Introduction
Rheumatoid arthritis (RA) is an inflammatory disease
characterized by intra-articular decreases in pH, aberrant
hyaluronan regulation and destruction of bone and cartilage
(1). It has been reported that
the severity of pain and joint damage correlates with the degree of
acidity in the synovial fluid from arthritic joints (2). Due to the fact that chondrocytes,
the only cell type present in articular cartilage, are important in
the pathogenesis of arthritis and are profoundly affected by local
pH (3), articular chondrocytes
were selected to examine the pathogenesis of RA in vitro in
the present study.
The acid-sensing ion channel (ASIC) is a member of
the degenerin/Na+ channel superfamily, and is an
insensitive cation channel activated by extracellular protons
(4). The ASIC family in mammals
includes four genes, encoding seven subtypes, in which ASIC1a is
the only subunit for the transport of Ca2+ (5-7).
In addition to the role of synaptic plasticity, the activation and
sensitization of ASIC1a is involved in acidosis-induced ischemic
brain damage caused by Ca2+ influx in neurons (8). Our previous studies have shown that
ASIC1a is involved in the injury of articular chondrocytes caused
by increased intracellular calcium ([Ca2+]i)
induced by acidosis (9,10). Furthermore, the inhibition of
ASIC1a was reported to confer a protective effect on articular
cartilage in adjuvant arthritis rats (10). Therefore, in the present study,
the role of ASIC1a in the acid-induced activation of articular
chondrocyte autophagy was further investigated.
Autophagy, a cellular self-digestion process, is an
essential, conserved, lysosomal degradation pathway that controls
the quality of the cytoplasm by eliminating protein aggregates and
damaged organelles (11). Low
levels of autophagic activity are commonly observed under normal
conditions, presumably preserving normal cellular homeostasis
(12). In addition to its vital
homeostatic role, this degradation pathway is involved in various
human disorders, including metabolic disease, neurodegenerative
diseases, cancer and inflammatory diseases (13-16). It has been reported that autophagy
can be induced by different extracellular or intracellular stress
and signals, including nutrient depletion, hypoxia, growth factor
deprivation, endoplasmic reticulum (ER) stress, the accumulation of
unfolded proteins, heat shock and microbial infection (17). A previous study indicated that
autophagy may protect cells from acidosis-induced cell damage
(18). In addition, autophagy was
reported to be activated in osteoarthritis models (19). However, whether autophagy can be
induced by acidic stimulation in rat articular chondrocytes in
vitro remains to be fully elucidated. Three autophagy-related
proteins, microtubule-associated protein 1 light chain 3II (LC3II),
uncoordinated-51 like kinase 1 (ULK1) and Beclin1, were selected as
markers of the extent of autophagy in the present study.
Additionally, it has been identified that influx of Ca2+
is closely associated with autophagy (20). The activation of
Ca2+-permeable ASIC1a was shown to be responsible for
acidosis-mediated ischemic brain injury caused by Ca2+
influx in neurons (7). Based on
these findings, the present study aimed to investigate whether the
inhibition of ASIC1a was involved in the activation of autophagy
through influencing Ca2+ influx.
Mammalian target of rapamycin (mTOR) is a
serine/threonine protein kinase that regulates cell growth,
proliferation, motility, survival, protein synthesis and
transcription. Substantial evidence indicates that mTOR functions
as a negative regulator of autophagy (21). In addition, rapamycin, an mTOR
inhibitor, has been shown to increase autophagy in several cell
types, including chondrocytes (22-24). Previous studies have indicated
that the calcium/calmodulin-dependent protein kinases, a family of
serine/threonine kinases responsive to intracellular
Ca2+ concentration, may have regulatory roles in
autophagy. CaMKKβ, an important member of the family, may function
as an upstream kinase for adenosine 5′-monophosphate
(AMP)-activated protein kinase (AMPK) and regulate autophagy in
response to elevations in cytosolic calcium through B-cell lymphoma
2 (25). It has been shown that
AMPK, by inducing tuberous sclerosis complex 1/2-Rheb inhibition of
mTOR, is also important in chondrocyte autophagy (26,27). Considering the aforementioned
results, these proteins may be involved in acid-induced
autophagy.
In the present study, in order to examine the
potential effect of ASIC1a on acid-induced autophagy and the
related underlying mechanisms, inhibition of ASIC1a was achieved
using small interfering (si) RNA technology or the inhibitor
psalmotoxin-1 (PcTX1). The expression levels of autophagy markers,
including LC3II, ULK1 and Beclin1, were evaluated using western
blot analysis and reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) analysis. In addition, intracellular
calcium ([Ca2+]i) was analyzed using a
Ca2+-imaging method. The protein expression levels of
CaMKKβ, AMPK and mTOR were also observed by western blot
analysis.
Materials and methods
Cell isolation and culture
A total of 10 male, 2-month-old Sprague-Dawley (SD)
rats (160-180 g) were purchased from Anhui Experimental Animal
Center of China [animal license no. SYXK (Wan) 2012-006]. They were
housed five per cage (43 cm long ×31 cm wide ×19 cm high) with
access to food and water ad libitum, and maintained under a
12:12 h light/dark cycle. The ambient temperature was maintained at
21-22°C with 50-60% relative humidity. All experiments performed on
animals were approved by the Animal Ethics Committee and complied
with the Principles of Laboratory Animal Use and Care of Animal
Ethics Committee of Anhui Medical University (Hefei, China;
LLSC20140039).
Rat articular cartilage chondrocytes were obtained
from the SD rats as described previously (28). Cartilage from the knee joint was
cut into small sections (~1 mm3) and initially digested
with 0.2% collagenase type II (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) in phosphate-buffered saline (PBS). Following
digestion, the cells were centrifuged at 323 × g for 15 min at 4°C
and washed three times with PBS. The freshly isolated chondrocytes
were plated at 2×104 cells/well in plastic dishes filled
with culture medium [Dulbecco’s modified Eagle’s medium (DMEM),
supplemented with 2 mM glutamine, 100 IU/ml penicillin, 100 mg/ml
streptomycin and 10% fetal calf serum]. The cultures were
maintained under sterile conditions at 37°C in a humidified air
atmosphere with 5% CO2 for up to 5 days.
Antibodies and reagents
Psalmotoxin-1 (PcTX1) was obtained from Alomone
Labs, Ltd. (Jerusalem, Israel). BAPTA-AM was obtained from Dojindo
Molecular Technologies, Inc. (Kumamoto, Japan). AMPK (cat. no.
bsm-33447R), phosphorylated (p-) AMPK (cat. no. bs-3027R) and
CaMKKβ (cat. no. bs-6253R) antibodies were obtained from
Biosynthesis Biotechnology Co., Ltd. (Beijing, China). mTOR (cat.
no. sc-1550-R), p-mTOR (cat. no. sc-293133) and β-actin (cat. no.
sc-517582) antibodies were obtained from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). ASIC1a (cat. no. SAB2108751) antibody
was obtained from EMD Millipore (Billerica, MA, USA). Lipofectamine
2000 and TRIzol reagent were purchased from Invitrogen Life
Technologies; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
DMEM, fetal calf serum, and Opti-MEM were purchased from Gibco;
Thermo Fisher Scientific, Inc.
siRNA-mediated suppression of ASIC1a
siRNA was utilized to suppress ASIC1a. The following
phosphorylated oligonucle-otides were used: ASIC1a (rat), forward
5′-CCC UUC AAC AUG CGU GAA UTT-3′ and reverse 5′-AUU CAC GCA UGU
UGA AGG GTT-3′; negative control, forward 5′-UUC UCC GAA CGU GUC
ACG UTT-3′ and reverse 5′-ACG UGA CAC GUU CGG AGA ATT-3′.
The expression levels were verified by RT-qPCR and western blot
analyses.
Acridine orange (AO) staining
Acidic vesicles (autophagolysosomes) were visualized
by supravital staining with AO (1 mM; Sigma; Merck KGaA). At the
indicated time points, cells mounted on microscope slides were
washed with PBS and placed in a trough with AO working solution (2
μg/ml). Following staining at 37°C for 15 min, the dishes
were washed gently with PBS and then examined under an inverted
fluorescent microscope (Olympus IX 83; Olympus Corporation, Tokyo,
Japan) with an emission wavelength of 405 nm. Depending on their
acidity, the autophagolysosomes appeared as orange/red fluorescent
cytoplasmic vesicles, whereas nuclei were stained green. The
accumulation of acidic vesicles was quantified as the red/green
fluorescence ratio.
[Ca2+]i
measurements
Intracellular Ca2+ imaging was performed
as previously described (29).
The cells were washed three times with D-Hank’s solution and
incubated with 4 μm Fluo-3-AM and 0.02% Pluronic F-127
(Biotium, Inc., Fremont, CA, USA) for 30 min at 37°C. Following
incubation, the cells were washed three times with Hank’s solution
at 25°C to remove the extracellular Fluo-3-AM. The cells were then
perfused, initially with D-Hank’s solution and PcTX1 and then with
buffer containing acid (pH 6.0). In order to eliminate the effects
of voltage-gated Ca2+ channels and Ca2+
release from intracellular stores, nimodipine (5 μM),
x-conotoxin MVIIC (3 μM) and 1 μM thapsigargin were
added to the extracellular fluid. The fluorescence of intracellular
FLU-3 was quantified by confocal laser scanning fluorescence
microscopy with an excitation wavelength of 488 nm and an emission
wavelength of 525 nm.
Quantitation of GFP-LC3-positive
cells
The rat articular chondrocytes were seeded at a
density of 3×105 cells/well into a 6-well plate
overnight. Subsequently, the GFP-LC3 plasmid (5 μl) was
transfected into cells with 5 μl Lipofectamine 2000
according to the manufacturer’s protocol. At 24 h
post-transfection, the cells were stimulated with acid for 3 h at
37°C, following which the cells were placed in DMEM supplemented
with 10% fetal calf serum and incubated for 4 h. The chondrocytes
were transfected with the GFP-LC3 plasmid and positive cells
expressed a green florescent punctate pattern, which indicated
autophagosome formation. Micrographs were captured on an Olympus
confocal laser scanning microscope (Olympus Corporation) and the
percentage of fluorescent cells was assessed.
RT-qPCR analysis
Total RNA was prepared using TRIzol reagent and
evaluated by a One Drop OD-1000 spectrophotometer (Nanjing Wuyi
Technology Co., Ltd., Nanjing, China). The primers were designed
and synthesized by Invitrogen (Thermo Fisher Scientific, Inc.),
according to the serial number from GenBank (Table I). Total RNA (500 ng) was reverse
transcribed using a first-strand cDNA kit (Fermentas; Thermo Fisher
Scientific, Inc.) into cDNA, according to the manufacturer’s
protocol and analyzed via qPCR using a SYBR-Green PCR Master Mix
(Takara Biotechnology Co., Ltd., Dalian, China) on a Step One
platform (Applied Biosystems; Thermo Fisher Scientific, Inc.). qPCR
was performed in a 25 μl volume for 35 cycles (40 sec at
95°C; 30 sec at 54°C; and 30 sec at 72°C). GAPDH was used as an
internal control for all samples. The relative amount of the target
gene was calculated using the 2−ΔΔCq method (30).
 | Table IPrimers of liver
fibrosis-related gene amplified by reverse
transcription-polymerase chain reaction. |
Table I
Primers of liver
fibrosis-related gene amplified by reverse
transcription-polymerase chain reaction.
| Primer | Primer
sequence | Product length
(bp) |
|---|
| ASICla | F:
5′-GGACACACAGATGGCTGATGAAA-3′
R: 5′-GTGTGTCCCCACACAGGCAAATA-3′ | 333 |
| Beclinl | F:
5′-CGTGGAGAAAGGCAAGATTGAAGA-3′
R: 5′-GTGAGGACACCCAAGCAAGACC-3′ | 146 |
| ULK1 | F:
5′-CCCAGCAACATCCGAGTCAAGA-3′
R: 5′-CAGGTCAGCCTTCCCATCGTAGT-3′ | 147 |
| GAPDH | F:
5′-CAACGGGAAACCCATCACCA-3′
R: 5′-ACGCCAGTAGACTCCACGACAT-3′ | 96 |
Western blot analysis
The cells were washed twice with ice-cold PBS and
lysed in buffer for 20-30 min on ice. The protein concentration was
measured using the Bradford assay. Equal quantities of protein
lysates (~50 μg) were separated on 10% SDS polyacrylamide
gels and electrophoretically transferred onto polyvinylidene
fluoride membranes. The membrane was blocked with 5% skim milk for
1 h at room temperature. The blots were probed with the appropriate
primary antibodies (ASIC1a, Beclin1, LC3, mTOR, p-mTOR, AMPKa1,
p-AMPKa1, CaMKKβ and β-actin; all 1:1,000) overnight at 4°C,
followed by incubation with horseradish
peroxidase-conjugated rabbit anti-mouse (1:10,000; cat. no.
ZB-2301; OriGene Technologies, Inc., Beijing, China) or goat
anti-rabbit IgG (1:10,000; cat. no. ZB-2301; OriGene Technologies,
Inc.) at 37°C for 2 h. The results were visualized using an ECL
assay kit (Pierce; Thermo Fisher Scientific, Inc.). Autoradiographs
were scanned using Image-Pro Plus 6.0 Imaging analysis software
(Media Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical analyses were performed using SPSS 16.0 software (SPSS,
Inc., Chicago, IL, USA). Comparisons among different treatment
groups were conducted using one-way analysis of variance followed
by LSD post hoc tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
Rat articular chondrocyte observation and
identification
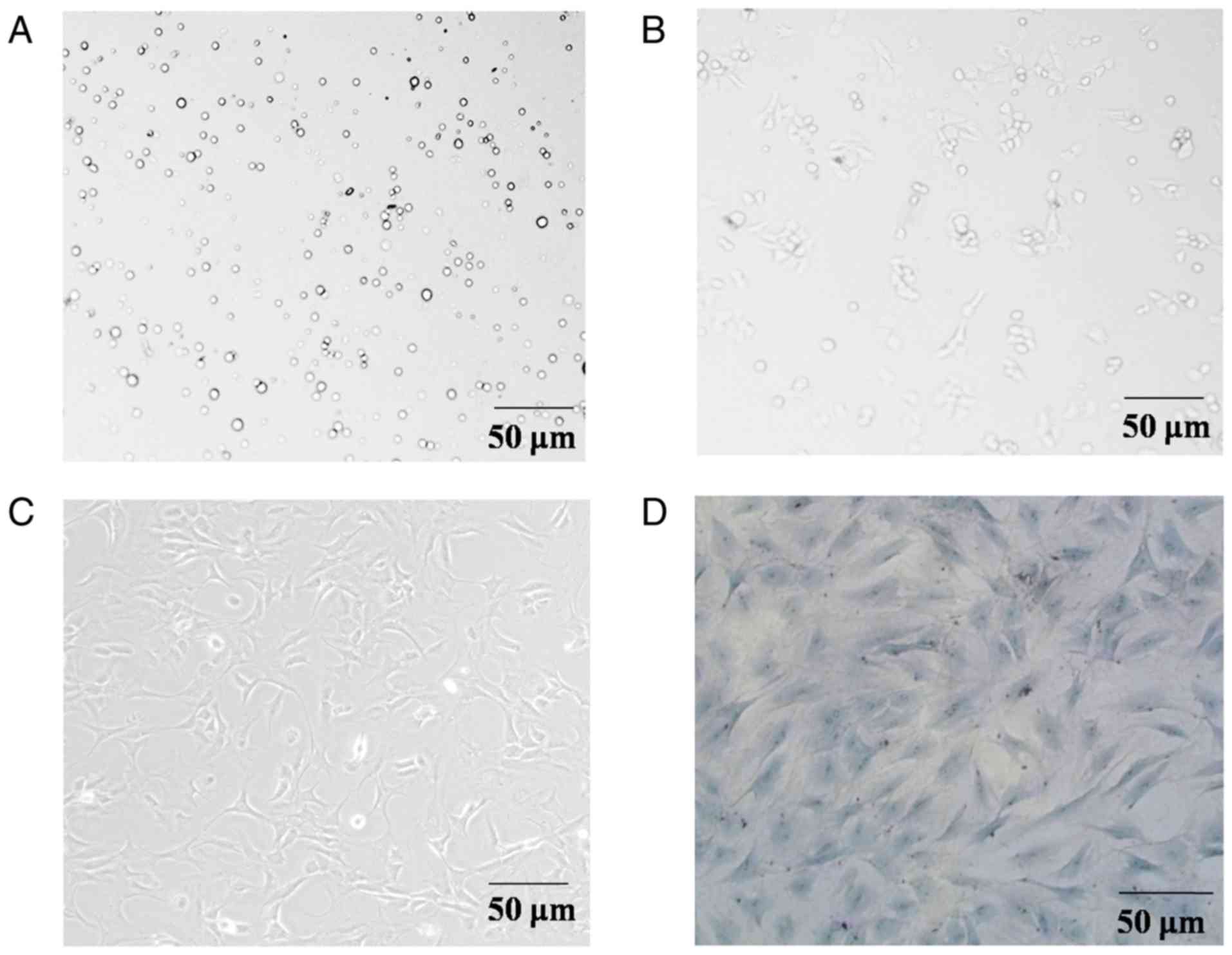
Primary rat articular chondrocytes were round or
polygonal in shape (Fig. 1A).
Following 24 h of culture, the majority of cells exhibited adherent
growth and long cytoplasmic shuttle translucent shapes (Fig. 1B). Following 72 h of cultivation,
the majority of cells were adherently extended to form protrusions
and joined into clusters (Fig.
1C). The cells were treated with toluidine blue, and the
results showed that the nuclei were stained dark blue, whereas the
cartilage cytoplasm and extracellular matrix were fuchsia on
account of their metachromatic property. In addition, the cells
exhibited a spindle shape and paving stone-like arrangement
(Fig. 1D). These results
demonstrated that the isolated cells were chondrocytes.
Extracellular acidification induces
articular chondrocyte autophagy
To investigate whether autophagy occurred in
response to extracellular acidification in the rat articular
chondrocytes, the cells were treated with acidic stimulation for
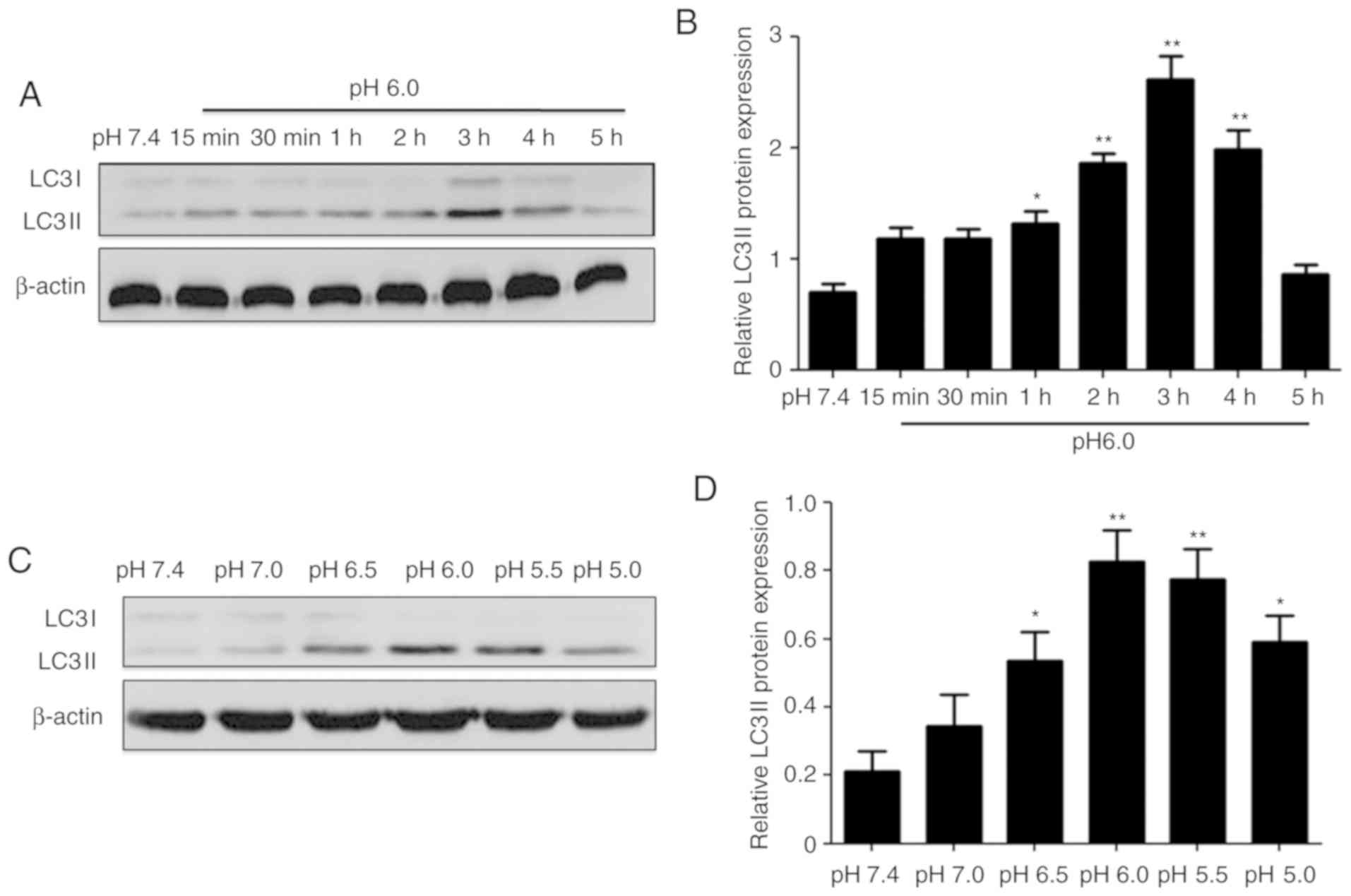
various pH and time periods. The protein expression of LC3II was
selected to represent the level of autophagy. The results showed
that extracellular acidification evidently increased the protein
levels of LC3II in a time-dependent (0-5 h; Fig. 2A and B) and pH-dependent (pH
7.4-5.0; Fig. 2C and D) manner.
These data suggested that acidic stimulation significantly
upregulated the expression of LC3II in chondrocytes, indicating
that extracellular acidification induced autophagy in rat articular
chondrocytes.
ASIC1a inhibition suppresses acid-induced
autophagy
As shown in Fig.
3A-C, extracellular acidification significantly increased the
protein levels of ASIC1a, whereas PcTX1 and ASIC1a RNA interference
(RNAi) reversed the promoting effect of extracellular acidification
on the protein (Fig. 3A and B)
and mRNA (Fig. 3C) expression
levels of ASIC1a in the articular chondrocytes.
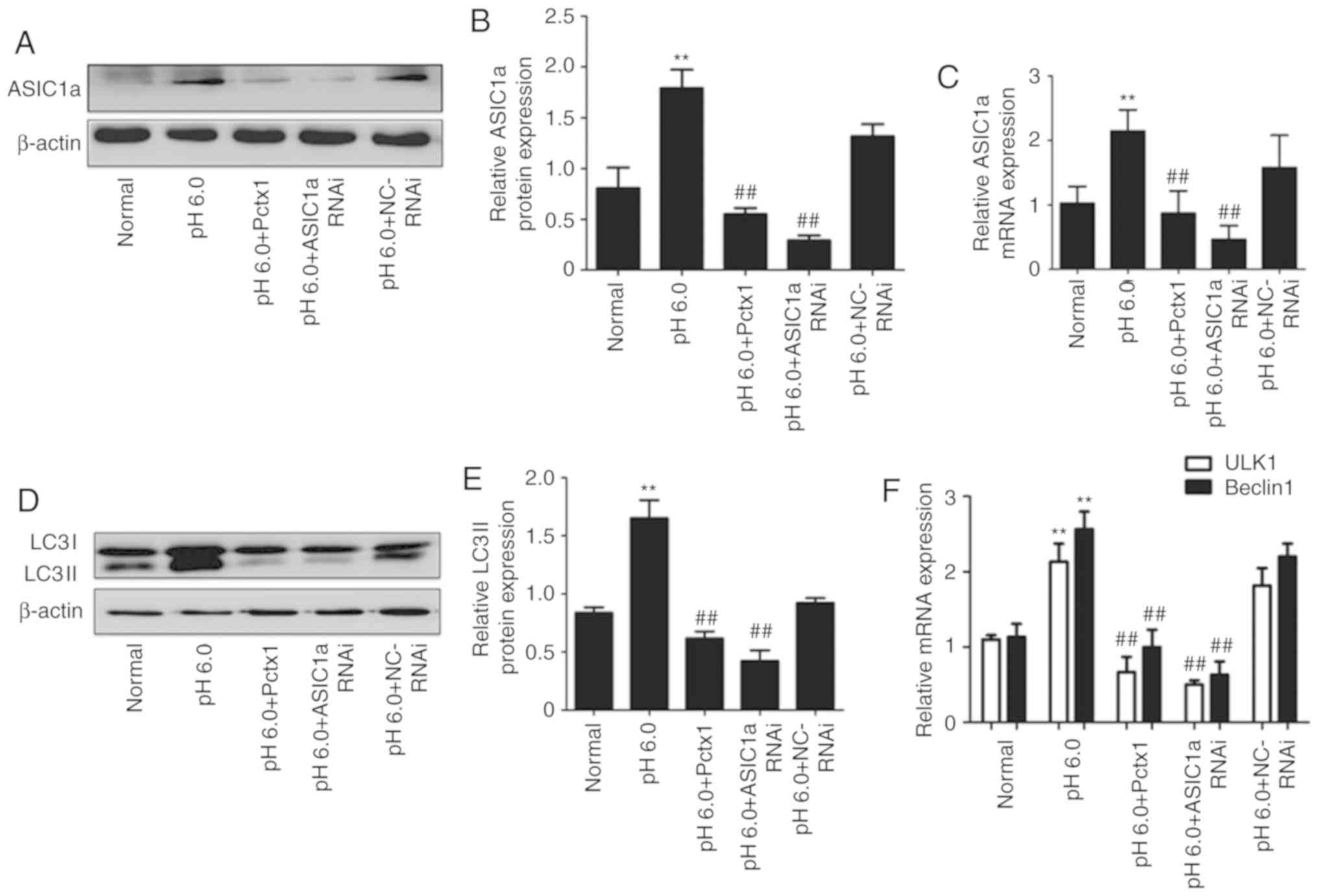 | Figure 3Effect of PcTX1 and small interfering
RNA technology on the expression of ASIC1a and the activation of
autophagy in rat articular chondrocytes. (A) Typical western blot
image of the protein expression of ASIC1a. (B) Statistical results
of the protein expression of ASIC1a. (C) Statistical results of the
mRNA expression of ASIC1a. (D) Typical western blot image of the
protein expression of LC3II. (E) Statistical results of the protein
expression of LC3II. (F) Statistical results of the mRNA expression
of Beclin1 and ULK1. (G) Articular chondrocytes were stained with
acridine orange (magnification, ×200). Left panels, nuclei (stained
green); middle panels, autophagolysosomes (stained orange-red); and
right panels, merged. (H) Statistical results of acridine orange.
Fluorescent microscopy demonstrated an increase in red fluorescence
in acid-treated chondrocytes, indicating the presence of
extracellular acidification as a marker of autophagy.
**P<0.01, vs. normal group; ##P<0.05,
vs. pH 6.0 group. ASIC1a, acid-sensing ion channel 1a; PcTX1,
psalmotoxin-1; LC3, microtubule-associated protein 1 light chain 3;
ULK1, uncoordinated-51 like kinase 1; RNAi, RNA interference; NC,
negative control. |
The results in Fig.
3D-F show the effect of PcTX1 and ASIC1a RNAi on the protein
and mRNA expression levels of autophagy markers, including LC3II
(Fig. 3D and E), Beclin1
(Fig. 3F) and ULK1 (Fig. 3F). Compared with those in the
normal group, the protein and mRNA expression levels of autophagy
markers LC3II, Beclin1 and ULK1 were upregulated in the pH 6.0
group. However, these changes were decreased in the PcTX1 and
ASIC1a RNAi groups, indicating that the inhibition of ASIC1a
suppressed acid-induced autophagy.
In addition, the state of autophagy was examined by
AO staining (Fig. 3G). The
results revealed that the autophgolysosomes appeared as orange/red
fluorescent cytoplasmic vesicles, whereas the nuclei were stained
green. The pH 6.0 group indicated a significant increase in
greenish-yellow fluorescence when compared with the normal group,
and the inhibition or silencing of ASIC1a by PcTX1 or siRNA
technology resulted in a decrease in punctate fluorescence
(Fig. 3H).
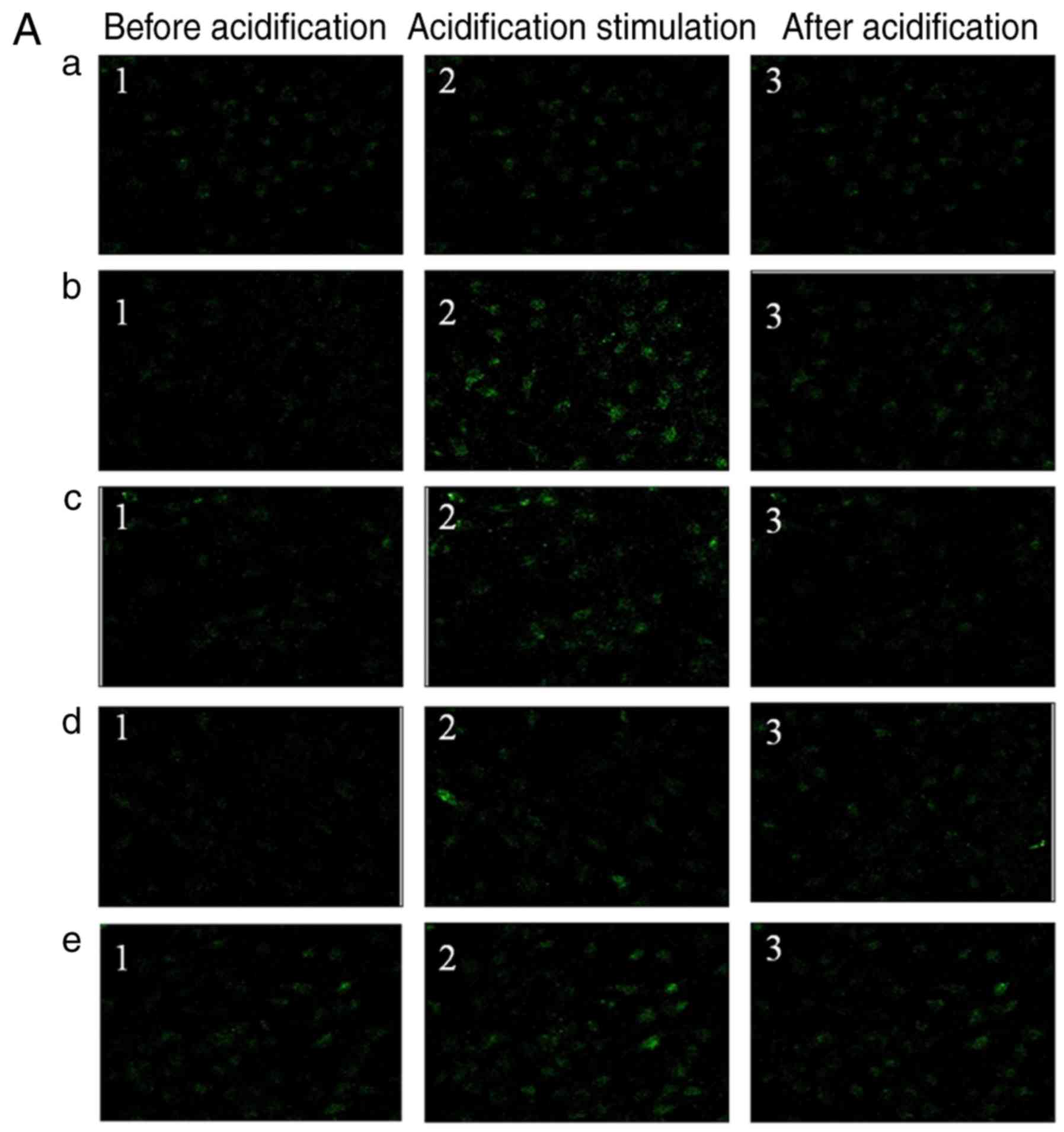
Knockdown of ASIC1a downregulates
intracellular [Ca2+]i in chondrocytes
incubated in an acidic environment
Changes in [Ca2+]i were
investigated in the articular chondrocytes incubated in an acidic
environment. In all experiments, 10 μM MK801, 5 μM
nimodipine, 3 μM x-conotoxin MVIIC and 1 μM
thapsigargin were added to inhibit the possible secondary
activation of glutamate receptors and voltage-gated Ca2+
channels and release of internal Ca2+ stores. As shown
in Fig. 4Aa-e and Ba-e,
[Ca2+]i was significantly elevated following
the application of extracellular acidification (pH 6.0) to
articular chondrocytes (Fig.
4Ba-e). However, silencing or inhibiting ASIC1a reduced the
intracellular Ca2+ concentration (Fig. 4A and B).
Ca2+ chelation inhibits
acid-induced autophagy
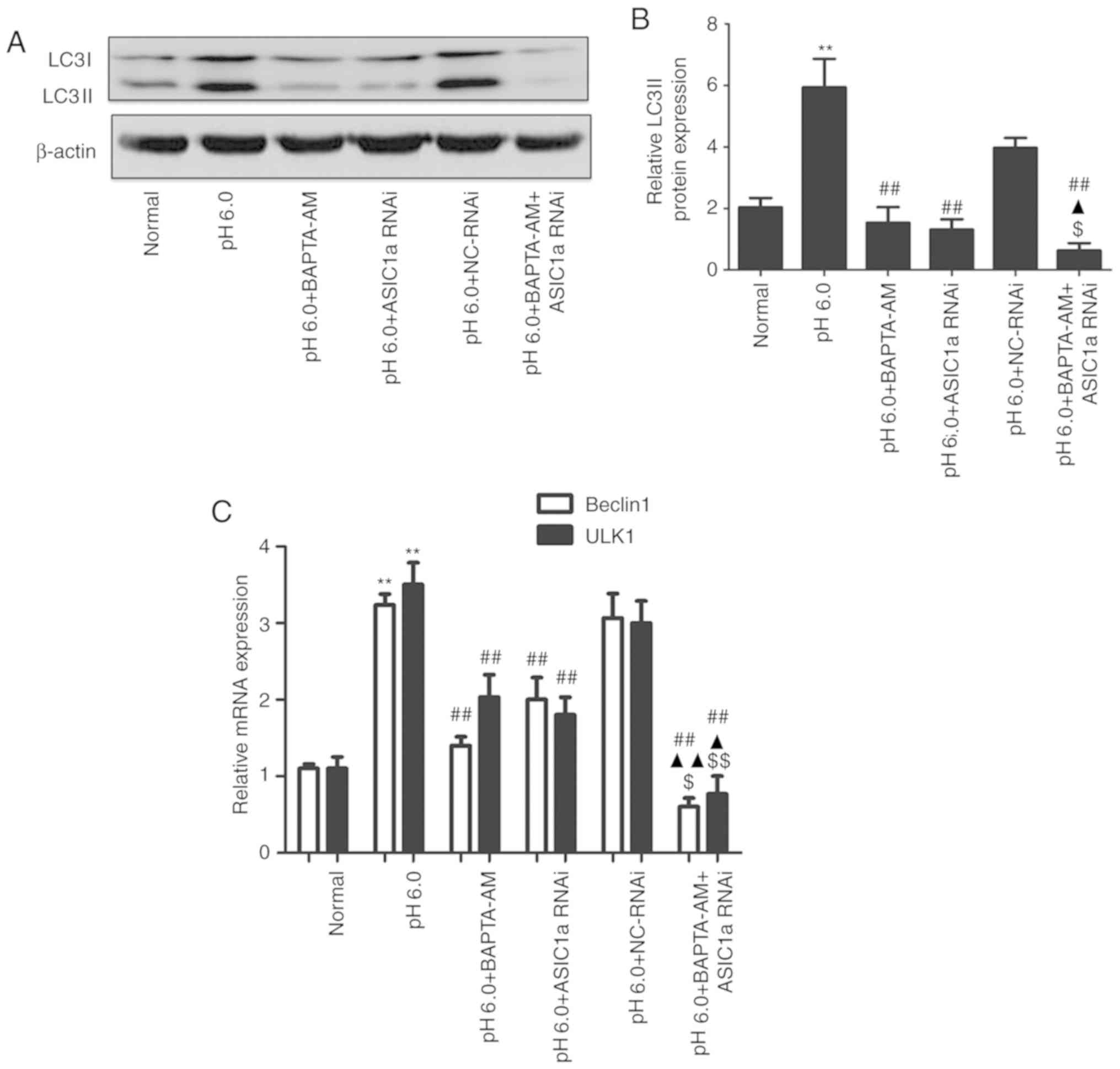
As indicated in Fig.
5A and B, compared with extracellular acidification (pH 6.0),
buffering the intracellular Ca2+ with cell-permeable
chelator BAPTA-AM eliminated the acid-induced increase in the
protein expression of LC3II. In addition, the levels of LC3II in
cells pretreated with siRNA against ASIC1a in combination with
BAPTA-AM were significantly lower compared with those in the cells
with BAPTA-AM or ASIC1a silencing alone. The mRNA expression levels
of Beclin1 and ULK1 (Fig. 5C)
followed the same trend as LC3II. The chondrocytes with
subsequently transfected with the GFP-LC3 plasmid. The results
showed that cells treated with acidic stimulation exhibited an
increase in fluorescent puncta, whereas the numbers of fluorescent
puncta decreased in the BAPTA-AM and siRNA ASIC1a groups.
The combined treatment resulted in a lower number of
LC3-positive fluorescent puncta (Fig. 5C). These results were consistent
with the protein and gene expression results, indicating that
ASIC1a and intracellular Ca2+ are required for
activation of the autophagic pathway in articular chondrocytes, and
that ASIC1a and elevated intracellular Ca2+ levels may
simultaneously serve critical roles in the regulation of
acid-induced autophagy.
CaMKKβ/AMPK/mTOR pathway is involved in
acid-induced activated articular chondrocyte autophagy
As shown in Fig.
6, compared with the normal group, increased protein levels of
CaMKKβ/β-actin (Fig. 6A and B)
and p-AMPK/AMPK (Fig. 6C and D)
were observed in the pH 6.0 group. By contrast, the protein levels
of p-mTOR/mTOR (Fig. 6E and F)
were lower than those in the normal group. These changes were
reversed in the BAPTA-AM and ASIC1a-siRNA groups. Following
combined treatment, the protein levels of CaMKKβ/β-actin and
p-AMPK/AMPK were decreased further, whereas the protein levels of
p-mTOR/mTOR were increased further.
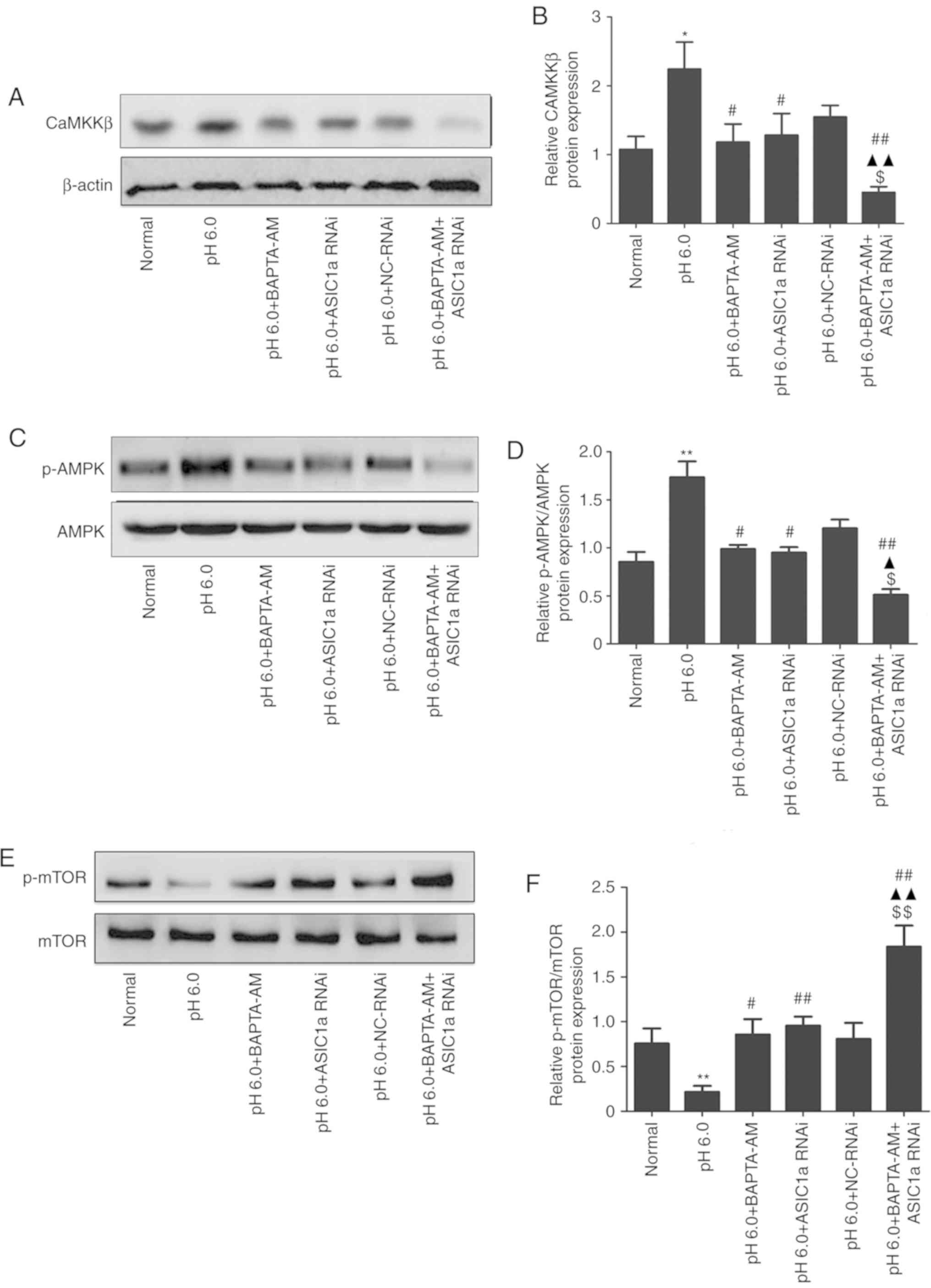 | Figure 6Effects of silencing the gene
expression of ASIC1a on acid-dependent activation of the
CaMKKβ/AMPK/mTOR pathway. (A) Typical western blot image of the
protein expression levels of CaMKKβ. (B) Statistical results of the
protein expression of CaMKKβ. (C) Typical western blot image of the
protein levels of p-AMPK/AMPK. (D) Statistical results of the
protein levels of p-AMPK/AMPK. (E) Typical western blot image of
the protein levels of p-mTOR/mTOR. (F) Statistical results of the
protein levels of p-mTOR/mTOR. *P<0.05 and
**P<0.01, vs. normal group, #P<0.05 and
##P<0.01, vs. pH 6.0 group; $P<0.01 and
$$P<0.05, vs. pH 6.0 + BAPTA-AM group;
▲P<0.01 and ▲▲P<0.05, vs. pH 6.0 +
ASIC1a RNAi group. ASIC1a, acid-sensing ion channel 1a; CaMKKβ,
Ca2+/calmodulin-dependent protein kinase kinase β; AMPK,
5′-monophosphate-activated protein kinase; mTOR, mammalian target
of rapamycin; p-, phosphorylated; RNAi, RNA interference; NC,
negative control. |
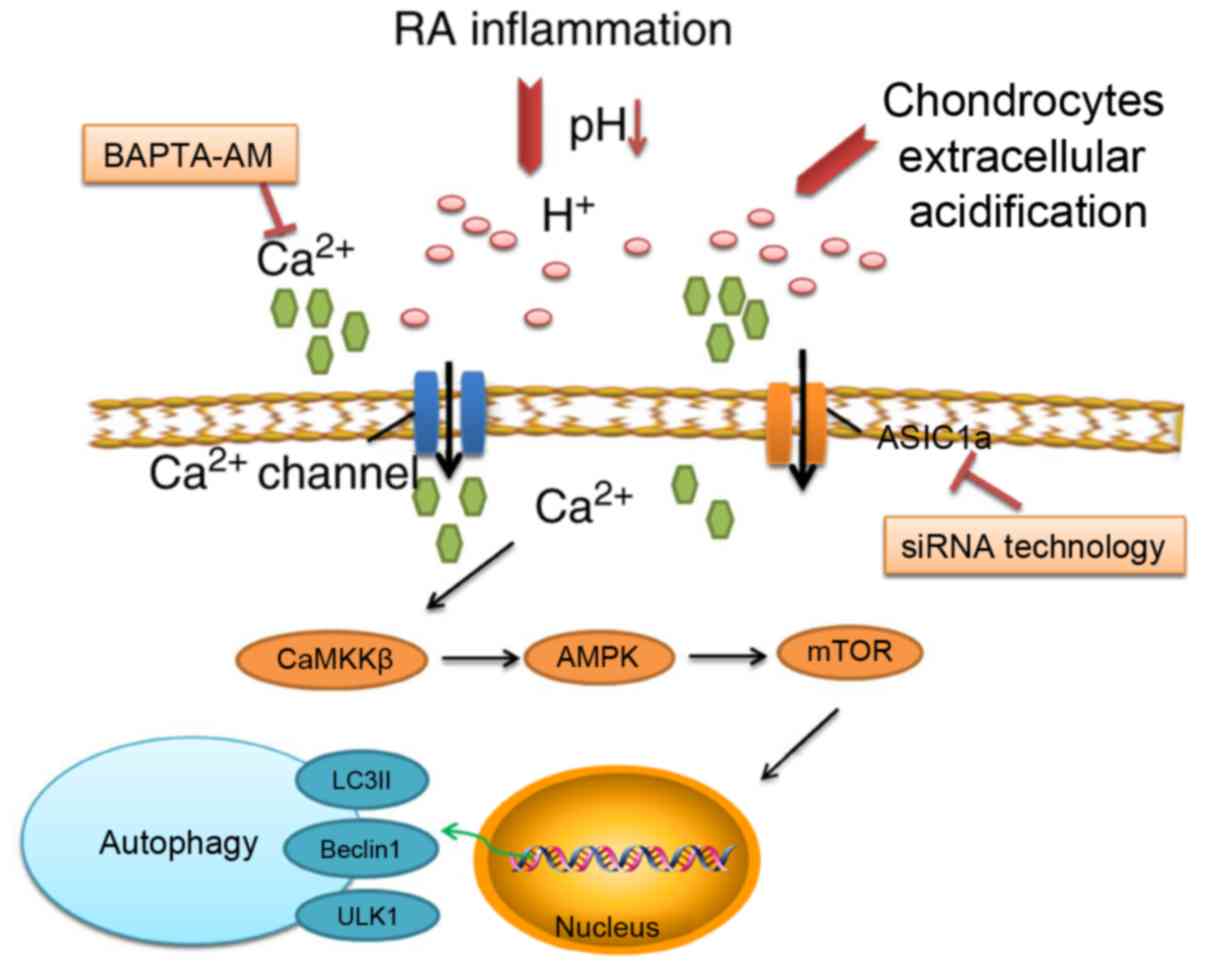
Simplified schematic representation of
the inhibition of ASIC1a-mediated signaling pathways in
autophagy
As shown in Fig.
7, the inhibition of ASIC1a attenuated the activation of
autophagy via elevated intracellular calcium levels and the
CaMKKβ/AMPK/mTOR signaling pathway.
Discussion
In the present study, it was demonstrated that
extracellular acidification induced the activation of autophagy in
a pH-and time-dependent manner in rat articular cartilage. Based on
these results, together with the fact that the inhibition of ASIC1a
had a protective effect on articular cartilage, the role of ASIC1a
in the acid-induced activation of autophagy was examined. The
results showed that inhibition of ASIC1a attenuated the activation
of autophagy via elevated intracellular calcium levels and the
CaMKKβ/AMPK/mTOR signaling pathway.
Chondrocytes, the only cell type present in
articular cartilage, have limited vascularity and exist in a low
oxygen microenvironment. They are critical in maintaining the
dynamic equilibrium between the synthesis and degradation of the
extracellular matrix. It has been reported that chondrocyte
metabolism is predominantly by anaerobic glycolysis, which produces
a large quantities of lactate molecules, rendering the pH of the
synovial tissue more acidic than the majority of other tissues
(31). As pH in the majority of
pathological conditions, including RA, tends to be ~5.5 (32), pH values of 7.0-5.0 were selected
to mimic the extracellular acidification of RA in the present
study. Based on the result that pH 6.0 induced the maximal level of
autophagy, as indicated by the expression of LC3II, the pH value of
6.0 was used to examined the effect of the inhibition of ASCS1a in
autophagy in the subsequent experiments.
Autophagy is a lysosomal degradation mechanism that
maintains cell homeostasis by transferring cell membranes into
lysosomes in double vesicles termed autophagosomes (33). Basal levels of autophagy maintain
intracellular homeostasis by removing damaged or toxic intrinsic
components (34). Autophagy is
stimulated under conditions of cellular stress. Under these
conditions, the recycling of its own material provides a cell with
building blocks that can be incorporated into newly synthesized
macromolecules for cellular anti-stress responses and energy
production to ensure survival. Autophagy is involved in various
pathological processes due to its role in these important cellular
functions (35). Although
essential for cellular homeostasis, the mechanisms regulating this
complex process, and the ramifications of any defects, remain to be
fully elucidated. Atg genes control the autophagic process, leading
to the induction and nucleation of autophagosomes and their
expansion and fusion with lysosomes. Among the Atg genes, Atg1,
Atg6 and Atg8 (ULK1, Beclin1 and LC3 in mammals, respectively) are
three major regulators of the autophagic pathway (36). In the autophagic pathway, the LC3
protein binds to phosphatidylethanolamine and is recruited to the
autophagosome membrane. This lipidated form of LC3 can be detected
as a band with an apparently lower molecular weight (LC3II)
compared with the non-lipidated, non-autophagic form (LC3I).
Therefore, the level of LC3II is an indication of the extent of
autophagy (37). In addition, the
autophagosomal proteins ULK1 and Beclin1, which initiate autophagy
and form autophagosomes, are considered to be markers of the extent
of autophagy, as described previously (38,39). Therefore, the mRNA and/or protein
expression levels of these three aforementioned markers were
measured in the present study. The results showed that the
inhibition of ASIC1a decreased the levels of these autophagy
markers, indicating that the inhibition of ASIC1a suppressed
acid-induced autophagy.
ASIC1a is a proton-gated ion channel for
Ca2+ transportation. It is expressed in the mammalian
nervous system and other tissues, in which it exerts
pathophysiological effects (40).
Our previous study indicated the presence of ASIC1a mRNA and its
protein in rat articular chondrocytes (28). In the present study, it was
observed that silencing or inhibiting ASIC1a attenuated the extent
of autophagy in rat articular chondrocytes, as indicated by the
decreased expression levels of LC3II, ULK1 and Beclin1. This
provides further evidence of an association between ASIC1a and
autophagy. Previous studies have also confirmed that the activation
or sensitization of calcium-permeable ASIC1a is responsible for the
acidosis-mediated cellular damage caused by intracellular
Ca2+ influx (41).
Consistently, in the present study, it was found that silencing or
inhibiting ASIC1a reduced the concentration of intracellular
Ca2+, again indicating that increased
[Ca2+]i, mediated via ASIC1a, may contribute
to acidosis-induced articular chondrocyte injury.
It is widely accepted that intracellular
Ca2+ signaling, as a versatile and dynamic secondary
messenger, is essential for important pathophysiological processes
in cells. Small changes in Ca2+ can affect the normal
physiological cell function. The role of Ca2+ signaling
in autophagy has been investigated extensively (42-44). However, the role of
Ca2+ signaling in the regulation of autophagy has been a
controversial issue, with reports suggesting both inhibitory
(45) and stimulatory (43) effects of Ca2+ on
autophagy. This discrepancy may be explained by the specific role
of different Ca2+ signals; a Ca2+ signal in
normal growth-promoting conditions, likely targeted towards
mitochondria, inhibits basal autophagy, whereas a different
Ca2+ signal under conditions of cellular stress can
stimulate autophagy (46). In the
present study, BAPTA-AM, a rapid intracellular Ca2+
chelating agent, was used to block Ca2+ channels
(47). Consistent with the
results of a previous study, which reported that the
BAPTA-AM-mediated chelation of intracellular Ca2+ is
involved in the regulation of autophagy (48), the results of the present study
indicated that the use of BAPTA-AM decreased the activation of
autophagy. Of note, when siRNA against ASIC1a was combined with
BAPTA-AM, the expression levels of LC3II, Beclin1 and ULK1 were
further reduced. Similar results were also identified in the
fluorescence images when the chondrocytes were transfected with the
GFP-LC3 plasmid. These results suggest that ASIC1a and
Ca2+ channels may have synergistic roles in affecting
the extent of autophagy.
A number of studies have demonstrated that mTOR is a
key mediator of growth factor signaling to autophagy (49,50). As the upstream regulatory factors
of mTOR, AMPK and CaMKKβ are also reported to be involved in the
progress of autophagy (25), the
CaMKKβ/AMPK/mTOR signaling pathway was evaluated in the present
study to examine the mechanisms of ASIC1a in autophagy. The results
showed that downregulated protein levels of p-mTOR/mTOR and
upregulated protein levels of CaMKKβ/β-actin and p-AMPK/AMPK were
reversed by the inhibition of ASIC1a, indicating that the
CaMKKβ/AMPK/mTOR signaling pathway may be involved in the role of
ASIC1a in autophagy.
The expression levels of LC3-II were significantly
decreased in the ASIC1a RNAi and BAPTA-AM groups. The expression of
LC3-II was also observed to be decreased further following combined
treatment with ASIC1a siRNA and BAPTA-AM in the present study.
These results indicated that silencing ASIC1a and the chelating of
Ca2+ by BAPTA-AM inhibited the activation of autophagy
induced by acidic stimulation. The mechanism underlying this effect
may be as follows: ASIC1a acts as a cation channel permeable to
Ca2+, but other channels also exist that can mediate
Ca2+ influx, including transient receptor potential
vanilloid channels (51) or
store-operated Ca2+ channels (52). By contrast, extracellular
acidification stimulation causes an elevation of intracellular
Ca2+ concentration, which may involve an influx from
extracellular Ca2+in addition to the release of
Ca2+ from an intracellular Ca2+ pool. The
concentration of BAPTA-AM used in the present study may have only
chelated a proportion of intracellular Ca2+. The
combination of BAPTA-AM and ASIC1a siRNA was more potent than
either treatment alone in reducing autophagy. These results suggest
that Ca2+ is an important factor in acid-induced
autophagy in articular chondrocytes, and ASIC1a may act as an
upstream regulator of autophagy by inhibiting the effects of
Ca2+.
In conclusion, the results of the present study
confirmed the presence of ASIC1a in articular chondrocyte autophagy
in an extracellular acidic environment. As a potential regulator,
ASIC1a induced an increase in intracellular calcium activated by
autophagy in acidic cells. In addition, the inhibition of ASIC1a
was found to attenuate the effects of activated autophagy through
the CaMKKβ/AMPK/mTOR signaling pathway, which provides evidence for
the involvement of ASIC1 in RA. This suggests that the role of
ASIC1a in chondrocyte autophagy is more complex than originally
thought, and may involve crosstalk with other survival strategies.
These results provide a basis for further investigation of this
potential regulator in chondrocyte autophagy.
Funding
This study was supported by the Natural Science
Foundation of China (grant no. 81271949).
Availability of data and materials
All data generated and analyzed during the present
study are included in this published article.
Authors’ contributions
WFG, YYX and FHC performed the experiments,
contributed to data analysis and wrote the manuscript. WFG, YYX,
JFG and FHC analyzed the data. FHC conceptualized the study design
and contributed experimental materials. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All experiments performed on animals were approved
by the Animal Ethics Committee and complied with the Principles of
Laboratory Animal Use and Care of Animal Ethics Committee of Anhui
Medical University (no. LLSC20140039).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
AO
|
acridine orange
|
|
ULK1
|
uncoordinated-51 like kinase 1
|
|
CaMKKβ
|
Ca2+/calmodulin-dependent
protein kinase kinase β
|
|
AMPK
|
5′-monophosphate-activated protein
kinase
|
|
mTOR
|
mammalian target of rapamycin
|
References
|
1
|
Scott DL, Wolfe F and Huizinga TW:
Rheumatoid arthritis. Lancet. 376:1094–1108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malemud CJ: Chondrocyte apoptosis in
rheumatoid arthritis: Is preventive therapy possible? Immunotherapy
(Los Angel). 1:1022015.
|
|
3
|
Chang J, Wang W, Zhang H, Hu Y, Wang M and
Yin Z: The dual role of autophagy in chondrocyte responses in the
pathogenesis of articular cartilage degeneration in osteoarthritis.
Int J Mol Med. 32:1311–1318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Waldmann R, Champigny G, Bassilana F,
Heurteaux C and Lazdunski M: A proton-gated cation channel involved
in acid-sensing. Nature. 386:173–177. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen CC, England S, Akopian AN and Wood
JN: A sensory neuron-specific, proton-gated ion channel. Proc Natl
Acad Sci USA. 95:10240–10245. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lingueglia E, de Weille JR, Bassilana F,
Heurteaux C, Sakai H, Waldmann R and Lazdunski M: A modulatory
subunit of acid sensing ion channels in brain and dorsal root
ganglion cells. J Biol Chem. 272:29778–29783. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xiong ZG, Chu XP and Simon RP:
Ca2+ -permeable acid-sensing ion channels and ischemic
brain injury. J Membr Biol. 209:59–68. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu W, Chen FH, Yuan FL, Zhang TY, Wu FR,
Rong C, Jiang S, Tang J, Zhang CC and Lin MY: Blockade of
acid-sensing ion channels protects articular chondrocytes from
acid-induced apoptotic injury. Inflamm Res. 61:327–335. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rong C, Chen FH, Jiang S, Hu W, Wu FR,
Chen TY and Yuan FL: Inhibition of acid-sensing ion channels by
amiloride protects rat articular chondrocytes from acid-induced
apoptosis via a mitochondrial-mediated pathway. Cell Biol Int.
36:635–641. 2012. View Article : Google Scholar
|
|
10
|
Yuan FL, Chen FH, Lu WG, Li X, Li JP, Li
CW, Xu RS, Wu FR, Hu W and Zhang TY: Inhibition of acid-sensing ion
channels in articular chondrocytes by amiloride attenuates
articular cartilage destruction in rats with adjuvant arthritis.
Inflamm Res. 59:939–947. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martínez-Borra J and López-Larrea C:
Autophagy and self-defense. Adv Exp Med Biol. 738:169–184. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galluzzi L, Morselli E, Vicencio JM, Kepp
O, Joza N, Tajeddine N and Kroemer G: Life, death and burial:
Multifaceted impact of autophagy. Biochem Soc Trans. 36:786–790.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He C, Zhu H, Li H, Zou MH and Xie Z:
Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances
cardiac autophagy and protects against cardiomyocyte apoptosis in
diabetes. Diabetes. 62:1270–1281. 2013. View Article : Google Scholar :
|
|
14
|
Michaud M, Martins I, Sukkurwala AQ,
Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot
G, et al: Autophagy-dependent anticancer immune responses induced
by chemotherapeutic agents in mice. Science. 334:1573–1577. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rubinsztein DC: The roles of intracellular
protein-degradation pathways in neurodegeneration. Nature.
443:780–786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vural A and Kehrl JH: Autophagy in
macrophages: Impacting inflammation and bacterial infection.
Scientifica (Cairo). 2014:8254632014.
|
|
17
|
Zhou XJ and Zhang H: Autophagy in
immunity: Implications in etiology of autoimmune/autoinflammatory
diseases. Autophagy. 8:1286–1299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wojtkowiak JW and Gillies RJ: Autophagy on
acid. Autophagy. 8:1688–1689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Caramés B, Hasegawa A, Taniguchi N, Miyaki
S, Blanco FJ and Lotz M: Autophagy activation by rapamycin reduces
severity of experimental osteoarthritis. Ann Rheum Dis. 71:575–581.
2012. View Article : Google Scholar :
|
|
20
|
Ureshino RP, Rocha KK, Lopes GS,
Bincoletto C and Smaili SS: Calcium signaling alterations,
oxidative stress, and autophagy in aging. Antioxid Redox Signal.
21:123–137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seldin MM, Lei X, Tan SY, Stanson KP, Wei
Z and Wong GW: Skeletal muscle-derived myonectin activates the
mammalian target of rapamycin (mTOR) pathway to suppress autophagy
in liver. J Biol Chem. 288:36073–36082. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu L, Feng Z, Cui S, Hou K, Tang L, Zhou
J, Cai G, Xie Y, Hong Q, Fu B and Chen X: Rapamycin upregulates
autophagy by inhibiting the mTOR-ULK1 pathway, resulting in reduced
podocyte injury. PLoS One. 8:e637992013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sarkar S, Ravikumar B, Floto RA and
Rubinsztein DC: Rapamycin and mTOR-independent autophagy inducers
ameliorate toxicity of polyglutamine-expanded huntingtin and
related proteinopathies. Cell Death Differ. 16:46–56. 2009.
View Article : Google Scholar
|
|
25
|
Høyer-Hansen M, Bastholm L, Szyniarowski
P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N,
Elling F, Rizzuto R, et al: Control of macroautophagy by calcium,
calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell.
25:193–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bohensky J, Leshinsky S, Srinivas V and
Shapiro IM: Chondrocyte autophagy is stimulated by HIF-1 dependent
AMPK activation and mTOR suppression. Pediatr Nephrol. 25:633–642.
2010. View Article : Google Scholar :
|
|
27
|
Srinivas V, Bohensky J and Shapiro IM:
Autophagy: A new phase in the maturation of growth plate
chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells
Tissues Organs. 189:88–92. 2009. View Article : Google Scholar :
|
|
28
|
Yuan FL, Chen FH, Lu WG, Li X, Wu FR, Li
JP, Li CW, Wang Y, Zhang TY and Hu W: Acid-sensing ion channel 1a
mediates acid-induced increases in intracellular calcium in rat
articular chondrocytes. Mol Cell Biochem. 340:153–159. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J,
Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ and Simon RP:
Neuroprotection in ischemia: Blocking calcium-permeable
acid-sensing ion channels. Cell. 118:687–698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T). method Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
31
|
Mathy-Hartert M, Martin G, Devel P,
Deby-Dupont G, Pujol JP, Reginster JY and Henrotin Y: Reactive
oxygen species down-regulate the expression of pro- inflammatory
genes by human chondrocytes. Inflamm Res. 52:111–118. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Razaq S, Wilkins RJ and Urban JP: The
effect of extracellular pH on matrix turnover by cells of the
bovine nucleus pulposus. Eur Spine J. 12:341–349. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mariño G, Madeo F and Kroemer G: Autophagy
for tissue homeostasis and neuroprotection. Curr Opin Cell Biol.
23:198–206. 2011. View Article : Google Scholar
|
|
35
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Caramés B, Taniguchi N, Seino D, Blanco
FJ, D’Lima D and Lotz M: Mechanical injury suppresses autophagy
regulators and pharmacologic activation of autophagy results in
chondroprotection. Arthritis Rheum. 64:1182–1192. 2012. View Article : Google Scholar
|
|
37
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apgp, is localized in autophago-some
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miki Y, Tanji K, Mori F, Utsumi J, Sasaki
H, Kakita A, Takahashi H and Wakabayashi K: Alteration of upstream
autophagy-related proteins (ULK1, ULK2, Beclin1, VPS34 and AMBRA1)
in lewy body disease. Brain Pathol. 26:359–370. 2016. View Article : Google Scholar
|
|
39
|
Liu W, Shang G, Yang S, Huang J, Xue X,
Lin Y, Zheng Y, Wang X, Wang L, Lin R, et al: Electroacupuncture
protects against ischemic stroke by reducing autophagosome
formation and inhibiting autophagy through the mTORC1-ULK1
complex-Beclin1 pathway. Int J Mol Med. 37:309–318. 2016.
View Article : Google Scholar :
|
|
40
|
Pandey AK, Hazari PP, Patnaik R and Mishra
AK: The role of ASIC1a in neuroprotection elicited by quercetin in
focal cerebral ischemia. Brain Res. 1383:289–299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Weng XC, Zheng JQ, Li J and Xiao WB:
Underlying mechanism of ASIC1a involved in acidosis-induced
cytotoxicity in rat C6 glioma cells. Acta Pharmacol Sin.
28:1731–1736. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Khan MJ, Rizwan Alam M, Waldeck-Weiermair
M, Karsten F, Groschner L, Riederer M, Hallström S, Rockenfeller P,
Konya V, Heinemann A, et al: Inhibition of autophagy rescues
palmitic acid-induced necroptosis of endothelial cells. J Biol
Chem. 287:21110–21120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang SH, Shih YL, Ko WC, Wei YH and Shih
CM: Cadmium-induced autophagy and apoptosis are mediated by a
calcium signaling pathway. Cell Mol Life Sci. 65:3640–3652. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Williams JA, Hou Y, Ni HM and Ding WX:
Role of intracellular calcium in proteasome inhibitor-induced
endoplasmic reticulum stress, autophagy, and cell death. Pharm Res.
30:2279–2289. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Khan MT and Joseph SK: Role of inositol
trisphosphate receptors in autophagy in DT40 cells. J Biol Chem.
285:16912–16920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Decuypere JP, Bultynck G and Parys JB: A
dual role for Ca(2+) in autophagy regulation. Cell calcium.
50:242–250. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Furuta A, Tanaka M, Omata W, Nagasawa M,
Kojima I and Shibata H: Microtubule disruption with BAPTA and
dimethyl BAPTA by a calcium chelation-independent mechanism in
3T3-L1 adipocytes. Endocr J. 56:235–243. 2009. View Article : Google Scholar
|
|
48
|
Pfisterer SG, Mauthe M, Codogno P and
Proikas-Cezanne T: Ca2+/calmodulin-dependent kinase
(CaMK) signaling via CaMKI and AMP-activated protein kinase
contributes to the regulation of WIPI-1 at the onset of autophagy.
Mol Pharmacol. 80:1066–1075. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Khan NM, Ansari MY and Haqqi TM: Sucrose,
but not glucose, blocks IL1- beta-induced inflammatory response in
human chondrocytes by inducing autophagy via AKT/mTOR pathway. J
Cell Biochem. 118:629–639. 2017. View Article : Google Scholar
|
|
50
|
Taneike M, Nishida K, Omiya S,
Zarrinpashneh E, Misaka T, Kitazume-Taneike R, Austin R, Takaoka M,
Yamaguchi O, Gambello MJ, et al: mTOR hyperactivation by ablation
of tuberous sclerosis complex 2 in the mouse heart induces cardiac
dysfunction with the increased number of small mitochondria
mediated through the down-regulation of autophagy. PLoS One.
11:e01526282016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hirata Y and Oku Y: TRP channels are
involved in mediating hypercapnic Ca2+ responses in rat
glia-rich medullary cultures independent of extracellular pH. Cell
Calcium. 48:124–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Takahashi K, Yokota M and Ohta T:
Molecular mechanism of 2- APB- induced Ca(2)(+) influx in external
acidification in PC12. Exp Cell Res. 323:337–345. 2014. View Article : Google Scholar : PubMed/NCBI
|