Introduction
Osteosarcoma (OS) remains the most common primary
malignant bone cancer affecting children and adolescents (1). Although the combination of modern
surgery and systemic chemotherapy has improved OS treatment
dramatically, no substantial change in survival has been seen over
the past 20 years (2). For this
reason, understanding the mechanisms underlying OS as well as
identifications of new molecular targets are of great
importance.
Mitogen-activated protein kinases (MAPK) are a
family of serine/threonine protein kinases widely conserved among
eukaryotes and plays essential roles in many cellular programs such
as cell proliferation, cell differentiation, cell movement and cell
death. The classical MAPK pathway consists of RAS, RAF, MEK and
ERK, sequentially relaying proliferative signals generated at the
cell surface receptors and through cytoplasmic signaling into the
nucleus (3). Under normal cellular
conditions, this pathway is stimulated by the binding of mitogens,
hormones, or neurotransmitters to receptor tyrosine kinases, which
upon dimerization triggers the activation of oncogenic RAS to
increase cellular RAS-GTP levels. Activated RAS then triggers the
formation of the ‘MAPK complex’ with downstream RAF, MEK1/2,
ERK1/2. Once activated, ERK1/2 regulates the expression of several
genes involved in cell proliferation, differentiation and survival
by phosphorylating nuclear transcription factors such as ETS,
ELK-1, MYC or indirectly by targeting intracellular signaling
molecules such as p90-RSK. The ERK1/2 also has profound effects on
the regulation of apoptosis by the post-translational
phosphorylation of apoptotic regulatory molecules including Bad,
Bim, Mcl-1, caspase 9 and Bcl-2 (3–5).
Constitutive ERK1/2 activation is often due to overexpression or
mutation of upstream receptor tyrosine kinases such as EGFR, PDGFR,
or VEGFR, increased expression of growth factor ligands, or
mutational activation of Ras and its downstream effectors (6).
The involvement of constitutive ERK1/2 activation in
the proliferation and tumoral progression has been largely reported
in many cancer cells including malignant melanomas (7), human hepatocellular carcinoma
(8), esophagogastric cancer
(9), breast cancer (10), renal cell carcinomas (11) and leukemia (12). The hyperactivation of ERK1/2 also
has been shown to promote resistance to chemotherapy drugs in many
cancer cells (4,5,13).
Therefore, ERK1/2 could be a good therapeutic target, as agents
inhibiting the action of ERK1/2 might prevent tumor cell
proliferation, promote apoptosis and reverse resistance to
therapy.
Although ERK1/2 was shown to be overexpressed in OS
cells and has been implicated as playing important roles in the
development of OS (14,15), the exact function of ERK1/2 in OS
growth and progression is not fully understood. Inhibition of
ERK1/2 has been shown to enhance the chemosensitivity of the cancer
cells towards the anticancer drugs (4,5). But
the relationship between ERK1/2 expression and response to
chemotherapy in human osteosarcoma cells has not been elucidated.
Small interfering RNA (siRNA) is a highly specific and efficient
tool to silence target genes (16). In the present study, we employed
siRNA targeting ERK1/2 to explore the potential of new therapeutic
targets in the treatment of OS. Our results suggest that knockdown
of ERK1/2 could inhibit OS proliferation and invasion, and could
significantly increase the chemosensitivity to cisplatin-induced
apoptosis in OS cells. Thus, ERK would be a good molecular target
for OS therapy.
Materials and methods
Cell culture and siRNA transfection
Human osteosarcoma cell line (U2-OS) was obtained
from the American Type Culture Collection (ATCC, Rockville, MD,
USA). Cells were cultured in Dulbecco’s modified Eagle’s medium
(Gibco RL, Grand Island, NY, USA) supplemented with 10% fetal
bovine serum, penicillin (100 U/ml) and streptomycin (100 μg/ml) in
a humidified 5% CO2 atmosphere. The human ERK1 and ERK2
specific siRNA were based on NCBI Reference Sequences (GenBank:
ERK1: NM_002746.2 and ERK2: NM_002745.4). ERK1/2 siRNA and
scrambled control siRNA (siCONTROL) were purchased from Cell
Signaling Technology (Beverly, MA, USA). All of the siRNA
transfections were performed using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA) in Opti-MEM (Invitrogen) according to the
manufacturer’s protocol with a final siRNA concentration of 100 nM.
The transfection reagent was removed after 12 h and the cells were
harvested after 48 h.
Real-time PCR analysis
Total RNA was isolated from transfected cells using
TRIzol reagent (Invitrogen). RNA was first retro-transcribed using
TaqMan® Reverse Transcription Kit (Applied Biosystems,
Foster City, CA, USA) and then real-time PCR was carried out using
and TaqMan SYBR Green Master Mix (Applied Biosystems). β-actin was
applied as an internal control. The following primers were used:
ERK1: 5′-CGCTACACGC AGTTGCAGTACA-3′ forward and 5′-AAGCGCAGCAGG
ATCTGGA-3′ reverse; ERK2: 5′-TGTTCCCAAATGCTG ACTCCAA-3′ forward and
5′-TCGGGTCGTAATACTGC TCCAGATA-3′ reverse; β-actin:
5′-GGCGGCACCACCATG TACCCT-3′ forward and 5′-AGGGGCCGGACTCGTCATA
CT-3′ reverse. The comparative Ct method was used to calculatethe
relative abundance of mRNA compared with that of β-actin expression
(17).
Western blot analysis
U2-OS cells were lysed in boiling buffer (10 mM)
containing 1% SDS and boiled for 5 min. After centrifugation at
20,800 × g for 5 min at 4°C, the supernatant was collected and
protein concentration was measured using the BCA assay (Pierce,
Rockford, IL, USA). A total of 30 μg of protein per sample was
resolved by 10% SDS-PAGE, followed by electro-transfer to PVDF
membranes (Millipore, Bedford, MA, USA). After transfer, the
membranes were blocked with 5% skim milk and probed with specific
primary antibodies overnight followed by 1 μg/ml horseradish
peroxidase conjugated secondary antibody. Antibodies against p44/42
MAPK (Erk1/2) antibody (1:1000 dilution) and phospho-p44/42 MAPK
(Erk1/2) (Thr202/Tyr204) antibody (1:1000 dilution) were from Cell
Signaling Technology (Beverly, MA, USA); antibodies for Bax (1:500
dilution), Bcl-2 (1:500 dilution), Mcl-1 (1:500 dilution), β-actin
(1:500 dilution) and anti-mouse and anti-rabbit antibodies linked
with horseradish peroxidase were from Santa Cruz (Santa Cruz, CA,
USA). Western blotting was performed as described previously
(18).
MTT assays for cell proliferation
Briefly, 3×103 cells per well were plated
in a 96-well plate. After 48 h of incubation, cells were treated
with ERK1/2 siRNA, scrambled control siRNA or with medium alone. At
various time-points, 3-(4,5-dimethyl-thiazol-2-yl)-2,
5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO, USA) was
added into each well at a final concentration of 5 mg/ml and
allowed to incubate at 37°C for 4 h. DMSO (150 μl) was then added
to stop the reaction and measured with an ELISA reader (Bio-Rad,
Hercules, CA, USA) at a wavelength of 570 nm. Viability of cells
was expressed relative to theoretical absorbance (A). Each
experiment was performed in triplicate.
Cell cycle analysis by flow
cytometer
U2-OS cells (5×105) were seeded into each
well of a 6-well culture plate and incubated overnight. Cells were
then transfected with the indicated siRNAs. After an additional
incubation for 48 h, cells were detached and fixed with 500 μl of
70% ethanol at −20°C for 2 h. Subsequently, the cells were washed
twice with PBS then stained with propidium iodide (50 μg/ml
propidium iodide and 100 μg/ml RNase A in PBS) at 37°C for 30 min.
Cell cycle analysis was performed on a FACScan flow cytometer
(Becton-Dickinson, San Jose, CA, USA).
Annexin V-propidium iodide (AnnV/PI)
staining apoptosis test
After siRNA transfection and cisplatin (10 μg/ml)
(Sigma) treatment, cells in each well were harvested and cell
apoptosis was detected by Annexin V-FITC/PI staining method. The
experiments were done in triplicate for each sample, and analyses
were performed using a FACScan flow cytometer (Becton-Dickinson) in
accordance with the manufacturer’s guidelines.
Invasion assay
Cell invasion assay was performed in BioCoat
transwell chambers (Corning Costar, Cambridge, MA, USA) with
uncoated porous inserts (pore size 8 μm) as previously described
(19). Matrigel™ Matrix (BD
Biosciences, San Jose, CA, USA) was diluted 1:5 using serum-free
DMEM medium and added to the transwell inserts. Cells at
1×104/ml from different groups were spread on the
transwell inserts pre-coated with Matrigel in 24-well plates and
cultured in serum-free DMEM. Medium containing 10% FBS was added to
the lower chamber as a chemoattractant. After 24-h incubation at
37°C in a 5% CO2 incubator, the insert was washed with
PBS, and cells on the upper surface of the insert were wiped off
using a cotton swab. Cells that migrated to the bottom surface of
the insert were fixed with methanol and stained by Giemsa. The
number of cells was counted under the microscope from randomly
selected five fields (magnification ×400).
Statistical analysis
Results are expressed as means ± standard deviation
(SD). Statistical analyses were performed using SPSS statistical
software (SPSS Inc., Chicago, IL, USA). The statistical
significance of the results was determined using the Student’s
t-test. Data were considered significant at P<0.05.
Results
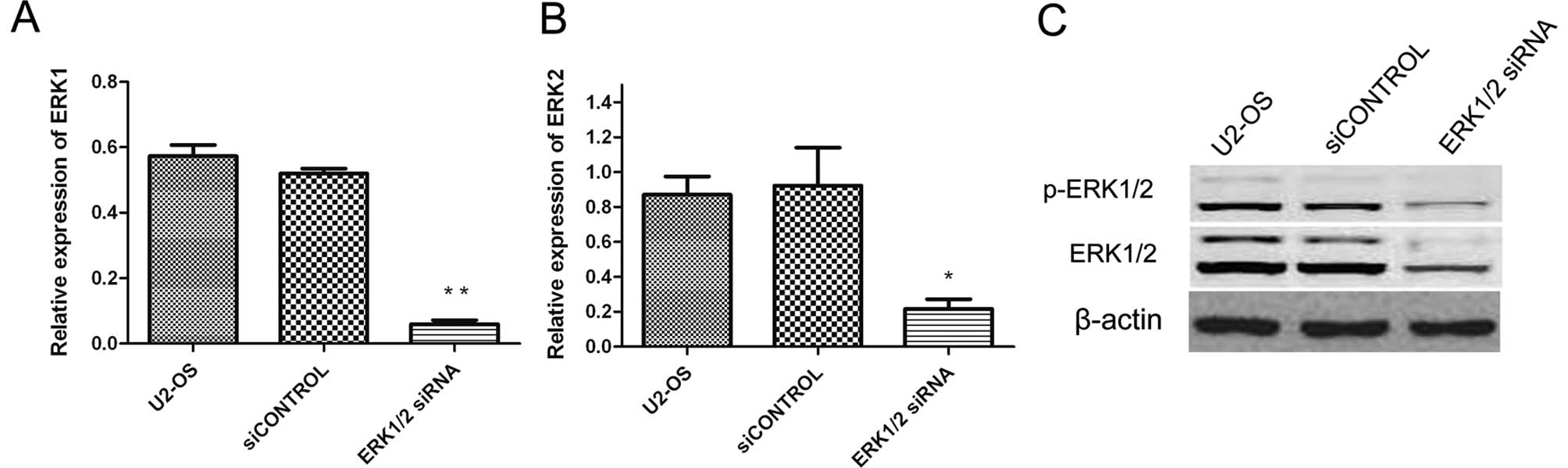
siRNA targeting ERK1/2 knocks down ERK1/2
expression
Since ERK1/2 levels are significantly higher in
tumor cells (including osteosarcoma) than that in normal cells, we
sought to determine whether the synthetic ERK1/2 siRNA could
inhibit the expression of ERK1/2 gene in U2-OS cells. Forty-eight
hours after siRNA transfection, ERK1/2 mRNA and protein were
measured by quantitative real-time PCR and Western blotting,
respectively. As shown in Fig. 1,
ERK1/2 siRNA could specifically and efficiently suppress ERK1/2
expression at both mRNA and protein levels compared to cells
treated with siCONTROL and the untreated cells.
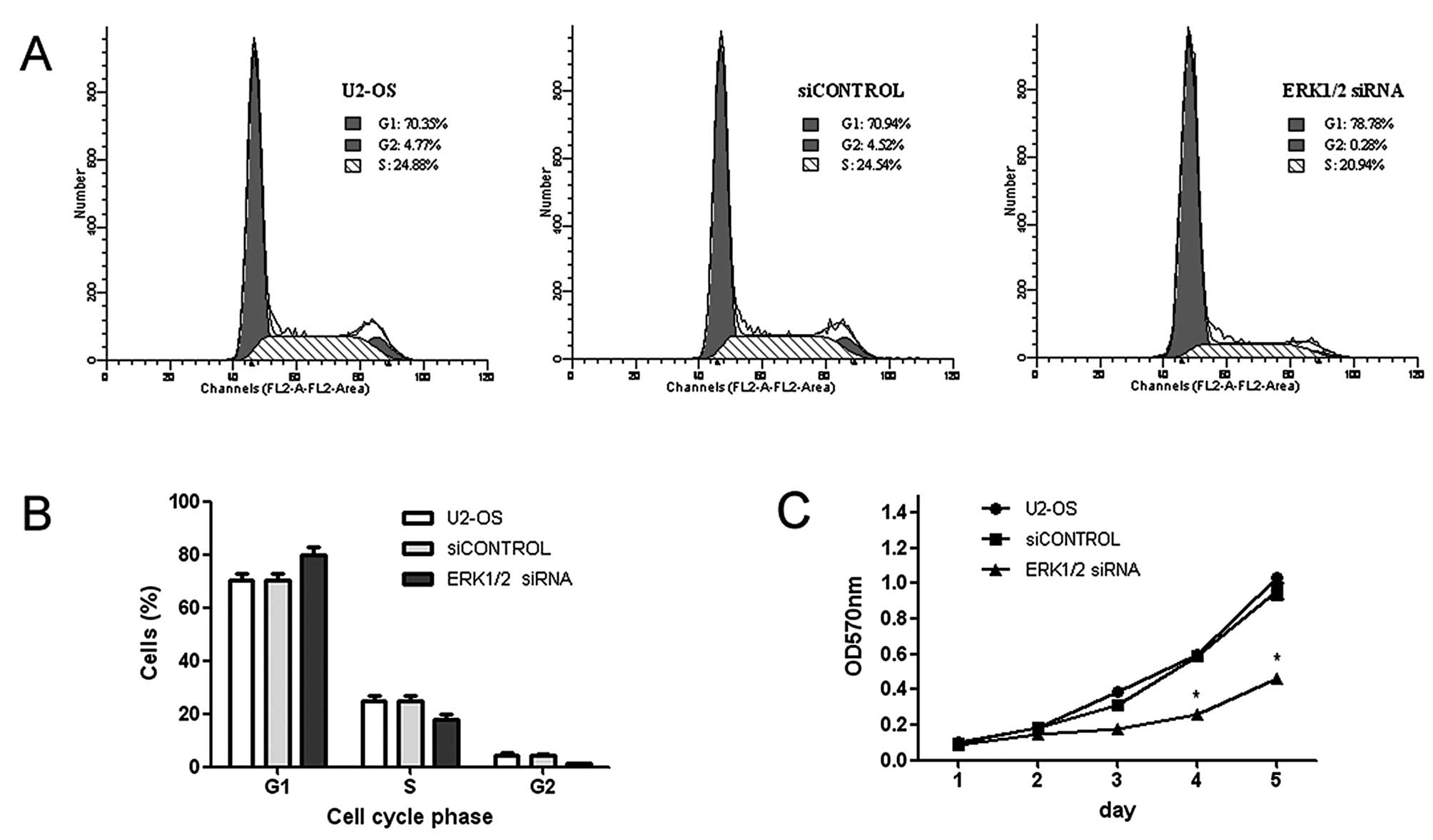
ERK1/2 knockdown inhibits osteosarcoma
cell growth
To determine whether ERK1/2 siRNA had an inhibitory
effect on U2-OS cell growth, we first performed determination of
cell proliferation with MTT assay. Fig. 2C showed that the growth curves for
ERK1/2 knockdown cells were significantly lower than those for
control cells for 5 days of incubation. Furthermore, the cell cycle
distribution of control and transfected cells was analyzed by flow
cytometry. As shown in Fig. 2A,
there was a significant increase in the percentage of cells in G1
phase after transfection with ERK1/2 siRNA. In detail, the
percentage of cells at G1 phase increased by 7.84% in the U2-OS
cells transfected with the ERK1/2 siRNA while the percentage of
cells at S phase decreased by 3.60% (Fig. 2A). The cells transfected with the
siCONTROL were set as the control group. This result indicated that
siRNA arrested the cells in G0 and G1 phases, delayed the
progression of cell cycle and inhibited cell proliferation.
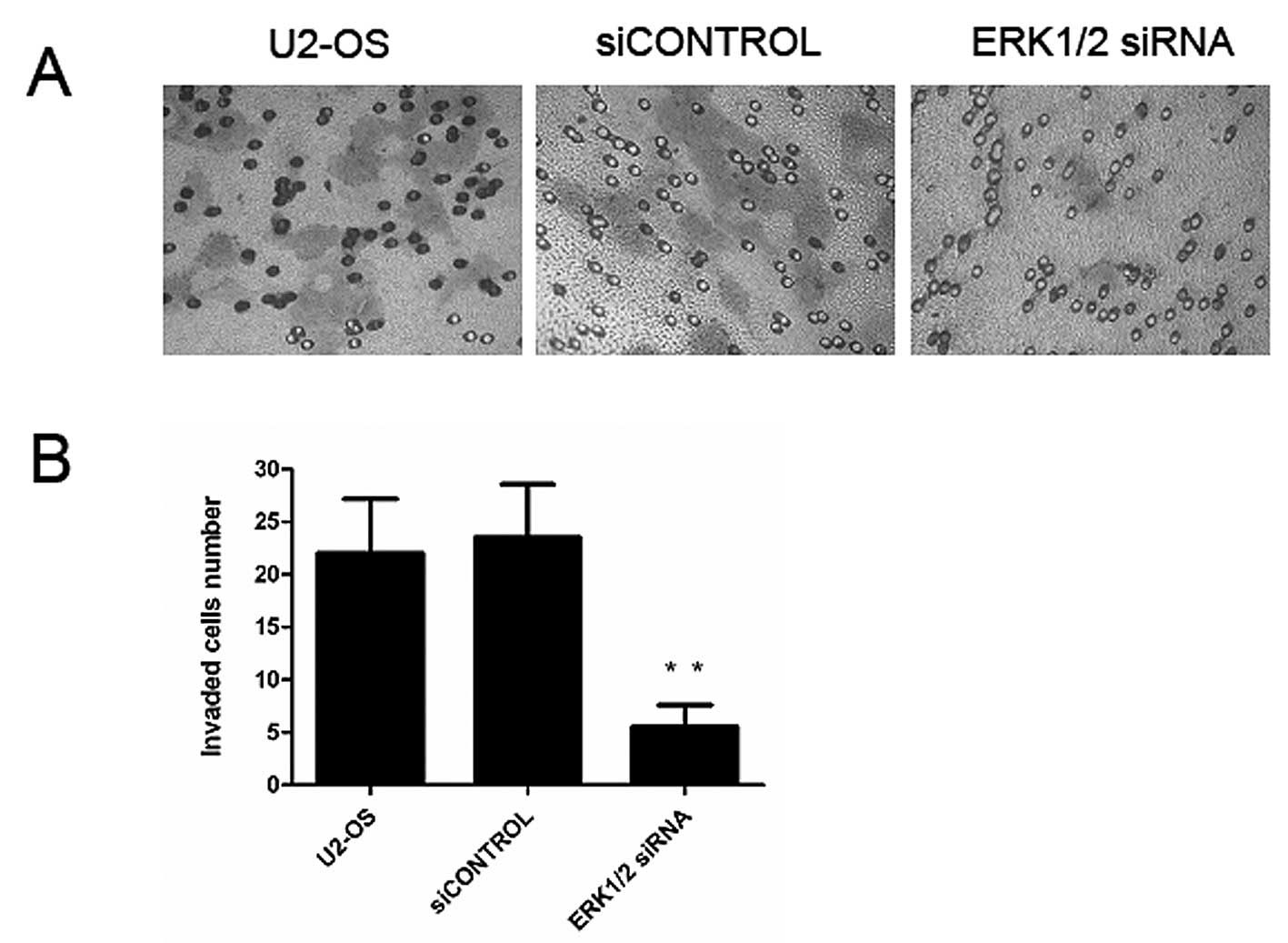
Downregulation of ERK1/2 inhibits
osteosarcoma cell invasion
Cancer cell migration and invasion play very
important roles in cancer metastasis. So, we further studied the
effect of ERK1/2 suppression on the invasion in U2-OS cells. The
results showed that the migratory ability was significantly reduced
through Matrigel coated chamber membranes (P<0.01, Fig. 3) compared with the untreated group,
or the siCONTROL group. These results indicate that silencing of
ERK1/2 plays an inhibitory effect on the invasion of U2-OS
cells.
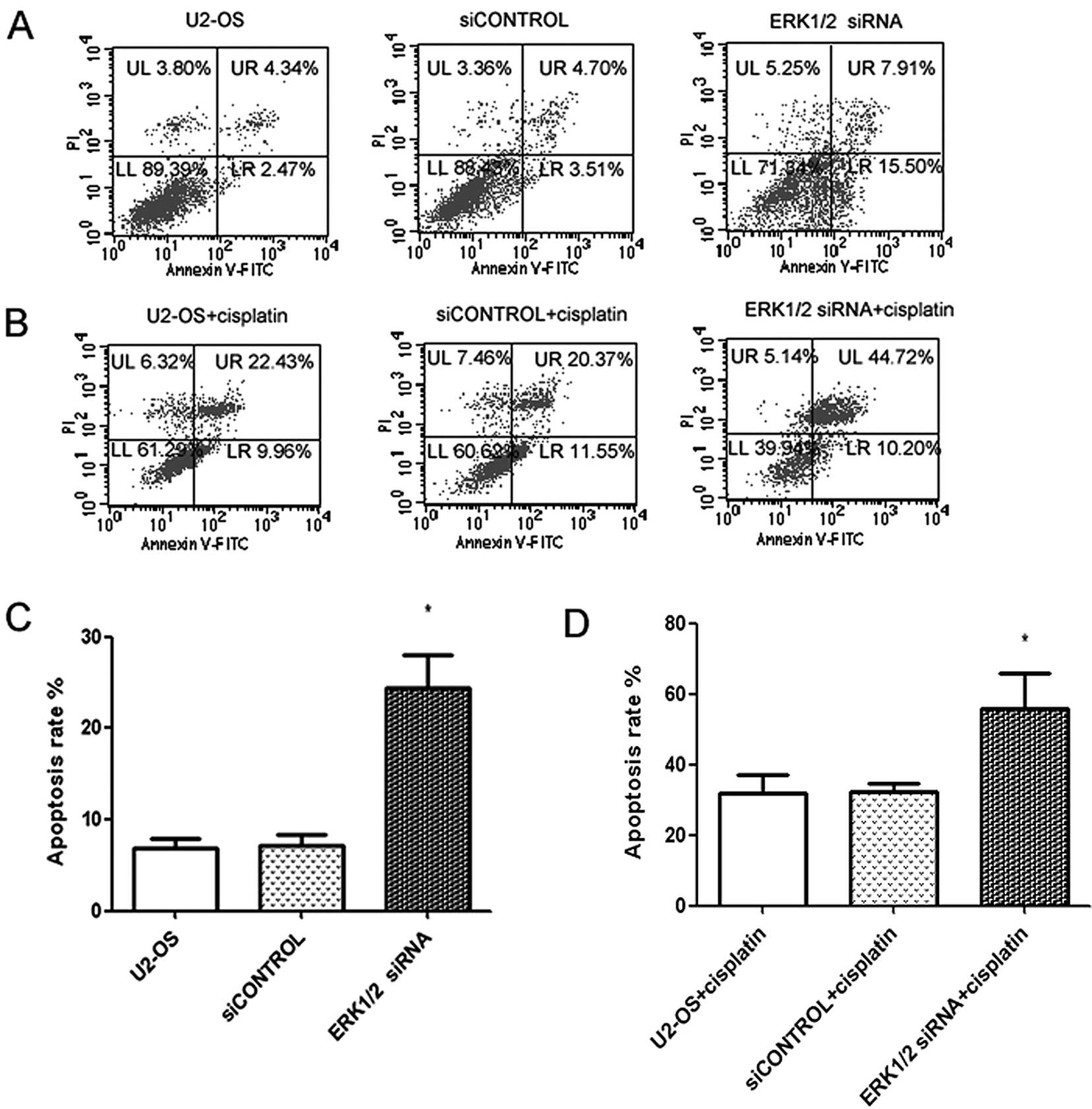
Inhibition of ERK1/2 increases
sensitivity of U2-OS cells to chemotherapy
Upregulation of the ERK1/2 gene has been reported to
be closely related to chemotherapy resistance, and inhibition of
the ERK1/2 gene has been shown to increase the sensitivity of tumor
cells to chemotherapeutic agents (20,21).
These exciting data prompted us to investigate the possibility of
the combination of cisplatin treatment and ERK1/2 depletion as a
clinical strategy for osteosarcoma chemotherapy. The effect of
ERK1/2 siRNA and cisplatin on cell apoptosis was examined in U2-OS
cell line by flow cytometric analysis. As shown in Fig. 4A and C, cell apoptosis was induced
after transfection with ERK1/2 siRNA at 48 h. However, cell
apoptosis was further enhanced when cells were treated with 10
μg/ml cisplatin (Fig. 4B and D),
suggesting that combining ERK1/2 inhibition with cisplatin
increased the incidence of apoptosis.
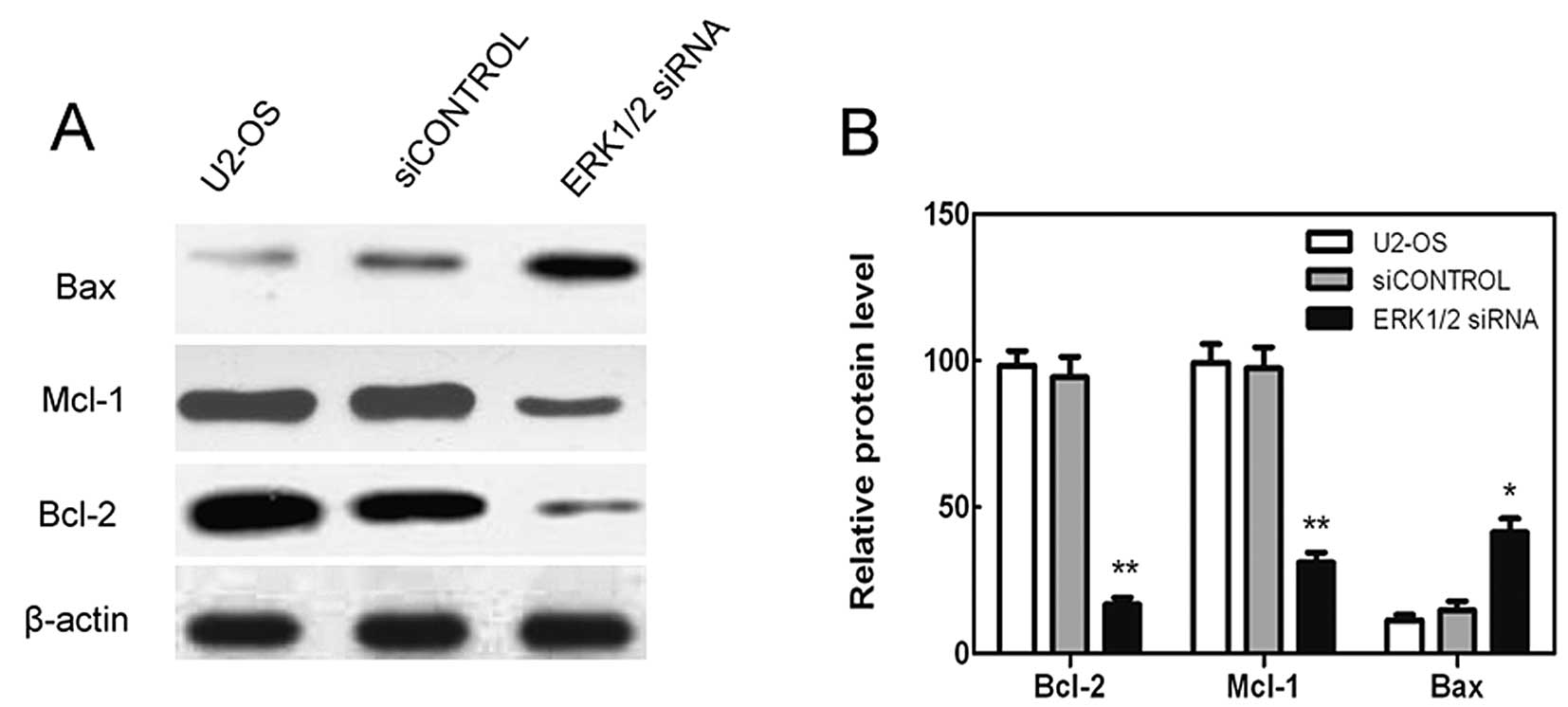
ERK1/2 knockdown decreases the expression
of anti-apoptotic proteins Bcl-2 and Mcl-1 while increases the
expression of proapoptotic protein Bax in U2-OS cells
In order to gain a better understanding of the
mechanism leading to cell death, the tumor cells were analyzed for
the alteration of different kinds of anti-apoptotic and
pro-apoptotic Bcl-2 family proteins. While ERK1/2 knockdown caused
downregulation of Bcl-2 and Mcl-1, increased levels of Bax were
evident (Fig. 5). Levels of Bcl-2,
Mcl-1 and Bax did not show a clear change as the cell was
transfected with the control siRNA (Fig. 5). Therefore, ERK1/2 knockdown
caused the increased cell death by activating pro-apoptotic Bcl-2
members, while inhibiting the anti-apoptotic ones.
Discussion
Osteosarcoma (OS) is the most frequent primary bone
sarcoma, accounting for approximately 20% of all primary sarcomas
in bone and approximately 5% of pediatric tumors overall (1). OS is characterized by aggressive
invasion, early metastasis and resistance to existing
chemotherapeutic agents or radiotherapy. Despite advances in
surgical and chemotherapeutic strategies, the prognosis of
osteosarcoma patients remains unfavorable (22). The failure to improve outcomes with
treatment intensification indicates the need for novel and
effective antitumor strategies, such as targeted therapy.
Constitutive activation of ERK1/2 has been
implicated in a wide variety of processes in the cancer cell during
the regulation of cell mortality, apoptosis, angiogenesis, invasion
and metastasis, and cell division cycle (4,23).
On the basis of the importance of ERK1/2 in tumor progression and
survival, ERK1/2 recently has been considered as a molecular target
for cancer chemotherapeutics (23–25).
Studies with genetic approaches including antisense
oligonucleotides and RNA interference (RNAi) have demonstrated that
inhibition of ERK1/2 signaling suppresses tumor growth and induces
apoptosis in vitro and in vivo (26–28).
Moreover, pharmacological agents including gefitinib and sorafenib
have also been demonstrated to be potential anticancer drugs due to
their inhibitory effects on ERK1/2 signaling (29,30).
Inhibiting specific gene expression by RNAi has
become an important method of cancer treatment (31,32).
Recently, ERK1/2 signal pathway was shown to be overexpressed in OS
cells and has been proposed as a novel marker of poor outcome in OS
(14,33,34).
To explore whether ERK1/2 could become a potential molecular target
for gene therapy of osteosarcoma, we employed RNAi technology to
downregulate ERK1/2 expression in a human cell line of
osteosarcoma, U2-OS. Comparing with control group, the mRNA and
protein expressions were decreased significantly in the cells
transfected with the ERK1/2 siRNA. These results indicated that
ERK1/2 siRNA could silence the expression of ERK1/2 effectively and
specifically in U2-OS cells.
We next examined the consequences of U2-OS cells
transfected with ERK1/2 siRNA. The proliferation rate was analyzed
by MTT and the cell cycle distribution was tested by flow
cytometry, then we found that ERK1/2 knocked down by siRNA
suppressed cell proliferation and induced an accumulation in G1
phase concomitant with a decrease in the S phase in U2-OS cells
in vitro. The results suggested that the observed
growth-inhibitory effect of ERK1/2 siRNA on U2-OS cells could be
mediated through modulation of cell cycle progression. Our results
are in line with others who showed that inhibition of ERK1/2
signaling induced cell cycle arrest in G0/G1 phase and reduced
proliferation in cancer cells (35,36).
These results manifested that ERK1/2 plays an important role both
in cell proliferation and cell cycle.
It is known that ERK1/2 phosphorylation and the
subsequent activation of myosin light chain kinase can modulate
focal contact dynamics in motile cells, promote migration and
invasion (5,7). We further studied the effect of
ERK1/2 suppression on the migration of U2-OS cells by mobility
assays. The results showed that the migratory ability was
significantly reduced through Matrigel coated chamber membranes
compared with the control group. Therefore, there is a strong
relationship between ERK1/2 and the invasion or migration ability
of human osteosarcoma cells. These results are consistent with
other recent reports that showed that inhibition of ERK1/2
signaling could reduce the migration and invasion of tumor cells
(37,38).
An important downstream effect of ERK1/2 activation
is the ERK1/2-dependent regulation of antiapoptotic Bcl-2 family
genes (6). Bcl-2 and its dominant
inhibitor Bax are key regulators of cell growth and apoptosis. The
anti-apoptotic Bcl-2 proteins protect cells from apoptosis, while
Bax accelerates cell death in response to certain apoptotic
stimuli. Treatment with ERK1/2 siRNA induced apoptosis (Fig. 4A and C) with corresponding decrease
in ERK1/2 activation, accompanied by the reduction of Bcl-2 and
Mcl-1 and upregulation of the levels of Bax protein in U2-OS cells
(Fig. 5). Such effects may be the
important mechanisms of action of ERK1/2 induced apoptosis and
suppressed the growth of the U2-OS cells. The displayed results
correspond with other recent reports that showed that inhibition of
ERK1/2 signaling was accompanied by growth inhibition and induction
of apoptosis (39,40). But understanding the detailed
molecular mechanisms needs further investigation.
Drug resistance is an important cause of treatment
failure and mortality in OS patients. Cisplatin is one of the most
commonly used anticancer drugs, but its application has been
limited by the presence of cellular resistance (41). Upregulation of ERK1/2 was reported
in multidrug resistant cancers cells (20). Numerous reports have shown
convincing data that the inhibition of ERK1/2 could sensitize tumor
cells to cisplatin-induced cell death (42–44).
However, the relationship between ERK1/2 expression and response to
chemotherapy in human osteosarcoma cells has not been disclosed.
Here, we postulate that inhibition of ERK1/2 expression via RNAi
could increase the chemosensitivity to cisplatin. As expected,
knockdown of ERK1/2 expression contributed to sensitizing
osteosarcoma cells to the anticancer drug cisplatin by increasing
apoptosis.
In this report, we have shown that downexpression of
ERK1/2 by siRNA inhibited the growth and invasion of U2-OS cells,
and found that the knockdown of ERK1/2 made cancer cells more
sensitive to cisplatin treatment and that the depletion of ERK1/2
enhanced cisplatin-induced apoptosis. Furthermore, we demonstrated
that the knockdown of ERK1/2 regulates the expression level of
Bcl-2, Mcl-1 and Bax. All these findings suggest that ERK1/2 may
have the wide therapeutic and/or adjuvant application in the
treatment of human osteosarcoma.
Acknowledgements
This study was supported by the Scientific and
Technological Project of Shandong Province, P.R. China
(2007GG30002029).
References
|
1
|
Mirabello L, Troisi RJ and Savage SA:
Osteosarcoma incidence and survival rates from 1973 to 2004: data
from the Surveillance, Epidemiology, and End Results Program.
Cancer. 115:1531–1543. 2009.
|
|
2
|
Akiyama T, Dass CR and Choong PF: Novel
therapeutic strategy for osteosarcoma targeting osteoclast
differentiation, bone-resorbing activity, and apoptosis pathway.
Mol Cancer Ther. 7:3461–3469. 2008.
|
|
3
|
Inamdar GS, Madhunapantula SV and
Robertson GP: Targeting the MAPK pathway in melanoma: why some
approaches succeed and other fail. Biochem Pharmacol. 80:624–637.
2010.
|
|
4
|
McCubrey JA, Steelman LS, Abrams SL, et
al: Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in
malignant transformation and drug resistance. Adv Enzyme Regul.
46:249–279. 2006.
|
|
5
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007.
|
|
6
|
Eisenmann KM, van Brocklin MW, Staffend
NA, Kitchen SM and Koo HM: Mitogen-activated protein kinase
pathway-dependent tumor-specific survival signaling in melanoma
cells through inactivation of the proapoptotic protein bad. Cancer
Res. 63:8330–8337. 2003.
|
|
7
|
Satyamoorthy K, Li G, Gerrero MR, et al:
Constitutive mitogen-activated protein kinase activation in
melanoma is mediated by both BRAF mutations and autocrine growth
factor stimulation. Cancer Res. 63:756–759. 2003.
|
|
8
|
Chen L, Shi Y, Jiang CY, Wei LX, Wang YL
and Dai GH: Expression and prognostic role of pan-Ras, Raf-1, pMEK1
and pERK1/2 in patients with hepatocellular carcinoma. Eur J Surg
Oncol. 37:513–520. 2011.
|
|
9
|
Barry OP, Mullan B, Sheehan D, et al:
Constitutive ERK1/2 activation in esophagogastric rib bone marrow
micrometastatic cells is MEK-independent. J Biol Chem.
276:15537–15546. 2001.
|
|
10
|
Sivaraman VS, Wang H, Nuovo GJ and Malbon
CC: Hyperexpression of mitogen-activated protein kinase in human
breast cancer. J Clin Invest. 99:1478–1483. 1997.
|
|
11
|
Oka H, Chatani Y, Hoshino R, et al:
Constitutive activation of mitogen-activated protein (MAP) kinases
in human renal cell carcinoma. Cancer Res. 55:4182–4187. 1995.
|
|
12
|
Gregorj C, Ricciardi MR, Petrucci MT, et
al: ERK1/2 phosphorylation is an independent predictor of complete
remission in newly diagnosed adult acute lymphoblastic leukemia.
Blood. 109:5473–5476. 2007.
|
|
13
|
Sridhar SS, Hedley D and Siu LL: Raf
kinase as a target for anticancer therapeutics. Mol Cancer Ther.
4:677–685. 2005.
|
|
14
|
Yang R, Piperdi S and Gorlick R:
Activation of the RAF/mitogen-activated protein/extracellular
signal-regulated kinase kinase/extracellular signal-regulated
kinase pathway mediates apoptosis induced by chelerythrine in
osteosarcoma. Clin Cancer Res. 14:6396–6404. 2008.
|
|
15
|
Shimo T, Matsumura S, Ibaragi S, et al:
Specific inhibitor of MEK-mediated cross-talk between ERK and p38
MAPK during differentiation of human osteosarcoma cells. J Cell
Commun Signal. 1:103–111. 2007.
|
|
16
|
Bass BL: Double-stranded RNA as a template
for gene silencing. Cell. 101:235–238. 2000.
|
|
17
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008.
|
|
18
|
Iwagaki A, Choe N, Li Y, Hemenway DR and
Kagan E: Asbestos inhalation induces tyrosine nitration associated
with extracellular signal-regulated kinase 1/2 activation in the
rat lung. Am J Respir Cell Mol Biol. 28:51–60. 2003.
|
|
19
|
Van Golen KL, Wu ZF, Qiao XT, Bao LW and
Merajver SD: RhoC GTPase, a novel transforming oncogene for human
mammary epithelial cells that partially recapitulates the
inflammatory breast cancer phenotype. Cancer Res. 60:5832–5838.
2000.
|
|
20
|
Lin JC, Chang SY, Hsieh DS, Lee CF and Yu
DS: Modulation of mitogen-activated protein kinase cascades by
differentiation-1 protein: acquired drug resistance of hormone
independent prostate cancer cells. J Urol. 174:2022–2026. 2005.
|
|
21
|
Zhang D, LaFortune TA, Krishnamurthy S, et
al: Epidermal growth factor receptor tyrosine kinase inhibitor
reverses mesenchymal to epithelial phenotype and inhibits
metastasis in inflammatory breast cancer. Clin Cancer Res.
15:6639–6648. 2009.
|
|
22
|
Bacci G, Longhi A, Versari M, Mercuri M,
Briccoli A and Picci P: Prognostic factors for osteosarcoma of the
extremity treated with neoadjuvant chemotherapy: 15-year experience
in 789 patients treated at a single institution. Cancer.
106:1154–1161. 2006.
|
|
23
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007.
|
|
24
|
Montagut C and Settleman J: Targeting the
RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 283:125–134.
2009.
|
|
25
|
Friday BB and Adjei AA: Advances in
targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase
cascade with MEK inhibitors for cancer therapy. Clin Cancer Res.
14:342–346. 2008.
|
|
26
|
Gailhouste L, Ezan F, Bessard A, et al:
RNAi-mediated MEK1 knock-down prevents ERK1/2 activation and
abolishes human hepatocarcinoma growth in vitro and in vivo. Int J
Cancer. 126:1367–1377. 2010.
|
|
27
|
Zeng P, Wagoner HA, Pescovitz OH and
Steinmetz R: RNA interference (RNAi) for extracellular
signal-regulated kinase 1 (ERK1) alone is sufficient to suppress
cell viability in ovarian cancer cells. Cancer Biol Ther.
4:961–967. 2005.
|
|
28
|
Koul S, Huang M, Chaturvedi L, Meacham RB
and Koul HK: p42/p44 mitogen-activated protein kinase signal
transduction pathway regulates interleukin-6 expression in PC3
cells, a line of hormone-refractory prostate cancer cells. Ann NY
Acad Sci. 1030:253–257. 2004.
|
|
29
|
Liu L, Cao Y, Chen C, et al: Sorafenib
blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and
induces tumor cell apoptosis in hepatocellular carcinoma model
PLC/PRF/5. Cancer Res. 66:11851–11858. 2006.
|
|
30
|
Janmaat ML, Rodriguez JA, Gallegos-Ruiz M,
Kruyt FA and Giaccone G: Enhanced cytotoxicity induced by gefitinib
and specific inhibitors of the Ras or phosphatidyl inositol-3
kinase pathways in non-small cell lung cancer cells. Int J Cancer.
118:209–214. 2006.
|
|
31
|
Lapteva N, Yang AG, Sanders DE, Strube RW
and Chen SY: CXCR4 knockdown by small interfering RNA abrogates
breast tumor growth in vivo. Cancer Gene Ther. 12:84–89. 2005.
|
|
32
|
Susa M, Iyer AK, Ryu K, et al: Inhibition
of ABCB1 (MDR1) expression by an siRNA nanoparticulate delivery
system to overcome drug resistance in osteosarcoma. PLoS One.
5:e107642010.
|
|
33
|
Pignochino Y, Grignani G, Cavalloni G, et
al: Sorafenib blocks tumour growth, angiogenesis and metastatic
potential in preclinical models of osteosarcoma through a mechanism
potentially involving the inhibition of ERK1/2, MCL-1 and ezrin
pathways. Mol Cancer. 8:1182009.
|
|
34
|
Do SI, Jung WW, Kim HS and Park YK: The
expression of epidermal growth factor receptor and its downstream
signaling molecules in osteosarcoma. Int J Oncol. 34:797–803.
2009.
|
|
35
|
Gysin S, Lee SH, Dean NM and McMahon M:
Pharmacologic inhibition of RAF-->MEK-->ERK signaling elicits
pancreatic cancer cell cycle arrest through induced expression of
p27Kip1. Cancer Res. 65:4870–4880. 2005.
|
|
36
|
Sutter AP, Maaser K, Gerst B, Krahn A,
Zeitz M and Scherubl H: Enhancement of peripheral benzodiazepine
receptor ligand-induced apoptosis and cell cycle arrest of
esophageal cancer cells by simultaneous inhibition of MAPK/ERK
kinase. Biochem Pharmacol. 67:1701–1710. 2004.
|
|
37
|
Shukla A, Hillegass JM, Macpherson MB, et
al: ERK2 is essential for the growth of human epithelioid malignant
mesotheliomas. Int J Cancer. 129:1075–1086. 2011.
|
|
38
|
Chen H, Zhu G, Li Y, et al: Extracellular
signal-regulated kinase signaling pathway regulates breast cancer
cell migration by maintaining slug expression. Cancer Res.
69:9228–9235. 2009.
|
|
39
|
Pellicano F, Simara P, Sinclair A, et al:
The MEK inhibitor PD184352 enhances BMS-214662-induced apoptosis in
CD34+ CML stem/progenitor cells. Leukemia. 25:1159–1167.
2011.
|
|
40
|
Trisciuoglio D, Iervolino A, Zupi G and
Del Bufalo D: Involvement of PI3K and MAPK signaling in
bcl-2-induced vascular endothelial growth factor expression in
melanoma cells. Mol Biol Cell. 16:4153–4162. 2005.
|
|
41
|
Zamble DB and Lippard SJ: Cisplatin and
DNA repair in cancer chemotherapy. Trends Biochem Sci. 20:435–439.
1995.
|
|
42
|
Mandic A, Viktorsson K, Heiden T, Hansson
J and Shoshan MC: The MEK1 inhibitor PD98059 sensitizes C8161
melanoma cells to cisplatin-induced apoptosis. Melanoma Res.
11:11–19. 2001.
|
|
43
|
Lu Y and Cederbaum A: The mode of
cisplatin-induced cell death in CYP2E1-overexpressing HepG2 cells:
modulation by ERK, ROS, glutathione, and thioredoxin. Free Radic
Biol Med. 43:1061–1075. 2007.
|
|
44
|
Wang J, Zhou JY and Wu GS: ERK-dependent
MKP-1-mediated cisplatin resistance in human ovarian cancer cells.
Cancer Res. 67:11933–11941. 2007.
|