Introduction
Despite the increasing level of modern medical care,
cancer remains difficult to be targeted effectively and
cancer-related mortality remains high worldwide. Metastasis, one of
the features of advanced cancer, is the predominant reason for the
high mortality. Therefore, potential inhibition of metastasis and
invasion of tumor cells has become the research direction and goal
for many researchers. In recent years, more and more transcription
factors, long non-coding RNAs (lncRNAs) and microRNAs (miRNAs)
associated with tumor metastasis have been revealed. Among them,
myocardin-related transcription factor A (MRTF-A) is a very
important transcription factor which can promote tumor
metastasis.
MRTF-A consists of 807 amino acid residues, and it
belongs to the family of serum amyloid P-component serum amyloid P
(SAP) proteins. MRTF-A can promote serum response factor (SRF)
protein binding to the conserved cis regulatory element CC(A/T)6GG
(known as CarG box), thus regulating the transcription of target
genes. It has an important role in the growth and development of
the organism (1-3). The activation of transforming growth
factor (TGF) β-related signaling pathways can be very effective to
induce MRTF-A translocation into the nucleus. Once there, MRTF-A
promotes the transcription of epithelial-mesenchymal transition
(EMT)-related molecules, such as α-smooth muscle actin (α-SMA), and
enhances the migration and metastasis of tumor cells (4-10).
Other studies have reported that the activation of
MRTF-A-mediated Rho associated coiled-coil containing protein
kinase (ROCK) signaling pathway can regulate the phosphorylation of
E-cadherin and decrease the adhesion ability of renal tubular
epithelial cells, resulting in the occurrence of EMT (11). In the present study, while
examining the role of MRTF-A in inducing the migration of breast
cancer cells, it was observed that the transcription factor nuclear
factor erythroid 2-like 1 (Nrf1) could regulate the above process.
Nrf1 (also known as NFE2L1, LCRF1 or TCF11) belongs to the nuclear
factor erythroid 2-related factor (NRF) family. It is ubiquitously
expressed and essential for maintaining cellular homeostasis, organ
integrity and oxidative stress during development and growth
(12-17).
There are multiple splicing isoforms for Nrf1 in
cells, such as the full-length Nrf1α, as well as the LCR-F1/Nrf1β,
Nrf1γ and Nrf1δ isoforms. To date, the specific biological function
of each isoform remains unclear. The present study aimed to explore
the possible mechanisms of Nrf1α and MRTF-A in regulating migration
and invasion of MDA-MB-231 breast cancer cells.
Materials and methods
Cell culture
MDA-MB-231 cells used in the present study were
purchased from American Type Culture Collection (cat. no. HTB-26;
Manassas, VA, USA). The cells were seeded in Dulbecco's modified
Eagle's medium-high glucose (DMEM-HG; Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at
37°C in humidified air with 5% CO2. Cos-7 cells
(American Type Culture Collection; cat. no. CRL-1651) and 293T
cells (American Type Culture Collection; cat. no. CRL-3216) were
cultured in DMEM containing 10% FBS, penicillin (100 U/ml) and
streptomycin (100 μg/ml).
Plasmid construction and standard
lentivirus production
Nrf1α (Gene ID, 4779) and MRTF-A (Gene ID, 57591)
were inserted into the lentivirus vector pCDH-CMV. The plasmids
pCDH-Nrf1α, pCDH-MRTF-A or pCDH-CMV (empty vector control) were
cotransfected with the psPAX2 and pMG2.G into 293T cells (at
~70-80% confluency) using Lipofectamine 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocols.
The medium was changed after 8 h, and 48 h later the supernatant
was collected and filtered. The target short hairpin (sh) RNAs
against Nrf1α gene and MRTF-A gene were inserted into the pLKO.1
vector. As a negative control, a non-targeting sequence that had no
significant homology to any mouse or human gene was inserted into
pLKO.1. pCDH-Nrf1α, shNrf1α and their corresponding controls were a
gift from Professor Jian Dong (North Carolina State University,
Raleigh, NC, USA). The shMRTF-A plasmid has been previously
described (18). shRNA lentiviral
particles were produced by co-transfecting 293T cells using
Lipofectamine 3000 with the lentivirus expression plasmids and
packaging plasmids. Silencing efficiency was detected using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analyses. miR-219 complementary sequence (AGA ATT GCG
TTT GGA CAA TCA) was inserted into pcDNA3.1(−) vector to silence
the function of miR-219 and pcDNA3.1(−) vector was used as a
control.
Lentivirus transduction of MDA-MB-231
cells
MDA-MB-231 cells were cultured in high DMEM
supplemented with 10% FBS and lentivirus was added at a
multiplicity of infection (MOI) of 5. Following overnight
incubation, the media containing the lentivirus was removed and
fresh media was added.
RT-qPCR
Total RNA, including miRNA, was extracted by using
the miRNA kit (Omega Bio-Tek, Inc., Norcross, GA, USA), according
to the manufacturer's protocol. The samples were
reverse-transcribed using M-MLV Reverse Transcriptase (Promega
Corporation, Madison, WI, USA). qPCR was performed in an StepOne
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Fast SYBR Green Master Mix was obtained from Applied
Biosystems (Thermo Fisher Scientific, Inc.). The relative
expression levels of MRTF-A and Nrf1α were normalized to GAPDH. The
primers for the qPCR analysis are listed in Table I. Amplification of U6 small
nuclear RNA served as an endogenous control to normalize miR-219
expression data. The primers for the miR-219 analysis are listed in
Table II. Thermocycling
conditions were as follows: 95°C for 5 min followed by 40 cycles of
95°C for 10 sec and 60°C for 30 sec, then a melting curve analysis
from 60 to 95°C every 0.2°C for 1.5 min was obtained. Each sample
was analyzed in triplicate and quantified using the
2−∆∆Cq method (19).
 | Table ISequences of primers used in reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Sequences of primers used in reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Primer | Sequence (5′-3′) |
|---|
| MRTF-A | Forward |
AAGGAACCACCTGGCTATGA |
| Reverse C |
TCCGCTCTGAATGAGAATGT |
| Nrf1 | Forward |
GCTGGACACCATCCTGAATC |
| Reverse |
GTAGGGTCGTCCGTTCTCAT |
| GAPDH | Forward |
TCAAGAAGGTGGTGAAGCAG |
| Reverse |
AGGTGGAGGAGTGGGTGTCG |
| Table IISequences of primers used in reverse
transcription-quantitative polymerase chain reaction analysis. |
Table II
Sequences of primers used in reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Primer | Sequence
(5′-3′) |
|---|
| miR-219 | Reverse
transcription |
CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAGAATTGC |
| Forward |
ACACTCCAGCTGGGTGATTGTCCAAACGC |
| Reverse |
TGGTGTCGTGGAGTCG |
| U6 | Reverse
transcription |
AACGCTTCACGAATTTGCGT |
| Forward |
CTCGCTTCGGCAGCACA |
| Reverse |
AACGCTTCACGAATTTGCGT |
Protein extraction and western
blotting
For western blot analysis, protein samples were
extracted from the cells with Protein Extraction Reagent (Pierce;
Thermo Fisher Scientific, Inc.). The concentrations of proteins
were determined using a bicinchoninic acid quantification kit
(Beyotime Institute of Biotechnology, Haimen, China). The proteins
(20 μg) were separated by SDS PAGE (10% gel) and transferred
onto a polyvinylidene difluoride membrane. The membrane was blocked
using 5% non fat milk at 25°C for 1 h, and then incubated with
primary antibodies overnight at 4°C. The antibodies used were as
follows: Anti-human GAPDH antibody (cat. no. 97166; 1:2,000, Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-human Nrf1
antibody (cat. no. 46743; 1:1,000, Cell Signaling Technology,
Inc.), anti-human MRTF-A antibody (cat. no. ab49311; 1:1,000,
Abcam, Cambridge, UK). Then, the membrane was incubated with
IRDyeTM-800 conjugated anti-mouse or anti-rabbit secondary
antibodies (cat. no. 115-005-146 and 115-005-144; 1:5,000, Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) at 25°C for
1 h at room temperature. The protein signals were visualized with
the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln,
NE, USA). GAPDH expression was used as an internal control. The
western blotting results were quantified using ImageJ software
(version 2.0; National Institutes of Health, Bethesda, MD,
USA).
Colony formation assay
Cells were transduced with Nrf1α-expressing
lentivirus or with shNrf1α lentivirus or their corresponding
controls, as indicated. Twenty-four hours later, transfected cells
were trypsinized, counted and replated at a density of 200 cells
per 6-cm dish. Ten days later, colonies resulting from the
surviving cells were fixed with 3.7% methanol, and stained with
0.1% crystal violet. Following capturing photos, the crystal violet
stain was washed with 33% acetic acid and the absorbance was
measured at 570 nm. Each assay was performed in triplicate.
Wound healing assay
Cells were subcultured in 6-well plates at a density
of 1×105 cells/well. Upon >80% confluence, the cell
monolayer was gently scraped with a yellow pipette tip to generate
a linear wound and washed twice with serum-free medium to remove
cell debris. Images were subsequently captured at 0 and 24 h. The
closure of the wounds was quantified by the distance of cells moved
into the wounded area. The experiment was repeated twice with
triplicate measurements in each experiment. The results were
quantified using ImageJ software (20).
Transwell invasion assay
The invasion assay was performed using transwell
chambers (Corning Incorporated, Corning, NY, USA), that had
Matrigel (50 μl; BD Biosciences, San Jose, CA, USA)
pre-coated polycarbonate membranes (8.0 μm pore size). A
total of 1×104 cells were suspended in 200 μl
FBS-free DMEM and added to the upper chamber. The lower chamber was
filled with 500 μl DMEM containing 10% FBS. Following
incubation for 24 h, cells on the lower surface of the membrane
were fixed in 4% paraformaldehyde and stained with 0.1% crystal
violet. Cells in four random microscopic fields (magnification,
×20) were counted in triplicates. After capturing photos, the
crystal violet was washed with 33% acetic acid and the absorbance
was measured at 570 nm.
Luciferase constructs, site-mutation, and
luciferase assay
The human miR-219 promoter was fused to the coding
sequence of the pGL-3 luciferase reporter vector (Promega
Corporation). The mutant (mut)-miR-219 promoter was identical to
the pGL-3-miR-219 promoter, except that the Nrf1α binding
antioxidant response element (ARE) site was cut down (the sequence
was AGTGGAAGC). The human Nrf1 promoter luciferase reporter
plasmids were also constructed in the same way. The 3′-untranslated
region (UTR) of the human MRTF-A was amplified from human genomic
DNA and individually inserted into the pmiGLO vector (Promega
Corporation). The primers for constructing the luciferase reporter
plasmids are listed in Table
III. Cells (2×105/well) were plated in 24-well
plates. Cos-7 cells were cotransfected with Nrf1α expression
plasmids (pcDNA3.1(-)-myocardin) or control vector (pcDNA3.1-) in
combination with miR-219-luc or mut-miR-219-luc. The transfection
of these plasmids was performed using Lipofectamine 3000 according
to the manufacturer's protocols. Cells were harvested 24 h
following transfection and luciferase activity was measured using
the Dual luciferase Assay System (Promega Corporation). Results
were expressed as a fold induction relative to the cells
transfected with the control vector (pcDNA3.1-) after normalization
to Renilla activity. In the results from the dual luciferase
assays, the columns represent the mean value of three independent
experiments and the error bars represent the standard
deviation.
| Table IIIPrimers used to generate the
luciferase reporter plasmids by polymerase chain reaction. |
Table III
Primers used to generate the
luciferase reporter plasmids by polymerase chain reaction.
| Plasmid name | Primer | Sequence
(5′-3′) |
|---|
| miR219
promoter-WT | F |
GGGACTCGAGTTGCCCAGTCCATCTTGGTTGTGTT |
| R |
AAGTCTCGAGTTTGAATAACGCCACGGGGCCATCA |
| miR219 promoter cut
down | F |
TAGGCTCGAGGCTCCAGAGGCCTTTGGTTTCCATG |
| R |
AAGTCTCGAGTTTGAATAACGCCACGGGGCCATCA |
| Nrf1
promoter-WT | F |
GCGCGCTAGCAATTCCATGAGTGGTTTGCTG |
| R |
ATTAGCTAGCGCTGCCTCCACAGCAGGCC |
| Nrf1 promoter-cut
CarG 1 | F |
GTGCTAGCCCGGGCTCGAGCGCAAGCACAAAATGGACTCG |
| R |
ACTTAGATCGCAGATCTCGAGCACAGCAGGCCCTAAGCCC |
| Nrf1 promoter-cut
CarG 1,2 | F |
GTGCTAGCCCGGGCTCGAGGGGGTCCTTTGGGCTGTTTC |
| R |
ACTTAGATCGCAGATCTCGAGGAGCTCGGAGCCTCCGCTTA |
| MRTF-A
3′UTR-WT | F |
ATTCGCTAGCAAGACGGGGTGGGGAAGGG |
| R |
GGGGTCTAGACAGCTGCTCTCCTCTGCCCTG |
| MRTF-A
3′UTR-MUT | F | TCCACATGGT
TGTGAGTCTTTGGGGGGGCA GCCCCTGCTT TTTCCC |
| R |
GGGAAAAAGCAGGGGCTGCCCCCCCAAAGACTCACAACCATGTGGA |
Chromatin immunoprecipitation (ChIP)
assay
A ChIP Assay kit (Merck KGaA, Darmstadt, Germany)
was used, following the manufacturer's instructions. Following
treatments as indicated, each experimental group was incubated with
1% formaldehyde to cross-link DNA-protein complexes. After washing
with ice-cold PBS for three times, cells were lysed in SDS lysis
buffer. Then, the lysate was sonicated to shear DNA to 200-1,000 bp
fragments. Anti-human Nrf1 antibody (cat. no. 46743; Cell Signaling
Technology, Inc.) or anti-human MRTF-A antibody (cat. no. ab49311;
Abcam) were used to immunoprecipitate the cross-linked proteins at
4°C overnight. Immunoglobulin G (cat. no. ab172730; Abcam) acted as
the negative control. The DNA was used as a template for PCR, where
the myocardin binding sites were utilized. The PCR products were
separated on 1% agarose gel. The PCR primer sequences are listed in
Table IV.
| Table IVSequences of primers used in
chromatin immunoprecipitation assay. |
Table IV
Sequences of primers used in
chromatin immunoprecipitation assay.
| Target region | Primer | Sequence
(5′-3′) |
|---|
| miR-219 promoter
ARE | F |
TTCAGCATGGTCTTCTCAG |
| R |
AACCAAAGGCCTCTGGAG |
| Nrf1 promoter CarG
box1 | F |
GTACTTAATCTGCAAACC |
| R |
TGAGTCATTAGTCCCTGT |
| Nrf1 promoter CarG
box2 | F |
AGATGGGACTGGAGAAAT |
| R |
GTAGAAACAGCCCAAAGG |
Statistical analysis
Quantitative data are expressed as mean ± standard
error of the mean. Statistical analysis of differences between two
groups was performed by Student's t-test. A one-way analysis of
variance followed by Tukey's test was used for comparing
differences among multiple groups. Statistical analysis was
performed with GraphPad Prism 5 (GraphPad Software, Inc., La Jolla,
CA, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
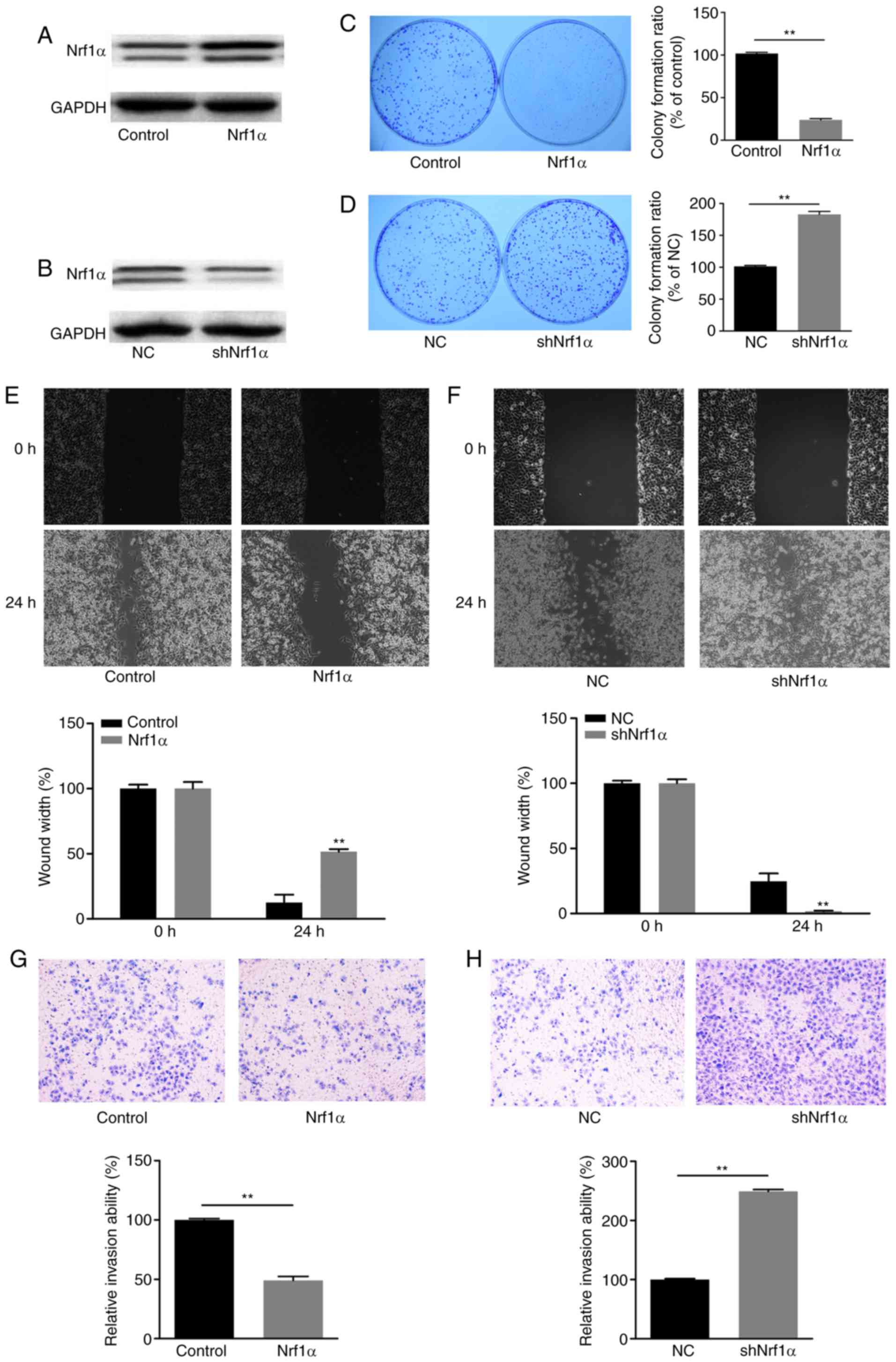
Nrf1α inhibits migration and invasion in
MDA-MB-231 breast cancer cells
To investigate the effect of Nrf1α in breast cancer
cells, the MDA-MB-231 breast cancer cell line was transduced to
overexpress Nrf1α (Nrf1α group), or to knockdown the expression of
Nrf1α (shNrf1α group). Western blot analysis was used to evaluate
the expression of Nrf1α in each group (Fig. 1A and B). Then, by using colony
formation, wound healing and transwell invasion assays, the growth,
migration and invasion capacities were measured in each cell group,
respectively. Notably, when Nrf1α was overexpressed, the
proliferation, migration and invasion ability of tumor cells was
significantly decreased compared with the control group (Fig. 1C, E and G). By contrast, when
endogenous Nrf1α was silenced by shRNA, the tumor cells exhibited
increased proliferation, migration and invasion compared with the
control group (Fig. 1D, F and H).
These findings suggested that Nrf1α was negatively associated with
the migration and invasion of MDA-MB-231 cells. Nrf1α expression
could inhibit migration and invasion of MDA-MB-231 breast cancer
cells.
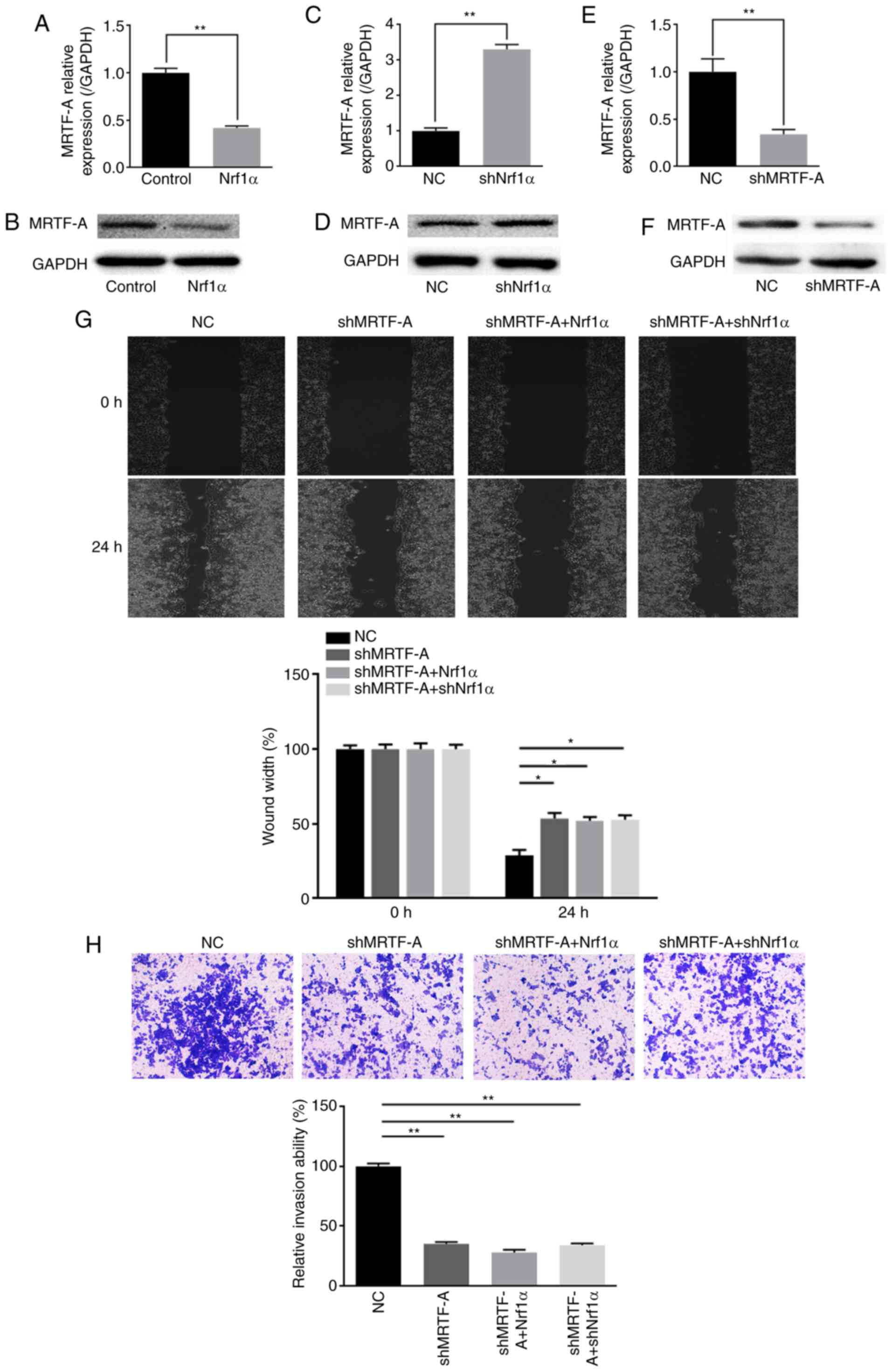
Nrf1α inhibits migration and invasion by
suppressing endogenous MRTF-A expression
Previous studies had demonstrated that MRTF-A
promotes the migration and invasion of tumor cells, including
breast cancer (21-23). As an important transcription
factor associated with tumor metastasis, MRTF-A was a focus of
study in our laboratory. While exploring the potential molecular
mechanisms by which Nrf1α regulates the function of tumor cells, a
regulatory relationship between Nrf1α and MRTF-A was discovered. As
presented in Fig. 2A-D, the
expression of endogenous MRTF-A was negatively associated with the
expression of Nrf1α, at both the mRNA and protein level.
Subsequently, the endogenous MRTF-A expression was silenced by
shRNA (Fig. 2E and F), and then
Nrf1α was overexpressed or knocked down on this setting. The
results demonstrated that, upon MRTF-A silencing, Nrf1α lost the
ability of regulating MDA-MB-231 cell migration and invasion
(Fig. 2G and H).
Nrf1α inhibits the expression of MRTF-A
via miR-219
The aforementioned results demonstrated that Nrf1α
inhibited the expression of MRTF-A to regulate migration and
invasion in breast cancer cells. However, the mechanism by which
Nrf1α regulated the expression of MRTF-A was unclear. It was
hypothesized that miRNAs may have participated in this process.
Through miRBase and TargetScan prediction programs analysis
(24,25), miR-219 was selected. Analysis of
miR-219 expression levels demonstrated that miR-219 was upregulated
when Nrf1α overexpressed (Fig.
3A), while when Nrf1α was silenced, miR-219 levels were
decreased (Fig. 3B). Next, the
miR-219 complementary sequence was used to silence the function of
miR-219 in the MDA-MB-231 cells, and then Nrf1α was overexpressed
or knocked down. RT-qPCR and western blot assays were used to
detect the expression of endogenous MRTF-A. The results
demonstrated that Nrf1α lost its function to regulate MRTF-A
expression, following miR-219 silencing (Fig. 3C and D). To further investigate
the molecular mechanism by which Nrf1α regulates miR-219,
luciferase assays were used to directly examine the effect of Nrf1α
on the transcriptional activity of the miR-219 promoter, which
contains a predicted ARE site. As illustrated in Fig. 3E, Nrf1α could bind to the ARE site
to activate the transcriptional activity of the miR-219 promoter.
In addition, ChIP assay confirmed the direct binding of Nrf1 to the
miR-219 promoter (Fig. 3F).
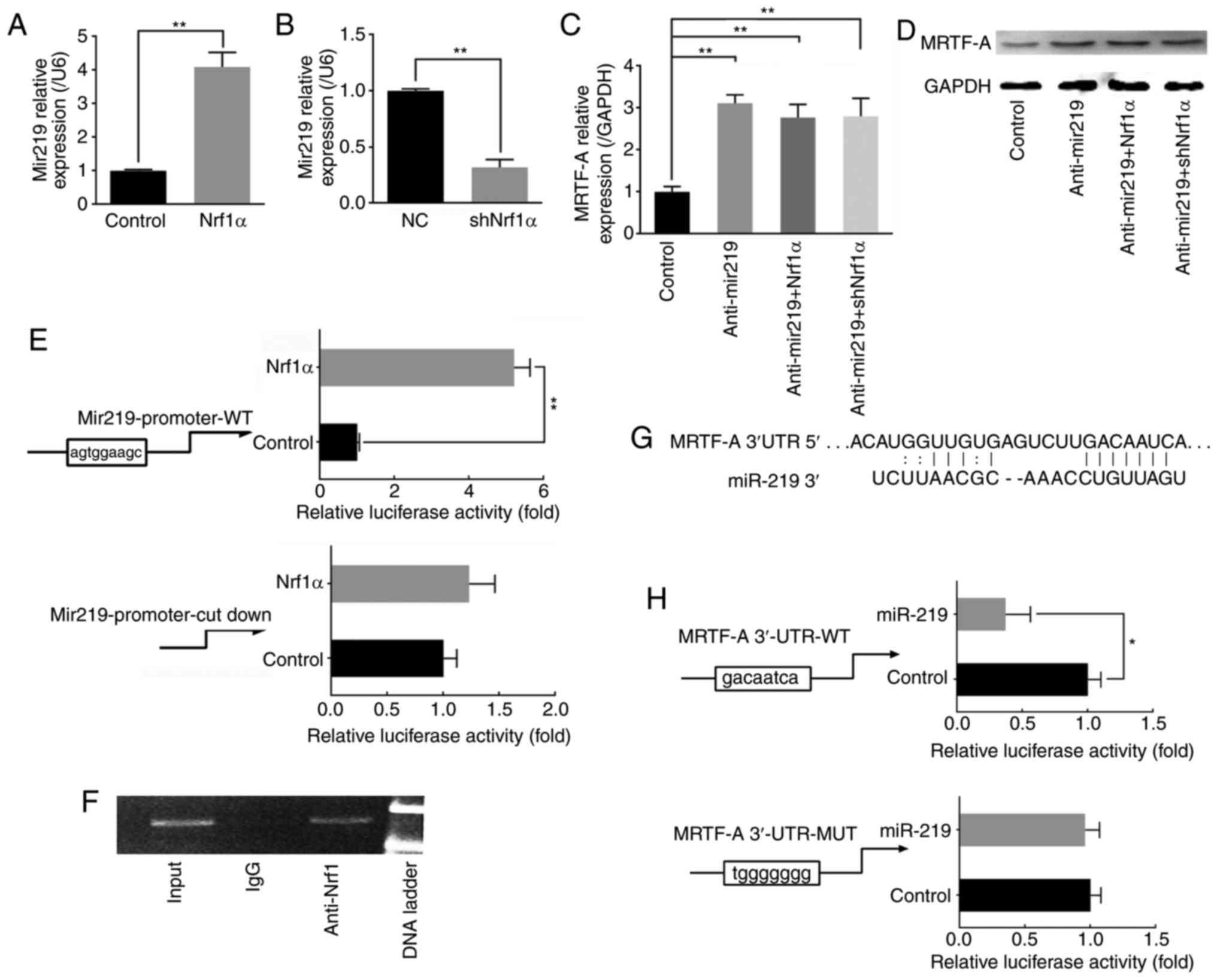 | Figure 3Nrf1α inhibits the expression of
MRTF-A via miR-219. (A) MDA-MB-231 cells were transduced with Nrf1α
or control empty vector for 24 h, and then miR-219 levels were
detected. (B) Endogenous Nrf1α was silenced by shRNA, and then the
levels of miR219 were detected. (C) Following knocking down
endogenous miR-219 in MDA-MB-231 cells, the effect of Nrf1α on mRNA
levels and (D) protein expression levels of MRTF-A was examined.
(E) Cos-7 cells were transfected with 0.8 μg Nrf1α and 0.2
μg miR219-luc or mut-miR219-luc expression plasmids, and
then a luciferase assay was performed. Empty vector pcDNA3.1
plasmid was used as a negative control. (F) Chromatin
immunoprecipitation assay was used to determine the sites on the
miR-219 promoter that Nrf1α directly bound in MDA-MB-231 cells. (G)
A predicted binding site for miR-219 was observed on the 3′-UTR of
MRTF-A. (H) Stable transfection of miR-219 into Cos-7 cells
resulted in decreased luciferase activities of the MRTF-A 3′-UTR.
n≥3. *P<0.05 and **P<0.01, with
comparisons indicated by lines. Nrf1α, nuclear factor erythroid
2-like 1; MRTF-A, myocardin-related transcription factor A; sh,
short hairpin; UTR, untranslated region; NC, negative control; WT,
wild-type; MUT, mutant. |
It is well known that the most common method by
which miRNAs regulate gene expression is to act on their 3′-UTR and
degrade their mRNA. As illustrated in Fig. 3G, binding sites of miR-219 were
predicted to exist in the 3′-UTR of MRTF-A. Therefore, the effect
of miR-219 on the MRTF-A 3′-UTR was examined by luciferase assay.
The results confirmed that miR-219 could bind to the 3′-UTR region
of MRTF-A directly, reducing the mRNA levels of MRTF-A (Fig. 3H).
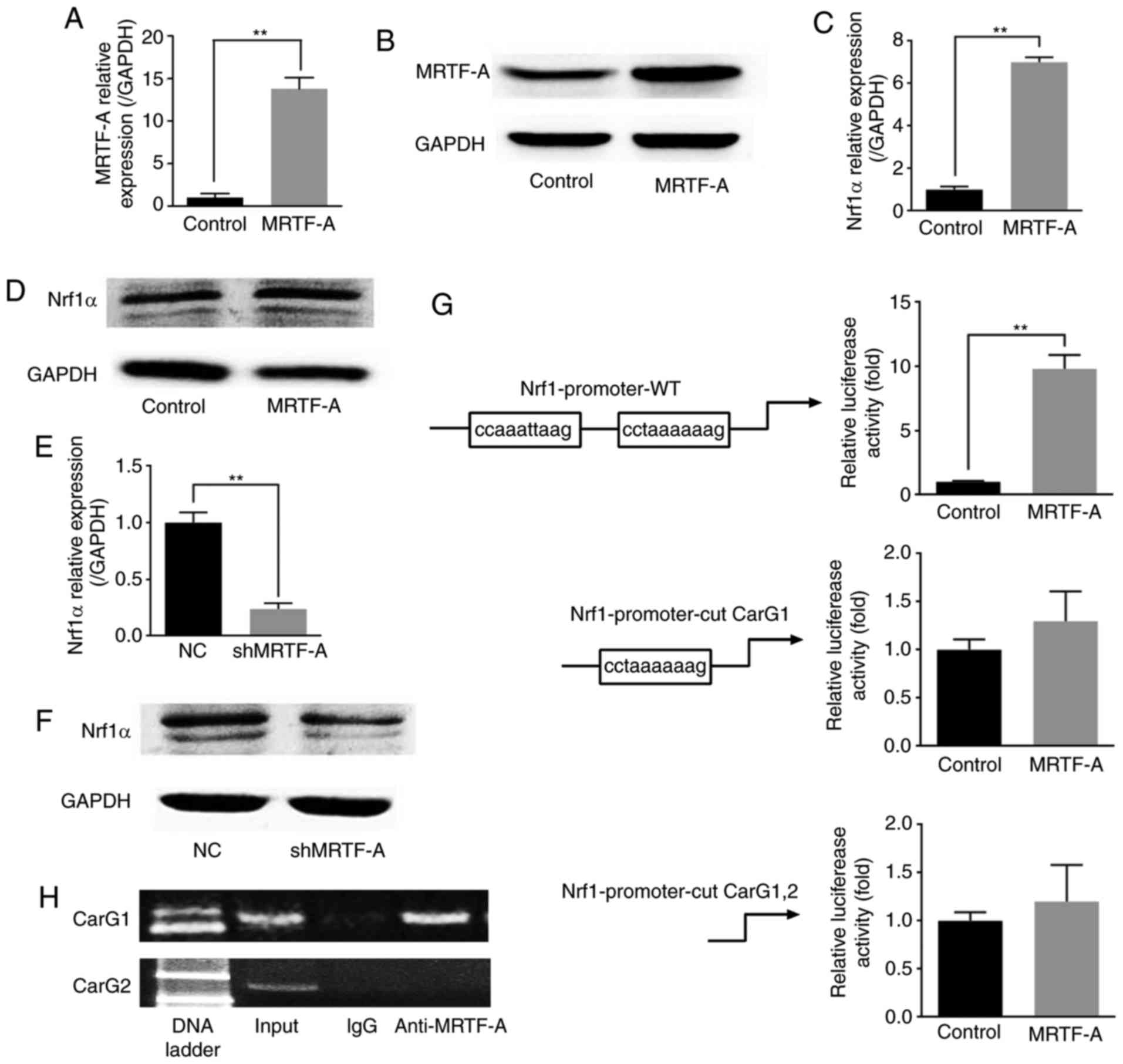
MRTF-A affects Nrf1α expression through
binding to the CarG box in the Nrf1α promoter
Previous studies have demonstrated that Nrf1α could
inhibit the migration and invasion of breast cancer cells via
MRTF-A. The present study revealed the potential molecular
interplay between these two factors in this process. Notably,
MRTF-A was demonstrated to negatively regulate the expression of
Nrf1α. To further explore the relationship between Nrf1α and
MRTF-A, a MRTF-A-overexpressing MDA-MB-231 line was established
(Fig. 4A and B). As presented in
Fig. 4C and E, the results of
RT-qPCR analysis indicated that the mRNA levels of Nrf1α presented
a positive correlation with the mRNA levels of MRTF-A, whether
MRTF-A was overexpressed or knocked down. The western blot assay
results also demonstrated that the protein expression of these two
factors was similar to the mRNA expression (Fig. 4D and F). As a strong drive factor
of containing the CarG locus genes, MRTF-A may activate Nrf1α
through this pathway. Two potential CarG boxes were observed on the
Nrf1α promoter region. The results from the luciferase assay
indicated that the transcriptional activity of Nrf1α promoter could
be upregulated by MRTF-A (Fig.
4G). However, the Nrf1α transcriptional activity was not
affected when the far CarG box (CarG 1) was removed, or when both
CarG boxes were removed (Fig.
4G). These results might indicate that MRTF-A affected the
Nrf1α transcriptional activity through binding to the far CarG box
(CarG 1). To further explore the mechanism of this regulation, ChIP
was used. The results demonstrated that MRTF-A was bound to the far
CarG box (CarG 1), but not the near CarG box (CarG 2), which was
consistent with the results from the luciferase assay (Fig. 4H).
Discussion
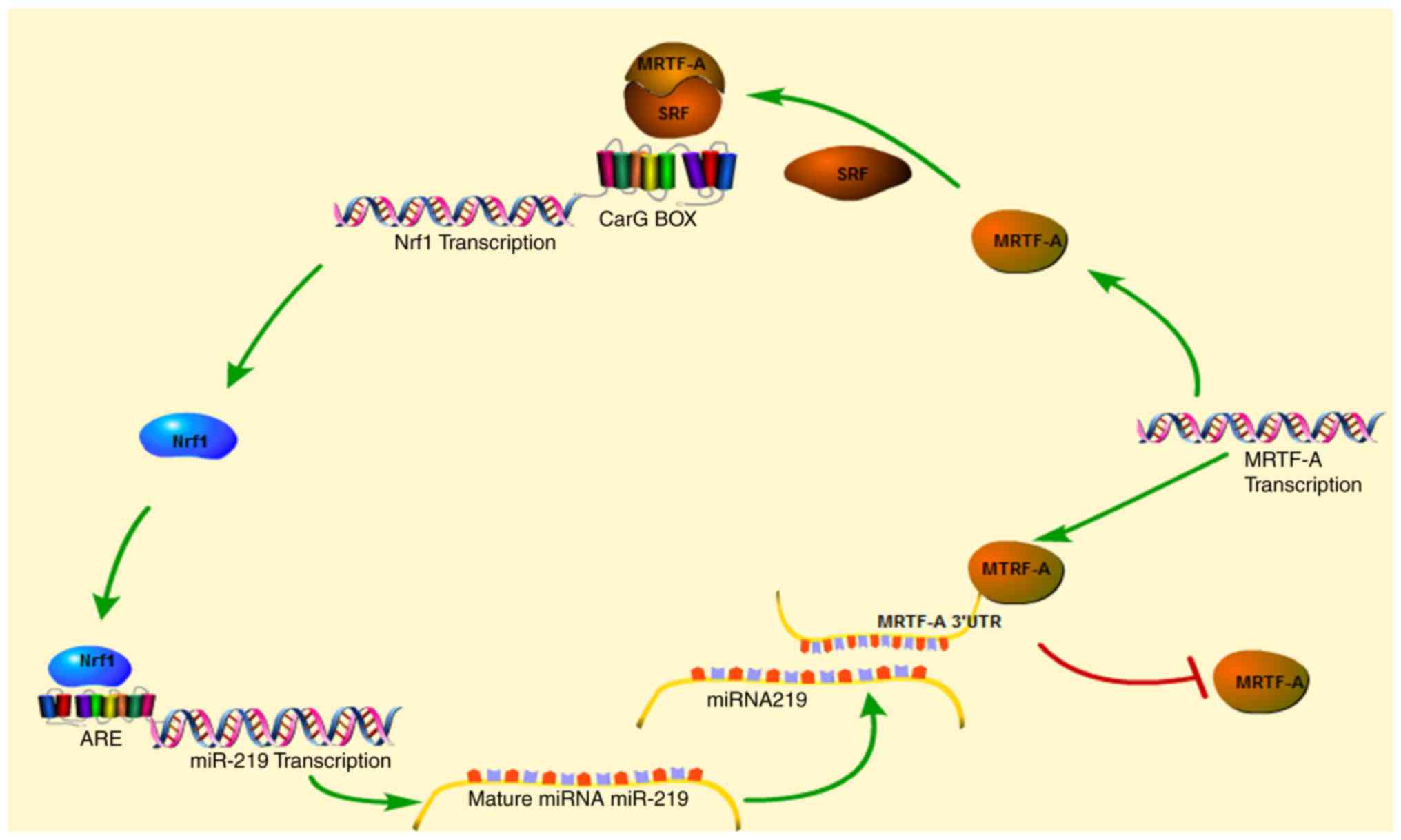
The present study demonstrated that Nrf1α regulated
migration and invasion of breast cancer cells by inhibiting the
expression of MRTF-A. Notably, MRTF-A can reverse activate the
expression of Nrf1α by forming a complex with SRF binding to the
CarG box in the promoter of Nrf1α. Thus, a regulation loop exists
between the two factors in the breast cancer cell line MDA-MB-231
(Fig. 5).
The transcription factor Nrf1 has an important role
in upregulating the antioxidant response by increasing glutathione
biosynthesis (13,14,16,26). Nrf1 is also known to regulate a
variety of antioxidant genes through the combination with ARE
(27). In the endoplasmic
reticulum (ER), Nrf1 is cleaved into many forms, such as Nrf1α,
LCR-F1/Nrf1β, Nrf1γ and Nrf1δ, and translocated from the ER to the
nucleus in response to ER stress (28,29). To date, the specific biological
function of each subtype remains unclear. Therefore, it is helpful
to explore the functional differences of each subtype. In the
present study, it was demonstrated that the migration and invasion
of breast cancer cells were inhibited following overexpression of
Nrf1α. By contrast, the ability of breast cancer cells to migrate
and invade was improved when the expression of Nrf1α was silenced.
Similar findings have been previously reported in HepG2 cells
following Nrf1α knock down (30).
This may suggest that the effect of Nrf1α in inhibiting migration
and invasion may be common in multiple types of cancer.
Subsequently, the present study attempted to reveal the molecular
mechanism by which Nrf1α inhibited migration and invasion in breast
cancer cells.
MRTF-A is an important transcription factor
associated with tumor migration and invasion. The present study
explored the hypothesis that a regulatory relationship may exist
between the two factors. Nrf1α could indeed inhibit the expression
of MRTF-A. Nrf1α, is known to positively regulate genes through ARE
sites. Therefore, it was speculated that miRNAs may exist that have
a role in the Nrf1α/MRTF-A regulation loop. Bioinformatics analysis
was used to discover potential miRNAs that may regulate MRTF-A
expression. In addition, the promoter regions of these miRNAs were
examined for the presence of ARE sites. Following these criteria,
miR-219 was identified as a potential target. Results from
luciferase and ChIP assays demonstrated that Nrf1α indeed regulated
MRTF-A expression via miR-219, which could directly bind to the
3′-UTR of MRTF-A.
In addition, MRTF-A was demonstrated to directly
upregulate the expression of Nrf1α by forming a complex with SRF
binding to the CarG box. Previous studies have also demonstrated
that MRTF-A is associated with cancer-related processes by the
SRF/MRTF-A signaling for the induction of target genes (31-35).
In conclusion, the present study demonstrated that a
regulation loop exists between Nrf1α and MRTF-A, and that this loop
controls the process of breast cancer cell migration and invasion.
Furthermore, the potential underlying mechanism was explored. The
present findings may provide a theoretical reference for the
clinical inhibition of tumor metastasis. Further confirmation of
these results with mouse models or patient tissues will be required
in future studies.
Funding
This study was financially supported by the National
Natural Science Foundation of China (grant nos. 31471282 and
31570764).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
YX, WX and TZ designed the experiments. YX, YL and
CL performed the experiments, analyzed and interpreted the data. YX
and YL were major contributors in writing the manuscript. The final
version of the manuscript has been read and approved by all
authors, and each author believes that the manuscript represents
honest work.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Liao XH, Wang N, Liu QX, Qin T, Cao B, Cao
DS and Zhang TC: Myocardin-related transcription factor-A induces
cardiomyocyte hypertrophy. IUBMB Life. 63:54–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parmacek MS: Myocardin-related
transcription factor-a: Mending a broken heart. Circ Res.
107:168–170. 2007. View Article : Google Scholar
|
|
3
|
Small EM, Thatcher JE, Sutherland LB,
Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara
K and Olson EN: Myocardin-related transcription factor-a controls
myofibroblast activation and fibrosis in response to myocardial
infarction. Circ Res. 107:294–304. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fan L, Sebe A, Péterfi Z, Masszi A,
Thirone AC, Rotstein OD, Nakano H, McCulloch CA, Szászi K, Mucsi I
and Kapus A: Cell contact-dependent regulation of
epithelial-myofibroblast transition via the Rho-Rho
kinase-phosphomyosin pathway. Mol Biol Cell. 18:1083–1097. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miano JM, Long X and Fujiwara K: Serum
response factor: Master regulator of the actin cytoskeleton and
contractile apparatus. Am J Physiol Cell Physiol. 292:C70–C81.
2007. View Article : Google Scholar
|
|
6
|
Sebe A, Masszi A, Zulys M, Yeung T,
Speight P, Rotstein O, Nakano H, Mucsi I, Szászi K and Kapus A:
Rac, PAK and p38 regulate cell contact-dependent nuclear
translocation of myocardin-related transcription factor. FEBS Lett.
582:291–298. 2008. View Article : Google Scholar
|
|
7
|
Zhao XH, Laschinger C, Arora P, Szászi K,
Kapus A and McCulloch CA: Force activates smooth muscle α-actin
promoter activity through the Rho signaling pathway. J Cell Sci.
120:1801–1809. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O'Connor JW, Riley PN, Nalluri SM, Ashar
PK and Gomez EW: Matrix rigidity mediates TGFβ1-induced
epithelial-myofibroblast transition by controlling cytoskeletal
organization and MRTF-A localization. J Cell Physiol.
230:1829–1839. 2015. View Article : Google Scholar
|
|
9
|
Fan WH and Karnovsky MJ: Increased MMP-2
expression in connective tissue growth factor over-expression
vascular smooth muscle cells. J Biol Chem. 277:9800–9805. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hinson JS, Medlin MD, Lockman K, Taylor JM
and Mack CP: Smooth muscle cell-specific transcription is regulated
by nuclear localization of the myocardin-related transcription
factors. Am J Physiol Heart Circ Physiol. 292:H1170–H1180. 2007.
View Article : Google Scholar
|
|
11
|
Tian YC, Fraser D, Attisano L and Phillips
AO: TGF-beta1-mediated alterations of renal proximal tubular
epithelial cell phenotype. Am J Physiol Renal Physiol.
285:F130–F142. 2007. View Article : Google Scholar
|
|
12
|
Farmer SC, Sun CW, Winnier GE, Hogan BL
and Townes TM: The bZIP transcription factor LCR-F1 is essential
for mesoderm formation in mouse development. Genes Dev. 11:786–798.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chan JY, Kwong M, Lu R, Chang J, Wang B,
Yen TB and Kan YW: Targeted disruption of the ubiquitous CNC-bZIP
transcription factor, Nrf-1, results in anemia and embryonic
lethality in mice. EMBO J. 17:1779–1787. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kwong M, Kan YW and Chan JY: The CNC basic
leucine zipper factor, nrf1, is essential for cell survival in
response to oxidative stress-inducing agents role for nrf1 in
gamma-gcs(l) and gss expression in mouse fibroblasts. J Biol Chem.
274:37491–37498. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Z, Chen L, Leung L, Yen TB, Lee C and
Chan JY: Liver-specific inactivation of the Nrf1 gene in adult
mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia.
Proc Natl Acad Sci USA. 102:4120–4125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ohtsuji M, Katsuoka F, Kobayashi A,
Aburatani H, Hayes JD and Yamamoto M: Nrf1 and Nrf2 play distinct
roles in activation of antioxidant response element-dependent
genes. J Biol Chem. 283:33554–33562. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsujita T, Peirce V, Baird L, Matsuyama Y,
Takaku M, Walsh SV, Griffin JL, Uruno A, Yamamoto M and Hayes JD:
Transcription factor Nrf1 negatively regulates the
cystine/glutamate transporter and lipid-metabolizing enzymes. Mol
Cell Biol. 34:3800–3816. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu Y, Luo Y, Wang ZY, Li X, Zheng P and
Zhang TC: MRTF-A can activate Nrf2 to increase the resistance to
doxorubicin. Oncotarget. 8:8436–8446. 2017.
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hermann MR, Jakobson M, Colo GP, Rognoni
E, Jakobson M, Kupatt C, Posern G and Fässler R: Integrins
synergise to induce expression of the MRTF-A-SRF target gene ISG15
for promoting cancer cell invasion. J Cell Sci. 129:1391–1403.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eisenach PA, Schikora F and Posern G:
Inhibition of arginyltransferase 1 induces transcriptional
activityof myocardin-related transcription factor A (MRTF-A) and
promotes directional migration. J Biol Chem. 289:35376–35387. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang WL, Lv W, Sun SZ, Wu XZ and Zhang
JH: miR-206 inhibits metastasis-relevant traits by degrading MRTF-A
in anaplastic thyroid cancer. Int J Oncol. 47:133–142. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Griffiths-Jones S, Saini HK, Van Dongen S
and Enright AJ: miRBase: Tools for microRNA genomics. Nucleic Acids
Res. 36:D154–D158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Edris B: A comparison of the oligomap and
targetscan algorithms for miRNA target analysis. BMI. 231:2011.
|
|
26
|
Chen L, Kwong M, Lu R, Ginzinger D, Lee C,
Leung L and Chan JY: Nrf1 is critical for redox balance and
survival of liver cells during development. Mol Cell Biol.
23:4673–4686. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gęgotek A and Skrzydlewska E: CNC proteins
in physiology and pathology. Postepy Hig Med Dosw (Online).
69:729–743. 2015.In Polish. View Article : Google Scholar
|
|
28
|
Zhang Q, Pi J, Woods CG and Andersen ME: A
systems biology perspective on Nrf2-mediated antioxidant response.
Toxicol Appl Pharmacol. 244:84–97. 2010. View Article : Google Scholar :
|
|
29
|
Zhang Y, Li S, Xiang Y, Qiu L, Zhao H and
Hayes JD: The selective post-translational processing of
transcription factor Nrf1 yields distinct isoforms that dictate its
ability to differentially regulate gene expression. Sci Rep.
5:129832015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ren Y, Qiu L, Lü F, Ru X, Li S, Xiang Y,
Yu S and Zhang Y: TALENs-directed knockout of the full-length
transcription factor Nrf1α that represses malignant behaviour of
human hepatocellular carcinoma (HepG2) cells. Sci Rep. 6:237752016.
View Article : Google Scholar
|
|
31
|
Scharenberg MA, Chiquet-Ehrismann R and
Asparuhova MB: Megakaryoblastic leukemia protein-1 (MKL1):
Increasing evidence for an involvement in cancer progression and
metastasis. Int J Biochem Cell Biol. 42:1911–1914. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Medjkane S, Perez-Sanchez C, Gaggioli C,
Sahai E and Treisman R: Myocardin-related transcription factors and
SRF are required for cytoskeletal dynamics and experimental
metastasis. Nat Cell Biol. 11:257–268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Descot A, Hoffmann R, Shaposhnikov D,
Reschke M, Ullrich A and Posern G: Negative regulation of the
EGFR-MAPK cascade by actin-MAL-mediated Mig6/Errfi-1 induction. Mol
Cell. 35:291–304. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Leitner L, Shaposhnikov D, Descot A,
Hoffmann R and Posern G: Epithelial protein lost in neoplasm α
(Eplin-alpha) is transcriptionally regulated by G-actin and
MAL/MRTF coactivators. Mol Cancer. 9:602010. View Article : Google Scholar
|
|
35
|
Yoshio T, Morita T, Tsujii M, Hayashi N
and Sobue K: MRTF-A/B suppress the oncogenic properties of v-ras-
and v-src-mediated transformants. Carcinogenesis. 31:1185–1193.
2010. View Article : Google Scholar : PubMed/NCBI
|