Introduction
Constitutive activation of the Wnt/β-catenin signal
pathway promotes uncontrolled cell growth and survival, and can
consequently drive cancer formation (1,2).
Dysregulated Wnt/β-catenin signaling is a common feature of many
malignant tumors of epithelial tissue origin (3–5). In
epithelial tumors, mutations in components of the β-catenin
destruction complex (such as APC, AXIN and GSK3β) or in the
β-catenin gene were shown to contribute to the cytosolic
accumulation of β-catenin and the activation of the Wnt/β-catenin
pathway (6,7). MicroRNAs (miRNAs) are single-stranded
non-coding RNAs of 21 to 23 nucleotides that repress translation or
induce cleavage of target mRNAs that are partially complementary to
the 3′ or 5′ untranslated regions (UTRs). MiRNAs have recently been
implicated in the regulation of tumorigenesis, differentiation,
proliferation and survival through the regulation of major cellular
pathways (8), especially in
epithelial tumors. The relationship between microRNAs and the
Wnt/β-catenin pathway in epithelial tumors has become a central
point of interest.
Our previous review on the Wnt/β-catenin pathway
described multiple genes involved in its regulation, with special
focus on the function of miRNAs. Several miRNAs have been found to
be regulators, either as oncogenes or tumor suppressor genes that
regulate the activity of the Wnt/β-catenin pathway (9). MiR-200a was reported to down-regulate
β-catenin-mediated transcription; however, little is known about
the mechanism involved in this activity. Here, we investigated
whether up- or down-regulation of miR-200a expression was
accompanied by changes in the activity of the Wnt/β-catenin signal
pathway in gastric adenocarcinoma SGC7901 cells and glioblastoma
U251 cells. We show that miR-200a can influence the biological
characteristics of SGC7901 and U251 cells by regulating the
down-stream targets of Wnt/β-catenin signaling. Furthermore, we
confirmed that CTNNB1 is a direct target of miR-200a. We determined
that miR-200a is an inhibitor of EMT in SGC7901 cells.
Materials and methods
Cell culture and transfection
Human stomach adenocarcinoma cell lines, SGC7901 and
U251, were obtained from the Laboratory of Neuro-Oncology, Tianjin
Neurological Institute. The cells were cultured in DMEM
supplemented with 10% fetal bovine serum (FBS). All cultures were
maintained at 37°C in a humidified atmosphere containing 5%
CO2. The miRNA mimic, miRNA inhibitor and negative
control were synthesized by GenePharma (Shanghai, China). For
transfection, trypsinized cells were plated in 6-well plates at
2-3×105 cells per well. MiRNA transfections were
performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
For each well, miRNA (100 pmol) in 250 μl of serum and
antibiotic-free medium was mixed with 5 μl of Lipofectamine 2000 in
250 μl of the same medium and allowed to stand at room temperature
for 20 min. The mixture was then added to cells and after 4 h the
medium was changed to complete medium.
The mimic and inhibitor sequences were: miR-200a
mimic sense: 5′-UAA CAC UGU CUG GUA ACG AUG U-3′; anti-sense:
5′-AUC GUU ACC AGA CAG UGU UAU U-3′; negative control sense: 5′-UUC
UCC GAA CGU GUC ACG UTT-3′; anti-sense: 5′-ACG UGA CAC GUU CGG AGA
ATT-3′; miR-200a inhibitor: 5′-ACA UCG UUA CCA GAC AGU GUU A-3′;
inhibitor negative control: 5′-CAG UAC UUU UGU GUA GUA CAA-3′.
Real-time PCR analysis
Total RNA was extracted using TRIzol Reagent
(Invitrogen) according to the standard protocol. A nanodrop
spectrophotometer (Gene) was used to detect the concentration of
total RNA. Total RNA (1 μg) was used to synthesize cDNA by reverse
transcription using MMLV reverse transcriptase (Promega Corp.,
Madison, WI, USA), according to the manufacturer’s instructions.
Real-time PCR analysis was performed to determine the abundance of
miR-200a in SGC7901 and U251 cells 48 h after transfection with
miR-200a mimic or inhibitor or scrambled negative control. The
expression of u6 was used as an internal control. We performed
qRT-PCR for 40 cycles, comprising 95°C for 10 min, 95°C for 15 sec,
65°C for 30 sec, 72°C for 30 sec and an extension at 72°C for 10
min.
Real-time PCR analysis was also performed to
determine β-catenin mRNA levels and data were normalized to GAPDH,
which was used as an internal control. Both reverse transcription
and qRT-PCR primers were purchased from GenePharma.
Plasmid construction
TOPflash and FOPflash reporters contain wild-type
(WT) and mutated TCF-4 consensus binding sites, respectively, and
are widely used to evaluate β-catenin-dependent signaling events
that drive the expression of TCF. These reporters have been
described previously (10). The
wild-type 3′ untranslated region (UTR) of the CTNNB1 gene,
containing predicted miR-200a target sites, and a mutated CTNNB1 3′
UTR in which the miR-200a target sites were mutated were inserted
into the XbaI and FseI sites of the pGL3 control
vector (GenScript, Nanjing, China) and were named pGL3-CTNNB1 and
pGL3-CTNNB1-mt, respectively.
Luciferase assays
Cells (0.5-1×105 cells/well) were plated
in 24-well plates 1 day prior to transfection. The miR-200a
mimic/inhibitor transfection was performed according to the
Lipofectamine 2000 instructions (Invitrogen), and 48 h after
reporter plasmid transfection, luciferase activity was measured
using a luciferase reporter assay system (Promega).
Western blot analysis
After transfection, cells were washed with ice-cold
phosphate-buffered saline (PBS) three times and were lysed for 30
min on ice in RIPA buffer in the presence of a proteinase inhibitor
cocktail, then centrifuged at 12,000 rpm for 15 min at 4°C.
Proteins were harvested and 40 μg from each sample was subjected to
SDS-PAGE separation, and then transferred to a PVDF membrane
(Millipore, USA). The membrane was incubated with primary
antibodies against β-catenin, ZEB1, ZEB2 (Abcam; 1:1000 dilution),
E-cadherin, N-cadherin, Tcf-4, Fra-1, MMP-9 and Cyclin D1 (Santa
Cruz; 1:1000 dilution), followed by incubation with an
HRP-conjugated secondary antibody (Zhongshan Bio, Beijing, China).
Specific proteins were detected using a SuperSignal protein
detection kit (Pierce, USA). The membrane was stripped and
re-probed with a primary antibody against GAPDH (Santa Cruz; 1:1000
dilution).
Fluorescence microscopy
Twenty-four hours after transfection, cells were
plated on glass cover slips and 48 h post transfection the cover
slips were washed extensively in phosphate buffered saline (PBS)
and fixed with 4% paraformaldehyde in PBS. After additional
washing, the cells were permeabilized with 1% Triton X-100 in PBS
for 10 min. The cover slips were then washed and blocked with 1%
BSA for 30 min. Cells were incubated in the appropriate primary
antibodies (β-catenin or TCF-4) overnight at 4°C. Samples were then
washed and incubated with species-specific secondary
rhodamine-labeled antibodies (TRITC) in PBS (1:100 dilution) for 60
min. Nuclei were stained with DAPI at RT for 10 min and cover slips
mounted with Antifade solution prior to imaging on a confocal
microscope (Leica microsystems, Heidelberg, Germany).
Wound healing assay
Cell culture and transfection conditions were
optimized to ensure a homogeneous and viable cell monolayer prior
to wounding. One day before transfection, equal numbers of SGC7901
cells (2×105) or U251 cells (1×105) were
seeded in 6-well plates. When cell confluence reached about 90%,
approximately 24 h post-transfection, an artificial homogenous
wound was made onto the monolayer using a sterile plastic 200 μl
micropipette tip. After wounding, debris was removed by washing
cells with PBS. At different time points, cells that migrated into
the wounded area or cells with extended protrusions from the wound
border were photographed at ×200 magnification under a light
microscope.
Transwell cell migration assay
The top chamber of a transwell chamber was incubated
with 60 μl Matrigel diluted with DMEM (1:2, Matrigel: DMEM) at 37°C
for 30 min. The Matrigel solidified and acted as the extracellular
membrane (ECM) for tumor cell invasion analysis. Transfected cells
were trypsinized, adjusted to 5×105/ml in DMEM, and 100
μl of the resuspended cell solution was added to the top chamber
above the Matrigel. The bottom chamber was filled with 600 μl of
chemoattractant solution. The transwell plate was assembled and
incubated at 37°C, in a 5% CO2 incubator. After 24 h,
the top chamber was removed, and the Matrigel and unmigrated cells
were gently scraped with a wet cotton swab. Cells were stained by
crystal violet for 3 min, and washed with PBS to remove excess
stain. Finally, cells were counted under a light microscope. The
average number of migrated cells per field was quantified under
high power (x200).
Flow cytometry
Forty-eight hours after transfection, cells were
trypsinized and collected by centrifugation, washed in PBS and
fixed with 75% ethanol overnight at 4°C. Cells were then washed
twice with PBS, and incubated with 200 μl RNase A (1 mg/ml) at 37°C
for 30 min. Cells were then stained in the dark with 800 μl
propidium iodide staining solution for 30 min at 4°C. Analysis was
performed on a FACSCalibur flow cyto-meter (Bio-Rad, USA).
MTT assay
Cells were seeded in a 96-well plate at a density of
3000 cells per well, and 24 h before transfection, cells were
incubated with 20 μl MTT solution (5 mg/ml). At 24, 48, 72, 96,
120, 144 and 168 h following transfection at 37°C for 4 h, the
solution was aspirated, and 200 μl DMSO was added to each well. The
Optical density (OD) was measured at a wavelength of 570 nm. The
data are presented as the mean ± SD, which are derived from
triplicate samples of at least three independent experiments.
Statistical analysis
A commercially available software package, SPSS16.0,
was used for statistical analysis. One-way analysis of variance
(ANOVA) and the χ2 test was used to analyze the
significance between groups. The LSD method of multiple comparisons
with parental and control vector groups was used when ANOVA showed
statistical significance. Statistical significance was determined
at the level of p<0.05.
Results
Modulation of miR-200a expression by a
mimic and an inhibitor
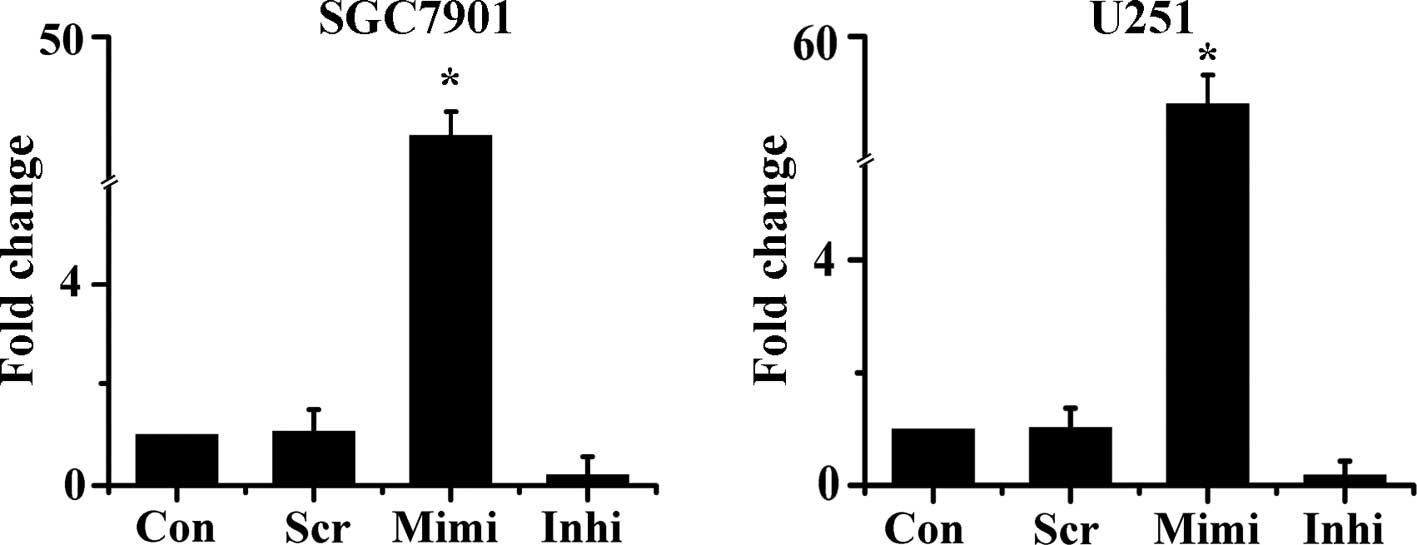
To monitor the expression of miR-200a in target
cells, the miR-200a mimic, inhibitor and scrambled control were
delivered into SGC7901 and U251 cells. The level of miR-200a
expression was then examined 48 h after transfection by qRT-PCR.
Expression of miR-200a was up-regulated by approximately 40-fold in
cells transfected with the miR-200a mimic. Meanwhile, in cells
transfected with the miR-200a inhibitor, miR-200a expression was
reduced by about 80% compared with control (Fig. 1). These results were used as the
basis of the subsequent experiments.
Relationship between miR-200a expression
and activity of the β-catenin/Wnt signaling pathway
Recent studies have reported that miR-200a targets
the mRNA of the E-cadherin repressor proteins, ZEB1 and ZEB2. This
results in an increase in the level of E-cadherin available for
binding to β-catenin and induces formation of the cell-cell
adhesion complex (11). We
considered that miR-200a may play an important role in regulating
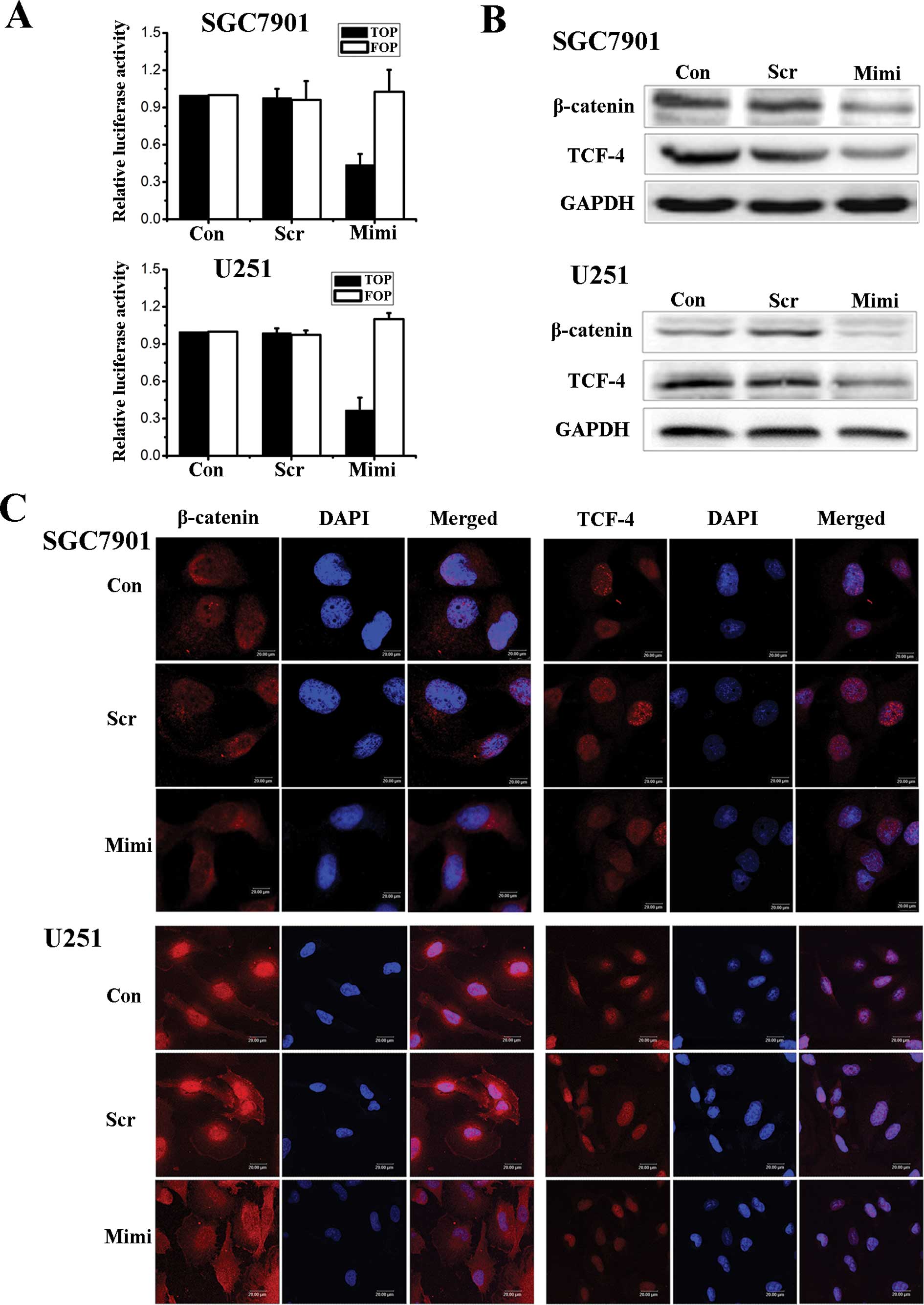
the activity of the Wnt/β-catenin pathway. In an effort to
determine the relationship between the expression of miR-200a and
the activity of the β-catenin/Wnt pathway, we first employed
TOPflash and FOPflash reporters, which are widely used to evaluate
β-catenin-dependent signaling activity, to evaluate the effects of
miR-200a on Wnt/β-catenin signaling in SGC7901 and U251 cells. The
luciferase activity of the cells changed as we hypothesized
(Fig. 2A). Wnt/β-catenin signaling
was inhibited when the level of miR-200a was up-regulated. We then
used Western blot assays to investigate the expression levels of
β-catenin and TCF-4 proteins (Fig.
2B). Fluorescence microscopy of β-catenin and TCF-4 showed that
the location of β-catenin in cells shifts from nuclear to
cytoplasmic when the expression of miR-200a increased. At the same
time, TCF-4 levels decreased in the nucleus (Fig. 2C).
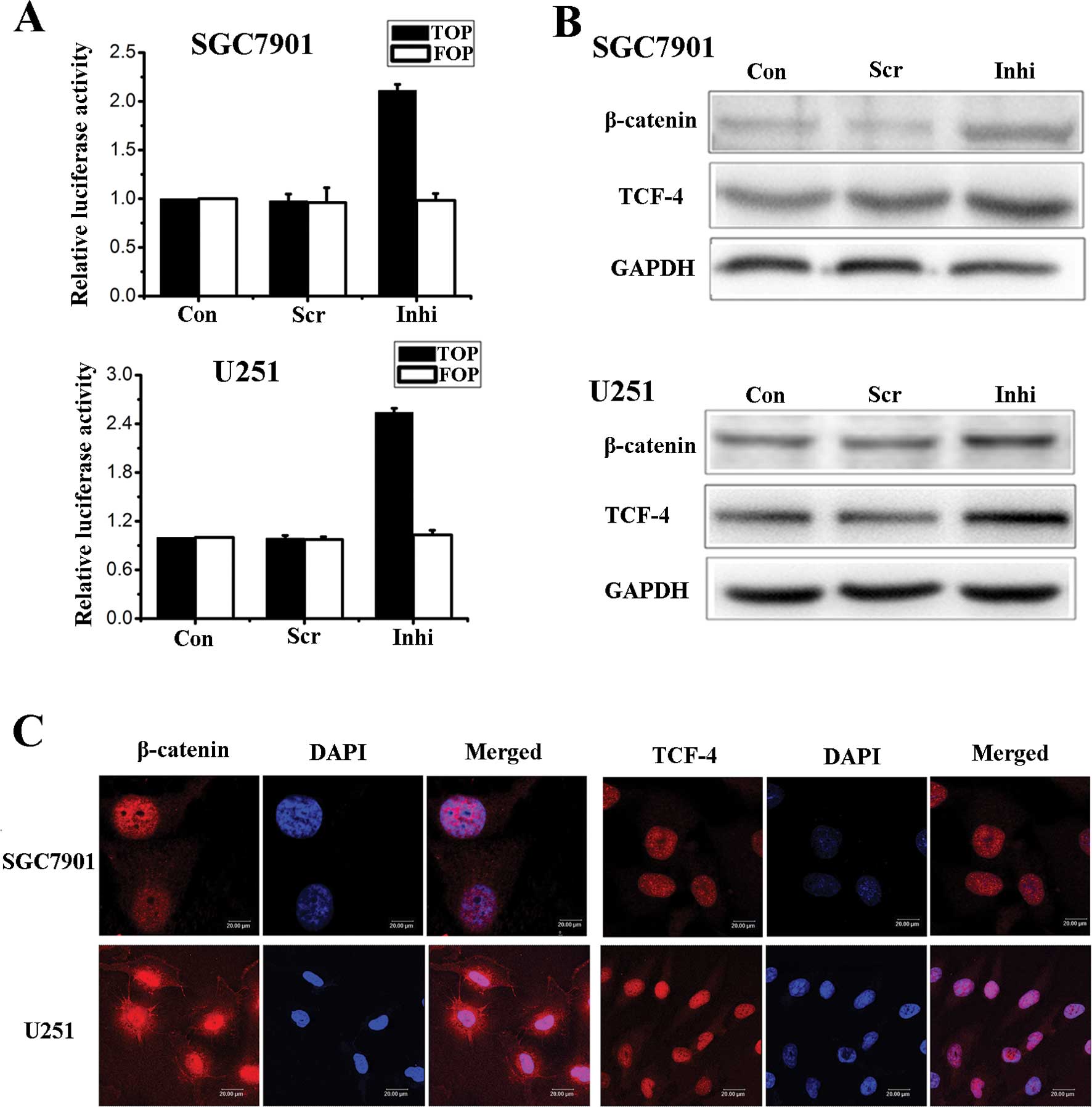
We also used the miR-200a inhibitor to down-regulate
the expression of miR-200a in SGC7901 and U251 cells. Using
luciferase assays, Western blot analysis and fluorescence
microscopy we showed that Wnt/β-catenin signaling activity was
negatively correlated with the level of miR-200a (Fig. 3).
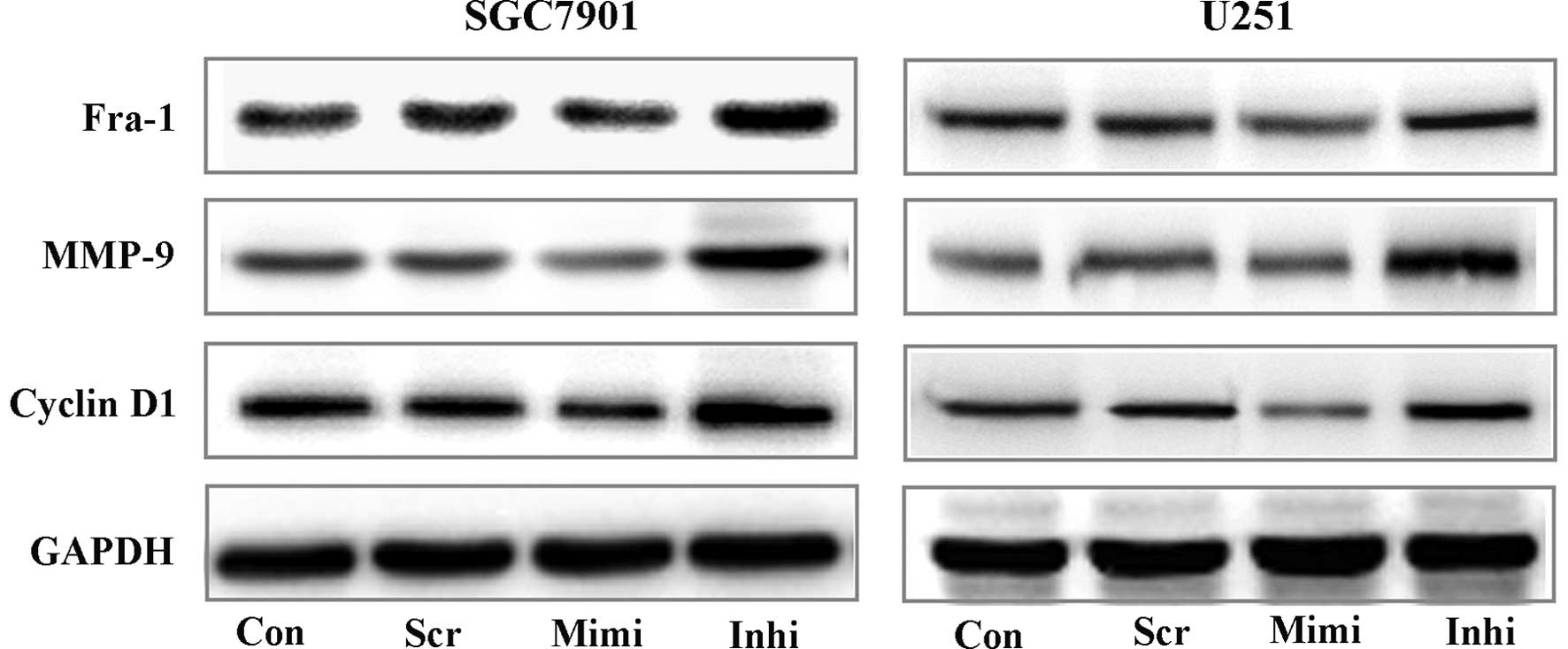
It is well known that the Wnt/β-catenin pathway has
essential functions in the regulation of cell growth and
differentiation. Here, we used Western blot assays to investigate
the expression of some downstream targets of Wnt/β-catenin
signaling, such as Fra-1, Cyclin D1 and MMPs (12–14)
(Fig. 4). These results showed an
important correlation between miR-200a expression and activity of
β-catenin/Wnt signaling. We therefore postulate that miR-200a
affects the biological activity of tumor cells.
Regulation of tumor cell activity by
miR-200a
We next investigated a functional outcome for the
miR-200a-mediated suppression of β-catenin/Wnt signaling. The
expression level of miR-200a clearly influenced the biological
activity of SGC7901 and U251 cells. Therefore, we investigated two
major biological activities of tumor cells, namely migration and
invasion potential and growth ability. Migration and invasion
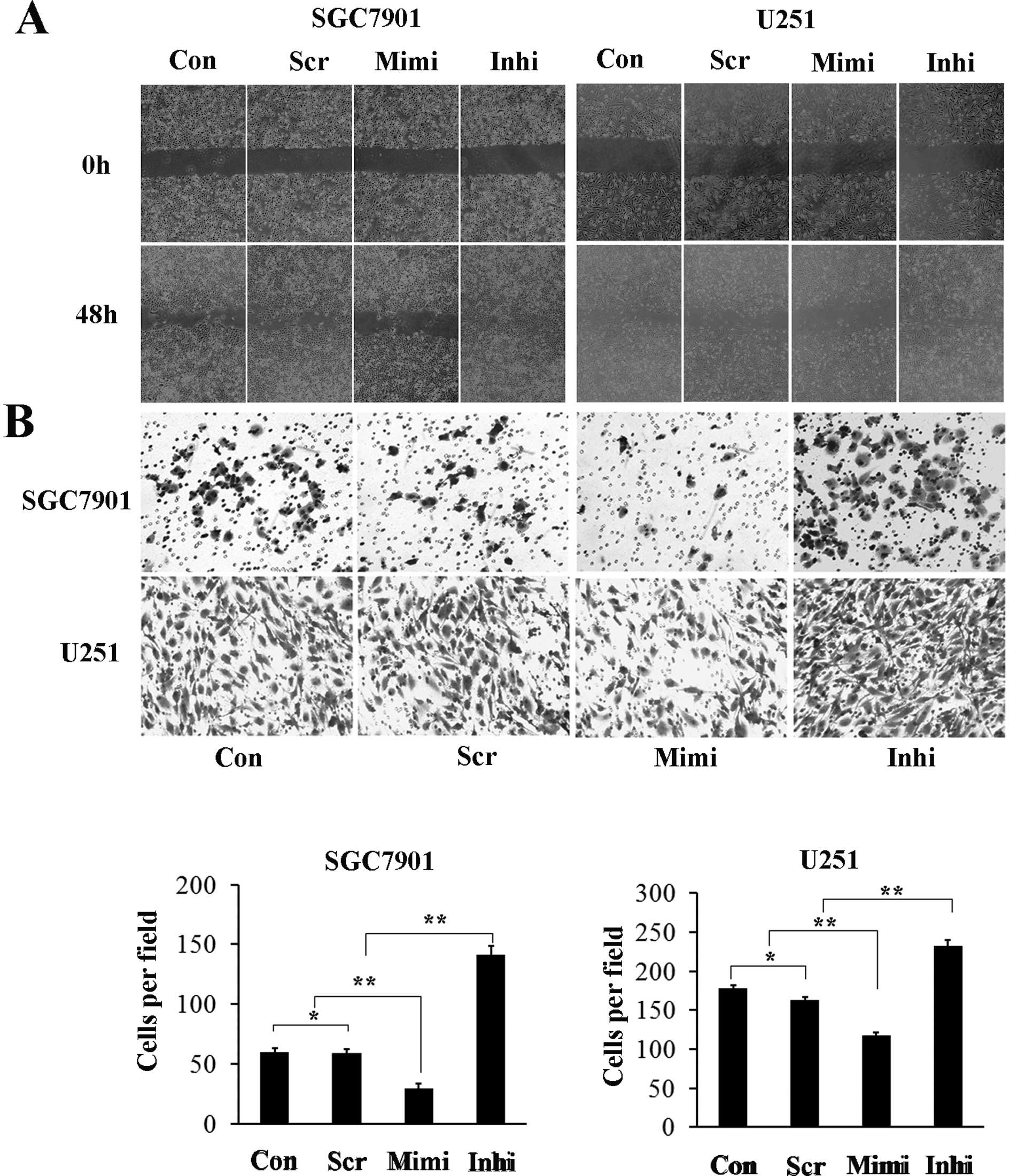
potential are important biological characteristics of malignant
tumor cells. Representative micrographs of wound healing assay and
of transwell filters are shown in Fig.
5. The number of cells invading through the matrigel in the
miR-200a mimic group was significantly decreased (29.3±4.1), while
in the miR-200a inhibitor group it was increased (141.0±7.5)
compared to control (59.7±3.0) and scrambled control (58.7±3.8)
groups. The invasion activity was inhibited by approximately 40% in
the miR-200a mimic group (117.7±8.5) and increased (232.3±13.3) in
the miR-200a inhibitor group compared with the control (178.3±11.3)
and scrambled control (163.0±14.0) groups (Fig. 4B). These results suggest that high
levels of miR-200a inhibit the migration and invasion capacity of
SGC7901 and U251 cells, while low levels of miR-200a has the
opposite effects.
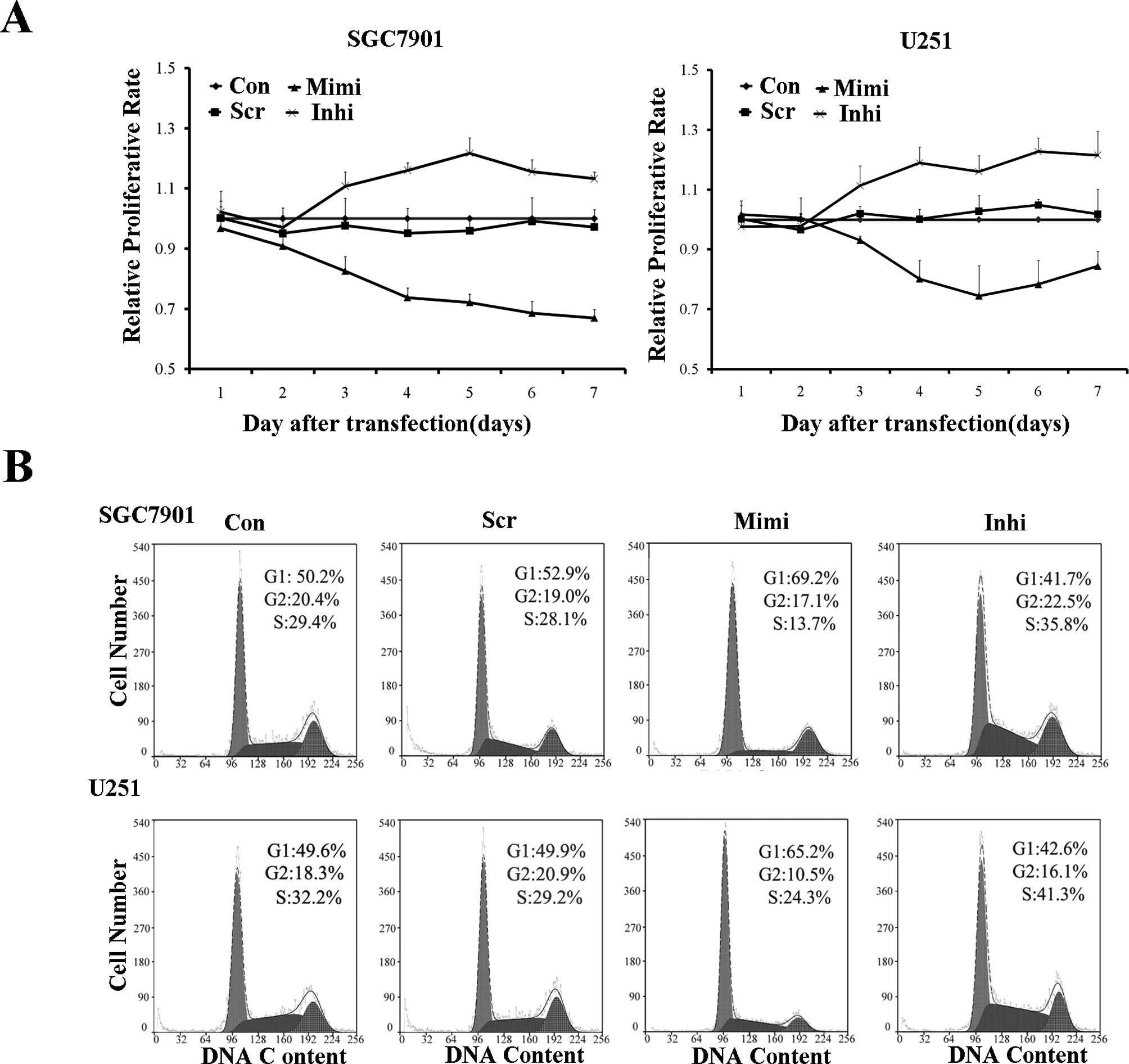
The proliferation rate of variously transfected
SGC7901 and U251 cells was measured using the MTT assay. The
miR-200a mimic group proliferated at a significantly lower rate
than the other groups. Cell cycle analysis confirmed these results.
The miR-200a mimic led to G0/G1 entry, while the miR-200a inhibitor
blocked G0/G1 entry (Fig. 6).
Target validation of miR-200a
MicroRNA-200a, a member of the miR-200 family,
stands out as an inhibitor of EMT. Direct evidence for the
EMT-inhibitory actions of miR-200a has been revealed in several
cancer cell lines, including nasopharyngeal carcinoma, endometrial
serous adenocarcinomas, bladder cancer and meningiomas (15–18).
The up-regulation of miR-200a in NRK52E cells was shown to
down-regulate the expression of TGF-β2, via direct interaction with
the 3′ UTR of TGF-β2 (19), which
can induce EMT and reduce the invasiveness in meningiomas (18). Up-regulation of miR-200a also
decreased cellular invasion and metastasis in nasopharyngeal
carcinoma cells (15). As
described above, miR-200a affects the phenotype of the SGC7901 and
U251 cells, although determination of the underlying mechanism
requires further investigation.
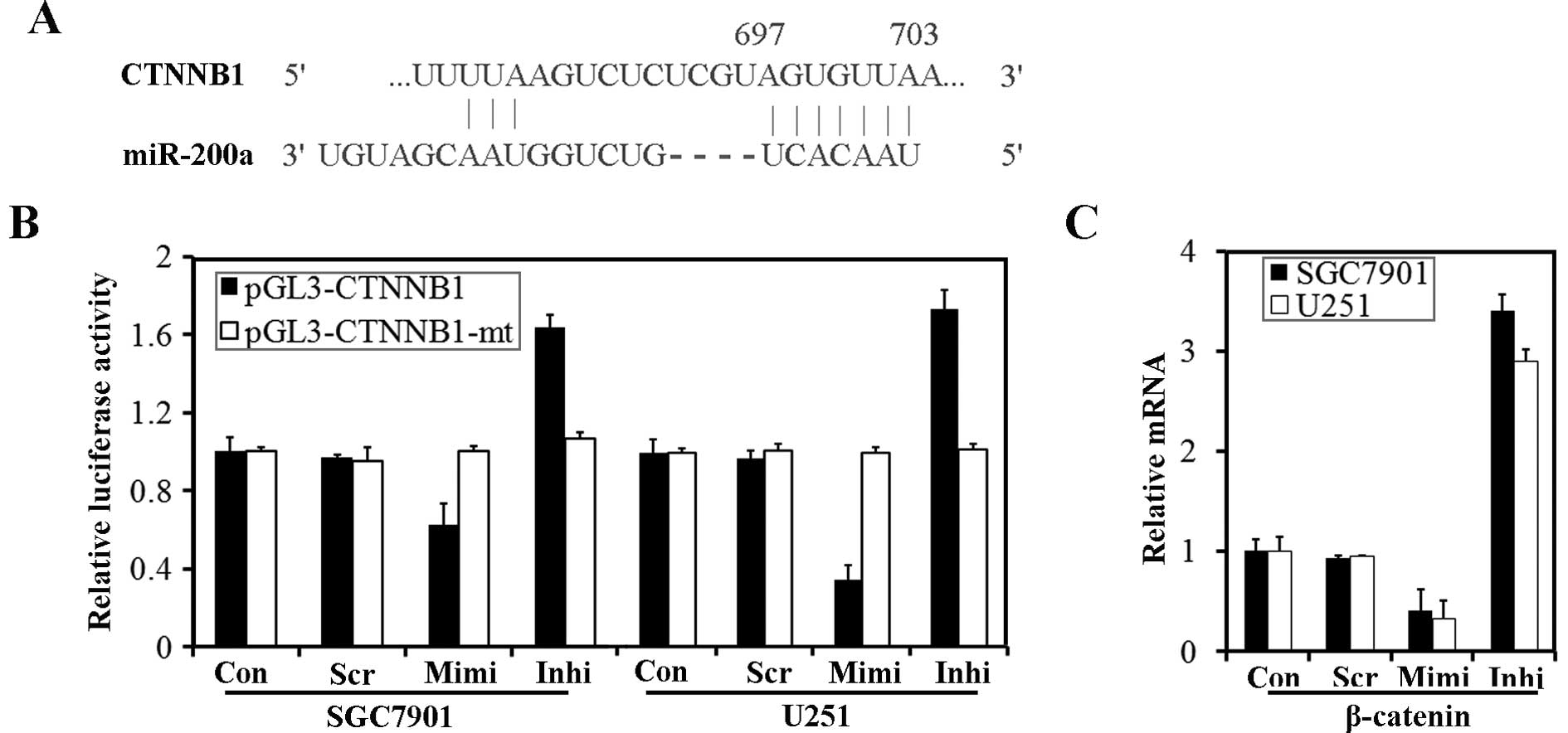
CTNNB1 (the gene which encodes β-catenin) has been
suggested to be a target of miR-200a (18,20).
According to TargetScanHuman 5.1, the 3′ UTR of CTNNB1 contains
predicted seed regions for miR-200a and miR-141 (Fig. 7A). To determine whether endogenous
miR-200 could target the 3′ UTR of CTNNB1 in SGC7901 and U251
cells, the 3′ UTR of CTNNB1 and a mutated CTNNB1 3′ UTR were cloned
into a modified pGL-3 control vector, placing it downstream of the
luciferase coding sequence. We delivered pGL3-CTNNB1 and
pGL3-CTNNB1-mt into cells transfected with the miR-200a mimic or
inhibitor. Luciferase assays revealed that over-expression of
miR-200a could significantly reduce luciferase activity, while
down-regulation of miR-200a caused an enhancement of luciferase
activity. However, transfection of pGL3-CTNNB1-mt had no effect on
luciferase activity of the cells (Fig.
7B). We have identified β-catenin, encoded by CTNNB1, as a
target protein for miR-200a. β-catenin mRNA was also down-regulated
by transfection of the miR-200a mimic (Fig. 7C).
Increasing miR-200a levels induces
mesenchymal to epithelial transition (MET) in SGC7901 cells
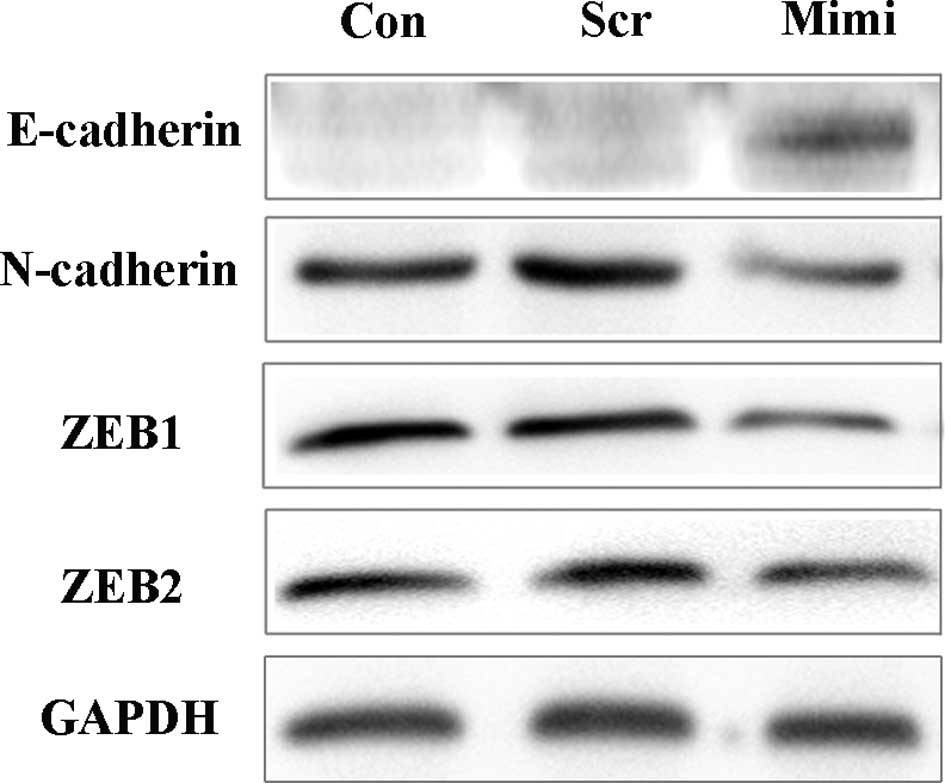
Recently, ZEB1 and ZEB2 have both been suggested to
be targets of miR-200a, and miR-200a was demonstrated to cause
changes in E-cadherin expression (16,21,22).
To determine whether the expression of E-cadherin is also under the
control of miR-200a in SGC7901 cells, we used the miR-200a mimic to
over-express miR-200a, and Western blot analysis was performed to
detect the expression of ZEB and cadherin/catenin complexes
(Fig. 8). Over-expression of
miR-200a reduced ZEB1, ZEB2 and N-cadherin protein levels, while
E-cadherin protein levels were increased, consistent with earlier
reports (15,16,21,22).
Discussion
This study highlights two different mechanisms by
which miR-200a regulates the activity of β-catenin. First we showed
that miR-200a can inhibit the activity of the Wnt/β-catenin
signaling pathway. Then we found that miR-200a can regulate the
expression of β-catenin through not only direct interaction with
the 3′ UTR of CTNNB1, but also via interaction with the
cadherin/catenin complex. In an effort to further investigate the
effect of miR-200a on the phenotype of tumor cells, we used
epithelial tumor cell lines, SGC7901 and U251, and found that
over-expression of miR-200a significantly inhibited SGC7901 and
U251 cell growth, invasion and induced G0/G1 phase arrest, while
reduced expression of miR-200a promoted tumor cell growth and
invasion. The induction of apoptosis by inhibiting the activity of
the Wnt/β-catenin signaling pathway in tumor cells was not detected
in our studies and further investigations are required to explain
the role of miR-200a in tumor cells.
As the most common primary brain tumor, gliomas
account for more than 70% of all primary central nervous system
neoplasms. Accumulating evidence suggests that aberrant activation
of Wnt/β-catenin signaling is involved in glioma development and
progression (23). We demonstrated
that up-regulation of miR-200a down-regulated the expression of
β-catenin and affected the activity of the Wnt/β-catenin signaling
pathway. Furthermore, at the protein level, our results indicate
that miR-200a also regulates some downstream targets of
Wnt/β-catenin signaling, such as Fra-1, Cyclin D1, and MMPs. Yue
et al (24) showed that
knockdown of β-catenin by siRNA in human U251 glioma cells
inhibited cell proliferation and invasive ability and induced
apoptotic cell death; however, in our studies, apoptosis in U251
cells was undetectable after miR-200a-induced β-catenin
down-regulation. It has also been reported that the Wnt/β-catenin
signaling pathway regulates the early and late stages of apoptosis
in other cancer cells (25,26),
so our studies did not conclusively demonstrate the mechanisms of
miR-200a action in U251 cells, and additional mechanisms are
possible, such as regulation through direct target genes other than
β-catenin, or even through other signaling pathways.
We identified for the first time in gastric
adenocarcinoma SGC7901 cells that over-expression of miR-200a
reduced levels of ZEB1, ZEB2 and N-cadherin and increase E-cadherin
levels. ZEB1 and ZEB2 are transcription factors that activate EMT
by binding to E-box elements present in the E-cadherin promoter,
suppressing transcription. While E-cadherin has a widely
acknowledged role in cell-cell adhesion, it also functions as an
invasion suppressor protein. During EMT E-cadherin is
down-regulated, while N-cadherin is induced and β-catenin is
released from junctional complexes and is translocated to the
nucleus (27–29). The cadherin-associated protein
β-catenin also has the potential to regulate cell motility or
invasion. Therefore, via regulating the expression of E-cadherin by
targeting ZEB1 and ZEB2, miR-200a can inhibit the invasive
potential of tumor cells. In addition, using luciferase reporter
assays, we demonstrated that the expression of β-catenin was
regulated directly by miR-200a in SGC7901 cells. MiR-200a could
also regulate tumor cell invasive ability by directly targeting
β-catenin. Although β-catenin was originally identified as an
integral component of the cadherin adhesion protein complex, it is
also an essential intracellular mediator for the Wnt/β-catenin
signaling pathway. So it is reasonable for us to consider that the
effect of miR-200a on tumor cell growth might be attributed to
β-catenin regulation. We used a new luciferase reporter assay
called TCF-responsive reporter, or TOPflash, to investigate
β-catenin/TCF-dependent transcriptional activity, and we found a
satisfactory correlation between miR-200a expression and
Wnt/β-catenin signaling pathway activity. In conclusion, we found
that in SGC7901 cells miR-200a regulated both the EMT of the cells
and the activity of the Wnt/β-catenin signaling pathway.
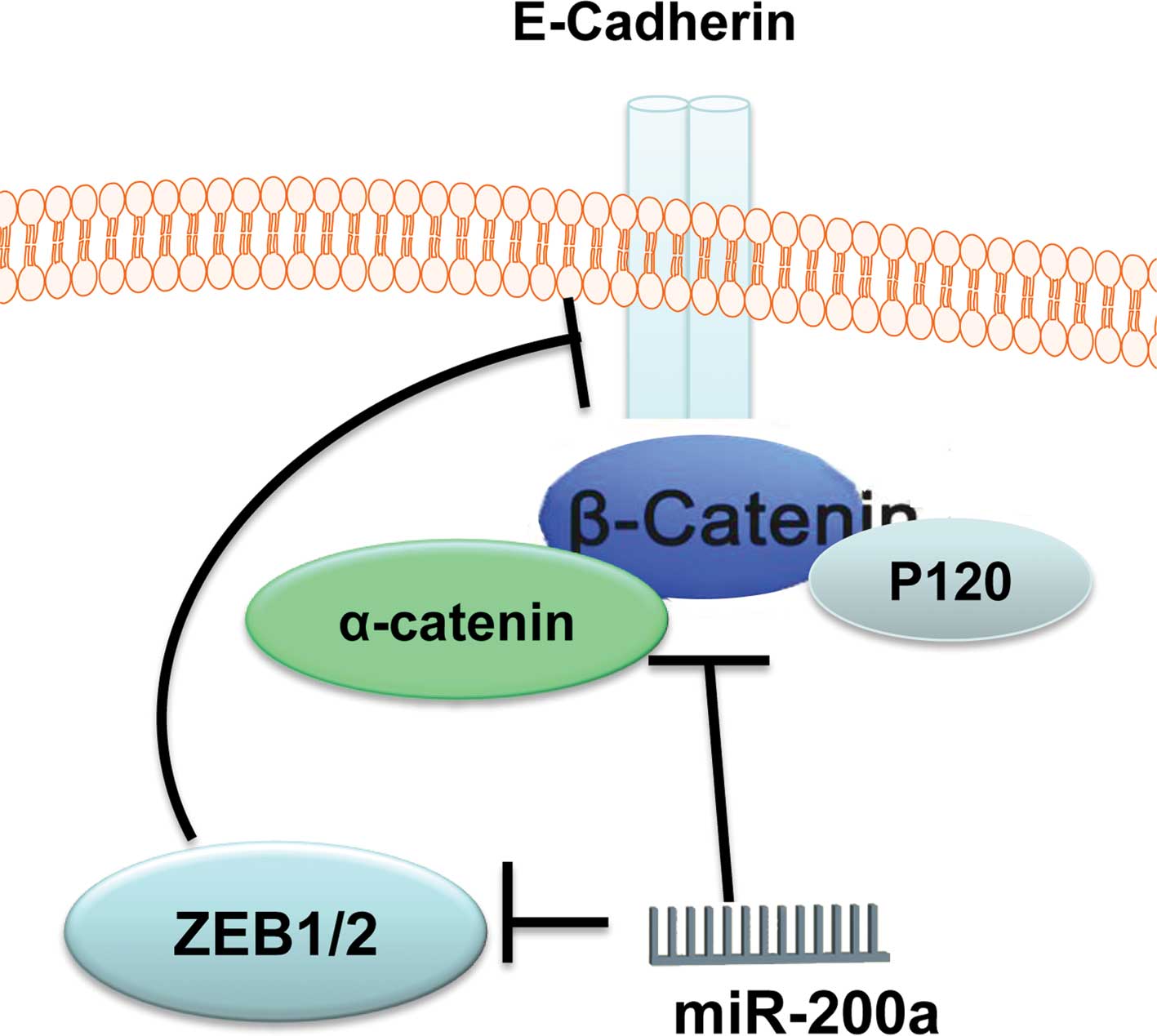
In summary, β-catenin is an important functional
target for miR-200a in gastric adenocarcinoma SGC7901 cells and
glioblastoma U251 cells. MiR-200a regulates the activity of
β-catenin through two kinds of mechanism (Fig. 9), and up-regulation of miR-200a is
a potential therapeutic strategy for glioma and gastric
adenocarcinoma.
Acknowledgements
This study was supported by the Chinese National
Natural Scientific Fund 81172356 and 81172406, Science and
Technology Fund of Tianjin Health Bureau (2010KY17), and by the
Natural Science Foundation of Tianjin (10JCZDJC18500), Tianjin
Municipal Science and Technology Commission (10SYSYJC28800). We
would like to thank Dr Daiming Fan for kindly providing SGC7901
gastric cancer cells and members of the Tianjin Laboratory of
Neuro-Oncology, Tianjin Neurological Institute for their technical
assistance.
References
|
1
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006.
|
|
2
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004.
|
|
3
|
Pinto D, Gregorieff A, Begthel H and
Clevers H: Canonical Wnt signals are essential for homeostasis of
the intestinal epithelium. Genes Dev. 17:1709–1713. 2003.
|
|
4
|
Yang LH, Xu HT, Han Y, et al: Axin
downregulates TCF-4 transcription via beta-catenin, but not p53,
and inhibits the proliferation and invasion of lung cancer cells.
Mol Cancer. 9:252010.
|
|
5
|
Sansom OJ, Reed KR, Hayes AJ, et al: Loss
of Apc in vivo immediately perturbs Wnt signaling, differentiation,
and migration. Genes Dev. 18:1385–1390. 2004.
|
|
6
|
Morin PJ, Sparks AB, Korinek V, et al:
Activation of beta-catenin-Tcf signaling in colon cancer by
mutations in beta-catenin or APC. Science. 275:1787–1790. 1997.
|
|
7
|
Ying Y and Tao Q: Epigenetic disruption of
the WNT/beta-catenin signaling pathway in human cancers.
Epigenetics. 4:307–312. 2009.
|
|
8
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
|
|
9
|
Huang K, Zhang JX, Han L, et al: MicroRNA
roles in beta-catenin pathway. Mol Cancer. 9:2522010.
|
|
10
|
Han L, Yang Y, Yue X, et al: Inactivation
of PI3K/AKT signaling inhibits glioma cell growth through
modulation of beta-catenin-mediated transcription. Brain Res.
1366:9–17. 2010.
|
|
11
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008.
|
|
12
|
Brabletz T, Herrmann K, Jung A, Faller G
and Kirchner T: Expression of nuclear beta-catenin and c-myc is
correlated with tumor size but not with proliferative activity of
colorectal adenomas. Am J Pathol. 156:865–870. 2000.
|
|
13
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999.
|
|
14
|
Wu B, Crampton SP and Hughes CC: Wnt
signaling induces matrix metalloproteinase expression and regulates
T cell transmigration. Immunity. 26:227–239. 2007.
|
|
15
|
Xia H, Cheung WK, Sze J, et al: miR-200a
regulates epithelial-mesenchymal to stem-like transition via ZEB2
and beta-catenin signaling. J Biol Chem. 285:36995–37004. 2010.
|
|
16
|
Adam L, Zhong M, Choi W, et al: miR-200
expression regulates epithelial-to-mesenchymal transition in
bladder cancer cells and reverses resistance to epidermal growth
factor receptor therapy. Clin Cancer Res. 15:5060–5072. 2009.
|
|
17
|
Hiroki E, Akahira J, Suzuki F, et al:
Changes in microRNA expression levels correlate with
clinicopathological features and prognoses in endometrial serous
adenocarcinomas. Cancer Sci. 101:241–249. 2010.
|
|
18
|
Saydam O, Shen Y, Wurdinger T, et al:
Downregulated microRNA-200a in meningiomas promotes tumor growth by
reducing E-cadherin and activating the Wnt/beta-catenin signaling
pathway. Mol Cell Biol. 29:5923–5940. 2009.
|
|
19
|
Wang B, Koh P, Winbanks C, et al: miR-200a
prevents renal fibrogenesis through repression of TGF-beta2
expression. Diabetes. 60:280–287. 2011.
|
|
20
|
Xia H, Ng SS, Jiang S, et al:
miR-200a-mediated downregulation of ZEB2 and CTNNB1 differentially
inhibits nasopharyngeal carcinoma cell growth, migration and
invasion. Biochem Biophys Res Commun. 391:535–541. 2010.
|
|
21
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008.
|
|
22
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008.
|
|
23
|
Sareddy GR, Panigrahi M, Challa S,
Mahadevan A and Babu PP: Activation of Wnt/beta-catenin/Tcf
signaling pathway in human astrocytomas. Neurochem Int. 55:307–317.
2009.
|
|
24
|
Yue X, Lan F, Yang W, et al: Interruption
of beta-catenin sup-presses the EGFR pathway by blocking multiple
oncogenic targets in human glioma cells. Brain Res. 1366:27–37.
2010.
|
|
25
|
Svedlund J, Auren M, Sundstrom M, et al:
Aberrant WNT/beta-catenin signaling in parathyroid carcinoma. Mol
Cancer. 9:2942010.
|
|
26
|
Li F, Chong ZZ and Maiese K: Winding
through the WNT pathway during cellular development and demise.
Histol Histopathol. 21:103–124. 2006.
|
|
27
|
Rosano L, Spinella F, Di Castro V, et al:
Endothelin-1 promotes epithelial-to-mesenchymal transition in human
ovarian cancer cells. Cancer Res. 65:11649–11657. 2005.
|
|
28
|
Vandewalle C, Comijn J, De Craene B, et
al: SIP1/ZEB2 induces EMT by repressing genes of different
epithelial cell-cell junctions. Nucleic Acids Res. 33:6566–6578.
2005.
|
|
29
|
Maeda M, Johnson KR and Wheelock MJ:
Cadherin switching: essential for behavioral but not morphological
changes during an epithelium-to-mesenchyme transition. J Cell Sci.
118:873–887. 2005.
|