Introduction
Gastric cancer is the second most common cause of
cancer-related death worldwide (1). Gastric adenocarcinoma has a poor
outcome since high percentage of cases present with advanced
disease. Chemotherapy has been considered to be useful treatment
for advanced gastric cancer, but its current 5-year survival rate
is less than 20% (1,2). Accordingly, the unmet need of
effective treatment has led to an intensive effort to examine
molecular regulators. Furthermore, based on the previous research
that gastric cancer results from accumulated genetic alterations,
which affect essential cellular functions for tumorigenesis,
investigations to find a good predictive biomarker for targeted
therapy have been undertaken in recent years in order to improve
present therapeutics (1,3).
The PI3K/AKT pathway is known to play a key role in
regulating various cellular processes, such as proliferation,
growth, apoptosis, cytoskeletal rearrangement and cell metabolism
(4,5). In gastric cancer, the PI3K/AKT
signaling is inappropriately activated through mutation or
alteration of many components of the PI3K pathway. Up to now, the
mechanisms observed widely for PI3K/AKT activation in gastric
cancer include somatic activating mutations and amplifications in
p110α (6–8), loss of the PTEN tumor suppressor
(8), and genetic amplifications of
AKT1 (9). Preclinical study of
human gastric cancer cell lines has demonstrated the
anti-proliferative effect of PI3K inhibition by LY294002 or mTOR
inhibition by everolimus and evidenced the synergistic efficacy
with 5-fluorouracil or sunitinib, indicating a role for the
PI3K/AKT pathway in gastric cancer carcinogenesis (10–12).
In addition to gastric adenocarcinoma, the PI3K/AKT pathway has
been an attractive target in clinical studies of various human
cancers.
Agents targeting PI3K/AKT pathway in clinical
development are pure PI3K inhibitors including NVP-BKM120, dual
PI3K-mTOR inhibitors, AKT inhibitors and mTOR inhibitors.
Isoform-specific PI3K inhibitors are also emerging.
According to previous studies, specific genetic
alterations, such as HER2 amplification and PIK3CA mutation, were
revealed as biomarkers for sensitivity to the PI3K inhibitor in
breast cancer (13). However,
cancers harboring KRAS mutations are likely to be insensitive to
single-agent PI3K inhibitors and showed synergism in combination
treatment with MEK inhibitors (14,15).
In other words, KRAS mutant cancers insensitive to single treatment
of PI3K inhibitors seem to induce at least one signaling mediator
in the alternate pathway, which contributes to resistance. Thus,
combined inhibition is required to suppress activation of other
pathways and feedback loop-induced activation of other oncogenic
signaling pathways, resulting in more potent induction of
apoptosis.
The STAT pathway is another possible inducible
pathway in response to PI3K inhibition and recently, STAT3 has been
reported as an essential molecule in RAS oncogenic transformation
(16). STATs are latent
transcription factors that are involved in cell proliferation,
survival, angiogenesis and immunosuppression (17). In diverse cancers including gastric
cancer, the STAT pathway, especially STAT3, is constitutively
activated and plays a major role in tumorigenesis (17,18).
Thereby, an effort for directly or indirectly targeting the STAT
signaling has been made to develop a new approach for effective
cancer therapy. For example, preclinical studies of inhibition of
STAT3 by STAT3 inhibitors or JAK2 inhibitors showed potent
anti-tumor activity in cancers including solid tumors as well as
myeloma (19,20).
In the present study, we characterized the antitumor
effects exerted by Class I PI3K single inhibition and combination
with STAT3 inhibition in gastric cancer cell lines for the first
time. Results indicate that NVP-BKM120, a pan-class I PI3K
inhibitor, is able to inhibit mTOR downstream activation, but
induces the phosphorylation of AKT and the activation of p-ERK or
p-STAT3 in KRAS mutant gastric cancer cells. The combination of
NVP-BKM120 and AG490, a STAT3 inhibitor, showed a synergism leading
to apoptosis, but this synergism was only observed in cells
harboring mutant KRAS.
Thus, our result suggests that dual inhibition of
PI3K and STAT3 signaling may be an effective therapeutic strategy
for KRAS mutant gastric cancer patients.
Materials and methods
Cell lines
Human gastric cancer cell lines (SNU-1, -5, -16,
-216, -484, -601, -620, -638, -668 and -719) were obtained from the
Korean Cell Line Bank (21) and
AGS was purchased from the American Type Culture Collection. All
cell lines were maintained in RPMI-1640 supplemented with 10% fetal
bovine serum (Hyclone Laboratories, Inc., Logan, UT, USA) and 10
μg/ml gentamicin (Cellgro, Herndon, VA, USA) at 37°C in a 5%
CO2 humidified atmosphere.
Reagents
NVP-BKM120, a pan-class I PI3K inhibitor, was
generously provided by Novartis Pharma AG (Basel, Switzerland)
(Fig. 1B). NVP-BKM120 inhibits
wild-type p110α (IC50 35 nM), with high selectivity
toward protein kinases and shows significant antitumor activity in
animal models (in the Novartis brochure). It has been in Phase I
clinical trials for solid tumors. AG490, a STAT3 inhibitor, was
purchased from Sigma-Aldrich. Stock solutions for both drugs were
prepared in dimethyl sulfoxide (DMSO) and stored at −20°C.
NVP-BKM120 and AG490 were diluted in DMSO prior to each experiment,
and the final concentration of DMSO was <0.1%.
Antibodies and Western blotting
Cells were grown in 100 mm dishes and treated with
NVP-BKM120 or AG490 for the indicated concentrations and time.
Cells were washed twice with ice-cold phosphate-buffered saline and
lysed in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.5, 1% NP-40,
0.1% sodium deoxycholate, 150 mM NaCl, 50 mM NaF, 1 mM sodium
pyrophosphate, 1 mM EDTA, and protease inhibitors). 20 μg of total
proteins were resolved using sodium dodecyl sulfate-polyacrylamide
gel electrophoresis. The resolved proteins were transferred to
nitrocellulose membranes and probed with antibodies. Lysates were
incubated with antibody overnight at 4°C. Detection was conducted
using an enhanced chemiluminescence system (Amersham Pharmacia
Biotech). Antibodies against p110α, p-AKT, p-p70S6K, p-4E-BP1,
4E-BP1, p-STAT3, p-ERK, and Cyclin D1 were purchased from Cell
Signaling Technology. Anti-p110β antibody was acquired from
Millipore. Anti-p27 [Kip1], anti-IRS-1 and anti-Cleaved PARP were
acquired from BD Transduction Laboratories. Cyclin B1, PTEN and
Actin antibodies were obtained from Santa Cruz Biotechnology.
Anti-α-tubulin antibody was purchased from Sigma-Aldrich.
Cell growth inhibition assay
Tetrazolium dye (MTT; Sigma-Aldrich) assays were
used to evaluate the growth inhibitory effects of NVP-BKM120,
AG490, or NVP-BKM120 plus AG490. The cells were seeded on 96-well
plates at a density of 1,000–3,000 cells per well, incubated for 24
h, and then treated for 72 h with drugs at 37°C. After drug
treatment, MTT solution was added to each well and incubated for 4
h at 37°C before the removal of the media. DMSO was then added and
mixed thoroughly for 20 min at room temperature. Cell viability was
determined by measuring absorbance at 540 nm in a microplate reader
(VersaMax, Molecular Devices). The drug concentrations required to
inhibit cell growth by 50% were determined through interpolation
from the dose-response curves (CalcuSyn, Biosoft). Four replicate
wells were used for each analysis, and at least three independent
experiments were conducted. The data from replicate wells are
presented as the mean numbers of remaining cells, with 95%
confidence intervals.
Cell cycle analysis
Cells were seeded in 60-mm dishes and treated with
NVP-BKM120, AG490 or NVP-BKM120 plus AG490 for 72 h. Floating and
adherent cells were collected by trypsinization and washed once
with PBS. Cells were incubated in 70% ethanol at −20°C overnight,
treated with 50 μg/ml RNase A, and stained with 50 μg/ml propidium
iodide. The cell DNA content (10,000 cells per experimental group)
was determined with a flow cytometer (FACS Canto™II,
Becton-Dickinson Biosciences) equipped with a ModFit LT program
(Verity Software House, Inc.). The experiments were repeated three
times.
Statistical analysis
An unpaired two-tailed t-test was used to determine
the significance of change in the levels of cell viability and cell
cycle analysis between the different treatment groups. Differences
between groups were considered statistically significant at
P<0.05.
Results
Anti-proliferative effects of NVP-BKM120
on human gastric cancer cell lines
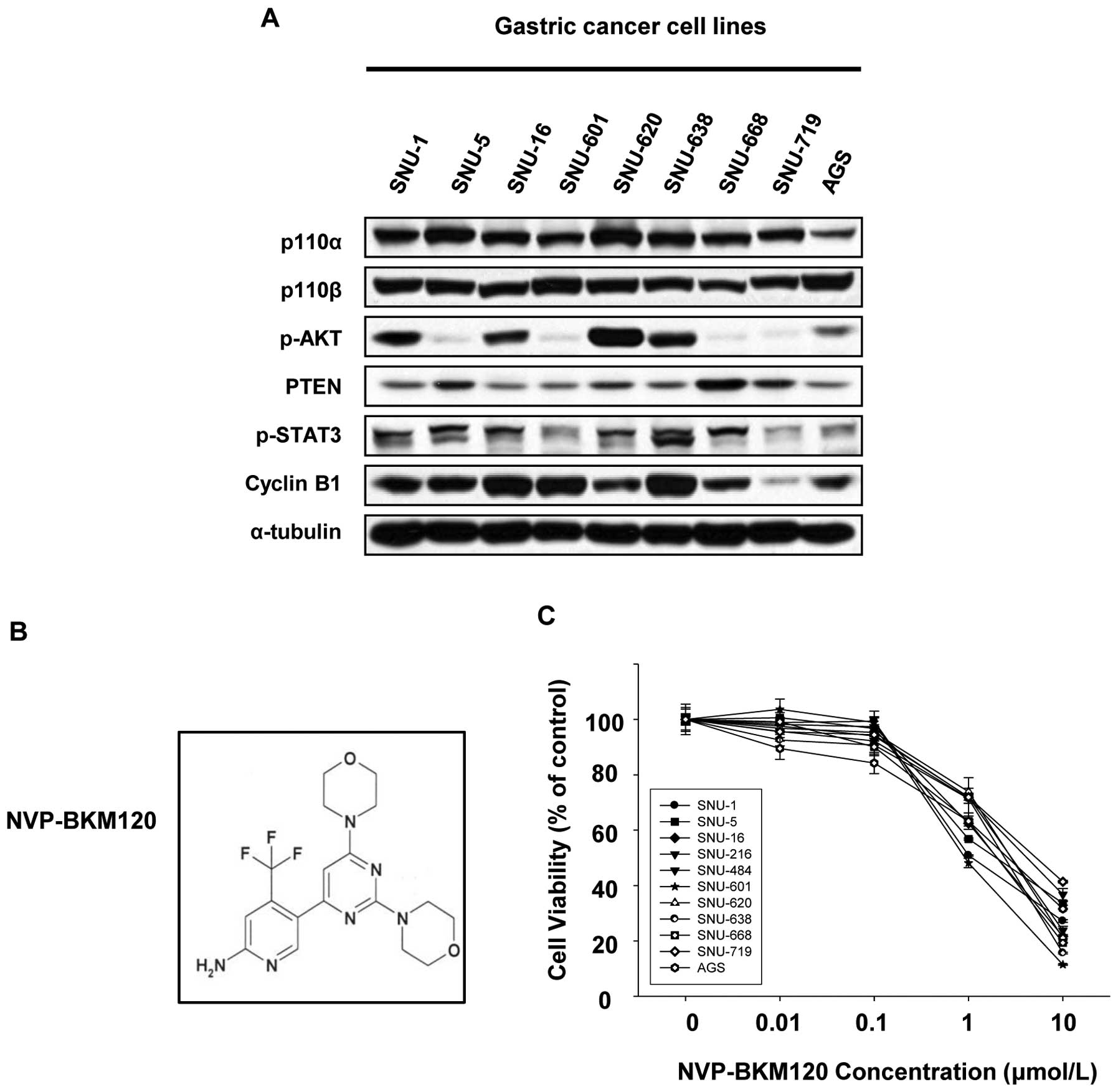
The baseline levels of p110α, p110β, PTEN,
phosphorylated AKT, ERK, and STAT3 in human gastric cancer cell
lines were measured by Western blotting. The levels of PTEN, p-AKT,
p-ERK and p-STAT3 were expressed to varying degrees in our panel of
gastric cancer cell lines and p110α and p110β were expressed to
similar degree in all cell lines (Fig.
1A).
To assess the anti-proliferative effects of a
pan-class I PI3K inhibitor, NVP-BKM120, on human gastric cancer
cells, we exposed 11 human gastric cancer cell lines with different
modulations of PI3K/AKT cascade (Table
I). Cell growth of all cell lines tested was effectively
suppressed by NVP-BKM120 with IC50 values ranging from
approximately 0.8 to 3 μmol/l (Table
I, Fig. 1C). In our panel of
gastric cancer cell lines, a correlation between the
anti-proliferative effect of NVP-BKM120 and the genetic alterations
including HER2 amplification, KRAS mutation, and both PIK3CA and
KRAS mutation at the same time, was not found (Fig. 1A). Additionally, the baseline
levels of Cyclin B1 was also irrelevant to the sensitivity to
NVP-BKM120 differently from a previous study reporting that cells
resistant to PI3K inhibitors expressed high levels of Cyclin B1
(15).
 | Table ISensitivity of NVP-BKM120 in human
gastric cancer cell lines. |
Table I
Sensitivity of NVP-BKM120 in human
gastric cancer cell lines.
| Cell lines | KRAS | Others | BKM120
IC50 (μmol/l) |
|---|
| SNU-601 | MT, G12D | | 0.816±0.063 |
| SNU-1 | MT, G12D | | 1.082±0.028 |
| SNU-668 | MT, Q61K | | 1.579±0.074 |
| AGS | MT, G12D | PIK3CA,
E453Ka | 1.741±0.117 |
| SNU-216 | WT | HER2
amplification | 2.692±0.082 |
| SNU-5 | WT | MET
amplification | 1.351±0.091 |
| SNU-638 | WT | MET
amplification | 2.282±0.053 |
| SNU-16 | WT | FGFR2
amplification | 1.573±0.001 |
| SNU-484 | WT | | 1.728±0.045 |
| SNU-620 | WT | | 2.939±0.001 |
| SNU-719 | WT | | 3.037±0.032 |
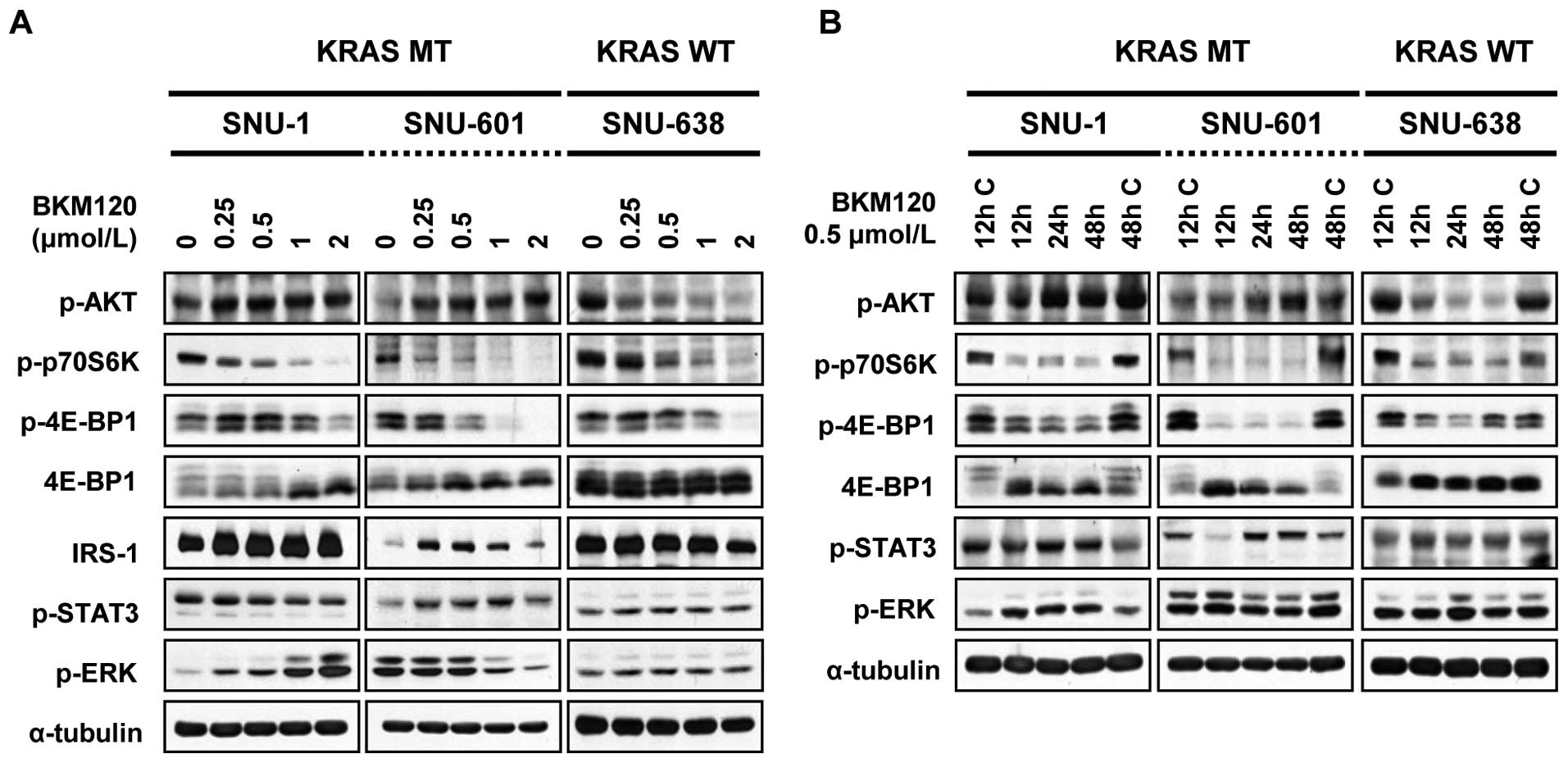
The effect of NVP-BKM120 on PI3K/AKT/mTOR
signaling in human gastric cancer cells
We next evaluated how inhibition of PI3K activity by
NVP-BKM120 affects downstream AKT and mTOR activities. Cells were
exposed to increased concentrations of NVP-BKM120 for 24 h and then
subjected to Western blotting of phosphorylated AKT, 4E-BP1,
p70S6K, ERK and STAT3. Dose- and time-response experiments revealed
that in SNU-1, SNU-601, and SNU-638 cells, inhibition of PI3K
activity resulted in an indirect downstream inhibition of AKT
(p70S6K, 4E-BP1, through mTOR) (Fig.
2). However, unlike SNU-638 cells, SNU-1 and SNU-601 cells,
which are KRAS mutants, showed increase of AKT phosphorylation in
both dose- and time-dependent manner. Presumably, we assumed that
it might be due to release of negative feedback loops of AKT
signaling. The best-known mechanism includes insulin receptor
substrate-1 (IRS-1) as an intermediary in the PI3K/AKT/mTOR
negative feedback loop (22,23).
Thus, we checked the dephosphorylation status of IRS-1 along with
the increasing level of AKT. As shown in Fig. 2A, NVP-BKM120 induced IRS-1 total
protein levels in SNU-1 and SNU-601 cell lines, which implies an
increase in IRS-1 stability and association with PI3K, but not in
SNU-638 cells.
Intriguingly, single treatment of NVP-BKM120 induced
an alternate pathway in KRAS mutant gastric cancer cell lines in a
cell line-specific manner. First, phosphorylation of ERK became
induced in SNU-1 cells but decreased in SNU-601 cells though both
cell lines are KRAS mutants. Second, p-STAT3 was increased in
SNU-601 cells in a dose- and time-dependent manner, but slightly
decreased in SNU-1 cells (Fig. 2).
In contrast to these cell lines, SNU-638 cells did not show any
induction of other pathways. This result coincides with previous
research with PI3K inhibitors that PI3K inhibitor single treatment
induces at least one signaling mediator in the alternate pathway
(24).
Combined inhibition of PI3K and STAT3 is
synergistic in human gastric cancer cells harboring mutated
KRAS
As Fig. 2 showed
different activation of the other pro-survival pathways in KRAS
mutants, we next studied the combination effect of NVP-BKM120 and
other inhibitors. Considering the compensatory relationship between
RAS/RAF/ERK and PI3K/AKT/mTOR pathways, KRAS mutant cancer cell
lines have shown a synergistic effect of PI3K and MEK inhibitors.
However, in our panel of gastric cancer cell lines while SNU-1
cells showed increase in phosphorylation of ERK, SNU-601 cells
showed its decrease along with PI3K inhibition. Also, SNU-601 cells
showed activation of STAT3. Since previous research demonstrated
that STAT3 is required in KRAS-driven oncogenic transformation
(16), we hypothesized that dual
inhibition of PI3K and STAT3 would be effective in KRAS mutant
gastric cancer cell lines. We used AG490 as a STAT3 inhibitor.
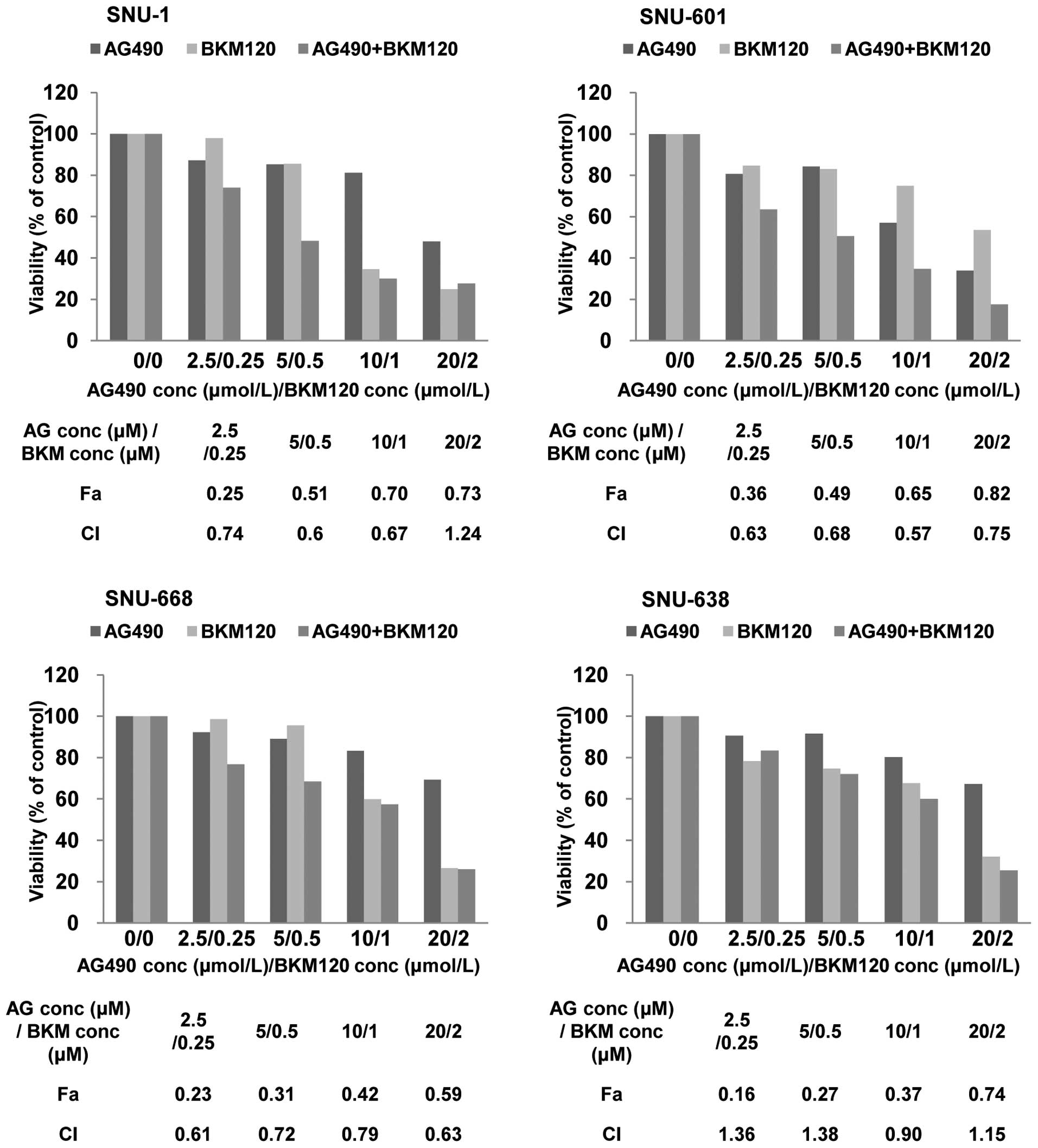
To characterize the level of the interaction
(synergistic, additive or antagonistic) between NVP-BKM120 and
AG490, combination index (CI) values were calculated based on the
Chou and Talalay median-effect principle (25). A CI is 1 for additive interactions,
greater than 1 for antagonistic interactions, and less than 1 for
synergistic interactions. As shown in Fig. 3, the combination of NVP-BKM120 and
AG490 induced synergistic killing of KRAS mutant gastric cancer
cells at different dose combinations and the synergistic effect was
especially distinctive at low dose combinations, contrast to KRAS
wild-type SNU-638 cells showing antagonistic effect at low dose
combinations. In aggregate, we found that PI3K inhibition by
NVP-BKM120 cooperated with AG490 in gastric cancer cells harboring
mutated KRAS.
The combination of NVP-BKM120 and AG490
induces apoptosis
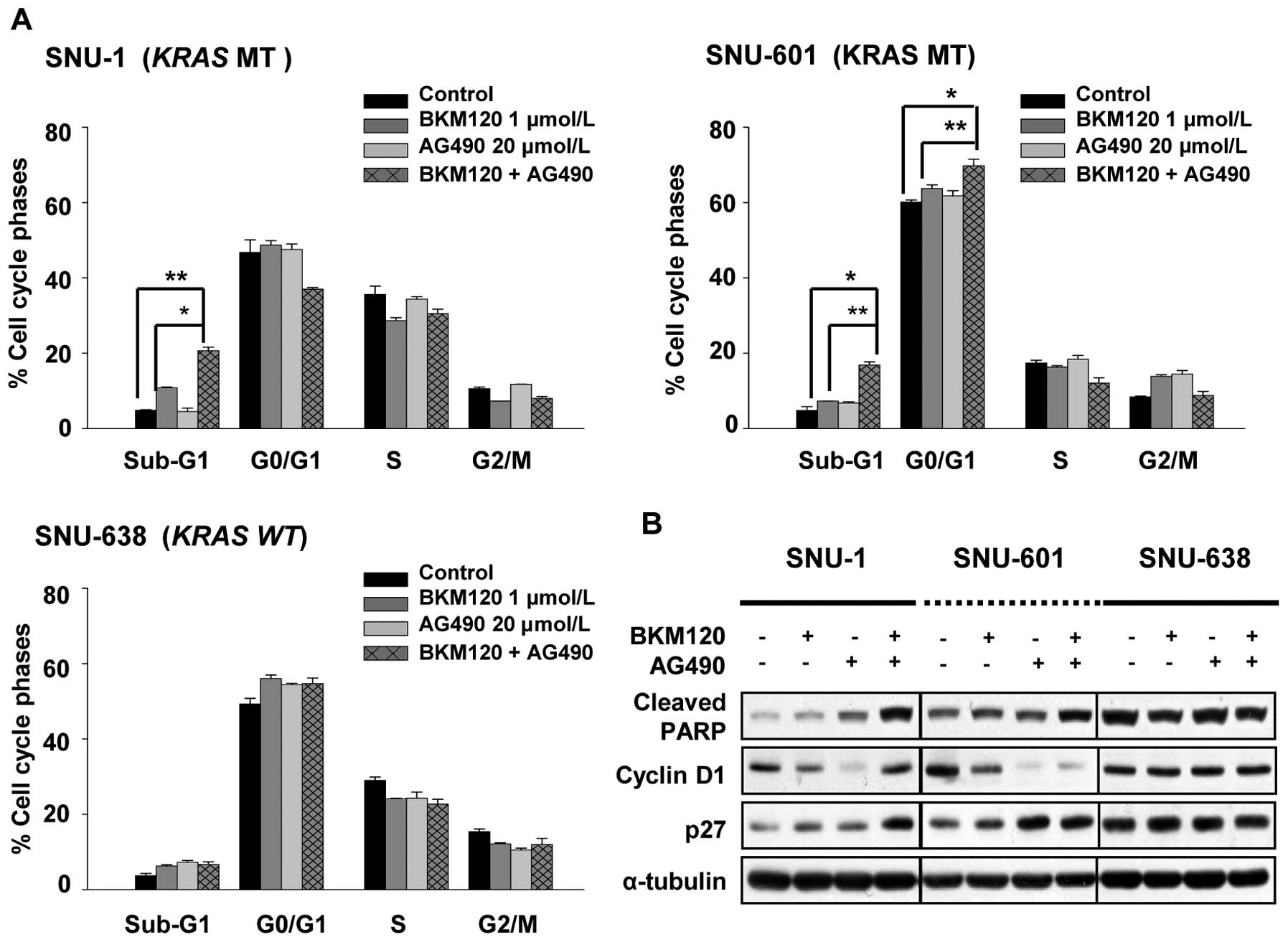
To confirm the synergistic interaction of NVP-BKM120
with AG490, we evaluated the cell cycle distribution in SNU-1,
SNU-601 and SNU-638 cells. We incubated SNU-1, SNU-601, and SNU-638
cells for 72 h in the presence of NVP-BKM120 alone, AG490 alone, or
combination of NVP-BKM120 and AG490 in concentrations as indicated
in Fig. 4A and did cell cycle
analysis using flow cytometry. We found that concurrent treatment
of NVP-BKM120 and AG490 leads to cell death in both SNU-1 and
SNU-601 cells, and in SNU-601 cells growth arrest in the G1 phase
of the cell cycle was detected as well.
We further investigated the relative expression
levels of cell-cycle related proteins by Western blotting (Fig. 4B). The combination of NVP-BKM120
and AG490 induced expressions of cleaved PARP and p27 and
down-regulated Cyclin D1 in SNU-601 cells.
The effect of combined inhibition of PI3K
and STAT3 on the signaling of human gastric cancer cells with
mutated KRAS
Because we could show that combined treatment of
NVP-BKM120 and AG490 caused synergistic inhibition of proliferation
and induction of apoptosis in KRAS mutant gastric cancer cell
lines, SNU-1 and SNU-601, we next examined their effects alone and
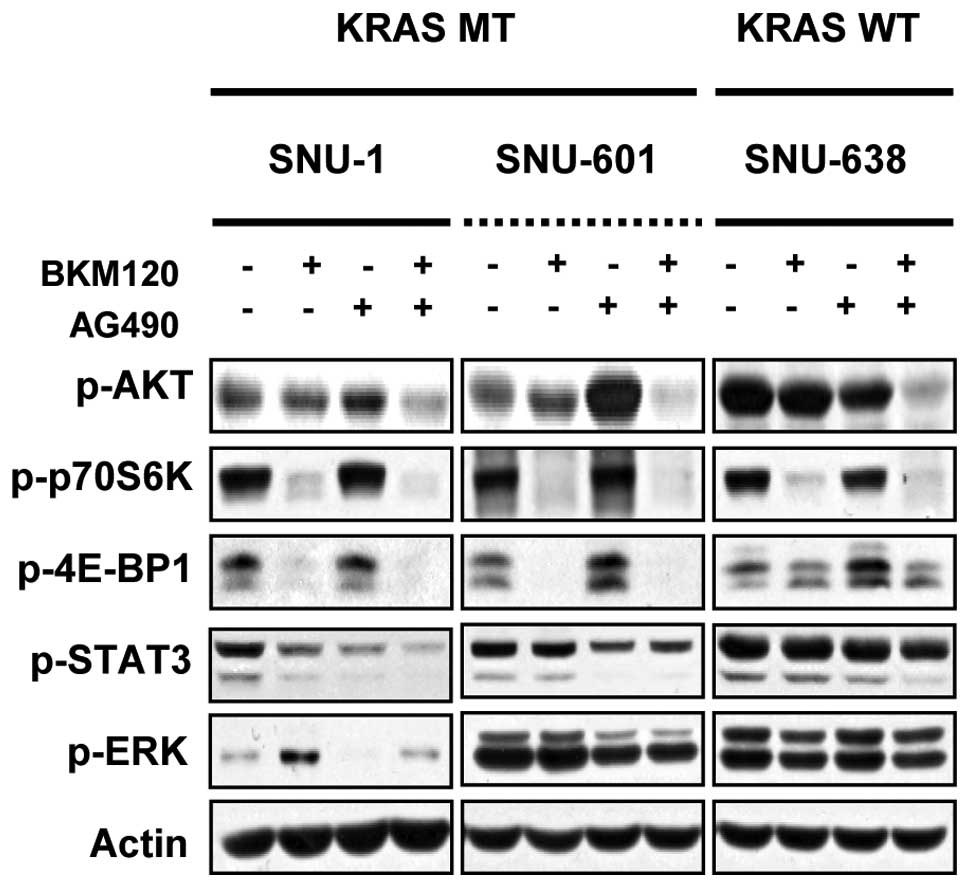
in combination on signaling pathways. As shown in Fig. 5, combined inhibition of PI3K and
STAT3 inhibited the phosphorylation of AKT and p70S6K in SNU-1,
SNU-601 and SNU-638 cells. The phosphorylation of 4E-BP1 and STAT3
were decreased only in SNU-1 and SNU-601 cells. In KRAS wild-type
SNU-638 cells phosphorylation of STAT3 was slightly decreased and
no significant change in expression levels of p-ERK and p-4E-BP1
was detected.
Discussion
The PI3K/AKT signaling axis is generally deregulated
by various genetic changes in solid tumors. The aberrant activation
of the PI3K/AKT pathway contributes to cell survival, protein
synthesis and cell metabolism. In gastric cancer, genetic
mutation/amplification of PIK3CA, AKT1 and KRAS, and loss of
heterozygosity of PTEN have been recognized so far (8,9).
However, it is not well understood how these changes qualitatively
or quantitatively affect PI3K signaling and whether gastric cancer
harboring these mutations is addicted to PI3K signaling and will be
sensitive to PI3K inhibitors.
In this study, we found that during PI3K inhibition,
AKT, ERK and/or STAT3 are activated as shown by the increased
levels of phosphorylation in gastric cancer cells. This is further
supported by previous studies on PI3K pathway that PI3K inhibitor
single treatment appears to be not sufficient as it induces at
least one signaling mediator in the alternate pathway. One possible
reason for the limited efficacy of single PI3K inhibition is the
presence of feedback loops. First, as shown in Fig. 2A, in SNU-1 and SNU-601 cells along
with PI3K inhibition by NVP-BKM120 single treatment, AKT is
activated through activated mTOR/S6K/IRS-1 negative feedback
mechanism. This is in accordance with recent studies that mTOR or
AKT inhibition increases PI3K activity and enhances AKT-independent
PI3K pathway (23,26,27).
Second, our data showed ERK or STAT3 activation after PI3K
inhibition alone in KRAS mutant cancer cell lines as a previous
study showed that inhibition of mTORC1 induces RAS pathway as well
(28). This compensatory
activation of other pro-survival pathways due to inhibition of the
PI3K pathway has been reported as prominent between the PI3K/AKT
and RAS/MAPK pathways. The PI3K/AKT and RAS/MAPK signaling pathways
influence each other rather than function independently, resulting
in active and complex crosstalk. For instance, it has been reported
that the interaction of RAS with p110α is required for RAS-driven
oncogenic transformation (29). In
this respect, in cancers, RAS activation may limit the activity of
single-agent PI3K inhibitors. One of the most typical resistant
mechanisms to PI3K inhibition is activating mutations in the
RAS/MAPK pathway (14,15,24,30,31).
Therefore, to combine inhibition of the PI3K/AKT pathway with
inhibition of the RAS/MAPK pathway is a possible approach in order
to overcome a compensatory interaction between these pathways
(14,24,32).
In contrast to the relation between PI3K and RAS
pathways, there has been only a few reports on interaction between
PI3K and STAT pathways. STAT3 is a latent cytoplasmic transcription
factor and its pathway is one of the key signaling pathways of
which deregulation drives tumorigenesis. STAT3 transmits signals to
the nucleus where STAT3 binds to specific DNA promoter and
regulates gene expression (17,33).
In many human cancers, STAT3 is constitutively activated and has
been described as a novel molecular target for cancer drug
discovery.
According to previous studies, mTOR as a serine
kinase positively activates STAT3 (34,35).
For RAS-dependent malignant transformation, activated STAT3 is
essential (16). In addition,
activated RAS/MAPK signaling can directly regulate mTOR via p90
ribosomal S6 kinase (RSK)-mediated phosphorylation of Raptor
independently of the PI3K/AKT pathway (36). Taken together, although a PI3K
inhibitor suppresses the PI3K/AKT/mTOR signaling pathway, oncogenic
RAS activates RAS/mTOR/STAT3 signaling (Fig. 6).
Therefore, we hypothesized that in KRAS mutant
gastric cancer cells, the blockade of both PI3K/AKT/mTOR and
KRAS/mTOR/STAT pathways using NVP-BKM120 and AG490 would be
synergistic. We used median-effect analysis of dose-response curves
to calculate the CI values and the results were represented as
synergy in KRAS-mutated gastric cancer cell lines, SNU-1, SNU-601
and SNU-668. The synergistic effect was demonstrated by induction
of apoptosis and by changes of signaling molecules along with
combination treatment of NVP-BKM120 and AG490 in SNU-1 and SNU-601
cells.
In case of SNU-668 cells, the effect of concurrent
PI3K and STAT3 inhibition on the expression of signaling molecules
might be stronger than SNU-1 and SNU-601 cells. Based on the recent
study a small reduction in PTEN gene expression can trigger cancer
susceptibility, the higher level of PTEN in SNU-668 would play an
enhanced role as a negative regulator of STAT3 and mTOR,
consequently resulting in more significant molecular change of
signaling (37). This is further
supported by our previous result that the SNU-668 cell line is less
sensitive to dual PI3K/mTOR inhibitor NVP-BEZ235, compared to other
gastric cancer cell lines (data not shown).
To our knowledge, this is the first study to show
that concurrent inhibition of PI3K and STAT signaling pathways is
synergistically effective in KRAS mutant gastric cancer cells.
Therefore, our study suggests the importance to select appropriate
patient subpopulations for clinical study. PI3K blockade in
combination with STAT3 inhibitors may benefit patients with gastric
cancer exhibiting oncogenic KRAS.
Acknowledgements
This study was supported in part by research grant
from Cancer Research Institute, Seoul National University
(cri-09-5) and by the BK21 project from the ministry of Education
and Human Resources Development. Yung-Jue Bang received research
funding from Novartis; all the other authors have no conflicts of
interest to disclose.
References
|
1
|
Hartgrink HH, Jansen EP, van Grieken NC
and van de Velde CJ: Gastric cancer. Lancet. 374:477–490. 2009.
View Article : Google Scholar
|
|
2
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
Statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
3
|
Wainberg ZA, Anghel A, Desai AJ, et al:
Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively
inhibits HER2-amplified human gastric cancer cells and is
synergistic with trastuzumab in vitro and in vivo. Clin Cancer Res.
16:1509–1519. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Engelman JA: Targeting PI3K signalling in
cancer: opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li VS, Wong CW, Chan TL, et al: Mutations
of PIK3CA in gastric adenocarcinoma. BMC Cancer. 5:292005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Velho S, Oliveira C, Ferreira A, et al:
The prevalence of PIK3CA mutations in gastric and colon cancer. Eur
J Cancer. 41:1649–1654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Byun DS, Cho K, Ryu BK, et al: Frequent
monoallelic deletion of PTEN and its reciprocal associatioin with
PIK3CA amplification in gastric carcinoma. Int J Cancer.
104:318–327. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Staal SP: Molecular cloning of the akt
oncogene and its human homologues AKT1 and AKT2: amplification of
AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci
USA. 84:5034–5037. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee KH, Hur HS, Im SA, et al: RAD001 shows
activity against gastric cancer cells and overcomes 5-FU resistance
by downregulating thymidylate synthase. Cancer Lett. 299:22–28.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Osaki M, Kase S, Adachi K, Takeda A,
Hashimoto K and Ito H: Inhibition of the PI3K-Akt signaling pathway
enhances the sensitivity of Fas-mediated apoptosis in human gastric
carcinoma cell line, MKN-45. J Cancer Res Clin Oncol. 130:8–14.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fuereder T, Jaeger-Lansky A, Hoeflmayer D,
et al: mTOR inhibition by everolimus counteracts VEGF induction by
sunitinib and improves anti-tumor activity against gastric cancer
in vivo. Cancer Lett. 296:249–256. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Brien C, Wallin JJ, Sampath D, et al:
Predictive biomarkers of sensitivity to the phosphatidylinositol 3'
kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin
Cancer Res. 16:3670–3683. 2010.
|
|
14
|
Engelman JA, Chen L, Tan X, et al:
Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D
and PIK3CA H1047R murine lung cancers. Nat Med. 14:1351–1356. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ihle NT, Lemos R Jr, Wipf P, et al:
Mutations in the phosphatidylinositol-3-kinase pathway predict for
antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is
a dominant predictor for resistance. Cancer Res. 69:143–150. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gough DJ, Corlett A, Schlessinger K,
Wegrzyn J, Larner AC and Levy DE: Mitochondrial STAT3 supports
Ras-dependent oncogenic transformation. Science. 324:1713–1716.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu H, Kortylewski M and Pardoll D:
Crosstalk between cancer and immune cells: role of STAT3 in the
tumour microenvironment. Nat Rev Immunol. 7:41–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
To KF, Chan MW, Leung WK, et al:
Constitutional activation of IL-6-mediated JAK/STAT pathway through
hypermethylation of SOCS-1 in human gastric cancer cell line. Br J
Cancer. 91:1335–1341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hedvat M, Huszar D, Herrmann A, et al: The
JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and
oncogenesis in solid tumors. Cancer Cell. 16:487–497. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nelson EA, Walker SR, Kepich A, et al:
Nifuroxazide inhibits survival of multiple myeloma cells by
directly inhibiting STAT3. Blood. 112:5095–5102. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ku JL and Park JG: Biology of SNU cell
lines. Cancer Res Treat. 37:1–19. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wan X, Harkavy B, Shen N, Grohar P and
Helman LJ: Rapamycin induces feedback activation of Akt signaling
through an IGF-1R-dependent mechanism. Oncogene. 26:1932–1940.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
O'Reilly KE, Rojo F, She QB, et al: mTOR
inhibition induces upstream receptor tyrosine kinase signaling and
activates Akt. Cancer Res. 66:1500–1508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sos ML, Fischer S, Ullrich R, et al:
Identifying genotype-dependent efficacy of single and combined
PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci
USA. 106:18351–18356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Okuzumi T, Fiedler D, Zhang C, et al:
Inhibitor hijacking of Akt activation. Nat Chem Biol. 5:484–493.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han EK, Leverson JD, McGonigal T, et al:
Akt inhibitor A-443654 induces rapid Akt Ser-473 phosphorylation
independent of mTORC1 inhibition. Oncogene. 26:5655–5661. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carracedo A, Ma L, Teruya-Feldstein J, et
al: Inhibition of mTORC1 leads to MAPK pathway activation through a
PI3K-dependent feedback loop in human cancer. J Clin Invest.
118:3065–3074. 2008.PubMed/NCBI
|
|
29
|
Gupta S, Ramjaun AR, Haiko P, et al:
Binding of ras to phosphoinositide 3-kinase p110α is required for
ras-driven tumorigenesis in mice. Cell. 129:957–968. 2007.
|
|
30
|
Wee S, Jagani Z, Xiang KX, et al: PI3K
pathway activation mediates resistance to MEK inhibitors in KRAS
mutant cancers. Cancer Res. 69:4286–4293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Halilovic E, She QB, Ye Q, et al: PIK3CA
mutation uncouples tumor growth and cyclin D1 regulation from
MEK/ERK and mutant KRAS signaling. Cancer Res. 70:6804–6814. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
She QB, Solit DB, Ye Q, O'Reilly KE, Lobo
J and Rosen N: The BAD protein integrates survival signaling by
EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor
cells. Cancer Cell. 8:287–297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: a leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yokogami K, Wakisaka S, Avruch J and
Reeves SA: Serine phosphorylation and maximal activation of STAT3
during CNTF signaling is mediated by the rapamycin target mTOR.
Curr Biol. 10:47–50. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou J, Wulfkuhle J, Zhang H, et al:
Activation of the PTEN/mTOR/STAT3 pathway in breast cancer
stem-like cells is required for viability and maintenance. Proc
Natl Acad Sci USA. 104:16158–16163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Carriere A, Cargnello M, Julien LA, et al:
Oncogenic MAPK signaling stimulates mTORC1 activity by promoting
RSK-mediated raptor phosphorylation. Curr Biol. 18:1269–1277. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Alimonti A, Carracedo A, Clohessy JG, et
al: Subtle variations in Pten dose determine cancer susceptibility.
Nat Genet. 42:454–458. 2010. View
Article : Google Scholar : PubMed/NCBI
|