Introduction
Central nervous system glioblastomas are among the
most aggressive and treatment-resistant cancers. The recent
discovery of self-renewing and uniquely tumorigenic brain tumor
stem cells (BTSCs) (1–4), also referred to as brain cancer
initiating cells, points to the presumption that this cancer stem
cell subpopulation intrinsically resistant to radio and
chemotherapeutic treatments might be responsible for the
phenotypical derivation of tumors and their recurrence.
Instead of specific markers, makers shared with
cancer stem cells (5,6) and BTSCs, such as AC133 (2), CD15 (7), and CD171 (8), have been documented. Among these,
AC133, an epitope of the CD133 protein, which is itself a pentaspan
glycoprotein identified firstly on hematopoietic stem cells, is the
best known (9–12). In vitro, in the presence of
EGF, FGF-2, and heparin, AC133 expressing cells isolated from human
glioblastoma regenerate, form neurosphere-like colonies and are
capable of generating cells that express markers of differentiated
neural cells (2). In xenograft
models using immunodeficient mice, they lead to cancers that are
phenotypically similar to the original tumors (2). AC133-positive cancer cells are also
particularly resistant to radiotherapy (13) and TRAIL-mediated apoptosis
(14). In addition, these cells
are capable of promoting tumor neovascularization by producing VEGF
(15). Finally, AC133
overexpression in human gliomas is associated with poor clinical
outcome (16).
Although these findings are in line with the
relevance of developing targeted strategies against BTSCs in
glioblastomas through AC133 recognition, other observations argue
for a more complex reality. Indeed, while less tumorigenic than
their AC133-positive counterparts (3), AC133-negative cells can lead to
tumors with a distinct phenotype (17). Moreover, the occurrence of BTSCs
does not exclude the existence of cellular networks in which
individually non-tumorigenic cell populations might cooperate to
produce tumors (18). In addition,
the tissue microenvironment might exert pivotal effects for tumor
development (18,19), and extrinsic cell modulators may
drive the expression of intrinsic markers.
In line with this, AC133 expression in glioblastoma
has been associated either with anatomical while not necessarily
functional perivascular niches (20) or hypoxic pseudopalissading necrotic
regions (21,22). Thus, it is not yet understood
whether AC133 incidence in those areas is due to improved survival
of AC133-positive cells or positive regulation of AC133 expression.
As oxygen is involved in the stem cell behavior (23) and tumor aggressiveness of human
glioblastoma (24), it is
important to consider its role on AC133 expression and
AC133-positive BTSC performance. As such, it has recently been
demonstrated that exposure to low oxygen tension (pO2)
allows for maintenance of the AC133 phenotype of non-sorted human
glioblastoma cells in vitro (25). In addition, sorted AC133-positive
human glioblastoma cells preserve their stem cell phenotype under
low oxygen tension in vitro (26).
Nonetheless, these studies did not address how cell
culture pO2 might affect the AC133 phenotype and the
behavior of cancer cells following implantation in animals. In our
study, we demonstrate that xenogenic experimental tumors, obtained
from non-sorted human glioblastoma cells cultured either at 3 or
21% O2, can significantly differ. In this context, we
investigate whether AC133 is an indicator of low oxygen tension or
of tumor aggressiveness. Finally, we discuss our data regarding the
relevance of biopsy-derived models for functional investigations or
for therapeutic targeting purposes.
Materials and methods
Patient tissue samples and human glioma
cell cultures
Specimens from patients undergoing biopsy for de
novo glioma were obtained from the Department of Neurosurgery
of the Angers CHU (France), and from the Department of Neurosurgery
of the Grenoble CHU (France), with institutional review board
approvals. Pathologic diagnosis established that GlioA, GlioB, and
GlioC tumor samples were grade IV WHO glioblastomas. Straight after
tissue dissociation as previously described (25), cells were plated on uncoated
plastic flasks at 2×104/ml of defined medium and
cultured at 37°C under an atmosphere containing 5% CO2
and either 3 or 21% O2. GlioA, GlioB and GlioC were
cultured in Dulbecco’s modified Eagle’s medium: Nutrient Mixture
F-12 (DMEM/F12, Biowhittaker, Verviers, Belgium) added with
Glutamax, B27 and N2 supplements (Invitrogen, Cergy Pontoise,
France), recombinant human EGF and FGF-2 (20 ng/ml each, R&D
Systems Europe, Lille, France), and heparin (5 μg/ml,
Sigma-Aldrich, Lyon, France). Growth factors and supplements were
added every 3 days for a period of 10–15 days, until new
dissociations with Versene (Lonza, Levallois-Perret, France) and
re-plating following initial culture setting. Under these permanent
conditions, cells grew and were maintained as floating
neurosphere-like colonies.
AC133 labeling and flow cytometry
Glioma cells exposed to different oxygen tensions
were collected and dissociated using Versene (Lonza). A total of
1.5×105 cells were incubated with 5 μg/ml AC133 antibody
(Miltenyi Biotech, Paris, France) or IgG1 isotype control
(BD-Biosciences, Le Pont-de-Claix, France) for 1 h at 4°C in PBS
containing 5% FBS and 0.02% sodium azide. Cells were then washed
three times in PBS containing 5% FBS and 0.02% sodium azide, and
incubated for 30 min at 4°C with FITC-conjugated goat anti-mouse
IgG F(ab’)2 fragment polyclonal antibody
(Dakocytomation, Trappes, France) at 20 μg/ml in PBS containing 5%
FBS and 0.02% sodium azide. Following three more washes in PBS
containing 5% FBS and 0.02% sodium azide, cells were re-suspended
in PBS containing 2% formaldehyde and 0.02% sodium azide. A BD
FACSCalibur™ fluorescent-activated flow cytometer and the BD
CellQuest™ software (BD-Biosciences) were used in order to proceed
to flow cytometry acquisition. Analysis was carried out using
WinMDI 2.9 software (Scripps Institute, La Jolla, CA, USA).
Treatment of human glioma cells with
cobalt dichloride (CoCl2)
GlioA, GlioB, and GlioC human glioblastoma cells
were dissociated in Versene (Lonza). They were then plated at
37.5×105 cells per ml in the aforementioned media and
incubated in the presence of vehicle alone (PBS) or 100–150 μM
CoCl2 for 24 h at 37°C, 5% CO2 and 3%
O2.
shRNA knockdown
Glioblastoma cells were stably transfected using
control transduction particles (SHC001V) or shRNA transduction
particles expressing siRNA against HIF-1α (IDs: TRCN0000003810,
TRCN0000003811 and TRCN0000010819), according to the manufacturer’s
instructions (Mission® pLKO.1-puro lentiviral particles,
Sigma-Aldrich). Cells were seeded at 5×103 in 96-well
plates in supplemented neurobasal medium, and infected with a
multiplicity of infection of 2. Puromycine (1 μg/ml, Sigma-Aldrich)
selected infected cells.
Q-PCR
Q-PCR analyses were carried out using a Chromo 4™
(Bio-Rad, Marnes-la-Coquette, France) and SYBR Green detection
(iQ-SYBR Supermix, Bio-Rad). Primers were designed using Primer3
software (http://frodo.wi.mit.edu/primer3/). The ΔCt method was
retained for quantification, and multiple genes were used for
normalization, as previously described (27).
Orthotopic xenograft assays
GlioA and GlioB human glioblastoma cells, grown at 3
or 21% O2, were dissociated in Versene, washed, and
resuspended at 50,000 cells in 5 μl Eagle’s minimum essential
medium (EMEM, Biowhittaker). SCID female mice (Charles River) were
anesthetized using xylazine (50 μg/g) (Rompun®, Bayer,
Puteaux, France) and Ketamine (10 μg/g) (Clorketam®,
Vétoquinol, Lure, France). Stereotactic implantation of the 5 μl
cell suspension was carried out into the right striatum using a
Hamilton syringe and a 32-gauge needle at the following
coordinates: 0.5 mm anterior from Bregma, 2 mm lateral from the
saggital suture, and 3 mm below dura. Cells were injected
progressively over 2.5 min, followed by 5 min of waiting, and
progressive needle removal from brain over 6 min. MRI was used to
monitor tumor growth. The Kaplan-Meier method was used to plot
animal survival. Animal care was carried out in line with relevant
European Community regulations (Official Journal of European
Community L358 12/18/1986).
Magnetic resonance imaging
Experiments were performed with a Bruker Avance DRX
300 (Bruker, Wiessembourg, France), equipped with a vertical super
wide bore magnet and shielded gradient insert. The resonant circuit
of the nuclear magnetic resonance (NMR) probe was a 38-mm diameter
birdcage. Rectal temperature was maintained at 37°C by using a
feedback-regulated heating pad. Brain lesion evolution was assessed
using T2-weighted images obtained using a rapid acquisition with
relaxation enhancement (RARE) (TR = 2000 ms; effective echo time =
31.7 ms; RARE factor = 8; FOV = 2.5 × 2.5 cm; matrix 128×128; nine
contiguous slices of 1.2 mm, four averages). In order to improve
tumor detection, FLAIR imaging was performed using a 600 ms
inversion pulse prior to the RARE pattern, providing enough time to
allow for the annulling of the normal parenchyma and therefore
tumor detection.
Immunohistochemistry
Brains from xenotransplanted mice were surgically
removed, snap-frozen in isopentane cooled at −35°C with liquid
nitrogen, and stored at −80°C before 10 μm transverse sections of
anterior brain were made using a Cryocut 3000 (Leica,
Rueil-Malmaison, France). After at least 24 h storage at −20°C and
30 min drying at room temperature, slides were fixed in −20°C cold
methanol for 10 min. Sections were then blocked with 10% normal
goat serum in PBS added with 4% bovine serum albumine for 30 min at
room temperature. Primary antibodies against CD133 (clone AC133 and
clone 293C3 both from Miltenyi Biotech) and the corresponding
negative isotype controls (mouse IgG1κ and mouse IgG2b, both from
BD Biosciences) were diluted in PBS containing 4% BSA and used at 5
μg/ml. They were applied overnight at 4°C. After washes in PBS, a
secondary biotinylated goat anti-mouse IgG antibody (Vector
Laboratories, Burlingame, USA) diluted in PBS containing 4% BSA was
applied at 15 μg/ml for 45 min at room temperature. After
additional washes in PBS, Alexa Fluor® 488 streptavidine
conjugates (Invitrogen, Cergy Pontoise, France) were applied in the
dark at 4 μg/ml for 45 min. Finally, labeled sections were washed
three times with PBS before mounting in fluorescent mounting medium
from Dakocytomation. All slides were examined under an Axioskop-2
Zeiss fluorescence microscope (Le Pecq, France). Images were
acquired through a Photometrics CoolSNAP ES camera equipped with a
QImaging CRI Micro Color 2 RGB Liquid Crystal filter and by using
the MetaVue™ imaging system (all from Roper Scientific, Evry,
France).
Statistical analysis
XLSTAT 2006 Version 2006.3 (Addinsoft Paris, France)
was used for data analysis. Statistical significance for each
experiment was determined by a Dunnett’s test. Alternatively, the
Gehan-Wilcoxon and the Mann and Whitney non-parametric tests were
used. The tests were considered as significant with p<0.05.
Results
Oxygen tension impacts the AC133
phenotype of human glioma cells in vitro
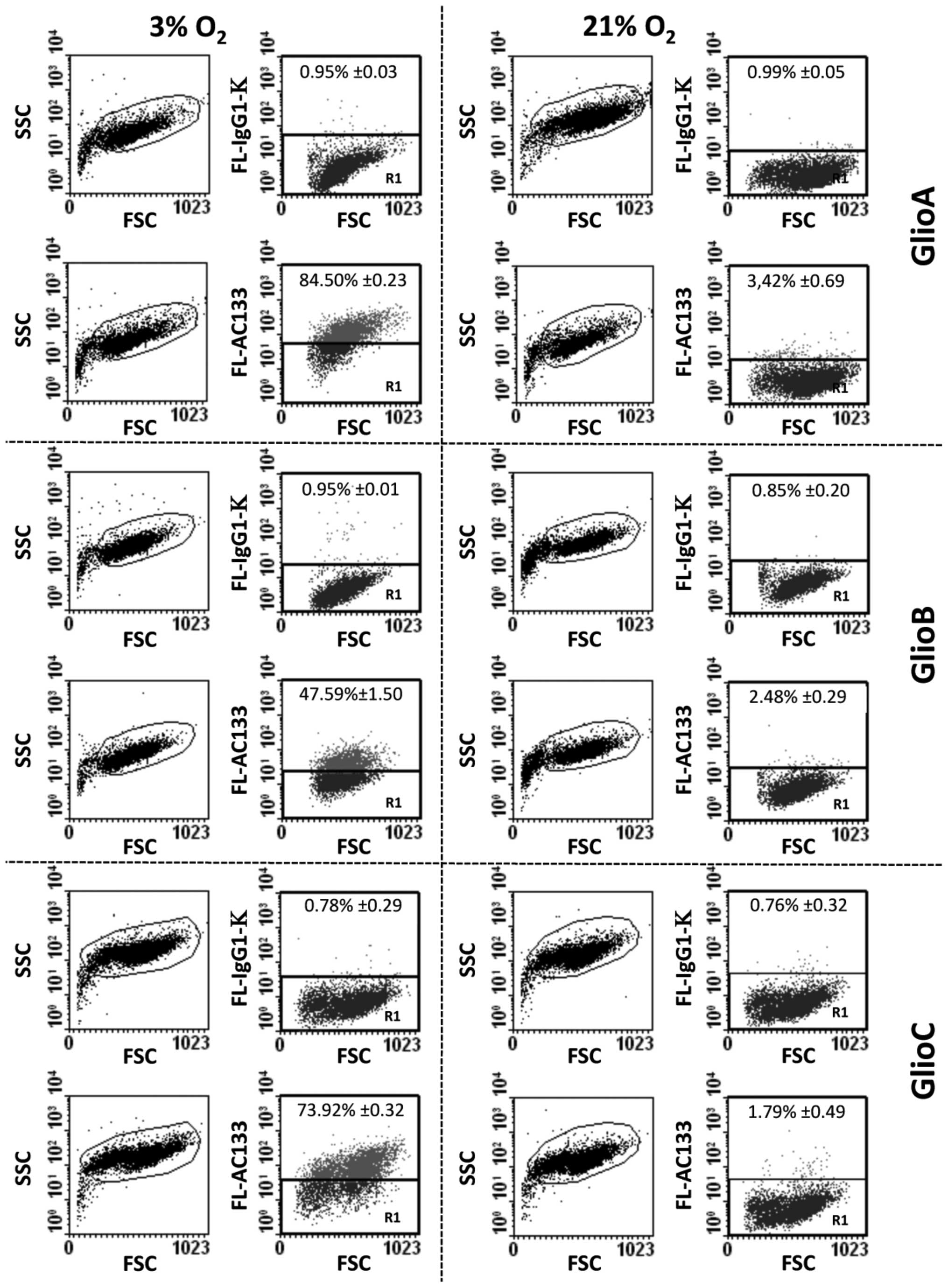
To determine the effect of oxygen pressure on AC133
expression in non-sorted primary human glioblastoma cells, GlioA,
GlioB, and GlioC were cultured either at 3% O2 or 21%
O2. High expression of AC133 was found in all cell lines
when maintained at low oxygen tension (from initial suspensions to
at least passage 30). As such, at matching cell passages flow
cytometry analysis revealed that the percentage of AC133-positive
cells was improved from 3 to 21% O2 condition (Fig. 1). Quantification of geomean
fluorescence intensity further indicated a mean reduction of AC133
expression per cell up to 99% between 21 and 3% O2
(Table I).
 | Table IGlioblastoma cells cultured at low
pO2 expressed improve levels of AC133 than those
cultured at high pO2.a |
Table I
Glioblastoma cells cultured at low
pO2 expressed improve levels of AC133 than those
cultured at high pO2.a
| GMFI at 3%
O2 (arbitrary units) | GMFI at 21%
O2 (arbitrary units) | Mean variation of
AC133 expression per cell |
|---|
| GlioA (passage
21) | 77.42±0.15 | 1.03±0.25 | −98.7% |
| GlioB (passage
14) | 16.70±0.56 | 2.08±0.15 | −87.6% |
| GlioC (passage
11) | 52.07±1.59 | 0.65±0.17 | −98.7% |
A role for HIF-1α in the regulation of
AC133 expression
Having established a role for oxygen tension in the
regulation of AC133, we next investigated whether HIF-1α a major
transcription factor regulated by oxygen tension, was involved in
this effect. HIF-1α has been described to be over-expressed in
various cancers including gliomas (28). It heterodimerizes with
constitutively expressed subunit HIF-1β to form HIF-1, a basic
helix-loop-helix structure that regulates the transcription by
specifically recognizing a short consensus HRE (hypoxia responsive
element) sequence in the promoter of hypoxia responsive genes. HRE
sequence is characterized by the presence of a consensus core CGTG
found in all known HIF-1α-responsive promoters (29). A multiple sequence alignment
ClustalW2 program revealed that the consensus core was present in
all CD133 promoters from P1 to P5. Interestingly, the analysis
showed that a sequence of 12 nucleotides present in P5 (known to be
functional in stem cells) TACGTGCTCTGG-nucleotides 5416–542
matched perfectly with that present in the [+656/+667] HRE sequence
of the human IGFBP-1 gene (30).
Hence, this sequence represents a potential target for the binding
of HIF-1 in glioblastoma cells.
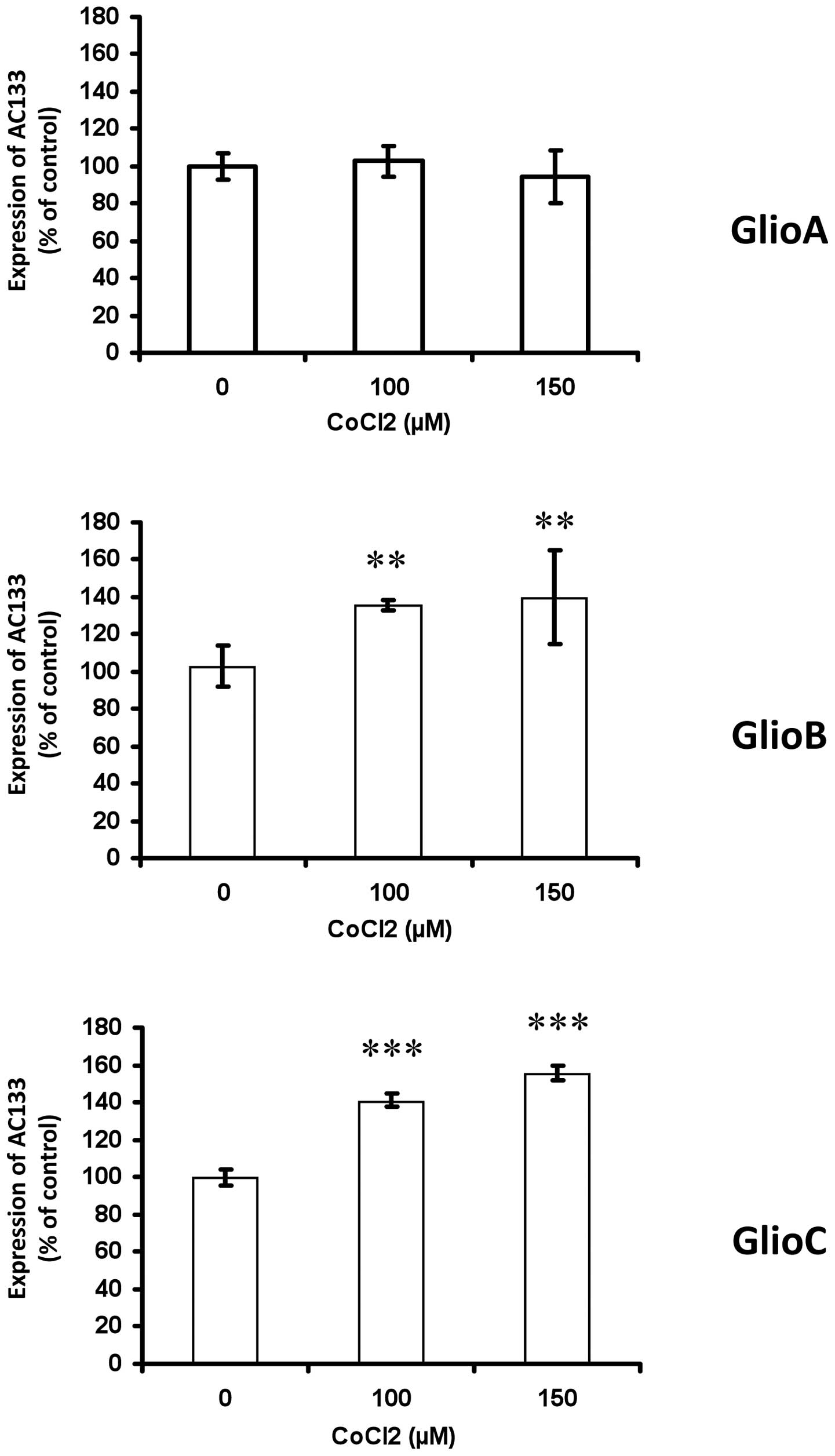
To determine the potential influence of HIF on
regulating the expression of AC133, cobalt chloride
(CoCl2), which inhibits the degradation of HIF (31), and the shRNA knockdown strategy
against HIF-1α were used. As HIF-1α stabilization has been shown to
increase from moderate to severe hypoxia while not induced under
ambient air (32), in order to try
getting its level maximal, the hypoxia-mimetic CoCl2 was
used already from the 3% O2 condition. When GlioA cells
were incubated for 24 h with 100 or 150 μM of CoCl2 in
low pO2 conditions, no significant change was observed
in AC133 expression as compared to control culture (Fig. 2). In contrast, CoCl2
treatment increased the expression of AC133 in GlioB and GlioC
(+36–41% for GlioB and +41–56% for GlioC) (Fig. 2). We further address the impact of
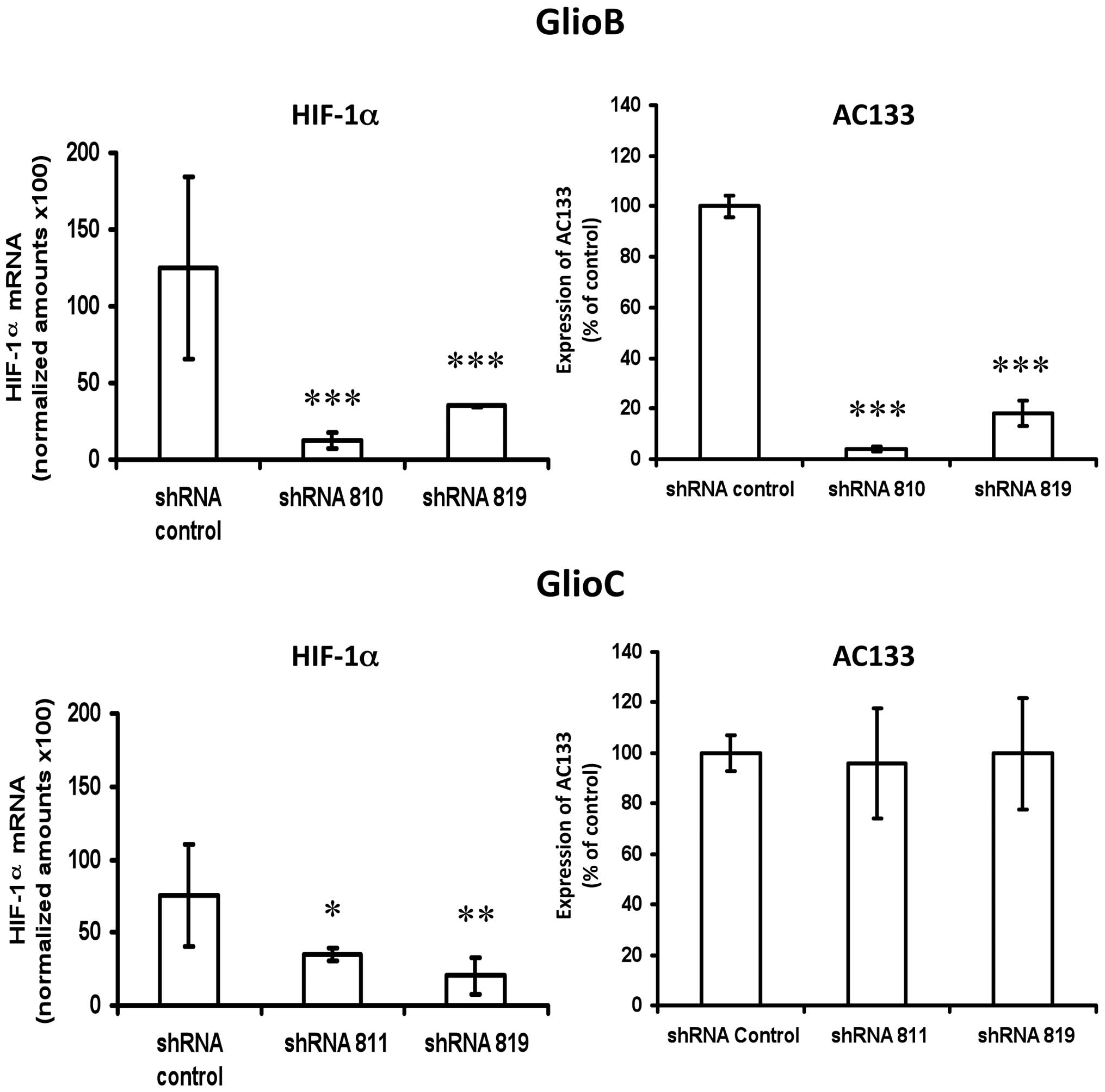
HIF-1α inhibition on CoCl2 responding glioblastoma cell
types. Transcriptional down-regulation of HIF-1α mRNA with a
lentiviral shRNA-based system performed on GlioB (knockdown
efficency of ~80% and GlioC (knockdown efficiency of ~65% (Fig. 3 left panels) was associated with a
80–90% reduction in AC133 expression for GlioB, but had no impact
on AC133 expression for GlioC (Fig.
3 right panels).
Human glioma cells exposed to different
oxygen tensions in vitro do not behave equally following orthotopic
transplantation in immunodepleted mice
Having established a role for oxygen tension and
HIF-1α in regulating AC133 in vitro, we wished to further
address whether tumor development and AC133 expression were
affected by the expansion of human glioblastoma cells under
different oxygen tension culture conditions. For this purpose, we
focused on the cell types for which tumors were detected through
MRI monitoring within 3 months after stereotactic injection of
glioblastoma cells in the right striatum of immunodepleted mice,
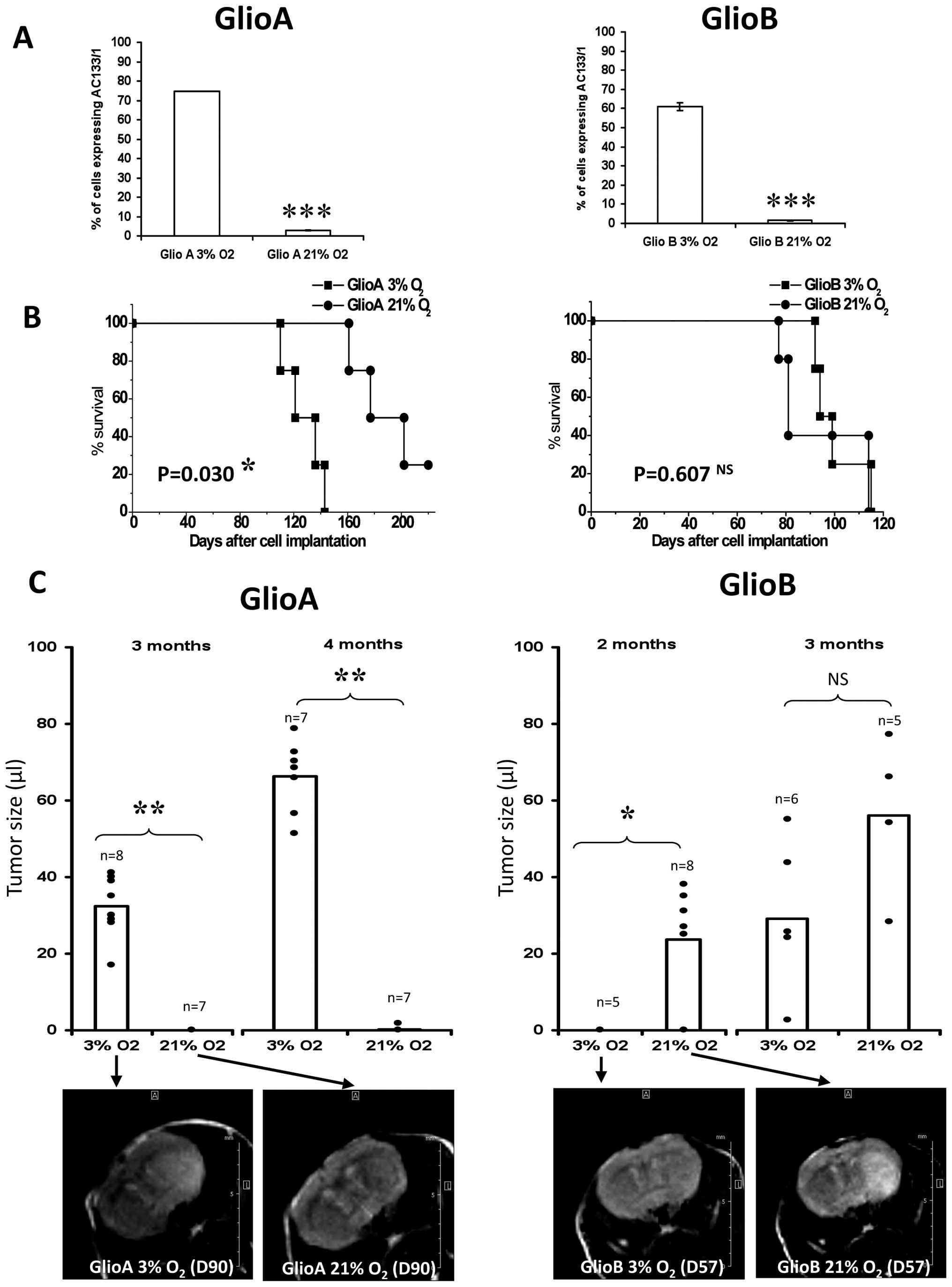
namely GlioA and GlioB (Fig. 4).
Kaplan-Meyer curves shown in Fig.
4A revealed that GlioA cell cultures at 3% O2 were
more aggressive than GlioA cells cultured at 21% O2. In
contrast, no significant differences in Kaplan-Meier curves were
observed on GlioB. However, tumors caused by the implantation of
GlioA cultured at 3% O2 prior to injection were detected
earlier than the tumors arising from GlioA cultured under 21%
O2 (Fig. 4B). Indeed,
mice injected with the cells cultured at 3% developed a detectable
tumor within 3 months post-injection (average tumor size 32±8 μl
(n=8)], whereas a similar size was observed 5 months post-injection
of GlioA cells initially cultured at 21% O2.
Interestingly, although no differences were observed
on Kaplan-Meier curves, GlioB cultured at 21 vs. 3% O2
appeared to differ on MRI images. When GlioB cells were cultured
in vitro at 21% O2 prior injection, brain tumor
occurred within 2 months [average tumor size 23±13 μl (n=8)],
whereas injection of cells cultured at 3% O2 reached
such a size after 3 months [average tumor size 34 ± 21 μl (n=6)]
(Fig. 4B). Examined together, the
data indicate that culture conditions are likely to exhibit a real
impact on tumor aggressiveness in vivo, underlying the fact
that the choice of culture parameters can modulate cell behavior
in vivo.
Extinction of AC133 expression of human
glioma cells exposed to low oxygen tension in vitro prevents in
vivo re-expression after orthotopic transplantation in
immunodepleted mice
In order to address human AC133 expression in mice
with tumor growth, a study was carried out on mice 24 h
post-injection of AC133 positive cells to validate AC133
immunohistochemical detection using the AC133 antibody or 293C3
antibody, both recognizing two different human epitopes of the
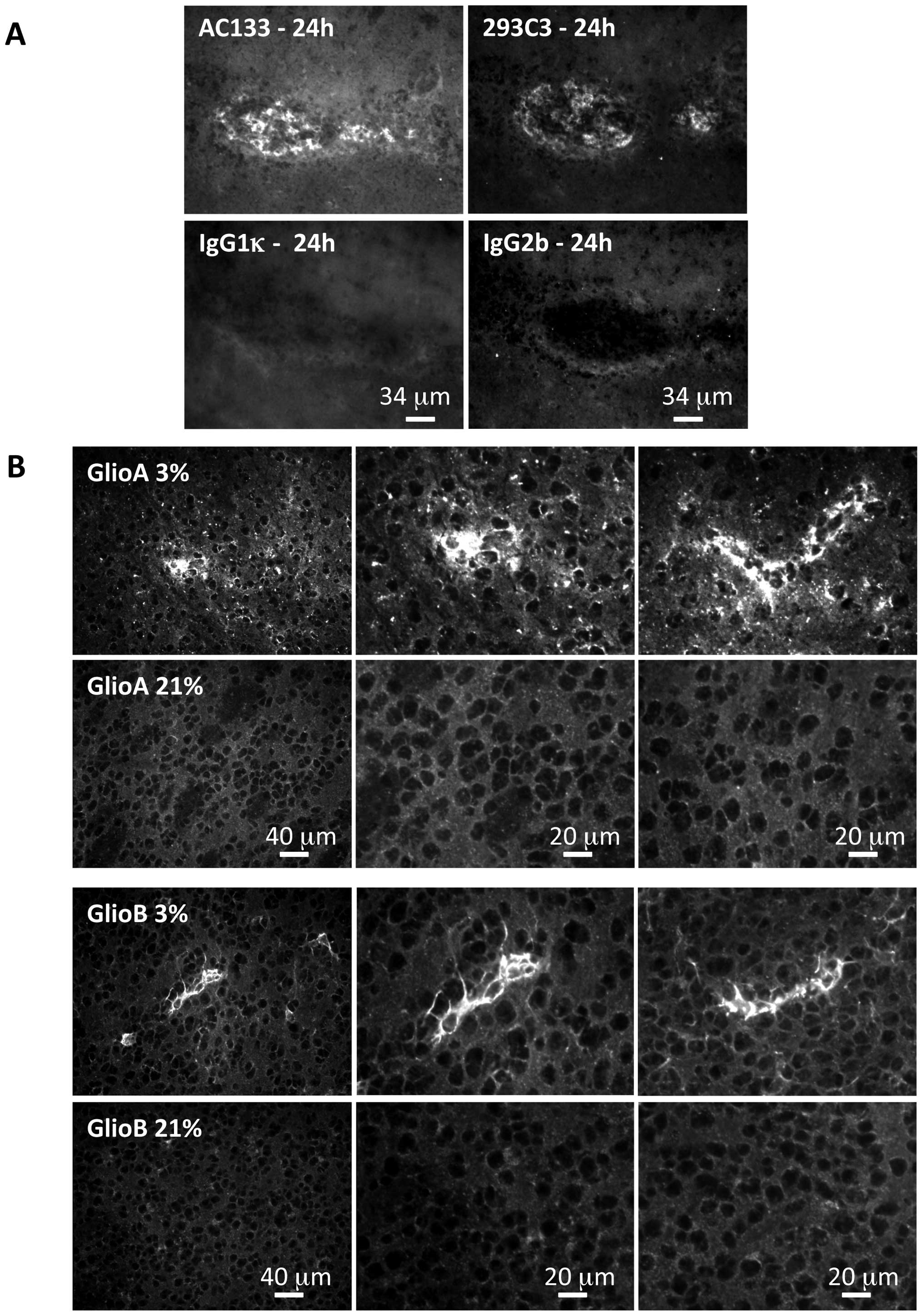
CD133 protein (Fig. 5A).
Applying this technique to brain tumors collected at
the end point of the experiment revealed that when the injected
cells were initially cultured at 3% O2, AC133 was still
detected, and this for both GlioA and GlioB cells (Fig. 5B). However, AC133 was detected in
limited clusters within the tumor, suggesting that not all the
tumor cells had kept the AC133 phenotype. The same approach on
tumors arising from cells cultured at 21% O2 did not
reveal any AC133 expression (Fig.
5B), indicating that neither GlioA nor GlioB cells grown in 21%
O2 before injection gave rise to AC133 cells in
vivo.
Discussion
Consequences of the 21% standard
pO2 culture condition on glioblastoma phenotypes
To address cancer cell behavior in vivo and
in vitro, cancer cell cultures are generally performed under
5% CO2 combined to classical atmospheric conditions of
approximately 21% O2 (160 mm Hg). However,
pO2 values do not exceed 12% O2 (95 mm Hg) in
the blood and vary from 1 to 5% (6–34 mm Hg) in normal tissues
including the brain (33).
Moreover, a characteristic feature of advanced solid tumors is to
display hypoxic tissue areas (pO2 ≤0.4% or 2.5 mm Hg)
due to insufficient vascularization, oversize tumor mass, and
necrosis (34). Thus, an
atmosphere containing 21% O2 should be physio-logically
considered hyperoxic. In the present study, although glioblastoma
cells were cultured as three-dimension neurospheres, a condition
that lowers pO2 due to the gradient of O2
diffusion from the external to the inner part of the spheres, our
data demonstrated that in vitro pO2 ranges
obtained at 3 versus 21% O2 resulted in distinct cell
behavior in vivo. Tumor aggressiveness was higher for GlioA
when cultured at 3 versus 21% O2. MRI detection of GlioB
grown at 3% was delayed when compared to GlioB grown at 21%, while
ultimately giving rise to similar adverse clinical effects.
Moreover, AC133, typically found on fresh human glioblastoma biopsy
specimens (21) or on short-term
primary glioblastoma cultures [(3,13);
our study]; was maintained after expansion in vitro at 3%
O2 while lost at 21% O2 and not re-expressed
after cell implantation in vivo. These combined findings
stressed that pO2 values obtained at 3% O2
preserve better the AC133 phenotype of glioblastoma cells than do
pO2 values obtained through the standard O2
atmospheric tension. Our results confirmed, therefore, that a low
pO2 (≤3% O2 or 24 mm Hg) should be considered
a basic condition to study glioblastoma cell behavior in their
current microenvironment. As such, the fact that pO2
irreversibly changes the phenotype of glioblastoma cell populations
is also reminiscent of the effects of serum and laminin on gene
expression profiles, expression of stem cell makers, and glioma
invasiveness (3,35,36).
As variations of pH, the traditional 21% O2 represents a
new environmental stress for glioblastoma cells that inevitably
triggers alterations of their differentiation, genetic and
epigenetic status, and survival. Considering tumor heterogeneity,
selection of glioblastoma cell clones will therefore be different
at 3 and 21% O2. As such, low oxygen tension is often
perceived as an obstacle for chemo- and radiotherapy due to the
induction of several resistance genes (13,36),
DNA repair or methylation (22),
miRNA expression (37), and
maintenance of stemness (38).
Conversely, high oxygen tension represents an oxidative stress that
may be associated with the selection of cells that are
well-equipped for reactive oxygen species detoxification (39).
Is AC133 a marker of BTSC non-chronic
exposure to high oxygen tension?
AC133 has initially been described as a marker of
hematopoietic stem cells (9,11),
while then associated with embryonic stem cells (40) and a variety of somatic stem cells
(41). AC133 was also recognized
as a putative cancer stem cell marker in blood, brain, colon,
prostate, lung, breast, liver, and skin cancers (12,41).
Although the BTSC hypothesis was strongly supported by recent data
(4,42,43),
the idea of a responsibility of cancer stem cells in glioblastoma
development remains to be documented (44) and does not exclude the role of
clonal selection (45). We
emphasize that if the hypothesis of brain cancer initiating cells
is correct, the loss of AC133 does not preclude their occurrence.
We did establish that GlioA and GlioB that do not contain high
AC133 expressing cells when cultured at 21% O2
self-renew in vitro and do form tumors in vivo. The
data give further significance to the originally established unique
ability of immunosorted AC133-positive cells to form brain tumors
(2,3,13,46)
and corroborate the fact that AC133-negative cells are also capable
of doing so (47,48). Thus, high AC133 expression is not a
marker of every cancer initiating cells within brain tumors.
Sorted AC133-positive cells have been described as
more aggressive than their AC133-negative counterparts (2,3,13).
Our data proved that when considering the full cancer cell
population, the major reduction in AC133 expression at high versus
low pO2 (87.6%, GlioB, Table I) as well as in AC133 positive cell
numbers (from 47.59 to 2.48%, GlioB, Fig. 1) allows for the development of
tumors that are similarly aggressive. Thus, AC133 does not appear
to be a general marker of tumor aggressiveness.
Low oxygen tension was associated with the stem
cell-like properties of AC133-positive glioblastoma cells (26). As we have confirmed here that this
also resulted in high levels of AC133, one might assume that AC133
expression constitutes a witness of low oxygen tension. The
presence of putative HRE in Prominin-1 promoters combined with the
modulation of AC133 expression by CoCl2 treatment and
HIF-1α shRNA knockdown supported this assertion. Previous data
obtained with siRNA against HIF-1α (49), or instead with an oxygen stable
HIF-1α construct (50), also
corroborated this, with a significant role for HIF-1α.
Interestingly, in contrast to what happen in glioblastoma, in
gastric, colorectal and lung cancer cell lines Matsumoto et
al established an inverse correlation between HIF-1α and CD133
expression thus indicating tissue specificities for the regulation
of CD133 by HIF-1α (51). However,
CoCl2 did not induce AC133 in GlioA O2, and
HIF-1α shRNAs were not able to reduce AC133 expression in GlioC.
Although constitutive expression of AC133 might be maximal in
GlioA, and HIF-2 is likely to compensate for the loss of HIF-1α in
GlioC (21,52), HIF-independent pathways may be
involved in the AC133 regulation by hypoxia. A variety of these
recognizable cell signals that translate to environmental
O2 changes have already been described, including:
reactive oxygen species (53),
thiol-based sensors (53), the
transcriptional co-activator PGC-1α (54), or mTOR inhibition via the
AMPK/TSC2/Rheb pathway (55).
Regardless of the signaling pathway involved in regulating AC133 by
pO2, we have established in our study that the loss of
AC133 at 21% O2 in vitro (data not shown) and
in vivo following glioma cell implantation in mouse brains
was irreversible. This lack of re-expression of AC133 therefore
supported the fact that AC133 is not a genuine marker of hypoxia in
glioblastoma. Indeed, low pO2 commonly involved in
glioblastoma growth and aggressiveness (23,24)
should be present within GlioA and GlioB tumors, which was
supported by a reduced vascularization observed using CD31 labeling
(data not shown). One-way regulation of AC133 by pO2
might be explained by the acquisition of a new pattern of
transcriptional activators or a new DNA methylation status of
glioblastoma cells at 21% (56,57).
As AC133 does not attest to the glioma cell
capability of forming tumors or to glioblastoma aggressiveness or
low oxygen tension, we propose that it represents a witness of
glioblastoma cell non-exposure to high oxygen tension. The presence
of AC133 positive glioblastoma cell populations that have also been
established at ambient oxygen setting could be explain in this
context by creation of hypoxic gradients within the growing glioma
spheres (58). This fact would be
attenuated by chronic exposure of cells to high oxygen tension
through sequential dissociation and re-plating. Hence, similarly to
developmental cues that lead to irreversible maturation of
early-to-late neural stem cell differentiation during development
such as FGF (59), high oxygen
tension may represent a component of the BTSC niche that drives an
early-to-late BTSC switch during gliomagenesis. If EGF receptor
expression represents a witness of the acquired phenotype for
neural stem cell maturation, loss of AC133 would be a witness of
BTSC maturation. To support this assertion, the loss of AC133
expression has been associated with cancer stem cell
differentiation in glioblastoma (60) and in colon cancer (61). Moreover, use of glioma cell
differentiation factors such as retinoic acid lead to
down-regulation of AC133 expression (62). In addition, transdifferentiation of
tumor cells into vessel formation was recently associated with
stemness phenotype and hypoxia in glioblastoma (63). Thus, irreversible AC133-loss may
also have an impact on this epithelial to mesenchymal transition
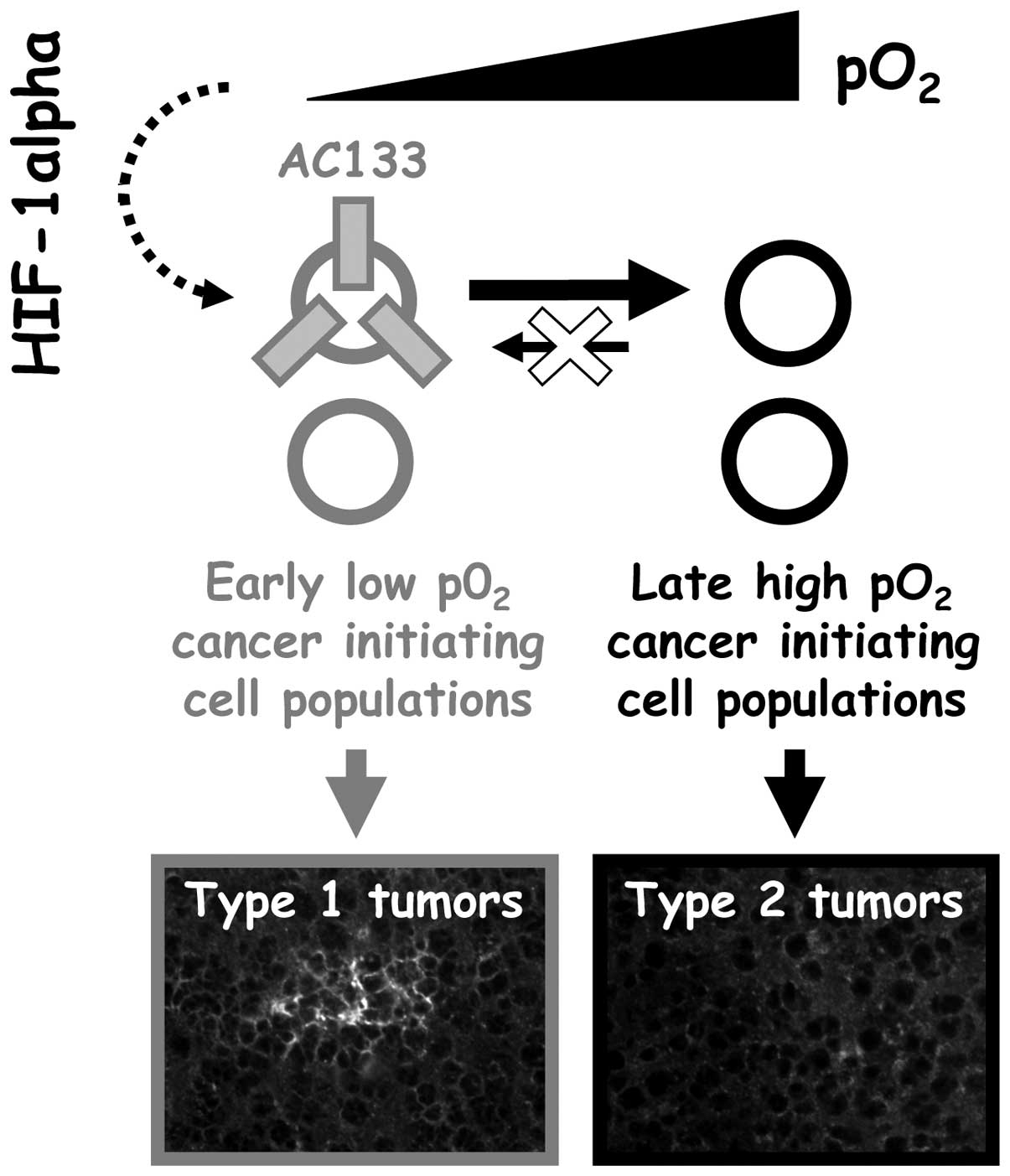
reciprocally. Two types of tumors could therefore be obtained from
non-sorted human glioblastoma cells expanded in vitro: type
1 tumors obtained from 3% O2-expanded cells (expressing
AC133) and type 2 tumors obtained from 21% O2-expanded
cells (no AC133 expression) (Fig.
6).
In conclusion, our present study underlines that
non-physiological oxygen tension alters subsequent in vitro
expansion and in vivo development of non-sorted human
glioblastoma cells. With the preservation of AC133 expression,
which can result from the prevention of AC133-positive cell death
or from continuous prominin-1 gene expression, the 3% O2
expansion condition mirrors much the biological reality. Thus, the
timing of environmental pO2 variations likely reflects a
changing pattern of plasma membrane protein expression during
glioblastoma growth that is associated with cell heterogeneity and
resistance. The fact AC133 was here associated with an early
glioblastoma phenotype suggests that identification of downstream
cancer initiating cell markers as well as evaluation of relative
anticancer drug sensitivity of type I and type II tumors (Fig. 6) would also be helpful in the
development of anti-glioblastoma strategies.
Acknowledgements
We would like to thank Catherine Guillet, Julien
Daligault, and Laurence Preisser (Service Commun de Cytométrie et
d’Analyse Nucléotidique, SCCAN, Angers, France) for their skillful
technical support. We are also grateful to Pierre Legras and Jérôme
Roux from the Service Commun d’Animalerie Hospitalo-Universitaire
(SCAHU, Angers, France). La Ligue Nationale Contre le Cancer
‘Equipe Labellisée 2007’ and Le Cancéropôle Grand-Ouest throughout
the ‘Réseau Gliome Grand-Ouest’ (REGGO) and the ‘Axe Cellules
Souches et Cancer’ supported this work. Erika Bourseau-Guilmain was
a fellow of the Conseil Général de Maine-et-Loire and the Ligue
Nationale Contre le Cancer. We also acknowledge the Comité
Départemental de Maine-et-Loire de la Ligue Contre le Cancer.
References
|
1
|
Ignatova TN, Kukekov VG, Laywell ED,
Suslov ON, Vrionis FD and Steindler DA: Human cortical glial tumors
contain neural stem-like cells expressing astroglial and neuronal
markers in vitro. Glia. 39:193–206. 2002.
|
|
2
|
Singh SK, Clarke ID, Terasaki M, et al:
Identification of a cancer stem cell in human brain tumors. Cancer
Res. 63:5821–5828. 2003.
|
|
3
|
Singh SK, Hawkins C, Clarke ID, et al:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004.
|
|
4
|
Jacques TS, Swales A, Brzozowski MJ, et
al: Combinations of genetic mutations in the adult neural stem cell
compartment determine brain tumour phenotypes. EMBO J. 29:222–235.
2010.
|
|
5
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008.
|
|
6
|
Mayol JF, Loeuillet C, Herodin F and Wion
D: Characterisation of normal and cancer stem cells: one
experimental paradigm for two kinds of stem cells. Bioessays.
31:993–1001. 2009.
|
|
7
|
Mao XG, Zhang X, Xue XY, et al: Brain
tumor stem-like cells identified by neural stem cell marker CD15.
Transl Oncol. 2:247–257. 2009.
|
|
8
|
Bao S, Wu Q, Li Z, et al: Targeting cancer
stem cells through L1CAM suppresses glioma growth. Cancer Res.
68:6043–6048. 2008.
|
|
9
|
Miraglia S, Godfrey W, Yin AH, et al: A
novel five-transmembrane hematopoietic stem cell antigen:
isolation, characterization, and molecular cloning. Blood.
90:5013–5021. 1997.
|
|
10
|
Yin AH, Miraglia S, Zanjani ED, et al:
AC133, a novel marker for human hematopoietic stem and progenitor
cells. Blood. 90:5002–5012. 1997.
|
|
11
|
Corbeil D, Roper K, Weigmann A and Huttner
WB: AC133 hematopoietic stem cell antigen: human homologue of mouse
kidney prominin or distinct member of a novel protein family?
Blood. 91:2625–2626. 1998.
|
|
12
|
Ferrandina G, Petrillo M, Bonanno G and
Scambia G: Targeting CD133 antigen in cancer. Expert Opin Ther
Targets. 13:823–837. 2009.
|
|
13
|
Bao S, Wu Q, McLendon RE, et al: Glioma
stem cells promote radioresistance by preferential activation of
the DNA damage response. Nature. 444:756–760. 2006.
|
|
14
|
Zobalova R, McDermott L, Stantic M,
Prokopova K, Dong LF and Neuzil J: CD133-positive cells are
resistant to TRAIL due to up-regulation of FLIP. Biochem Biophys
Res Commun. 373:567–571. 2008.
|
|
15
|
Bao S, Wu Q, Sathornsumetee S, et al: Stem
cell-like glioma cells promote tumor angiogenesis through vascular
endothelial growth factor. Cancer Res. 66:7843–7848. 2006.
|
|
16
|
Zeppernick F, Ahmadi R, Campos B, et al:
Stem cell marker CD133 affects clinical outcome in glioma patients.
Clin Cancer Res. 14:123–129. 2008.
|
|
17
|
Joo KM, Kim SY, Jin X, et al: Clinical and
biological implications of CD133-positive and CD133-negative cells
in glioblastomas. Lab Invest. 88:808–815. 2008.
|
|
18
|
Garcion E, Naveilhan P, Berger F and Wion
D: Cancer stem cells: Beyond Koch’s postulates. Cancer Lett.
278:3–8. 2008.
|
|
19
|
Scadden DT: The stem-cell niche as an
entity of action. Nature. 441:1075–1079. 2006.
|
|
20
|
Calabrese C, Poppleton H, Kocak M, et al:
A perivascular niche for brain tumor stem cells. Cancer Cell.
11:69–82. 2007.
|
|
21
|
Li Z, Bao S, Wu Q, et al:
Hypoxia-inducible factors regulate tumorigenic capacity of glioma
stem cells. Cancer Cell. 15:501–513. 2009.
|
|
22
|
Pistollato F, Abbadi S, Rampazzo E, et al:
Intratumoral hypoxic gradient drives stem cells distribution and
MGMT expression in glioblastoma. Stem Cells. 28:851–862. 2010.
|
|
23
|
Panchision DM: The role of oxygen in
regulating neural stem cells in development and disease. J Cell
Physiol. 220:562–568. 2009.
|
|
24
|
Evans SM, Judy KD, Dunphy I, et al:
Hypoxia is important in the biology and aggression of human glial
brain tumors. Clin Cancer Res. 10:8177–8184. 2004.
|
|
25
|
Platet N, Liu SY, Atifi ME, et al:
Influence of oxygen tension on CD133 phenotype in human glioma cell
cultures. Cancer Lett. 258:286–290. 2007.
|
|
26
|
McCord AM, Jamal M, Shankavarum UT, Lang
FF, Camphausen K and Tofilon PJ: Physiologic oxygen concentration
enhances the stem-like properties of CD133+ human
glioblastoma cells in vitro. Mol Cancer Res. 7:489–497. 2009.
|
|
27
|
Vandesompele J, De Preter K, Pattyn F, et
al: Accurate normalization of real-time quantitative RT-PCR data by
geometric averaging of multiple internal control genes. Genome
Biol. 3:Research00342002.
|
|
28
|
Zhong H, De Marzo AM, Laughner E, et al:
Overexpression of hypoxia-inducible factor 1alpha in common human
cancers and their metastases. Cancer Res. 59:5830–5835. 1999.
|
|
29
|
Wenger RH, Stiehl DP and Camenisch G:
Integration of oxygen signaling at the consensus HRE. Sci STKE.
2005:re122005.
|
|
30
|
Tazuke SI, Mazure NM, Sugawara J, et al:
Hypoxia stimulates insulin-like growth factor binding protein 1
(IGFBP-1) gene expression in HepG2 cells: a possible model for
IGFBP-1 expression in fetal hypoxia. Proc Natl Acad Sci USA.
95:10188–10193. 1998.
|
|
31
|
Yuan Y, Hilliard G, Ferguson T and
Millhorn DE: Cobalt inhibits the interaction between
hypoxia-inducible factor-alpha and von Hippel-Lindau protein by
direct binding to hypoxia-inducible factor-alpha. J Biol Chem.
278:15911–15916. 2003.
|
|
32
|
Vordermark D and Brown JM: Evaluation of
hypoxia-inducible factor-1alpha (HIF-1alpha) as an intrinsic marker
of tumor hypoxia in U87 MG human glioblastoma: in vitro and
xenograft studies. Int J Radiat Oncol Biol Phys. 56:1184–1193.
2003.
|
|
33
|
Csete M: Oxygen in the cultivation of stem
cells. Ann NY Acad Sci. 1049:1–8. 2005.
|
|
34
|
Vaupel P, Kelleher DK and Hockel M: Oxygen
status of malignant tumors: pathogenesis of hypoxia and
significance for tumor therapy. Semin Oncol. 28:29–35. 2001.
|
|
35
|
Lee J, Kotliarova S, Kotliarov Y, et al:
Tumor stem cells derived from glioblastomas cultured in bFGF and
EGF more closely mirror the phenotype and genotype of primary
tumors than do serum-cultured cell lines. Cancer Cell. 9:391–403.
2006.
|
|
36
|
Shervington A and Lu C: Expression of
multidrug resistance genes in normal and cancer stem cells. Cancer
Invest. 26:535–542. 2008.
|
|
37
|
Huang X, Le QT and Giaccia AJ:
MiR-210-micromanager of the hypoxia pathway. Trends Mol Med.
16:230–237. 2010.
|
|
38
|
Pistollato F, Chen HL, Rood BR, et al:
Hypoxia and HIF1alpha repress the differentiative effects of BMPs
in high-grade glioma. Stem Cells. 27:7–17. 2009.
|
|
39
|
Dringen R, Pfeiffer B and Hamprecht B:
Synthesis of the antioxidant glutathione in neurons: supply by
astrocytes of CysGly as precursor for neuronal glutathione. J
Neurosci. 19:562–569. 1999.
|
|
40
|
King FW, Ritner C, Liszewski W, et al:
Subpopulations of human embryonic stem cells with distinct
tissue-specific fates can be selected from pluripotent cultures.
Stem Cells Dev. 18:1441–1450. 2009.
|
|
41
|
Wu Y and Wu PY: CD133 as a marker for
cancer stem cells: progresses and concerns. Stem Cells Dev.
18:1127–1134. 2009.
|
|
42
|
Alcantara Llaguno S, Chen J, Kwon CH, et
al: Malignant astrocytomas originate from neural stem/progenitor
cells in a somatic tumor suppressor mouse model. Cancer Cell.
15:45–56. 2009.
|
|
43
|
Wang Y, Yang J, Zheng H, et al: Expression
of mutant p53 proteins implicates a lineage relationship between
neural stem cells and malignant astrocytic glioma in a murine
model. Cancer Cell. 15:514–526. 2009.
|
|
44
|
Hill RP: Identifying cancer stem cells in
solid tumors: case not proven. Cancer Res. 66:1890–1895. 2006.
|
|
45
|
Adams JM and Strasser A: Is tumor growth
sustained by rare cancer stem cells or dominant clones? Cancer Res.
68:4018–4021. 2008.
|
|
46
|
Piccirillo SG, Reynolds BA, Zanetti N, et
al: Bone morphogenetic proteins inhibit the tumorigenic potential
of human brain tumour-initiating cells. Nature. 444:761–765.
2006.
|
|
47
|
Ogden AT, Waziri AE, Lochhead RA, et al:
Identification of A2B5+CD133−
tumor-initiating cells in adult human gliomas. Neurosurgery.
62:505–514. 2008.
|
|
48
|
Wang J, Sakariassen PO, Tsinkalovsky O, et
al: CD133 negative glioma cells form tumors in nude rats and give
rise to CD133 positive cells. Int J Cancer. 122:761–768. 2008.
|
|
49
|
Soeda A, Park M, Lee D, et al: Hypoxia
promotes expansion of the CD133-positive glioma stem cells through
activation of HIF-1alpha. Oncogene. 28:3949–3959. 2009.
|
|
50
|
Bar EE, Lin A, Mahairaki V, Matsui W and
Eberhart CG: Hypoxia increases the expression of stem-cell markers
and promotes clonogenicity in glioblastoma neurospheres. Am J
Pathol. 177:1491–1502. 2010.
|
|
51
|
Matsumoto K, Arao T, Tanaka K, et al: mTOR
signal and hypoxia-inducible factor-1 alpha regulate CD133
expression in cancer cells. Cancer Res. 69:7160–7164. 2009.
|
|
52
|
Heddleston JM, Li Z, McLendon RE,
Hjelmeland AB and Rich JN: The hypoxic microenvironment maintains
glioblastoma stem cells and promotes reprogramming towards a cancer
stem cell phenotype. Cell Cycle. 8:3274–3284. 2009.
|
|
53
|
Lopez-Barneo J, Pardal R and Ortega-Saenz
P: Cellular mechanism of oxygen sensing. Annu Rev Physiol.
63:259–287. 2001.
|
|
54
|
Arany Z, Foo SY, Ma Y, et al:
HIF-independent regulation of VEGF and angiogenesis by the
transcriptional coactivator PGC-1alpha. Nature. 451:1008–1012.
2008.
|
|
55
|
Liu L, Cash TP, Jones RG, Keith B,
Thompson CB and Simon MC: Hypoxia-induced energy stress regulates
mRNA translation and cell growth. Mol Cell. 21:521–531. 2006.
|
|
56
|
Tabu K, Sasai K, Kimura T, et al: Promoter
hypomethylation regulates CD133 expression in human gliomas. Cell
Res. 18:1037–1046. 2008.
|
|
57
|
Yi JM, Tsai HC, Glockner SC, et al:
Abnormal DNA methylation of CD133 in colorectal and glioblastoma
tumors. Cancer Res. 68:8094–8103. 2008.
|
|
58
|
Wion D, Christen T, Barbier EL and Coles
JA: PO(2) matters in stem cell culture. Cell Stem Cell. 5:242–243.
2009.
|
|
59
|
Lillien L and Raphael H: BMP and FGF
regulate the development of EGF-responsive neural progenitor cells.
Development. 127:4993–5005. 2000.
|
|
60
|
Chen R, Nishimura MC, Bumbaca SM, et al: A
hierarchy of self-renewing tumor-initiating cell types in
glioblastoma. Cancer Cell. 17:362–375. 2010.
|
|
61
|
Kemper K, Sprick MR, De Bree M, et al: The
AC133 epitope, but not the CD133 protein, is lost upon cancer stem
cell differentiation. Cancer Res. 70:719–729. 2010.
|
|
62
|
Campos B, Wan F, Farhadi M, et al:
Differentiation therapy exerts antitumor effects on stem-like
glioma cells. Clin Cancer Res. 16:2715–2728. 2010.
|
|
63
|
Soda Y, Marumoto T, Friedmann-Morvinski D,
et al: Trans-differentiation of glioblastoma cells into vascular
endothelial cells. Proc Natl Acad Sci USA. 108:4274–4280. 2011.
|