Introduction
Intracerebral hemorrhage (ICH) is a fatal stroke
subtype, associated with high mortality worldwide, and it accounts
for 10–15% of all hospitalized stroke patients (1,2).
Blood-brain barrier (BBB) damage and angioedema occur in the early
stages of injury. BBB breakdown leads to brain injury progression
and long-term neurological deficits (3). Destruction of the BBB directly
aggravates vasogenic brain edema, and has been identified as an
important contributor of secondary brain damage (4). Endothelial cells are interconnected
by tight junctions (TJs) and mainly composed of zonula occludens-1
(ZO-1), occludin and claudin proteins (5). ZO-1 anchors the transmembrane
protein occludin to the actin cytoskeleton, which confers the
capacity of BBB to preclude permeation of blood substances
(6). Changes in TJ components
result in loss of BBB integrity and BBB decomposition (7). A variety of signal transduction
pathways mediate the opening of tight junctions, including
Ras-related C3 botulinum toxin substrate 1 (Rac1) (8). Therefore, regulating BBB
permeability and reducing the degradation of TJ proteins may allow
for a more effective ICH treatment strategy.
The endocannabinoid system has been studied as a
potential therapeutic target for neuroprotection (9). There are two major subtypes of
cannabinoid receptor, cannabinoid receptor 1 (CNR1) and cannabinoid
receptor 2 (CNR2) (10,11). CNR2 is highly expressed in immune
cells and mediates inflammatory responses (12). Previous studies have reported that
cannabinoid receptors serve an important regulatory role in normal
BBB physiology and protect BBB during ischemic stroke (13). Pharmacological data has suggested
that CNR2 agonists increase the presence of tight junction proteins
in membrane fractions in endothelial resistance and
neuroinflammation (14). JWH133
(a specific CNR2 agonist), which exhibits a very high affinity for
CNR2 (Ki=3.4 nmol/l) but low affinity for CNR1 (Ki=677 nmol/l), has
been reported to attenuate brain edema in rat models of germinal
matrix hemorrhage (15), and to
improve functional outcomes and reduce brain edema following
experimental subarachnoid hemorrhage in rats. In addition, JWH-133
treatment attenuates BBB damage in traumatic brain injury (16). These results indicate that CNR2
activation has potential as a practical and therapeutic
intervention.
Rac1 is an important and well-studied member of a
small-molecular-weight guanosine-5′-triphosphate (GTP) binding
protein located inside the cell membrane. It serves an important
role in cell proliferation and differentiation (17) and in regulation of cytoskeletal
organization in many cell types. Rac1 maintains and stabilizes the
barrier function of microvascular endothelial cells (18). Rac1 activation inhibits RhoA,
which may preserve BBB integrity and, therefore, reduce the
development of vasogenic brain edema after ICH (19). CNR2 concurrently or sequentially
affects TJ protein stability via the Rho GTPase pathway (20). CNR2 agonists reduce integrin
activation and lamellipodia formation in primary monocytes via
inhibition of small GTPases (RhoA and Rac1) and affects
cytoskeletal proteins (21).
However, whether CNR2 activation can prevent BBB disruption and
brain edema by inhibiting Rac1 activation in ICH, remains
unknown.
Therefore, the present study investigated the
effects of the CNR2 agonist, JWH133, on a rat model of ICH-induced
BBB damage, and the role of Rac1 in the neuroprotective process.
The present study demonstrated that JWH133, a selective CNR2
agonist, improved neurofunctional deficits, reduced brain edema and
alleviated BBB damage following ICH, and these effects were
reversed by AM630 (CNR2 antagonist) and NSC23766 (Rac1 antagonist)
treatment.
Materials and methods
Chemicals
JWH133 (CNR2 agonist), and AM630 (CNR2 antagonist)
were purchased from Tocris Bioscience (Bristol, UK). JWH133 and
AM630 were dissolved in dimethyl sulfoxide (DMSO) and then diluted
with sterile PBS. NSC23766 (Rac1 inhibitor) was purchased from
Abcam (Cambridge, UK) and also dissolved in DMSO. Rac1 Pull-down
Activation Assay kit was obtained from Cytoskeleton Inc. (Denver,
CO, USA). Rat anti-Rac1 monoclonal antibody (cat. no. PA1-091;
1:1,000) was purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). Anti-occludin (cat. no. sc-133256; 1:800),
anti-ZO-1 (cat. no. sc-33725; 1:800) and anti-claudin-5 (cat. no.
sc-374221; 1:800) antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). An antibody targeting
β-actin (cat. no. 3700; 1:1,000) was purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Goat anti-rabbit (cat. no.
sc-2004; 1:2,000) and goat anti-mouse secondary antibodies (cat.
no. sc-2005; 1:2,000) were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA).
Animals and treatments
A total of 180 male Sprague-Dawley rats (275–300 g)
were purchased from the Laboratory Animal Center, Third Military
Medical University (Chongqing, China). Rats were housed in a light
and temperature controlled environment at 20–22°C. Food and water
were available ad libitum. Experimental protocols were
approved by the Medical Ethics Committee of the Third Military
Medical University.
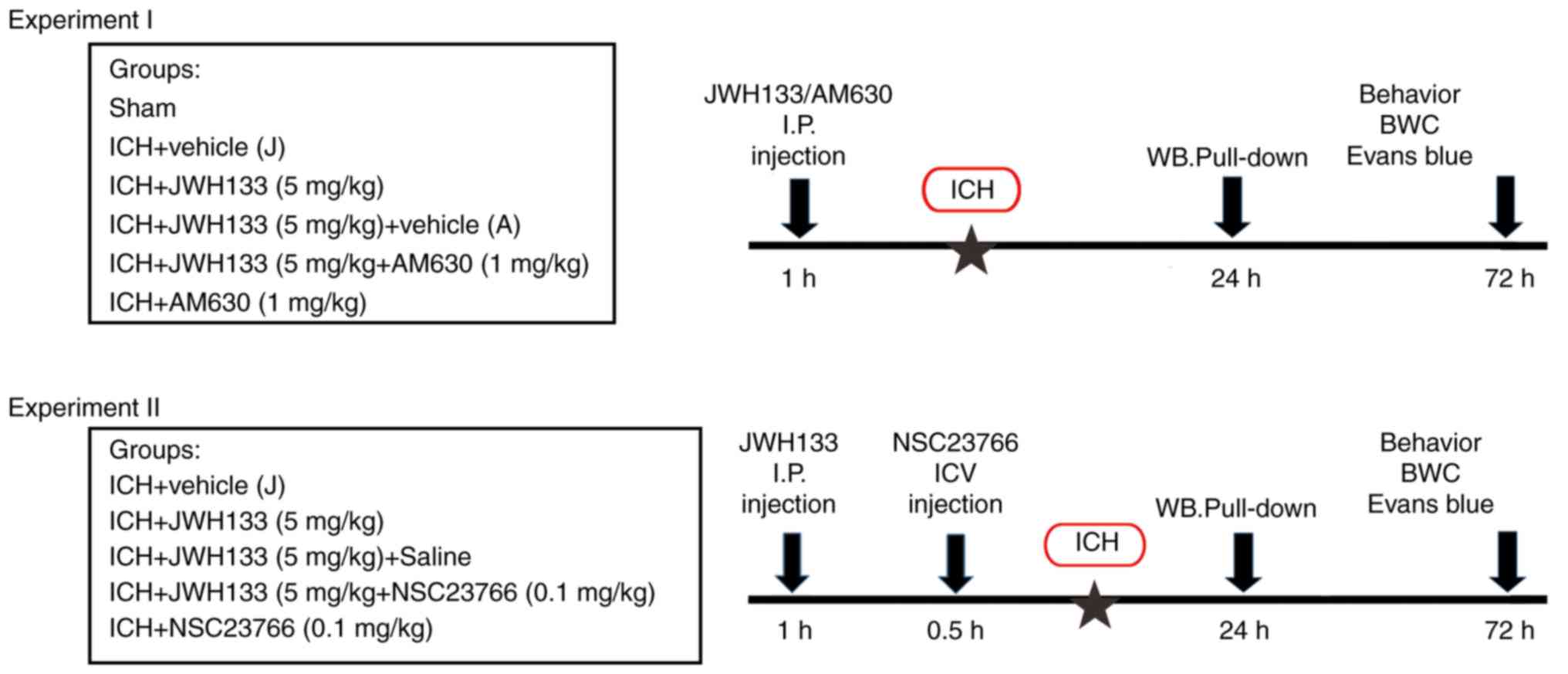
The experiments were divided into two parts. In
experiment I, animals were randomly divided into the following
groups (15 rats per group): Sham-operated (Sham group), ICH+vehicle
(Vehicle group), ICH+JWH133 (JWH133 group), ICH+JWH133+vehicle
(JWH133+vehicle group), ICH+ JWH133+AM630 (JWH133+AM630 group) and
ICH+AM630 (AM630 group). JWH133 (5.0 mg/kg) and AM630 (1.0 mg/kg)
were intraperitoneally (i.p.) injected 1 h prior to ICH. In
experiment II, animals were randomly divided into the following
groups (15 rats per group): Sham-operated (Sham group), ICH+vehicle
(Vehicle group), ICH+JWH133 (JWH133 group), ICH+J WH133+vehicle (J
WH133+vehicle group), ICH+JWH133+NSC23766 (JWH133+NSC23766 group)
and ICH+NSC23766 (NSC23766 group). JWH133 (5.0 mg/kg) was injected
i.p. at 1 h prior to IHC, and NSC23766 (0.1 mg/kg) was administered
intracerebroventricularly (ICV) at 30 min prior to ICH. Animals
were sacrificed 24 and 72 h following surgery. The control rats
received an equal volume of vehicle. The physiological parameters
of rats were measured at 24 and 72 h following the ICH procedure.
The experimental design is presented in Fig. 1.
Rat ICH model
The rat ICH model was established as previously
described (22). Briefly, animals
were anesthetized with an intraperitoneal injection of
pentobarbital (40 mg/kg) and placed on a stereotaxic apparatus. A
1-mm cranial burr hole was drilled, and a needle was inserted into
the right basal ganglia under stereotactic guidance (coordinates:
0.2-mm anterior, 3.5-mm lateral to the midline and 5.5-mm ventral),
and 100 µl of autologous arterial blood extracted from the femoral
artery was slowly infused at a rate of 5 µl/min using a
microinfusion pump. After the injection was complete, the needle
was left in place for 20 min before withdrawal. Control animals
were injected with 100 µl of normal saline. Bone wax was used to
close the burr hole, and the skin incision was closed with sutures.
In order to avoid infection, all procedures were performed under
aseptic conditions.
Western blot analysis
Rats were sacrificed at 24 and 72 h post-ICH, and
brain tissues surrounding the hemorrhagic region were isolated.
Total protein was isolated from brain tissues using RIPA lysis
buffer (Beyotime Institute of Biotechnology, Beijing, China). The
protein concentrations were determined by bicinchoninic acid (BCA)
assay. Equal amounts of protein (40 µg) were separated on 10%
sodium dodecyl sulfate-polyacrylamide gels and transferred into
PVDF membranes (EMD Millipore, Billerica, MA, USA). The membrane
was blocked in 5% non-fat milk at room temperature for 1 h and
incubated overnight at 4°C with the following primary antibodies:
Anti-occludin, anti-ZO-1, anti-claudin-5 and β-actin. Then the
membranes were incubated with secondary antibody for 1 h at room
temperature. (1:2,000; Santa Cruz Biotechnology, Inc.) and
immunoblots were visualized using an ECL Plus chemiluminescence kit
(Amersham; GE Healthcare, Chicago, IL, USA). Blot bands were
semi-quantitatively analyzed using ImageJ (version 4.0; National
Institutes of Health, Bethesda, MD, USA).
Functional behavioral test
The sensorimotor Garcia (23) and Corner test (24) were conducted in a blinded fashion,
and used to assess neurofunctional deficits in rats at 24 and 72 h
post-ICH. The Garcia Test was modified to examine spontaneous
activity, axial sensation, vibrissae touch and limb symmetry, as
well as the animal's ability to turn laterally, outstretch
forelimbs and climb. The worst performance was awarded 0 points,
and the best performance, 3 points, for each subtest. The sum point
score was calculated to determine neurological function (maximum
score, 21).
The corner test was conducted as described by Li
et al (24). The rat was
placed between two boards, each with dimension of 30×20×1
cm3. The edges of the two boards were connected at an
angle of 30°. A small opening along the joint between the two
boards encouraged entry into the corner. The rat was placed between
the two angled boards facing the corner and half way to the corner.
When the rat entered far into the corner both sides of the
vibrissae were stimulated together. Rearing forwards and upwards
was observed, followed by turning to face the open end. The turns
in each direction were recorded for each test. A total of 10 trials
were performed for each rat, with a break of ≥30 sec between
trials. The % of right turns was calculated. Only turns involving
full rearing along either board were included.
Analysis of the brain water content
The brain water content was examined in rats at 24
and 72 h post-ICH, according to a previous study (25). Animals were anesthetized with
pentobarbital (40 mg/kg; i.p.), sacrificed and the brains were
surgically removed. The cerebrum was divided into 5 parts:
Ipsilateral cortex (Ipsi-CX) and contralateral cortex (Cont-CX),
ipsilateral basal ganglia (Ipsi-BG) and contralateral basal ganglia
(Cont-BS), and cerebellum (cerebel). Each part was immediately
weighed using an analytical microbalance (APX-60; Denver
Instrument, Bohemia, NY, USA) to measure the wet weight, and then
they were dried at 100°C for 24 h to measure the dry weight. The
brain water content was calculated using the formula: Brain water
content (%) = (wet weight−dry weight)/wet weight ×100.
Evans blue staining
Evans blue staining was performed at 24 and 72 h
post-ICH to evaluate blood-brain barrier permeability, as
previously described by Ma et al (26). The rats were anesthetized and
received femoral vein injection of 2% Evans blue solution (5 ml/kg;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), allowing the dye to
circulate for 3 h prior to transcranial perfusion with PBS pH 7.4.
Following transcranial perfusion, the brains were surgically
removed, divided into right and left brain hemispheres and stored
at −80°C until use. Brain specimens were homogenized in PBS,
sonicated and centrifuged at 12,000 × g at 4°C for 30 min. The
supernatant was collected and an equal amount of trichloroacetic
acid (50%) was added. The supernatant was incubated overnight at
4°C and centrifuged at 15,000 × g for 30 min at 4°C. The absorbance
of the sample was measured at 620 nm using a spectrophotometer
(Multiskan GO; Thermo Fisher Scientific, Inc.) and quantified
according to a standard curve.
Rac1-GTPase pull-down assay
Rac1 activation assays were performed as described
previously (27). In brief, brain
tissues surrounding the hemorrhagic region underwent homogenization
in RIPA buffer (Santa Cruz Biotechnology, Inc.) supplemented with
protease and phosphatase inhibitors (Sigma-Aldrich; Merck KGaA) at
4°C. The samples were incubated in a 4°C water bath for 1.5 h.
Samples were centrifuged for 10 min at 10,000 × g at 4°C and the
supernatants were collected. The protein concentration was
determined using a BCA kit. A portion of each supernatant was added
to Pak-GST-conjugated beads (10 µl) for detection of total Rac1
GTPases. The solution was continuously shaken for 2 h at 4°C, and
centrifuged for 1 min at 3,000 × g at 4°C. The samples were washed
three times with lysis buffer by centrifugation at 4,500 rpm at 4°C
for 5 min. Proteins bound to the beads were rinsed with distilled
water and the Rac1-GTPase activity was determined by western
blotting analysis.
Statistical analysis
The data was analyzed using SPSS 13.0 software (SPSS
Inc., Chicago, IL, USA). Data were expressed as the mean ± standard
deviation. One-way analysis of variance was used to compare the
differences among the multiple groups of data, followed by Tukey's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
JWH133 treatment reduces neurofunctional
deficit and brain edema at 24 and 72 h following ICH
A modified method of autologous blood injection was
used to establish the ICH model, as described by Yang et al
(22). Neurofunctional deficits
and brain water content were evaluated at 24 and 72 h following
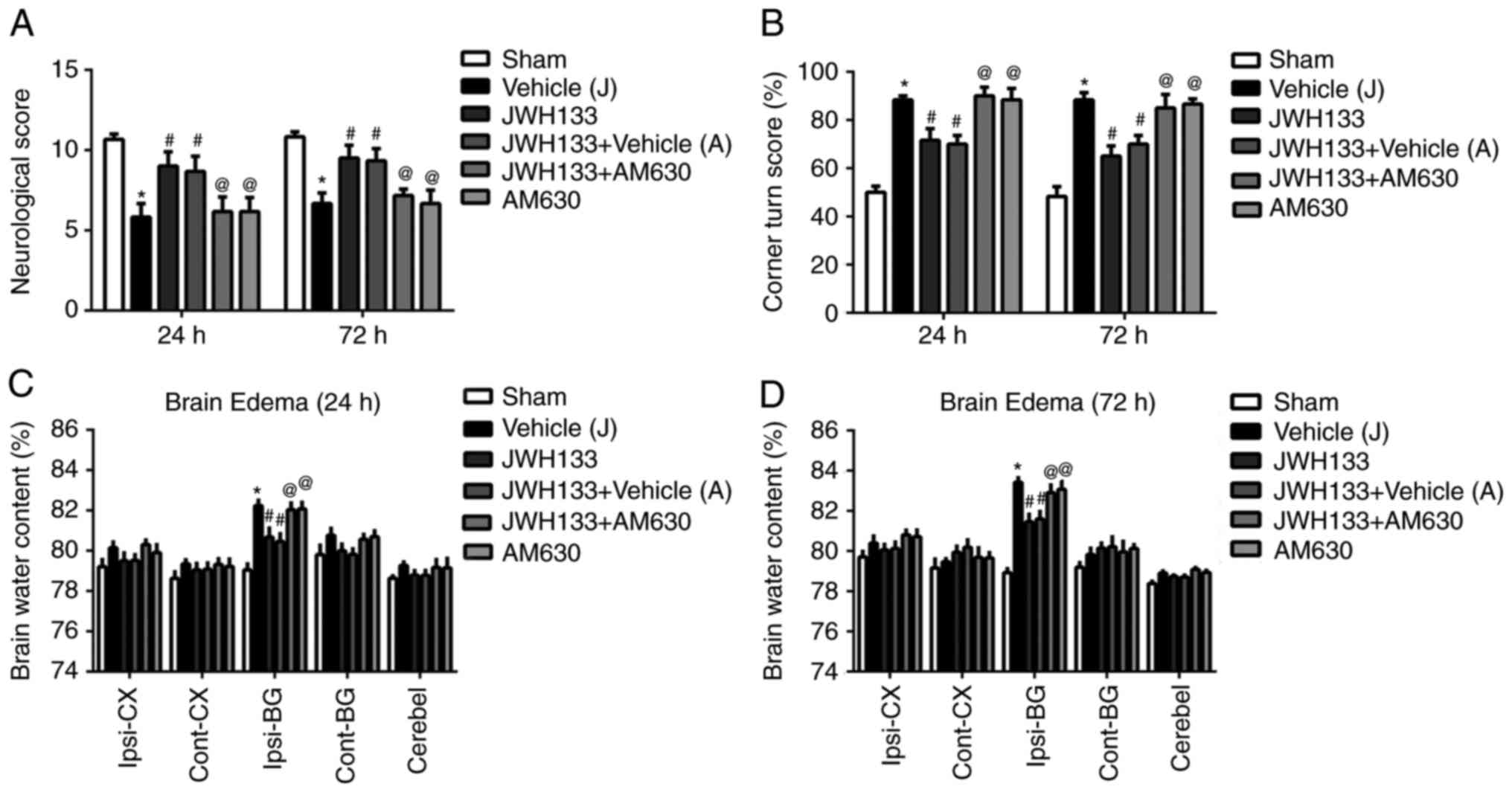
surgery. As illustrated in Fig. 2A
and B, rats performed significantly lower scores during the
Garcia and Corner tests at 24 and 72 h following surgery. In
addition, the results demonstrated increased brain water content in
the ipsilateral basal ganglia of the brain in the vehicle group,
compared with the sham group (Fig. 2C
and D). JWH133 administration post-surgery significantly
ameliorated the neurofunctional deficits and reduced the brain
water content compared with the vehicle group (Fig. 2). However, the selective CNR2
antagonist, AM630, reversed this treatment effect (Fig. 2).
JWH133 protects against ICH-induced BBB
destruction
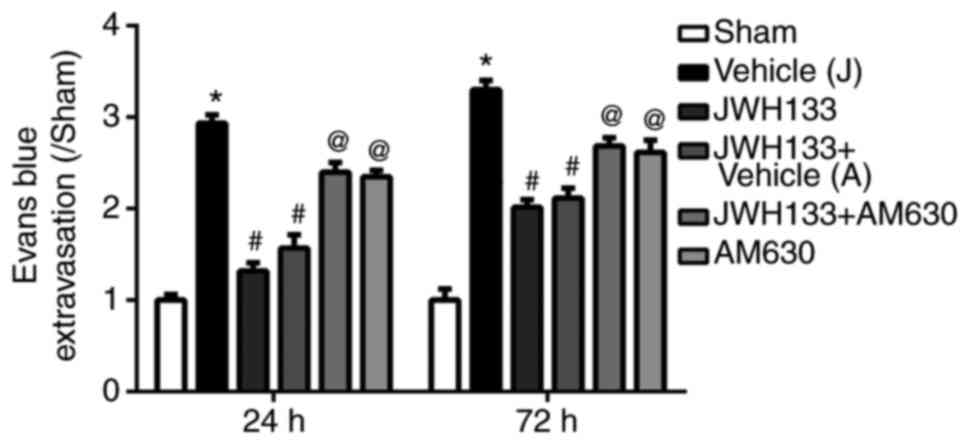
Evans blue staining was used to detect BBB integrity
at 24 and 72 h following ICH. The results demonstrated that Evans
blue leakage was significantly increased at 24 and 72 h post-ICH in
the vehicle group compared with the sham group (Fig. 3). JWH133 treatment significantly
reduced Evans blue leakage at 24 and 72 h post-ICH compared with
the vehicle group (Fig. 3).
However, the effect of JWH133 on decreasing the Evans blue leakage
was reversed by AM630 treatment (Fig.
3).
JWH133 administration upregulates
GTP-Rac1 protein expression following ICH
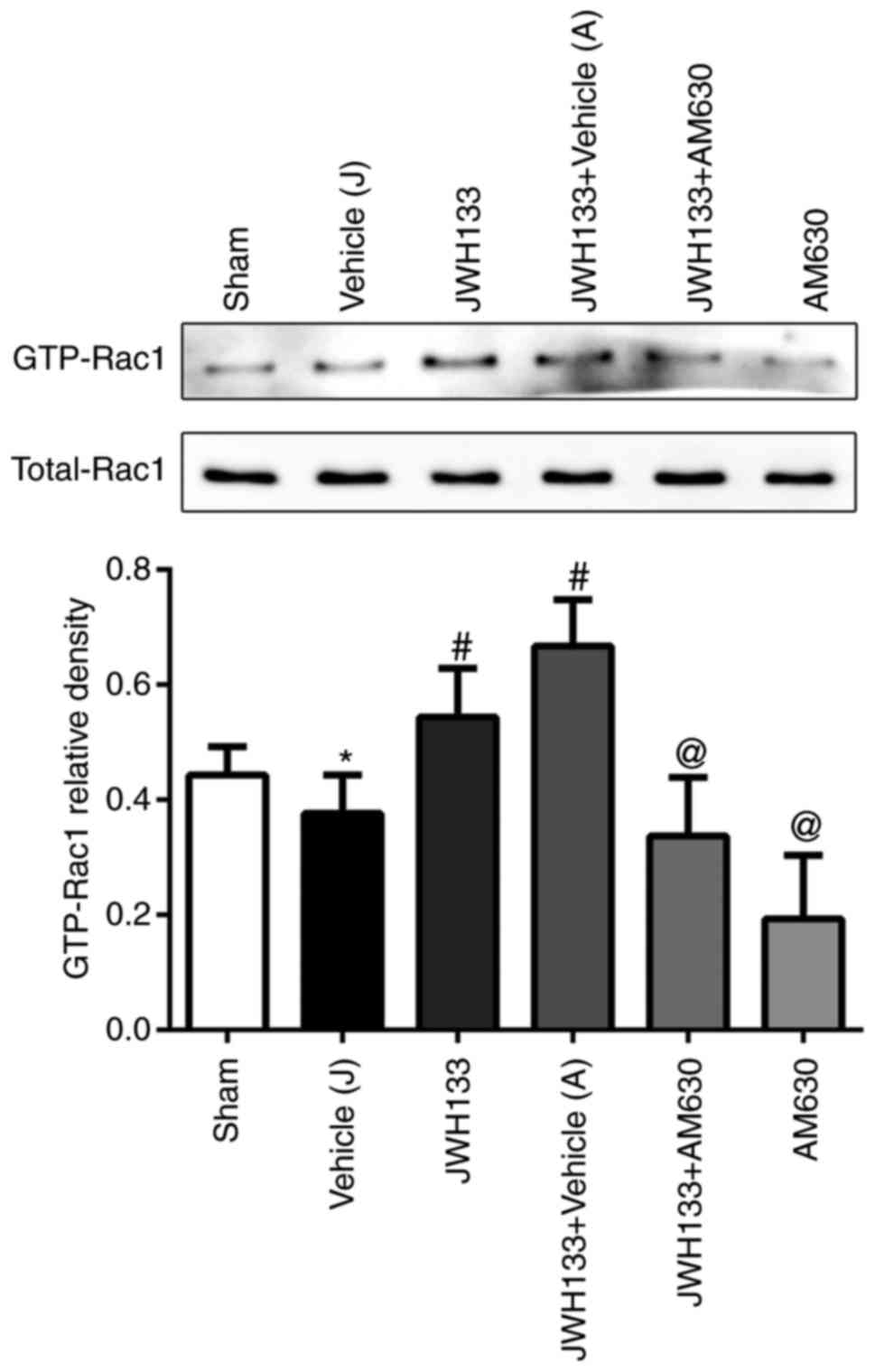
To elucidate the potential mechanism of JWH133 on
the CNR2/Rac1/TJ signaling pathway following ICH, a pull-down
method was used to analyze the effect of JWH133 on the expression
of GTP-Rac1 protein at 24 h post-ICH. As illustrated in Fig. 4, the GTP-Rac1 protein levels were
decreased in the ipsilateral basal ganglia at 24 h post-ICH
compared with the sham group. However, the GTP-Rac1 protein
expression levels were higher in the JWH133-treated group and
JWH133+vehicle group, compared with the vehicle group (Fig. 4). The combination treatment of
JWH133 with AM630 reversed the GTP-Rac1 protein upregulation,
compared with the JWH133 group (Fig.
4).
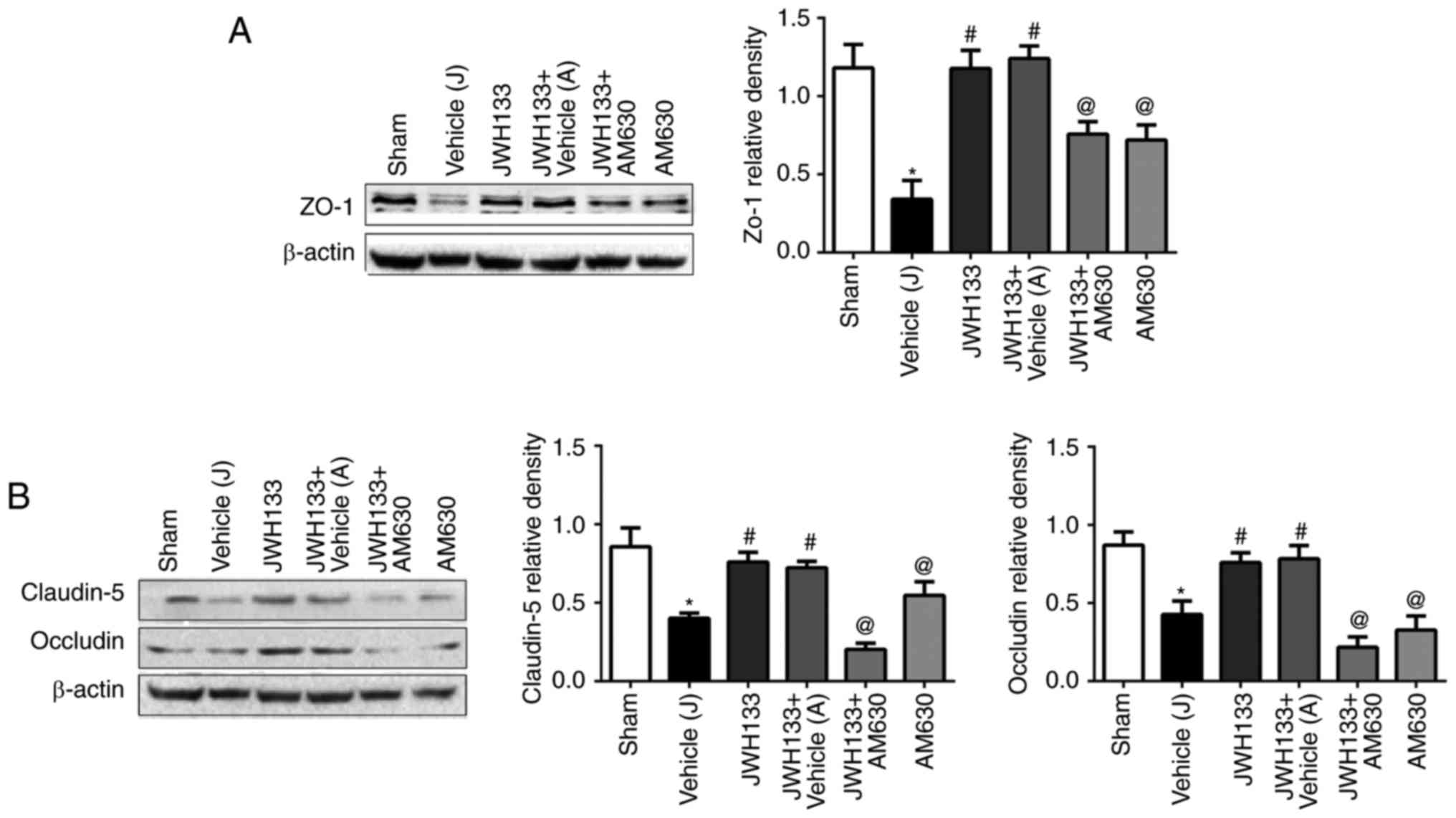
JWH133 administration prevents
ICH-induced TJ-related protein attenuation
The expression levels of the TJ proteins, ZO-1,
claudin-5 and occludin, were measured by western blotting at 24 h
post-ICH. As illustrated in Fig.
5, the results demonstrated that the protein expression levels
of ZO-1, claudin-5 and occludin in the ipsilateral basal ganglia of
rats were significantly decreased at 24 h post-ICH compared with
the sham group. JWH133 administration significantly upregulated the
protein expression levels of ZO-1, claudin-5 and occludin (Fig. 5). The combination treatment of
JWH133 with AM630 reversed ZO-1, claudin-5 and occludin protein
levels compared with the JWH133 group (Fig. 5).
Rac1 inhibition reverses the
neuroprotective effect of JWH133
In order to confirm that the neuroprotection effect
of JWH133 was mediated by Rac1, the Rac1 inhibitor, NSC23766, was
injected in the rats at 0.5 h prior to ICH. As illustrated in
Fig. 6A, NSC23766 administration
significantly decreased the JWH133-induced upregulation of GTP-Rac1
protein expression. Next, neurofunctional deficits, brain water
content, and Evans Blue permeability were detected at 24 and 72 h
post-ICH. NSC23766 administration reversed the protective effect of
JWH133 treatment, as demonstrated by significantly lower scores in
the Garcia and Corner test (Fig. 6B
and C), as well as significantly increased brain water content
in the ipsilateral basal ganglia of the brain (Fig. 6D and E). In addition, the
combination treatment of JWH133 with NSC23766 increased Evans Blue
leakage compared with the JWH133 group (Fig. 6F).
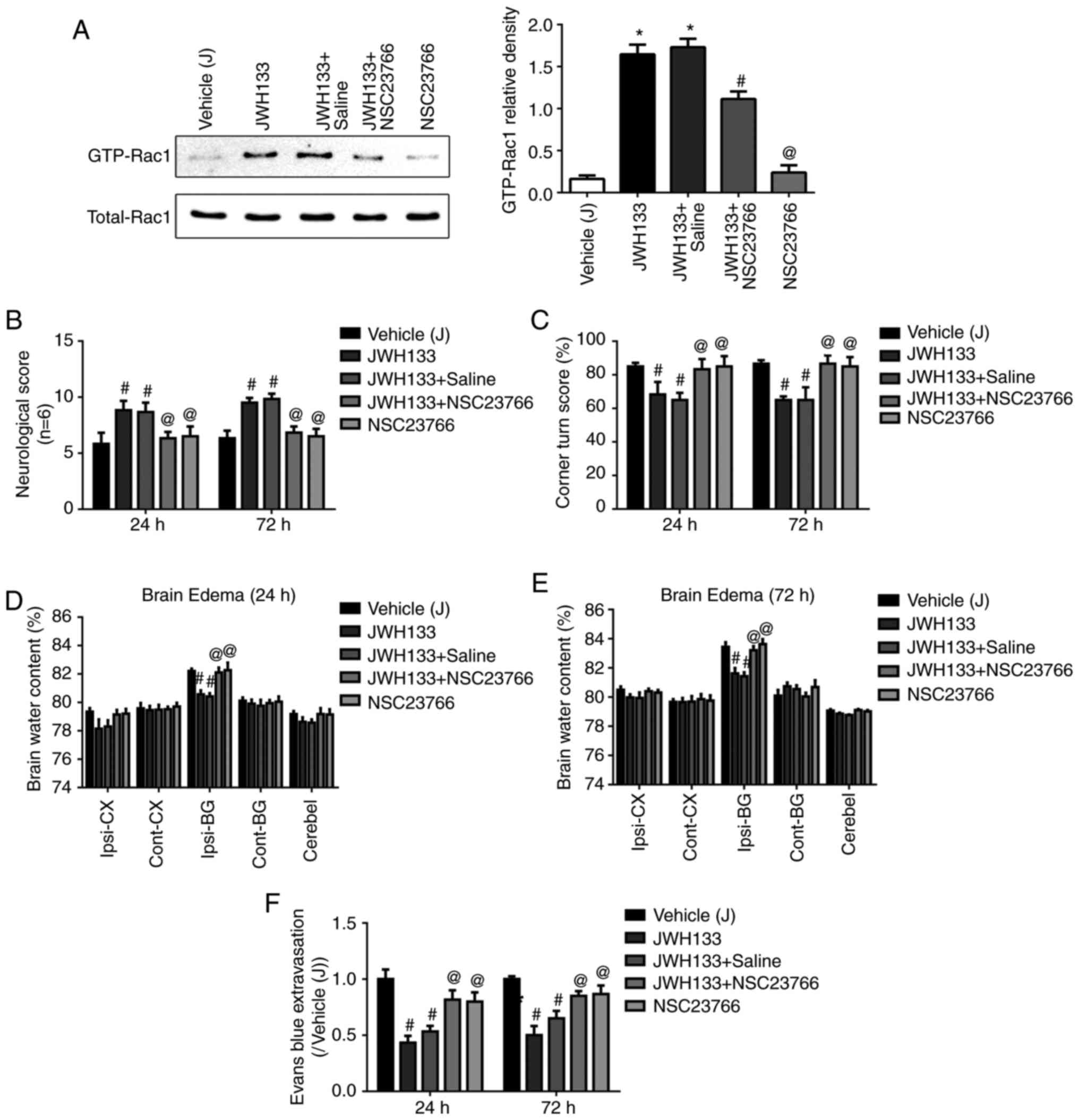 | Figure 6Rac1 inhibition reverses the
neuroprotective effect of JWH133. (A) Effects of NSC23766 and
JWH133 on Rac1-GTPase activity in rats at 24 h following ICH. (B)
Effects of NSC23766 and JWH133 on Garcia test scores, (C) Corner
test scores, (D and E) brain water content, and (F) Evans blue
extravasation in rats at 24 and 72 h following ICH. Data were
expressed as mean ± standard deviation (n=15).
*P<0.05 vs. sham; #P<0.05 vs. Vehicle;
@P<0.05 vs. JWH133. Rac1, Ras-related C3 botulinum
toxin substrate 1; ICH, intracerebral hemorrhage; Ipsi-BG,
ipsilateral basal ganglia; Ipsi-CX, ipsilateral cortex; Cont-BG,
contralateral basal ganglia; Cont-CX, contralateral cortex;
Cerebel, cerebellum. |
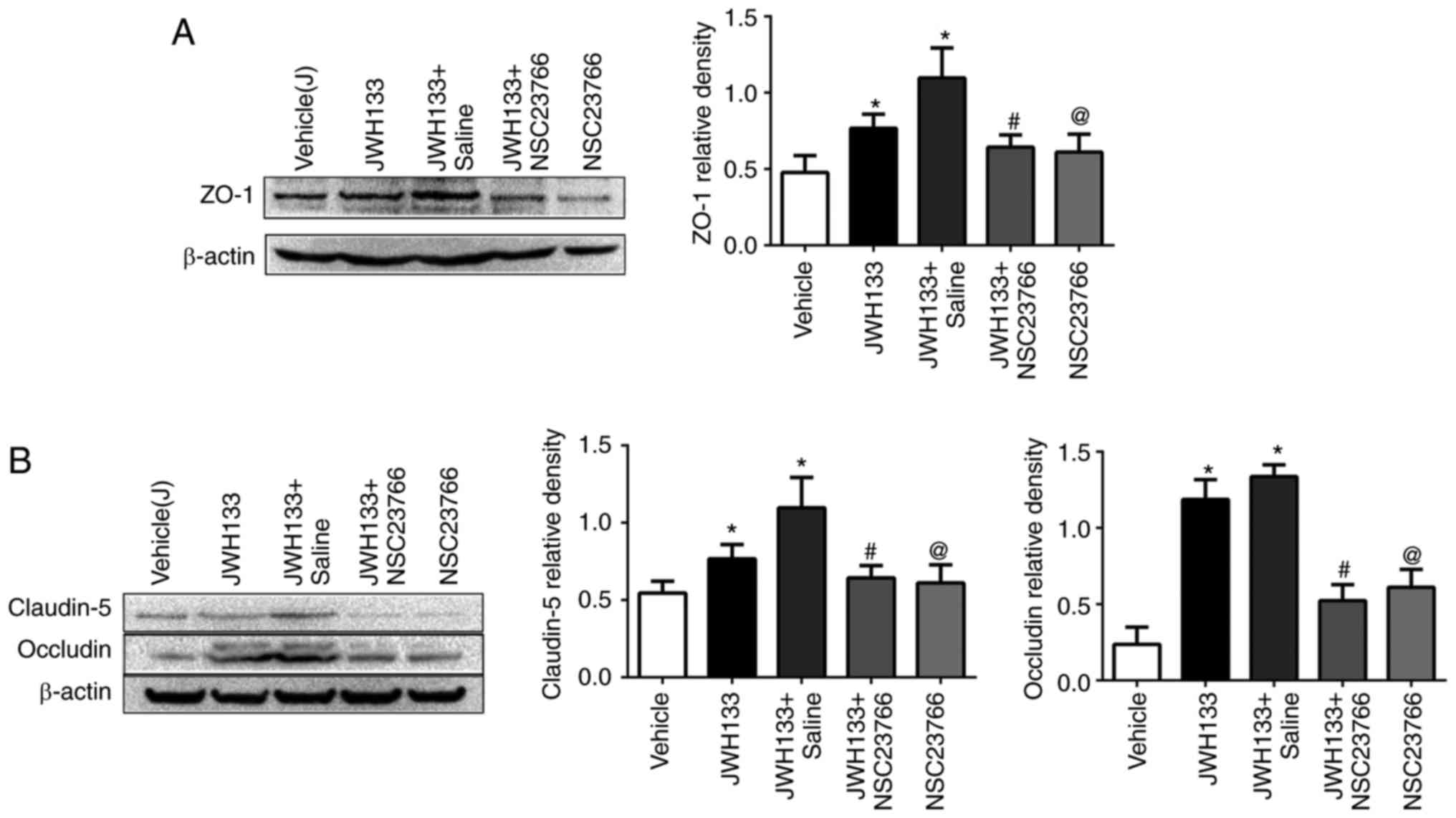
Rac1 inhibition reverses the effect of
JWH133 on TJ-related protein expression post-ICH
The results demonstrated that JWH133 treatment
increased ZO-1, claudin-5 and occludin protein expression levels at
24 h post-ICH, compared with the vehicle group (Fig. 7). However, NSC23766
administration, alone or in combination with JWH133, efficiently
decreased ZO-1, claudin-5 and occludin protein expression compared
with the JWH133 group (Fig.
7).
Discussion
In the present study, autologous blood was infused
into the caudate nucleus of rats to establish a ICH model, similar
to models that are widely used to study the mechanisms of brain
injury. This rat model was then used to investigate the effects of
CNR2 activation in ICH. The results demonstrated that JWH133
administration attenuated neurofunctional deficits and brain edema
in rats at 24 and 72 h following experimental ICH. In addition,
JWH133 treatment decreased BBB permeability at 24 and 72 h
following experimental ICH. These results suggested that
cannabinoid receptor agonists may serve a protective role following
ICH. However, whether CNR2 activation can prevent BBB disruption
and brain edema by inhibiting Rac1 activation in ICH has not been
clarified.
In addition, in the present study, the expression
levels of GTP-Rac1 and the tight junction proteins ZO-1, occludin
and claudin-5 were detected at 24 h following treatment with JWH133
alone or in combination with AM630 or NSC23766. The results
demonstrated that JWH133 treatment increased the expression levels
of ZO-1, occludin and claudin proteins, and induced activation of
Rac1 (GTP-Rac1) at 24 h following ICH. Combination treatment with
the CNR2 antagonist, AM630, or the Rac1 inhibitor, NSC23766,
reversed all the treatment effects of JWH133. These results suggest
that the neuroprotection was mediated by CNR2-induced activation of
the Rac1 pathway. However, the expression of GTP-Rac1 and the tight
junction proteins at 72 h post-treatment of JWH133 alone or
combination of AM630 or NSC23766 require further study.
Increased BBB permeability and consequent vasogenic
edema formation occurred following ICH. BBB disruption results from
ICH by influx of blood-borne cells and substances into the brain
parenchyma. This leads to early brain edema and sensorimotor
dysfunction (28,29) and ultimately results in high
morbidity and mortality rates (30). It has been previously reported
that CNR2 activation has an important role in preventing brain
edema and neuroinflammation. CNR2 agonists diminish
inflammation-induced activation of brain endothelial cells and BBB
disruption in conjunction with increased TJ protein expression
(31). A previous study reported
that JWH133 treatment increased ZO-1 expression and attenuated
neurological outcome and brain edema, protecting the BBB following
subarachnoid hemorrhage (14). In
addition, JWH133 attenuated BBB dysfunction and preserved TJ
protein expression in the rodent brain following LPS-induced
encephalitis (32). The present
findings are consistent with previous reports in which CNR2
activation by JWH133 significantly reduced BBB permeability and
increased ZO-1, occludin and claudin expression.
Rac1 is a small GTPase that belongs to the Ras
superfamily. Rac1 has been previously reported to maintain and
stabilize barrier functions of microvascular endothelial cells
(18). CNR2 agonists attenuate
the activation of ras homolog family member A (RhoA) and Rac1
during migration (33). CNR2
agonists reduce integrin activation and lamellipodia formation in
primary monocytes via inhibition of small GTPases (RhoA and Rac1)
and effects on cytoskeletal proteins (34). JWH133 also activates the
extracellular signal-regulated kinase (ERK) and phosphoinositide
3-kinase (PI3K) pathways. A previous study has reported that JWH133
induced a greater and sustained activation of ERK1/2 kinase in
lipopolysaccharide (LPS)/interferon (IFN)-γ-induced interleukin
(IL)-12p40 release from murine macrophages (35). JWH-133 treatment activates ERK1/2
and signal transducer and activator of transcription (STAT)-3 in a
mouse model of ischemia/reperfusion (36). JWH-133 induced PI3K/AKT activity
in osteoclast precursors and osteoblasts of breast cancer-induced
osteolysis (37). JWH133
increased oligodendrocyte progenitor cell proliferation by
activating PI3K/AKT (38). In the
present study, it was demonstrated that administration of JWH133
increased the GTP-Rac-1/total-Rac-1 protein expression ratio, and
the barrier protective effect of JWH133 was reversed by NSC23766
(Rac1 inhibitor). These findings suggest that Rac1 participates in
the improvement of BBB destruction and brain edema by JHH133 in
experimental ICH. In the present study, only one selective CNR2
agonist, JWH133, was selected to investigate the effects on a rat
model of ICH-induced BBB damage and its potential mechanism. The
effects of other CNR2 agonists, such as AM1241, on a rat model of
ICH require further investigation.
In conclusion, the present results demonstrated that
the CNR2 agonist JWH133 attenuated neurofunctional deficits and
brain edema, and improved the BBB dysfunction induced by ICH,
through the Rac1 pathway. Therefore, it is likely that targeting
CNR2 may be a clinically feasible approach in the future to protect
against ICH.
Funding
The present study was supported by the National Key
Basic Research Program of China, 973 Program (grant no.
2014CB541000) and the National Natural Science Foundation of China
(grant no. 31071036).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
ZW wrote the manuscript and interpreted the data. YL
and SC analyzed the data and revised the manuscript. RL searched
the literature and collected the data. GC designed the study. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Experimental protocols involving animals were
approved by the Medical Ethical Committee of the Third Military
Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Broderick JP, Adams HP Jr, Barsan W,
Feinberg W, Feldmann E, Grotta J, Kase C, Krieger D, Mayberg M,
Tilley B, et al: Guidelines for the management of spontaneous
intracerebral hemorrhage: A statement for healthcare professionals
from a special writing group of the stroke council, American Heart
Association. Stroke. 30:905–915. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qureshi AI, Mendelow AD and Hanley DF:
Intracerebral haemorrhage. Lancet. 373:1632–1644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sweeney MD, Sagare AP and Zlokovic BV:
Blood-brain barrier breakdown in Alzheimer disease and other
neurodegenerative disorders. Nat Rev Neurol. 14:133–150. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guan JX, Sun SG, Cao XB, Chen ZB and Tong
ET: Effect of thrombin on blood brain barrier permeability and its
mechanism. Chin Med J (Engl). 117:1677–1681. 2004.
|
|
5
|
Ballabh P, Braun A and Nedergaard M: The
blood-brain barrier: An overview: Structure, regulation, and
clinical implications. Neurobiol Dis. 16:1–13. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bazzoni G and Dejana E: Endothelial
cell-to-cell junctions: Molecular organization and role in vascular
homeostasis. Physiol Rev. 84:869–901. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luh C, Kuhlmann CR, Ackermann B,
Timaru-Kast R, Luhmann HJ, Behl C, Werner C, Engelhard K and Thal
SC: Inhibition of myosin light chain kinase reduces brain edema
formation after traumatic brain injury. J Neurochem. 112:1015–1025.
2010. View Article : Google Scholar
|
|
8
|
Kahles T, Luedike P, Endres M, Galla HJ,
Steinmetz H, Busse R, Neumann-Haefelin T and Brandes RP: NADPH
oxidase plays a central role in blood-brain barrier damage in
experimental stroke. Stroke. 38:3000–3006. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu JY and Chen C: Endocannabinoids in
synaptic plasticity and neuroprotection. Neuroscientist.
21:152–168. 2015. View Article : Google Scholar
|
|
10
|
Devane WA, Dysarz FR III, Johnson MR,
Melvin LS and Howlett AC: Determination and characterization of a
cannabinoid receptor in rat brain. Mol Pharmacol. 34:605–613.
1988.PubMed/NCBI
|
|
11
|
Matsuda LA, Lolait SJ, Brownstein MJ,
Young AC and Bonner TI: Structure of a cannabinoid receptor and
functional expression of the cloned cDNA. Nature. 346:561–564.
1990. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haugh O, Penman J, Irving AJ and Campbell
VA: The emerging role of the cannabinoid receptor family in
peripheral and neuro-immune interactions. Curr Drug Targets.
17:1834–1840. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chi OZ, Barsoum S, Grayson J, Hunter C,
Liu X and Weiss HR: Effects of cannabinoid receptor agonist WIN
55,212-2 on blood-brain barrier disruption in focal cerebral
ischemia in rats. Pharmacology. 89:333–338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fujii M, Sherchan P, Krafft PR, Rolland
WB, Soejima Y and Zhang JH: Cannabinoid type 2 receptor stimulation
attenuates brain edema by reducing cerebral leukocyte infiltration
following subarachnoid hemorrhage in rats. J Neurol Sci.
342:101–106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tao Y, Tang J, Chen Q, Guo J, Li L, Yang
L, Feng H, Zhu G and Chen Z: Cannabinoid CB2 receptor stimulation
attenuates brain edema and neurological deficits in a germinal
matrix hemorrhage rat model. Brain Res. 1602:127–135. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Amenta PS, Jallo JI, Tuma RF and Elliott
MB: A cannabinoid type 2 receptor agonist attenuates blood-brain
barrier damage and neurodegeneration in a murine model of traumatic
brain injury. J Neurosci Res. 90:2293–2305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gerhard R, John H, Aktories K and Just I:
Thiol-modifying phenylarsine oxide inhibits guanine nucleotide
binding of Rho but not of Rac GTPases. Mol Pharmacol. 63:1349–1355.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang B, Krafft PR, Ma Q, Rolland WB,
Caner B, Lekic T, Manaenko A, Le M, Tang J and Zhang JH: Fibroblast
growth factors preserve blood-brain barrier integrity through RhoA
inhibition after intracerebral hemorrhage in mice. Neurobiol Dis.
46:204–214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Persidsky Y, Heilman D, Haorah J,
Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K and
Ikezu T: Rho-mediated regulation of tight junctions during monocyte
migration across the blood-brain barrier in HIV-1 encephalitis
(HIVE). Blood. 107:4770–4780. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang J, Chen Q, Guo J, Yang L, Tao Y, Li
L, Miao H, Feng H, Chen Z and Zhu G: Minocycline attenuates
neonatal germin al-matrix-hemorrhage-induced neuroinflammation and
brain edema by activating cannabinoid receptor 2. Mol Neurobiol.
53:1935–1948. 2016. View Article : Google Scholar
|
|
22
|
Yang F, Wang Z, Zhang JH, Tang J, Liu X,
Tan L, Huang QY and Feng H: Receptor for advanced glycation
end-product antagonist reduces blood-brain barrier damage after
intracerebral hemorrhage. Stroke. 46:1328–1336. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garcia JH, Wagner S, Liu KF and Hu XJ:
Neurological deficit and extent of neuronal necrosis attributable
to middle cerebral artery occlusion in rats. Statistical
validation. Stroke. 26:627–635. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Blizzard KK, Zeng Z, DeVries AC,
Hurn PD and McCullough LD: Chronic behavioral testing after focal
ischemia in the mouse: Functional recovery and the effects of
gender. Exp Neurol. 187:94–104. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Manaenko A, Chen H, Kammer J, Zhang JH and
Tang J: Comparison Evans Blue injection routes: Intravenous versus
intraperitoneal, for measurement of blood-brain barrier in a mice
hemorrhage model. J Neurosci Methods. 195:206–210. 2011. View Article : Google Scholar
|
|
26
|
Ma Q, Huang B, Khatibi N, Rolland WN II,
Suzuki H, Zhang JH and Tang J: PDGFR-alpha inhibition preserves
blood-brain barrier after intracerebral hemorrhage. Ann Neurol.
70:920–931. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheng CC, Lai YC, Lai YS, Hsu YH, Chao WT,
Sia KC, Tseng YH and Liu YH: Transient knockdown-mediated
deficiency in plectin alters hepatocellular motility in association
with activated FAK and Rac1-GTPase. Cancer Cell Int. 15:292015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang L, Fan W, Cai P, Fan M, Zhu X, Dai Y,
Sun C, Cheng Y, Zheng P and Zhao BQ: Recombinant ADAMTS13 reduces
tissue plasminogen activator-induced hemorrhage after stroke in
mice. Ann Neurol. 73:189–198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vose LR, Vinukonda G, Jo S, Miry O,
Diamond D, Korumilli R, Arshad A, Zia MT, Hu F, Kayton RJ, et al:
Treatment with thyroxine restores myelination and clinical recovery
after intra-ventricular hemorrhage. J Neurosci. 33:17232–17246.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou Y, Wang Y, Wang J, Anne Stetler R and
Yang QW: Inflammation in intracerebral hemorrhage: From mechanisms
to clinical translation. Prog Neurobiol. 115:25–44. 2014.
View Article : Google Scholar
|
|
31
|
Yang MC, Zhang HZ, Wang Z, You FL and Wang
YF: The molecular mechanism and effect of cannabinoid-2 receptor
agonist on the blood-spinal cord barrier permeability induced by
ischemia-reperfusion injury. Brain Res. 1636:81–92. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aijaz S, Balda MS and Matter K: Tight
junctions: Molecular architecture and function. Int Rev Cytol.
248:261–298. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kurihara R, Tohyama Y, Matsusaka S, Naruse
H, Kinoshita E, Tsujioka T, Katsumata Y and Yamamura H: Effects of
peripheral cannabinoid receptor ligands on motility and
polarization in neutrophil-like HL60 cells and human neutrophils. J
Biol Chem. 281:12908–12918. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rom S, Zuluaga-Ramirez V, Dykstra H,
Reichenbach NL, Pacher P and Persidsky Y: Selective activation of
cannabinoid receptor 2 in leukocytes suppresses their engagement of
the brain endothelium and protects the blood-brain barrier. Am J
Pathol. 183:1548–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Correa F, Mestre L, Docagne F and Guaza C:
Activation of cannabinoid CB2 receptor negatively regulates
IL-12p40 production in murine macrophages: Role of IL-10 and ERK1/2
kinase signaling. Br J Pharmacol. 145:441–448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Montecucco F, Lenglet S, Braunersreuther
V, Burger F, Pelli G, Bertolotto M, Mach F and Steffens S: CB
cannabinoid receptor activation is cardioprotective in a mouse
model of ischemia/reperfusion. J Mol Cell Cardiol. 46:612–620.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sophocleous A, Marino S, Logan JG, Mollat
P, Ralston SH and Idris AI: Bone cell-autonomous contribution of
type 2 cannabinoid receptor to breast cancer-induced osteolysis. J
Biol Chem. 290:22049–22060. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gomez O, Sanchez-Rodriguez MA,
Ortega-Gutierrez S, Vazquez-Villa H, Guaza C, Molina-Holgado F and
Molina-Holgado E: A basal tone of 2-arachidonoylglycerol
contributes to early oligodendrocyte progenitor proliferation by
activating phosphatidylinositol 3-Kinase (PI3K)/AKT and the
mammalian target of rapamycin (MTOR) pathways. J Neuroimmune
Pharmacol. 10:309–317. 2015. View Article : Google Scholar : PubMed/NCBI
|