
H2O2-induced VSMCs. VSMCs were transfected
with miR-26a mimics for 2 h, followed by treatment with 200
µM H2O2 for 6 h, and the cells were
harvested for western blot analysis. (A) Protein expression levels
of AKT, p-AKT, p-mTOR and mTOR were detected by western blot
analysis. (B) Protein bands were semi-quantitatively analyzed using
ImageJ software, normalized to β-actin density. Data are presented
as the mean ± standard deviation of three independent experiments.
**P<0.01, vs. H2O2 group.
VSMCs, vascular smooth muscle cells; H2O2,
hydrogen peroxide; miR, microRNA; mTOR, mammalian target of
rapamycin; p-, phosphorylated. |
Akt/mTOR signaling pathway is required
for mediating the effects of miR-26a on the inhibition of
H2O2-induced VSMC injury
To investigate whether the Akt/mTOR signaling
pathway is critically involved in the function of miR-26a in the
inhibition of H2O2-induced VSMC injury, the
activity of the Akt/mTOR signaling pathway was inhibited with
API-2, which is a potent inhibitor of all three AKT isoforms,
exhibiting a specific activity on mutant AKT1 (29,30). The results showed no significant
difference in the protein level of PTEN between the miR-26a mimics
group and miR-26a mimics + API-2 group, however, the protein levels
of p-AKT were decreased by treatment with API-2 (Fig. 7A). The overexpression of miR-26a
attenuated the reduction in viability of the
H2O2-treated VSMCs, whereas API-2 partly
abrogated the promoting effects of the miR-26a mimics on the cell
viability (Fig. 7B).
Subsequently, it was demonstrated that miR-26a mimics inhibited the
activity of caspase 3, apoptosis and ROS production, and its
inhibitory effect was reversed following treatment with API-2
(Fig. 7C–E). These data suggested
that the Akt/mTOR signaling pathway was required for mediating the
protective effects of miR-26a on the inhibition of
H2O2-induced VSMC injury.
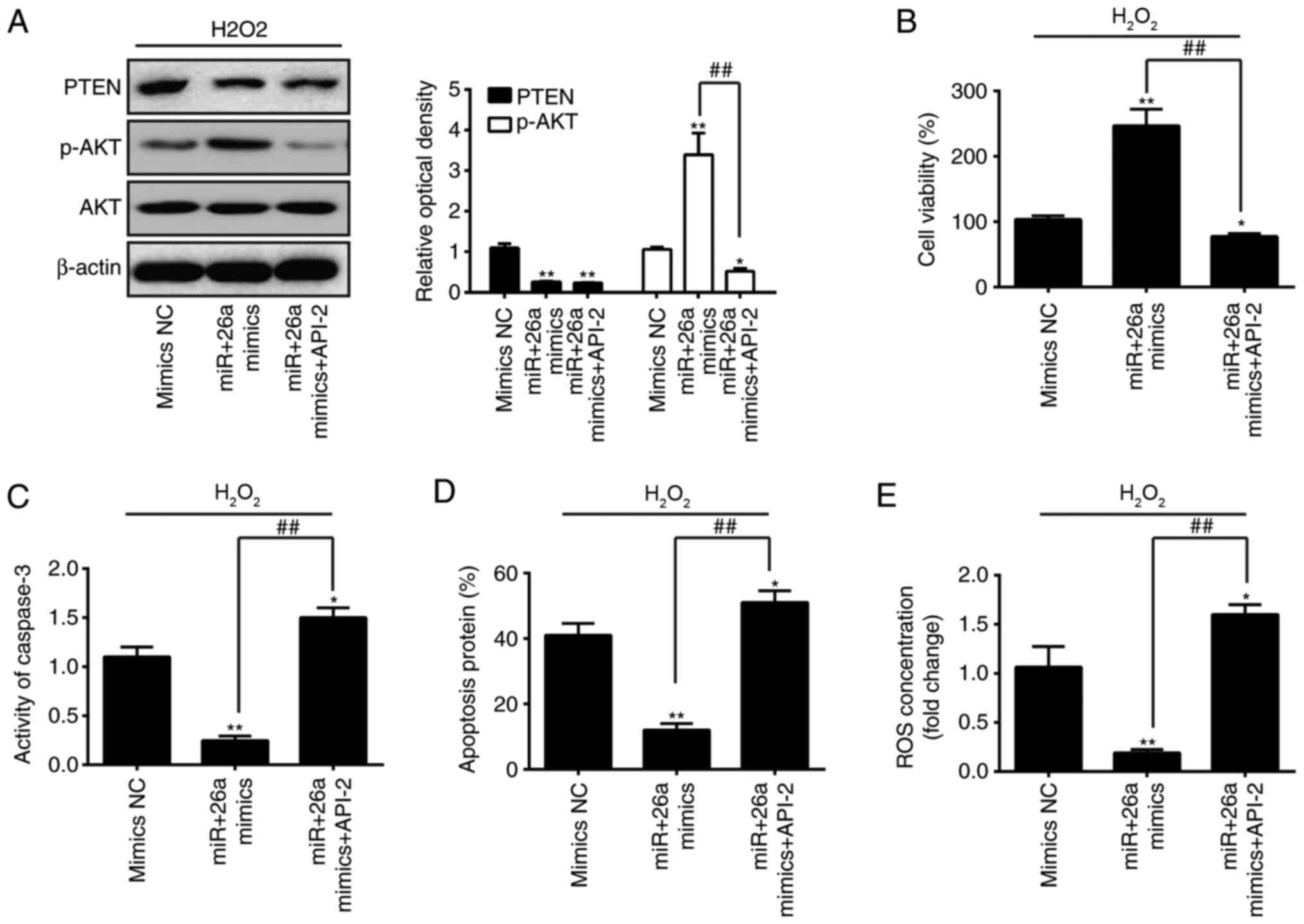 | Figure 7Akt/mTOR signaling pathway is
required for mediating the effects of miR-26a on the inhibition of
H2O2-induced VSMC injury. VSMCs were
transfected with miR-26a mimics for 2 h, followed by treatment with
200 µM H2O2 and API-2 for 6 h, and the
cells were harvested for analysis. (A) Protein expression levels of
p-AKT and PTEN were detected by western blot analysis. (B) Cell
viability was assessed using a Cell Counting Kit-8 assay. (C)
Activity of caspase-3 was measured using a commercial kit. (D)
Apoptosis was detected by flow cytometry. (E) Production of ROS was
detected using 2′,7′-DCF diacetate. Data are presented as the mean
± standard deviation of three independent experiments.
*P<0.05 and **P<0.01, vs. mimics group;
##P<0.01, vs. miR-26a mimics group. VSMCs, vascular
smooth muscle cells; H2O2, hydrogen peroxide;
miR, microRNA; PTEN, phosphatase and tensin homolog; mTOR,
mammalian target of rapamycin; p-, phosphorylated; ROS, reactive
oxygen species; NC, negative control. |
Discussion
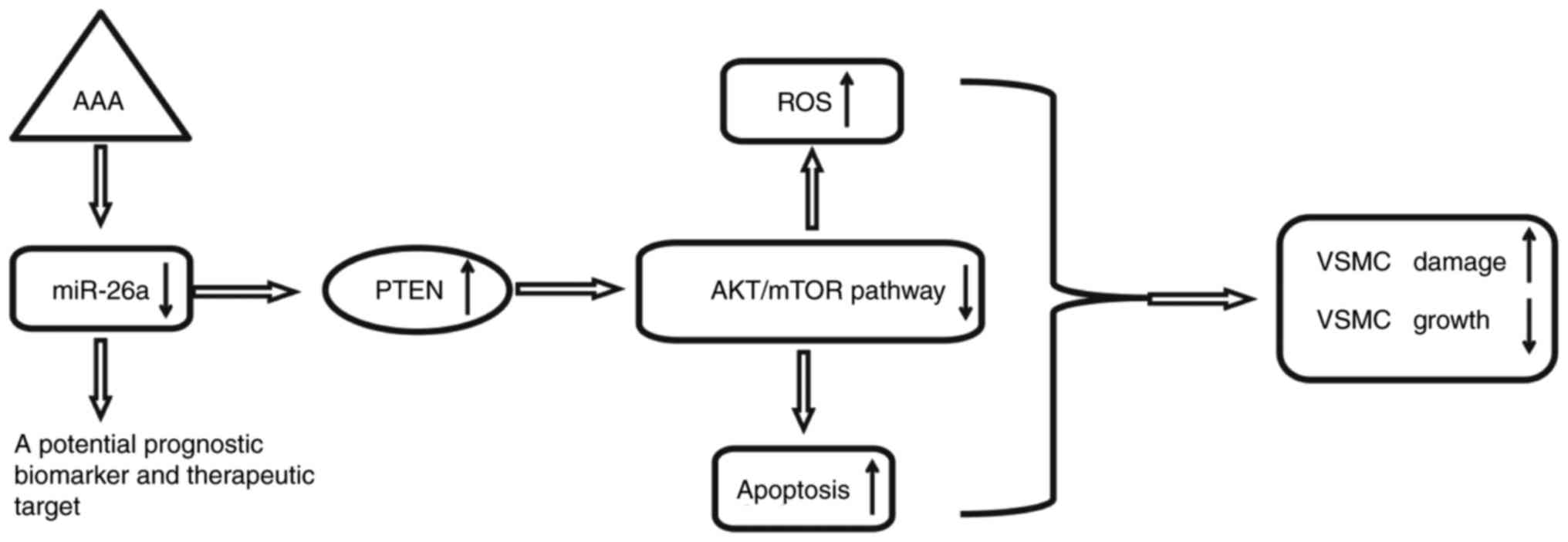
In the present study, it was shown that miR-26a was
significantly downregulated in the peripheral blood of patients
with AAA. Using an H2O2-induced VSMC injury
model, it was found that the overexpression of miR-26a protected
VSMCs against H2O2-induced ROS accumulation
and apoptosis. Notably, the data indicated that the overexpression
of miR-26a exerted protective effects by reactivating the
PTEN/AKT/mTOR signaling pathway in the
H2O2-induced VSMCs (Fig. 8). These findings suggested that
miR-26a may be an effective therapeutic target for the treatment of
AAA in clinical practice.
Several studies have demonstrated that miRNAs are
aberrantly expressed in AAA aortic tissues and are involved in
several pathophysiological processes, including inflammation,
oxidation and apoptosis (31,32). For example, Maegdefessel et
al found that miR-21 is a key modulator of VSMC injury in an
AAA murine model, and lentiviral overexpression of miR-21 inhibited
the development of AAA and protected from aneurysm expansion
(33). Nakao et al showed
that miR-33 was increased in human AAA tissues and the inhibition
of miR-33 suppressed the formation of AAA via c-Jun n-terminal
kinase inactivation (34). Kim
et al found that miR-205 was induced in the abdominal aortic
endothelium in an AAA mouse model, and silencing miR-205
effectively prevented the development of AAA through decreased
aortic MMP activity and inflammation (35). These previous findings suggest
that miRNAs may be an attractive therapeutic target for AAA.
Therefore, the present study performed an miRNA microarray
profiling assay on the peripheral blood of patients with AAA to
screen for the miRNAs involved in AAA, and found large numbers of
miRNAs were significantly deregulated; in particular, miR-26a was
one of the miRNAs exhibiting the most marked down regulation. These
results suggested a potential clinical relevance of miR-26a in
human AAA.
miR-26a has been reported to act as a novel
regulator of VSMC function, with marked effects on cell
proliferation and apoptosis (22,36). Another study reported the
regulatory effects of miR-26a on cellular oxidative stress
(37). Therefore, it was
hypothesized that miR-26a may affect the progression of AAA through
the regulation of cell apoptosis and oxidative stress, which has
not been investigated previously. In the present study, using an
H2O2-induced VSMC injury model, it was
observed that H2O2 treatment of VSMCs led to
significant decrease in cell viability and significant increases in
apoptosis and ROS production in a dose-dependent manner.
Accordingly, treatment of VSMCs with H2O2
significantly decreased the expression of miR-26a in a
dose-dependent manner, which provided support for the possible role
of miR-26a in AAA. The overexpression of miR-26a exerted a
protective effect against H2O2-induced
injury, consolidating the functional roles of miR-26a in AAA.
To elucidate the potential mechanism of the
protection role of miR-26a against
H2O2-induced injury, bioinformatics analysis
was performed to predicate the putative targets of miR-26a, and
PTEN was identified as the potential target of miR-26a. PTEN, a
negative regulator of the AKT/mTOR signaling pathway, has been
shown to regulate cell proliferation, survival and growth of VSMCs,
in addition to its role in tumor cells (38,39). Notably, a previous study showed
that miR-26a suppressed the proliferation and metastasis of
different gastric cancer cells via targeting PTEN (40). Yu et al found that miR-26a
inhibited proliferation and migration by targeting the PTEN gene in
HaCaT keratinocytes (41).
However, whether PTEN is a functional target of miR-26a in VSMCs
remained to be elucidated. In the present study, PTEN was validated
as a target gene of miR-26a in VSMCs. In addition, the protective
effects of miR-26a mimics on H2O2-induced
VSMC injury were abrogated by the overexpression of PTEN,
suggesting that miR-26a protected against
H2O2-induced VSMC injury via suppressing the
expression of PTEN. However, the possible molecular mechanism
requires further investigation to be fully understood.
It is well known that the PTEN gene negatively
regulates the PI3K/AKT/mTOR pathway. Activation of the
PI3K/Akt/mTOR signaling pathway is implicated in VSMC function
(phenotype), including cell migration (42), proliferation (43), calcification (44) and apoptosis (45). In a porcine pancreatic
elastase-induced AAA model, activation of the PI3K/Akt has also
been demonstrated to mediate a protective effect against AAA
(46). Previous studies have
reported that miRNAs are important in the regulation of VSMC
function through activating the PI3K/Akt signaling pathway. For
example, Jiang et al showed that the miR-21-mediated
activation of the PI3K/Akt signaling pathway may contribute to VSMC
proliferation (47). Of note,
Jiang et al demonstrated that miR-26a was involved in the
Toll-like receptor 9 mediated growth and migration of lung cancer
cells through the PI3K-Akt signaling pathway (48). In order to elucidate the
mechanisms underlying the miR-26a-associated protection from VSMC
injury, the effects of aberrantly expressed miR-26a on key kinases
in the Akt/mTOR signaling pathway were examined in the present
study. The data showed that the Akt/mTOR signaling pathway was
suppressed in H2O2-treated VSMCs, and the
overexpression of miR-26a restored the activity of this signaling
pathway, suggesting that the protective effect of miR-26a on
H2O2-induced VSMC injury may mediated by the
Akt/mTOR signaling pathway.
In conclusion, the present study revealed that the
expression of miR-26a was downregulated in the peripheral blood of
patients with AAA, and the overexpression of miR-26a inhibited
H2O2-induced VSMC injury by reactivating the
PTEN/AKT/mTOR signaling pathway. These findings support the
hypothesis that enhancing the expression of miR-26a may be a
desirable therapeutic approach for the treatment of AAA, however
this requires further experiments in animals prior to clinical
application.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors’ contributions
JP and XH performed the experiments and wrote the
manuscript. JP, XH, PL and LZ analyzed the data. LZ designed the
study and contributed experimental materials. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Ethics Committee of the First Hospital of Hebei Medical University.
All experiments were performed in accordance with the ethical
guidelines of the Institutional Animal Care and Use Committee of
the First Hospital of Hebei Medical University. Informed consent
was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Davis FM, Rateri DL and Daugherty A:
Mechanisms of aortic aneurysm formation: Translating preclinical
studies into clinical therapies. Heart. 100:1498–1505. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nordon IM, Hinchliffe RJ, Loftus IM and
Thompson MM: Pathophysiology and epidemiology of abdominal aortic
aneurysms. Nat Rev Cardiol. 8:92–102. 2011. View Article : Google Scholar
|
|
3
|
Golledge J and Norman PE: Current status
of medical management for abdominal aortic aneurysm.
Atherosclerosis. 217:57–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Heart disease and stroke statistics-2014 update: A report from the
American Heart Association. Circulation. 129:e28–e292. 2014.
View Article : Google Scholar
|
|
5
|
Moxon JV, Parr A, Emeto TI, Walker P,
Norman PE and Golledge J: Diagnosis and monitoring of abdominal
aortic aneurysm: Current status and future prospects. Curr Probl
Cardiol. 35:512–548. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin YC, Huang YC, Chen SC, Liaw CC, Kuo
SC, Huang LJ and Gean PW: Neuroprotective effects of ugonin K on
hydrogen peroxide-induced cell death in human neuroblastoma SH-SY5Y
cells. Neurochem Res. 34:923–930. 2009. View Article : Google Scholar
|
|
7
|
Schramm A, Matusik P, Osmenda G and Guzik
TJ: Targeting NADPH oxidases in vascular pharmacology. Vascul
Pharmacol. 56:216–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Morimoto K, Hasegawa T, Tanaka A, Wulan B,
Yu J, Morimoto N, Okita Y and Okada K: Free-radical scavenger
edaravone inhibits both formation and development of abdominal
aortic aneurysm in rats. J Vasc Surg. 55:1749–1758. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yajima N, Masuda M, Miyazaki M, Nakajima
N, Chien S and Shyy JY: Oxidative stress is involved in the
development of experimental abdominal aortic aneurysm: A study of
the transcription profile with complementary DNA microarray. J Vasc
Surg. 36:379–385. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shang T, Liu Z, Zhou M, Zarins CK, Xu C
and Liu CJ: Inhibition of experimental abdominal aortic aneurysm in
a rat model by way of tanshinone IIA. J Surg Res. 178:1029–1037.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song Q, Gou WL and Zhang R: FAM3A protects
HT22 cells against hydrogen peroxide-induced oxidative stress
through activation of PI3K/AKT but not MEK/ERK pathway. Cell
Physiol Biochem. 37:1431–1441. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin N, Hatton ND, Harrington MA, Xia X,
Larsen SH and Rhoades RA: H(2)O(2)-induced egr-1, fra-1, and c-jun
gene expression is mediated by tyrosine kinase in aortic smooth
muscle cells. Free Radic Biol Med. 29:736–746. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li PF, Dietz R and von Harsdorf R:
Differential effect of hydrogen peroxide and superoxide anion on
apoptosis and proliferation of vascular smooth muscle cells.
Circulation. 96:3602–3609. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu XR, Cao L, Li T, Chen LL, Yu YY, Huang
WJ, Liu L and Tan XQ: Propofol attenuates
H2O2-induced oxidative stress and apoptosis
via the mitochondria- and ER-medicated pathways in neonatal rat
cardiomyocytes. Apoptosis. 22:639–646. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li T and Cho WC: MicroRNAs: Mechanisms,
functions and progress. Genomics Proteomics Bioinformatics.
10:237–238. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu J, Wang J, Li X, Liu X, Yu X and Tian
Y: MicroRNA-145 mediates the formation of angiotensin II-induced
murine abdominal aortic aneurysm. Heart Lung Circ. 26:619–626.
2017. View Article : Google Scholar
|
|
17
|
Maegdefessel L, Azuma J, Toh R, Merk DR,
Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ,
et al: Inhibition of microRNA-29b reduces murine abdominal aortic
aneurysm development. J Clin Invest. 122:497–506. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iaconetti C, De Rosa S, Polimeni A,
Sorrentino S, Gareri C, Carino A, Sabatino J, Colangelo M, Curcio A
and Indolfi C: Down-regulation of miR-23b induces phenotypic
switching of vascular smooth muscle cells in vitro and in vivo.
Cardiovasc Res. 107:522–533. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lai Z, Lin P, Weng X, Su J, Chen Y, He Y,
Wu G, Wang J, Yu Y and Zhang L: MicroRNA-574-5p promotes cell
growth of vascular smooth muscle cells in the progression of
coronary artery disease. Biomed Pharmacother. 97:162–167. 2018.
View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Yang C, Zhao L, Yuan W and Wen J:
Cordycepin induces apoptotic cell death and inhibits cell migration
in renal cell carcinoma via regulation of microRNA-21 and PTEN
phosphatase. Biomed Res. 38:313–320. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang Y, Vater C, Jacobi A, Liebers C, Zou
X and Stiehler M: Salidroside exerts angiogenic and cytoprotective
effects on human bone marrow-derived endothelial progenitor cells
via Akt/mTOR/p70S6K and MAPK signalling pathways. Br J Pharmacol.
171:2440–2456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leeper NJ, Raiesdana A, Kojima Y, Chun HJ,
Azuma J, Maegdefessel L, Kundu RK, Quertermous T, Tsao PS and Spin
JM: MicroRNA-26a is a novel regulator of vascular smooth muscle
cell function. J Cell Physiol. 226:1035–1043. 2011. View Article : Google Scholar :
|
|
24
|
Adam M, Raaz U, Spin JM and Tsao PS:
MicroRNAs in abdominal aortic aneurysm. Curr Vasc Pharmacol.
13:280–290. 2015. View Article : Google Scholar
|
|
25
|
Zhang Y, Qin W, Zhang L, Wu X, Du N, Hu Y,
Li X, Shen N, Xiao D, Zhang H, et al: MicroRNA-26a prevents
endothelial cell apoptosis by directly targeting TRPC6 in the
setting of atherosclerosis. Sci Rep. 5:94012015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen C, Cheng G, Yang X, Li C, Shi R and
Zhao N: Tanshinol suppresses endothelial cells apoptosis in mice
with atherosclerosis via lncRNA TUG1 up-regulating the expression
of miR-26a. Am J Transl Res. 8:2981–2991. 2016.PubMed/NCBI
|
|
27
|
Yang P, Peairs JJ, Tano R and Jaffe GJ:
Oxidant-mediated Akt activation in human RPE cells. Invest
Ophthalmol Vis Sci. 47:4598–4606. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Byeon SH, Lee SC, Choi SH, Lee HK, Lee JH,
Chu YK and Kwon OW: Vascular endothelial growth factor as an
autocrine survival factor for retinal pigment epithelial cells
under oxidative stress via the VEGF-R2/PI3K/Akt. Invest Ophthalmol
Vis Sci. 51:1190–1197. 2010. View Article : Google Scholar
|
|
29
|
Saura C, Roda D, Roselló S, Oliveira M,
Macarulla T, Pérez-Fidalgo JA, Morales-Barrera R, Sanchis-García
JM, Musib L, Budha N, et al: A first-in-human phase I study of the
ATP-competitive AKT inhibitor ipatasertib demonstrates robust and
safe targeting of AKT in patients with solid tumors. Cancer Discov.
7:102–113. 2017. View Article : Google Scholar :
|
|
30
|
Lin J, Sampath D, Nannini MA, Lee BB,
Degtyarev M, Oeh J, Savage H, Guan Z, Hong R, Kassees R, et al:
Targeting activated Akt with GDC-0068, a novel selective Akt
inhibitor that is efficacious in multiple tumor models. Clin Cancer
Res. 19:1760–1772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu G, Huang Y, Lu X, Lu M, Huang X, Li W
and Jiang M: Identification and characteristics of microRNAs with
altered expression patterns in a rat model of abdominal aortic
aneurysms. Tohoku J Exp Med. 222:187–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pahl MC, Derr K, Gabel G, Gäbel G,
Hinterseher I, Elmore JR, Schworer CM, Peeler TC, Franklin DP, Gray
JL, Carey DJ, et al: MicroRNA expression signature in human
abdominal aortic aneurysms. BMC Med Genomics. 5:252012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maegdefessel L, Azuma J, Toh R, Deng A,
Merk DR, Raiesdana A, Leeper NJ, Raaz U, Schoelmerich AM, McConnell
MV, et al: MicroRNA-21 blocks abdominal aortic aneurysm development
and nicotine-augmented expansion. Sci Transl Med. 4:122ra1222012.
View Article : Google Scholar
|
|
34
|
Nakao T, Horie T, Baba O, Nishiga M,
Nishino T, Izuhara M, Kuwabara Y, Nishi H, Usami S, Nakazeki F, et
al: Genetic ablation of MicroRNA-33 attenuates inflammation and
abdominal aortic aneurysm formation via several anti-inflammatory
pathways. Arterioscler Thromb Vasc Biol. 37:2161–2170. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim CW, Kumar S, Son DJ, Jang IH,
Griendling KK and Jo H: Prevention of abdominal aortic aneurysm by
anti-microRNA-712 or anti-microRNA-205 in angiotensin II-infused
mice. Arterioscler Thromb Vasc Biol. 34:1412–1421. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang X, Dong M, Wen H, Liu X, Zhang M, Ma
L, Zhang C, Luan X, Lu H and Zhang Y: MiR-26a contributes to the
PDGF-BB-induced phenotypic switch of vascular smooth muscle cells
by suppressing Smad1. Oncotarget. 8:75844–75853. 2017.PubMed/NCBI
|
|
37
|
Suh JH, Choi E, Cha MJ, Song BW, Ham O,
Lee SY, Yoon C, Lee CY, Park JH, Lee SH and Hwang KC: Up-regulation
of miR-26a promotes apoptosis of hypoxic rat neonatal
cardiomyocytes by repressing GSK-3β protein expression. Biochem
Biophys Res Commun. 423:404–410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moon SK, Kim HM and Kim CH: PTEN induces
G1 cell cycle arrest and inhibits MMP-9 expression via the
regulation of NF-kappaB and AP-1 in vascular smooth muscle cells.
Arch Biochem Biophys. 421:267–276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang J and Kontos CD: Inhibition of
vascular smooth muscle cell proliferation, migration, and survival
by the tumor suppressor protein PTEN. Arterioscler Thromb Vasc
Biol. 22:745–751. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ding K, Wu Z, Wang N, Wang X, Wang Y, Qian
P, Meng G and Tan S: MiR-26a performs converse roles in
proliferation and metastasis of different gastric cancer cells via
regulating of PTEN expression. Pathol Res Pract. 213:467–475. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu N, Yang Y, Li X, Zhang M, Huang J, Wang
X and Long X: MiR-26a inhibits proliferation and migration of HaCaT
keratinocytes through regulating PTEN expression. Gene.
594:117–124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee GL, Wu JY, Yeh CC and Kuo CC: TLR4
induces CREB-mediated IL-6 production via upregulation of F-spondin
to promote vascular smooth muscle cell migration. Biochem Biophys
Res Commun. 473:1205–1210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hu WJ, Zhang Z and Dai M: Paeonol affects
proliferation activity of rat vasular endothelial cells induced by
lipopolysaccharide and co-cultured with smooth muscle cells via
inhibiting pathway of PI3K/AKT-NF-κB signaling. Zhongguo Zhong Yao
Za Zhi. 41:2298–2302. 2016.In Chinese. PubMed/NCBI
|
|
44
|
Chang X, Zhang B, Lihua L and Feng Z: T3
inhibits the calcification of vascular smooth muscle cells and the
potential mechanism. Am J Transl Res. 8:4694–4704. 2016.PubMed/NCBI
|
|
45
|
Cui L, Bai Y, Zhang J, Zhang S and Xu J:
Effects of extracellular acid stimulation on rat vascular smooth
muscle cell in Gas6/Axl or PI3K/Akt signaling pathway. Clin Exp
Hypertens. 38:451–456. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang S, Kan X, Li Y, Li P, Zhang C, Li G,
Du J and You B: Deficiency of γδT cells protects against abdominal
aortic aneurysms by regulating phosphoinositide 3-kinase/AKT
signaling. J Vasc Surg. 67:899–908. 2018. View Article : Google Scholar
|
|
47
|
Jiang Q, Han Y, Gao H, Tian R, Li P and
Wang C: Ursolic acid induced anti-proliferation effects in rat
primary vascular smooth muscle cells is associated with inhibition
of microRNA-21 and subsequent PTEN/PI3K. Eur J Pharmacol.
781:69–75. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jiang DS, Wang YW, Jiang J, Li SM, Liang
SZ and Fang HY: MicroRNA-26a involved in Toll-like receptor
9mediated lung cancer growth and migration. Int J Mol Med.
34:307–312. 2014. View Article : Google Scholar : PubMed/NCBI
|