Introduction
Statins, the 3-hydroxy-3-methylglutaryl coenzyme A
(HMGCoA) reductase inhibitors, are a class of drugs that inhibit
the rate-limiting step in the cholesterol biosynthetic pathway.
Cholesterol is an important structural component of cell membranes
and the physiological requirements derive by endogenous synthesis
or exogenous supply. Increases in lipid levels lead to
atherosclerosis and narrowing of the blood vessels, which in turn
may affect the blood supply to the heart, brain and peripheral
circulation, leading to morbidity or mortality (1).
Statins, by inhibiting cholesterol biosynthesis,
emerged as principal agents in lowering the incidence of
cardiovascular disease. However, it must be considered that any
compound leading to depletion of cholesterol, which is the main
structural component of cell membranes, affects various cellular
events and impairs homeostasis. Statins, potent inhibitors of
cholesterol synthesis, act by inhibiting 3-hydroxy-3-methylglutaryl
CoA (HMG-CoA) reductase, which catalyzes the conversion of HMG-CoA
to mevalonate (2). In addition to
the cholesterol-lowering property, many biological effects of
statins can be derived from cholesterol-independent pleiotropic
mechanisms, which are likely a consequence of blocking
intracellular signaling (3).
The role of statins extends beyond its
lipid-lowering effects, as they are known to improve endothelial
functions and participate in plaque stabilization, immune
modulation and antioxidant activity and also act as
anti-inflammatory and anticancer agents. These properties, together
with a high safety profile, have made statins more attractive and
continue to be a widely prescribed drug.
Their pleiotropic or cholesterol-independent effects
at the cellular and molecular levels are highly related to numerous
cellular functions, such as proliferation and differentiation.
Treatment with simvastatin, mevastatin, atorvastatin or pravastatin
induces morphological change and decrease cell proliferation. It
has been observed that the use of simvastatin was more effective in
cancer cells and embryonic stem cells (ESCs), in relation to normal
cells. In ESC, the loss of self-renewal by simvastatin was
characterized by marked down-regulation of several genes with
function of ESC markers such as alkaline phosphatase, Oct4, Nanog,
Rex-1 and SSEA-1. Simvastatin effects were selectively reversed by
either mevalonate or its metabolite, geranylgeranyl pyrophosphate
(GGPP), but not by cholesterol or farnesyl pyrophosphate (4).
Besides their use in the treatment of lipid
disorders, statins have been studied for their anti-carcinogenic
effects in several models, including carcinomas of the colon and
rectum, prostate, breast, lung and skin (5,6).
Many studies have shown the anti-proliferative and pro-apoptotic
effects of statins to a greater degree in malignant than in
non-malignant cells (7,8). Statins also can trigger different
tumor cells to undergo apoptosis in vitro and suppress tumor
growth (9,10).
In addition, substantial experimental and clinical
evidence suggests that statins exhibit anticancer effects mediated
by apoptosis and cell cycle arrest (11) through various signaling pathways.
It has been hypothesized that statin-induced apoptosis is mediated
by regulating BCL-2 family members involved in mitochondrial
apoptosis pathway of various cells types (8,10,12,13).
Moreover, statin attenuates the p53 stability response to DNA
damage probably by phosphorylation of Mdm2. The tumor suppressor
p53 is a key regulator of apoptosis, which has pro-apoptotic
activity. Under stress conditions, p53 is stabilized and acts as a
transcription factor that may increase the expression of
pro-apoptotic target genes, such as Puma, Noxa, Bax and Bid
(14). On the other hand,
cytoplasmic p53 interacts with BCL-2 family member BCL-2 or BCL-XL,
which results in activation and translocation of Bax and Bid to
mitochondrial outer membrane. Moreover, p53 also translocates to
the mitochondria to activate the mitochondrial apoptosis pathway
(14–16). However, the molecular links between
pro-apoptotic function of p53 and mitochondrial dysfunction in
statin-induced apoptosis are not well understood.
Survivin is involved in apoptosis, and seems to be
induced by simvastatin, the smallest member of the inhibitor of
apoptosis protein (IAP) family. Survivin plays an important role
not only in inhibiting apoptosis but also in regulating mitosis.
Moreover, this gene is highly expressed in transformed cells and in
most human cancers, including lung, breast, pancreatic and colon
carcinomas, soft tissue sarcomas, brain tumors and hematologic
malignancies (17).
Based on the above, we investigated the role of
simvastatin in cancer cell growth inhibition showing the capacity
of this drug to induce apoptosis and demonstrate that the induction
of this process implicate the transcription up-regulation of Bax
and down-regulation of BCL-2, two genes with important roles in the
determination of programmed cell death.
Materials and methods
Cell culture
The MCF7 human breast cancer cells, SAEC human
normal small airway epithelial cells, HepG2 human hepatocellular
carcinoma cells, NCI-N87 human gastric cancer (NCI gastric cells)
and NCiH12299 human non-small cell lung carcinoma (NCH lung) cells
were purchased from American Type Culture Collection; the cells
were grown at sub-confluent culture in Dulbecco’s modified Eagle
medium or RPMI supplemented with L-glutamine, 100 U/ml penicillin,
10 μg/ml streptomycin and 10% fetal bovine serum, in 5%
CO2 incubator at 37°C.
Simvastatin treatment
Simvastatin (Calbiochem-Merck Co., Darmstadt,
Germany) carboxylate forms represent a lipophilic
3-hidroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor
that blocks Ras function through the inhibition of farnesylation
and inhibit glucose-induced Ca2+ channels in rat islet β
cells and cell proliferation of human smooth muscle cells. This
drug is soluble in dimethyl-sulfoxide (DMSO) and at a minor rate in
ethanol.
In our experiments, simvastatin was dissolved in
DMSO prepared in a 20-mM stock solution stored frozen at −20°C. For
the experiment, the cells were plated and added with 20 μM
simvastatin for 24–72 h in normal culture conditions. At the end of
treatment, cells were washed with PBS and harvested by scraping in
TRIzol reagent (Invitrogen, Carlsbad, CA, USA).
RNA and cDNA synthesis
Total mRNA was extracted from simvastatin-treated
cells according to Chomczynski and Sacchi method, using TRIzol
reagent (Invitrogen) according to the manufacturer’s instructions
and the integrity of purified RNA was verified using agarose gel
electrophoresis.
For cDNA synthesis, total mRNA was extracted from
simvastatin-treated cells according to Chomczynski and Sacchi; 2 μg
of total RNA in a final volume of 25 μl was reverse-transcribed by
Avian myeloblastosis virus (AMV) reverse transcriptase (Gibco-BRL)
according to the manufacturer’s instruction, in the presence of
random hexamer primers (Promega Corp., Madison, WI, USA) at 37°C
per 60 min. The cDNA was controlled by PCR with housekeeping GAPDH
or actin primers.
DNA fragmentation analysis
The internucleosomal DNA fragmentation, a typical
biochemical apoptotic event, was evaluated by agarose gel
electrophoresis; 1×106 cells were incubated in the
standard culture medium at 37°C for different time in the presence
of 20 μM simvastatin. At the end of the treatment, the cells were
harvested by a cell scraper, centrifuged, washed in PBS and
suspended in 100 μl TNE buffer (150 mM sodium chloride, 10 mM EDTA
and 10 mM Tris-HCl pH 8). The cell suspensions were lysed with 3
volumes of lysis buffer (0.2% SDS and 50 μg/ml RNAse in TNE) and
the lysate was incubated at 37°C for 1 h. High-molecular weight
genomic DNA was extracted from lysates and analyzed using
electrophoresis on 1.5% agarose gel in Tris-acetate-EDTA (TAE)
(18). The induction of apoptosis
by simvastatin was verified with the more sensible technique of
TUNEL.
TUNEL analysis
Cleavage of genomic DNA occurring during apoptosis
may yield double-stranded, low-molecular weight DNA fragments as
well single-strand nicks in high-molecular weight DNA. Those DNA
strand breaks can be identified by labeling free 3′-OH termini
using an enzymatic terminal deoxynucleotidyl transferase (TdT),
which catalyzes tailing of fluorescein-labeled dUTP in the TUNEL
(TdT-mediated dUTP nick end labeling) reaction. For TUNEL analysis,
the Roche in situ cell death fluorescein detection kit
(Roche Diagnostics, Mannheim, Germany) according to the
manufacturer procedures was used. In brief, the plated cells were
maintained in the presence of 20 μM simvastatin for 72 h, after the
treatment cells were washed with PBS, fixed with 2% PFA,
permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate
solution, added with TUNEL reaction mix and incubated in humidified
chamber at 37°C for 60 min in the dark.
PCR analysis
Three samples of the different cancer cell types
were cultured for 48 h in DMEM 10% FCS containing 20 μM
simvastatin. Control cell samples without simvastatin addition were
maintained in identical culture conditions. Total RNA and cDNA
synthesis was performed according to the procedure described. PCR
analysis of both Bax and BCL-2 gene expression was performed using
a GeneAmp PCR System 9700 (Applied Biosystems) and hot start Taq
Gold (Applera). Actin was used as a housekeeping control gene. The
sequences of primers utilized were as follows: Bax forward, 5′-CCA
GCT CTG AGC AGA TCA TG-3′ and reverse, 5′-TGC TGG CAA AGT AGA AAA
GG-3′; BCL-2 forward, 5′-GAC TTC GCC GAG ATG TCC AG-3′ and reverse,
5′-CAG GTG CCG GTT CAG GTA CT-3′; Actin forward, 5′-GAC TAC CTC ATG
AAG ATC CT-3′ and reverse, 5′-GCT TGC TGA TCC ACA TCT GC-3′. PCR
conditions were as follows: initial denaturation at 95°C for 10 min
followed by 35 cycles: 95°C for 45 sec, 54°C for 45 sec and 72°C
for 45 sec with a final extension at 72°C for 10 min. The
amplification products were analyzed on a 1% agarose gel in 0.5X
Tris Borate EDTA (TBE) buffer to control the amplicon lengths.
Real-time PCR
Real-time PCR analysis of BCL-2 and Bax gene
expression was performed using the iCycler® apparatus
(Bio-Rad, Hercules, CA) with sequence-specific primer pairs for the
genes tested. The housekeeping gene actin was used for the
normalization.
The primers used were the following: β-actin
forward, 5′-GAC TAC CTC ATG AAG ATC CT-3′ and reverse, 5′-GCT TGC
TGA TCC ACA TCT GC-3′; hBax forward, 5′-CCA GCT CTG AGC AGA TCA
TG-3′ and reverse, 5′-TGC TGG CAA AGT AGA AAA GG-3′; and hBCL-2
forward, 5′-GAC TTC GCC GAG ATG TCC AG-3′ and reverse, 5′-CAG GTG
CCG GTT CAG GTA CT-3′. The cDNA was serially diluted and every
dilution was run at least in triplicate. The real-time PCR analysis
was performed as follows: initial denaturation step, 95°C for 3 min
followed by 50 cycles of denaturation at 95°C for 1 sec; annealing,
10 sec at 50°C; and elongation, 8 sec at 72°C. The IQ SYBR-Green
SuperMix (Bio-Rad) was used for real-time PCR monitoring of the
amplification. Briefly, amplification was performed in a total
volume of 30 μl; the reaction mix was performed with 15 μl of 2X IQ
SYBR-Green SuperMix, 0.5 μl of each primer (16 μM) and 2 μl of cDNA
(or water as control, was always included). The real-time PCR
products were run on 2% agarose gel in TAE (standard
Tris-acetate-EDTA electrophoretic buffer). The amplicons of
expected size were extracted, purified and controlled for sequences
by Biogem DNA Sequencing Core (Biogem, Naples, Italy). The sequence
analysis do not show any differences from Bax and BCL-2 gene
sequences already deposited in GenBank. Results were evaluated by
iCycler iQ Real-Time Detection System Software®
(Bio-Rad). Data were calculated on the basis of the threshold cycle
(Ct) value. The expression of the analyzed genes was first
normalized with respect to β-actin. The relative amount of mRNA on
the y-axis in the graph was expressed as the inverse of Ct
normalized values and multiplied by 100 (1/Ct × 100). The value
obtained was proportional to mRNA expression.
Western blot analysis of Bax and BCL-2 in
simvastatin-treated MCF7 cells
MCF7 breast cancer cells and SAEC human normal small
airway epithelial cells were collected in 1.5-ml tubes, washed with
PBS and lysed in 0.4 ml of lysis buffer (0.06 M Tris-HCl, pH 6.8,
10% glycerol, 2% SDS, 5% β-mercaptoethanol and 0.0025% bromophenol
blue). DNA was sheared by a needle and the solution was heated at
95°C for 5 min and centrifuged at 15000 g for 2 min. The total
protein was quantified and loaded on SDS-polyacrylamide gel, run at
40 mA and transferred to nitrocellulose by electroblotting. Filters
were blocked with 5% non-fat milk in TBS/Tween-20 buffer (137 mM
NaCl, 20 mM Tris-HCl, pH 7.4, 0.1% Tween-20) before incubation with
antibodies against Bax (mouse monoclonal dilution 1:200; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), BCL-2 (mouse monoclonal
dilution 1:200; Santa Cruz Biotechnology) or β-actin (mouse
monoclonal dilution 1:30000; Sigma-Aldrich, Munich, Germany),
followed by horseradish peroxidase-conjugated anti-mouse-IgG
secondary antibody (dilution 1:30000; Promega Corp.) dissolved in
1% non-fat milk in TBS/Tween-20. Immune complex were detected by
the Super Signal West Pico Chemiluminescent Kit (Pierce, Rockford,
IL, USA) and exposed to X-ray film (Kodak, Wiesbaden, Germany).
Statistics
Each experiment was repeated three times. Results
are expressed as mean ± SEM and the effects were compared with
untreated control cells on the same plate. Paired t-test were used
to analyze the effect of simvastatin; P<0.05 was considered
significant.
Results
DNA laddering
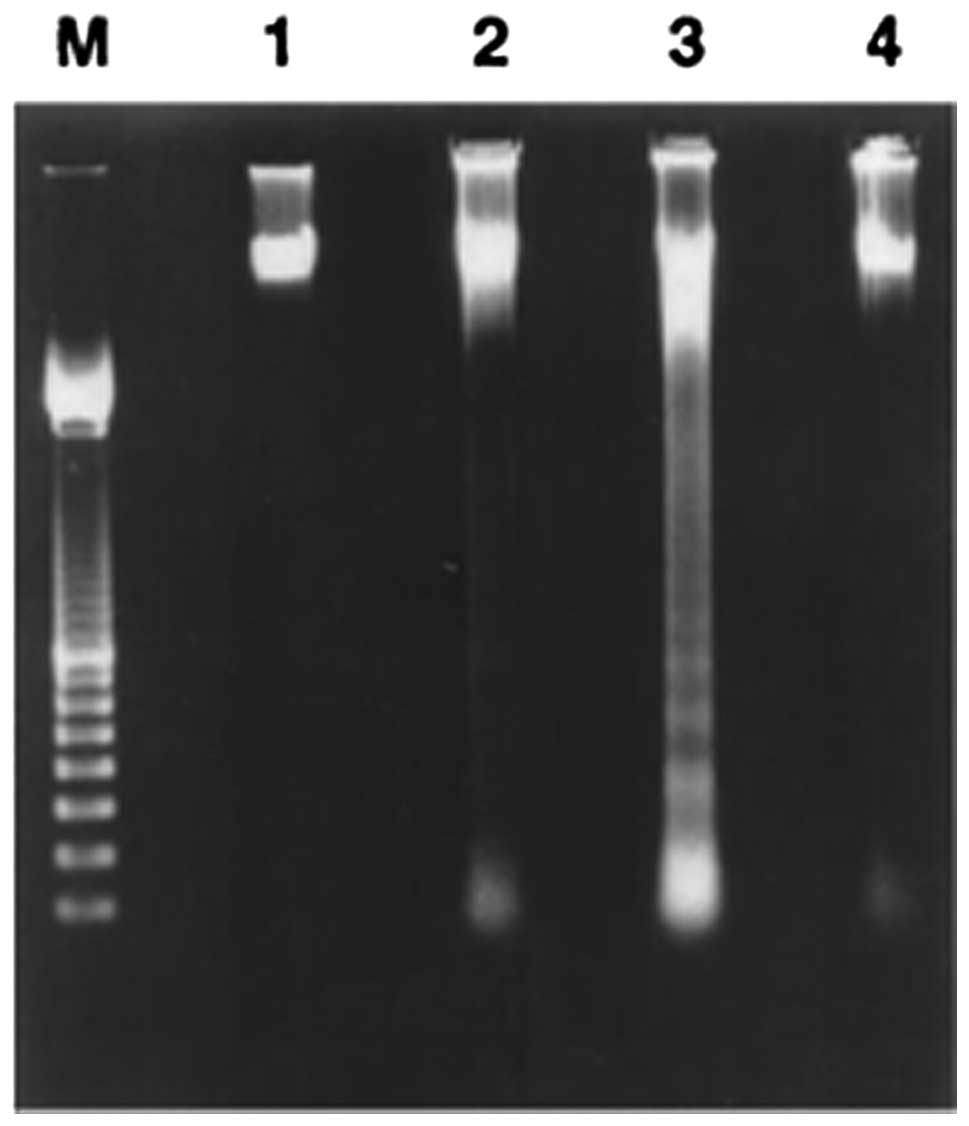
Simvastatin treatment induces DNA fragmentation.
Internucleosomal DNA fragmentation is a classic biochemical event
occurring in cells undergoing apoptosis. DNA degradation,
characterized by a typical electrophoretic ladder, was found in
breast cancer MCF7 cells treated for 1 or 3 days with simvastatin
20 μM, whereas it did not occur in cells without treatment
(Fig. 1) or in SAEC human normal
small airway epithelial cells treated with the same simvastatin
concentration for 3 days. The induction of the apoptotic event was
time-dependent and is minimal after 1 day of treatment but reached
maximum intensity in cancer cells treated with 20 μM simvastatin
for 3 days (Fig. 1). In
situ DNA fragmentation also was demonstrated by the TUNEL
assay, which showed the occurrence of DNA fragmentation (presence
of green nuclei) in several cancer cell types but not in fibroblast
cells treated for 2 days with 20 μM of simvastatin (Fig. 2).
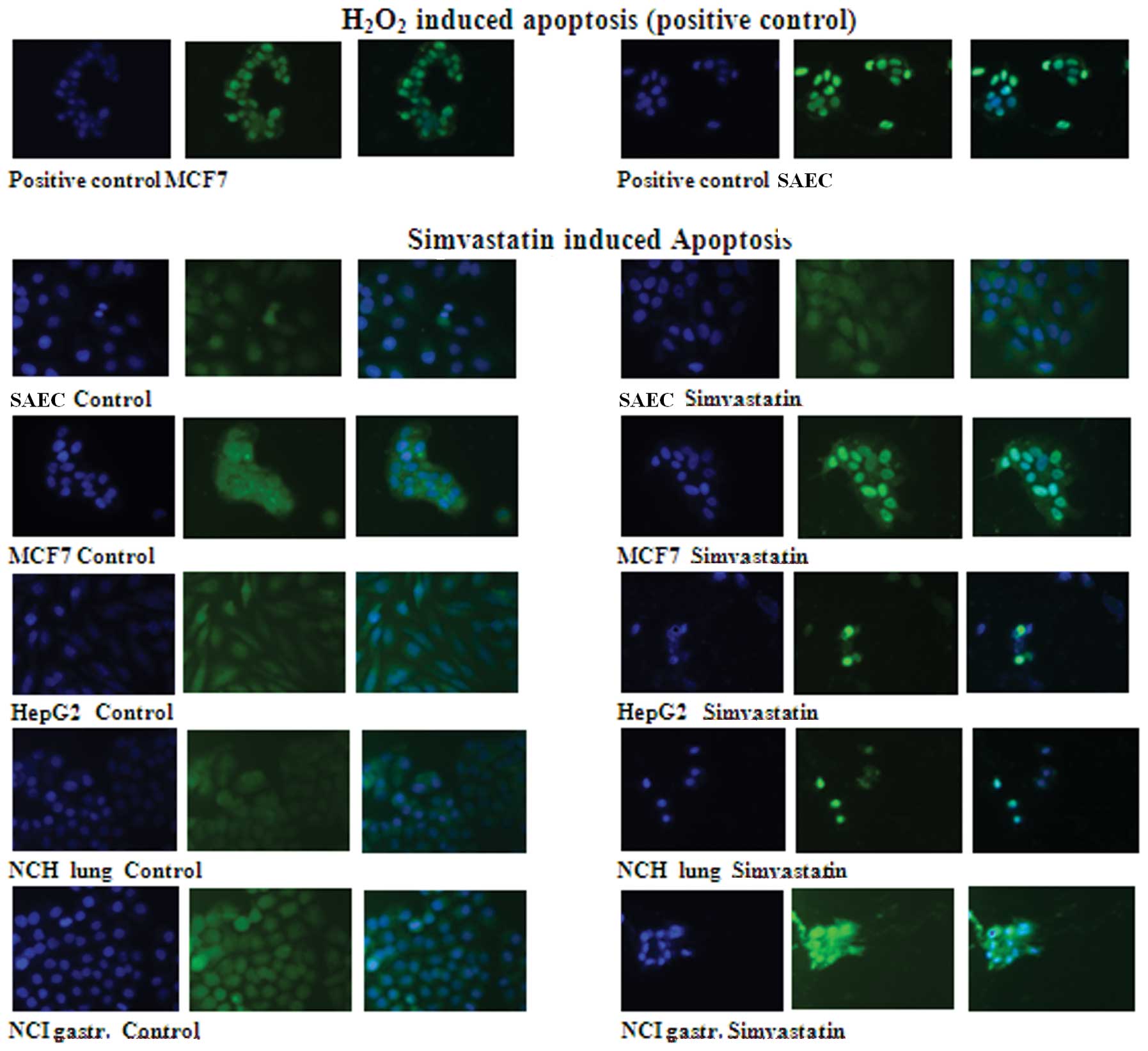
TUNEL apoptosis analysis
The TUNEL analysis clearly showed that 72-h
treatment with 20 μM simvastatin induces a well-defined apoptosis
in five different types of human cancer cells but not in
non-tumoral human SAEC that do not show apoptotic signs either when
the drug treatment was prolonged >72 h. As positive control of
TUNEL reaction, SAEC normal epithelial and breast cancer MCF7
cells, in which the apoptosis was induced by 30-min treatment with
hydrogen peroxide, was used.
For all cell lines treated with simvastatin, the
nuclei DAPI coloration (blue) and the fluorescein (green)
coloration of apoptotic nuclei were visible, and the composite
image was observed (Fig. 2).
The composite coloration clearly appears as the
fluorescein is localized only in the nuclei, showing that 72-h
treatment with 20 μM simvastatin induces strong apoptosis in cancer
cells but not in normal cells. SAEC control cells after 3 days of
treatment did not show apoptotic coloration in the nuclei. On the
contrary, the apoptosis induction with a strong green coloration
localized in the inner nuclei is clearly evident in MCF7 breast
cancer cells. Similar results appear in hepatocellular carcinoma
HepG2 cells, lung carcinoma NCH lung cells and gastric cancer NCI
gastric cells; in these cancer cell lines, there was a high
mortality after 72-h drug treatment and only a minor number of
cells remain attached on the cell culture plate. In these residual
cells, a very strong nuclei green coloration due to the
well-evident apoptosis induction was observed (Fig. 2).
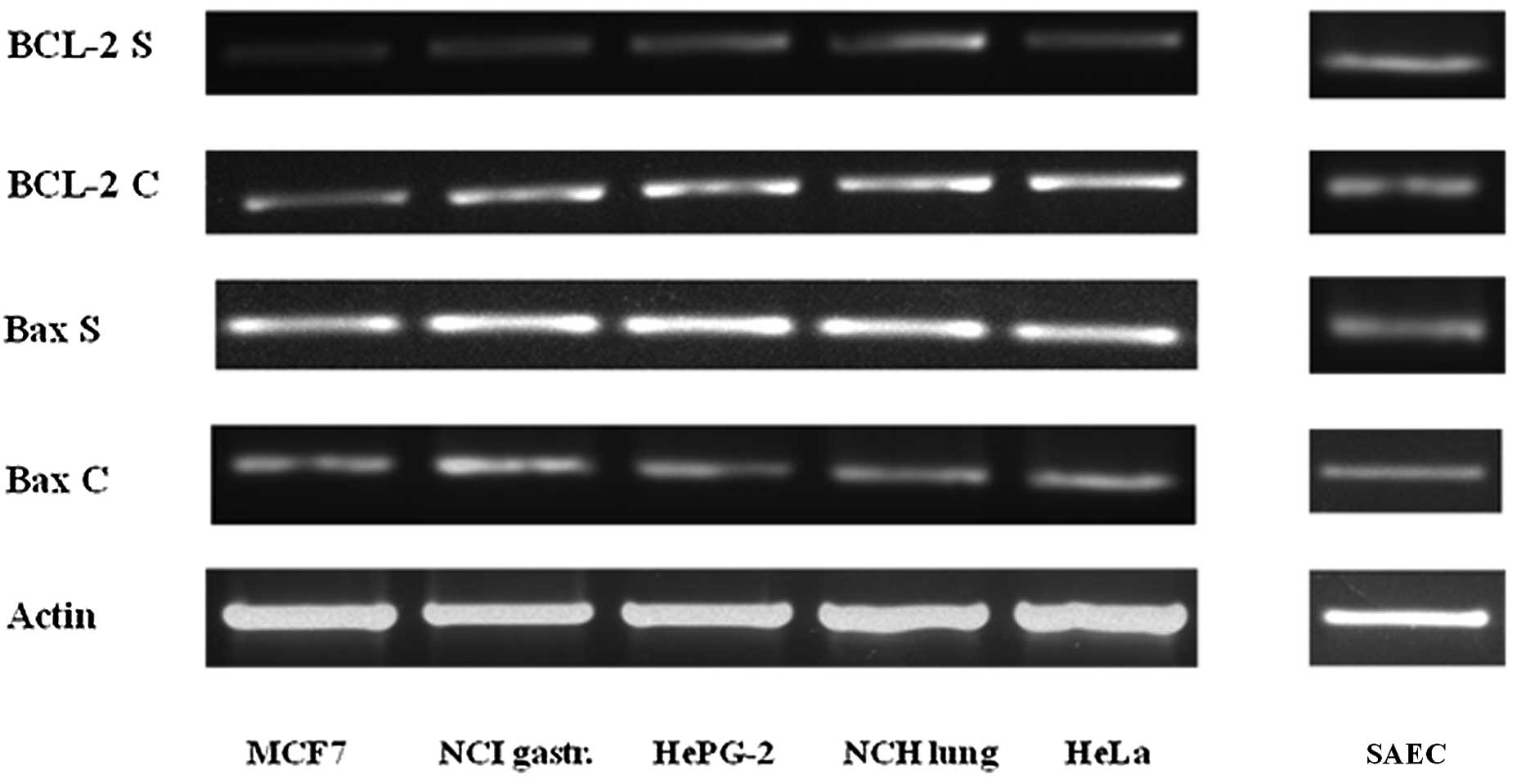
PCR analysis
The Bax and BCL-2 PCR analysis demonstrate that the
simvastatin treatment, in five different types of cancer cells,
induce apoptotic death, characterized by an increase in the
expression of the pro-apoptotic gene Bax and, at the same time, a
decrease in the expression of the anti-apoptotic gene BCL-2
compared to the untreated control cells. In the SAEC non-cancerous
cells, there was no difference in the expression of these two genes
after the same simvastatin treatment as shown in Fig. 3.
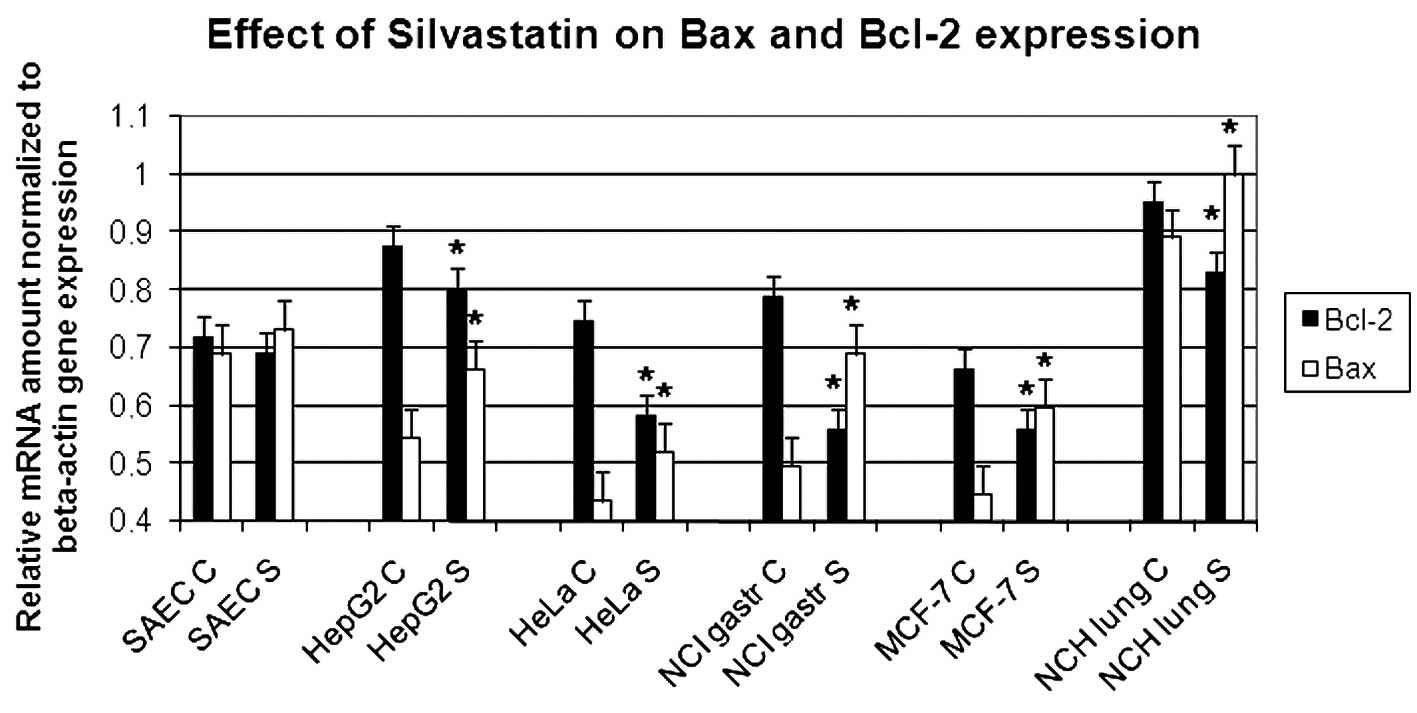
Real-time PCR analysis
The variation of apoptotic gene expression, already
demonstrated in the semi-quantitative PCR analysis, is clearly
confirmed by real-time amplification as shown in Fig. 4.
In all the cancer cell lines analyzed, the
simvastatin treatment appears to reduce anti-apoptotic BCL-2
expression and increase the transcription of Bax pro-apoptotic
gene. The values have been normalized with respect to actin. The
graph represents three independent experiments with each bar as
mean ± SD. P<0.05 compared to control group.
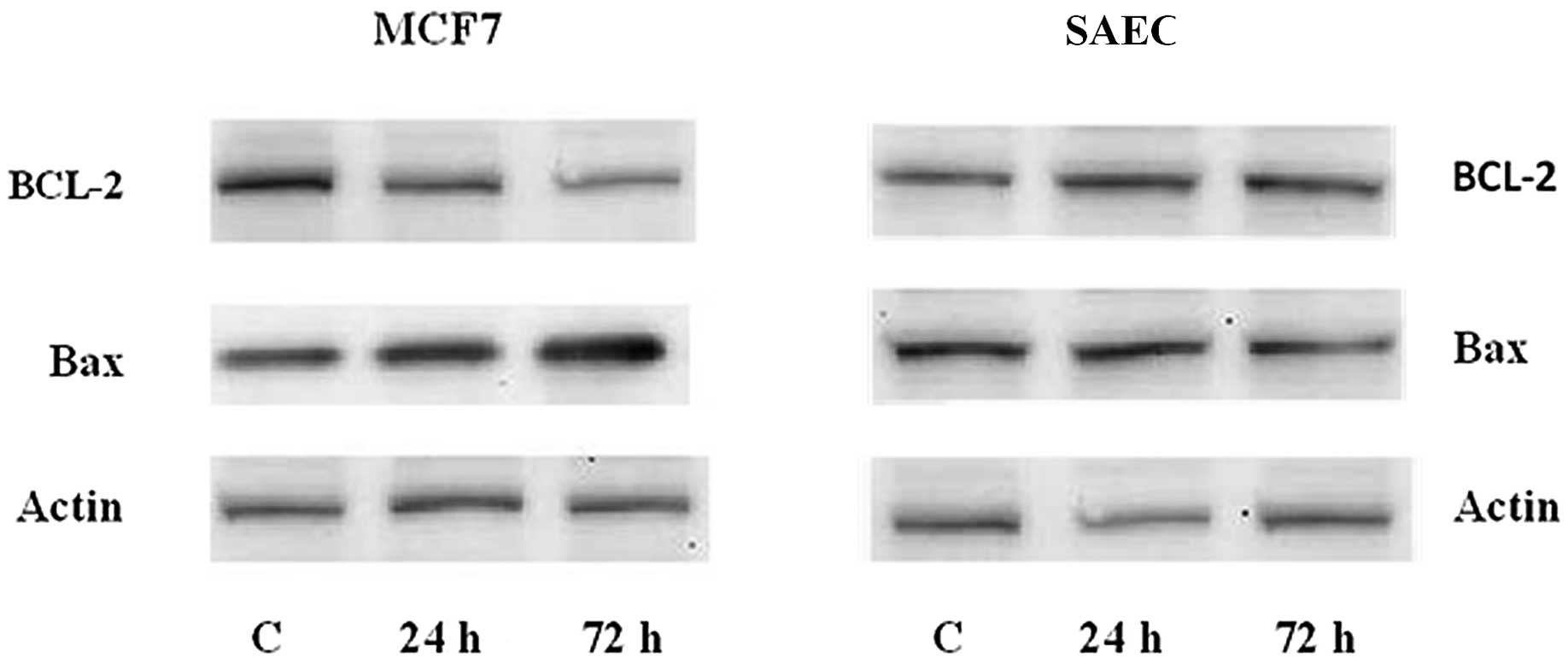
Western blot analysis of Bax and BCL-2 in
simvastatin-treated MCF7 cells
The induction of apoptosis during the simvastatin
treatment was evinced in the variation of the expression of
proteins responsible for the regulation of apoptosis mechanism. We
have analyzed Bax and BCL-2, the two principal genes involved in
the apoptosis control, and have demonstrated that at the basis of
apoptosis induction, there is an up-regulation of Bax protein,
which has a well-known pro-apoptotic effect, and a down-regulation
of BCL-2, which is known to protect the cells from apoptosis. In
this experiment, we have demonstrated that in MCF7 breast cancer
cells, the quantity of apoptotic proteins vary, increasing the time
of simvastatin presence in the cell culture. On the contrary, in
non-malignant SAEC cells, there are no variations in Bax and BCL-2
protein quantity (Fig. 5).
Discussion
Many studies show statins to be beneficial as
anticancer agents (19). Their
anti-tumor effects may be due to the involvement of important
biological processes, such as inhibition of cell proliferation,
promotion of apoptosis, inhibition of angiogenesis, prevention of
metastasis, improvement of immunity or possibly targeting the
cancer stem cell population (20).
In the present study, we investigated the
anti-proliferative effects of simvastatin on several types of
cancer cells. According to our results, the anticancer cell growth
inhibition is due to the deregulation of apoptosis induction.
Apoptosis plays a key role in the pathogenesis of cancers and the
genes relating to this process are focus of interest in the studies
of cancer onset and progression. It is well-known that Bax and
BCL-2 are transcriptional targets for the tumor suppression protein
p53 (17), which is responsible
for the induction of cell cycle arrest and/or apoptosis in response
to DNA damage (21).
The progression of cancer mainly depends on the
balance between the pro-apoptotic protein such as Bax and
anti-apoptotic protein such as BCL-2 (22). Moreover, p53-independent
mitochondrial-mediated apoptosis has been reported following
lovastatin exposure in a mouse mammary carcinoma (23).
It has been proposed that simvastatin regulates
BCL-2 protein levels through the induction of ET-1, which involves
the transcription factor NFATc3 binding to the BCL-2 promoter. This
hypothesis is based on the evidence that exogenous ET-1
significantly increased BCL-2 protein levels in neuroblastoma cells
and this effect was attenuated in the presence of ETA/B receptor
antagonists (24).
In our study, we have demonstrated that the
incubation of several lines of human cancer cells, of different
histology, with 20 μM simvastatin caused a significant increase in
Bax expression and a decrease in BCL-2, both at mRNA and protein
levels, and as a consequence of this gene expression deregulation,
we observed strong apoptosis induction, after addition of
simvastatin, in all the cancer cell lines examined. As determined
with the TUNEL analysis, the clearest results were observed in MCF7
breast cancer cells and in NCI gastric cancer cells with clear
fluorescence inside the cell nuclei. In lung carcinoma and in
hepatic carcinoma cells, apoptotic death was very strong and the
small number of cells that remain attached to the plate showing a
very strong fluorescein coloration in the nuclei. On the contrary,
the non-cancerous fibroblasts are not sensitive to simvastatin and
do not show any sign of apoptosis, even after 3 days of exposure to
the drug. This different sensitivity between the normal and cancer
cells may have important implications on the possibility to use
this drug, already widely used in the therapy of
hypercholesterolemia, as protection against cancer progression or
cancer prevention.
In conclusion, these studies indicate that statins
are able to induce apoptosis in different cancer cell lines and the
mechanism at the basis of induction of this important process
involves the regulation of Bax and BCL-2 gene expression. The
confirmation of these effects with experiments on animals in
vivo, to verify if, at therapeutic or at higher doses,
simvastatin may induce a regression in tumor mass, preventively
induced in mice with syngenic cancer cells or with carcinogenic
chemicals, is important. These experiments are ongoing in our
laboratory.
There is an increasing interest in cancer protection
and in all the drugs that at low dose, alone or in combination with
different modes of action and low toxicity function as
chemopreventive agents. Therefore, we are also studying other
molecules that are generally used for the treatment of well-known
pathology and which present interesting effects on cancer cell
proliferation.
Acknowledgements
The authors are grateful to Mr. F. Moscatiello for
skillful technical assistance. This study was financially supported
by the Institute of Genetics and Biophysics A. Buzzati Traverso
CNR. No additional external funding was received for this
study.
References
|
1
|
Gauthaman K, Fong CY and Bongso A:
Statins, stem cells, and cancer. J Cell Biochem. 106:975–983. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liao JK and Laufs U: Pleiotropic effects
of statins. Ann Rev Pharmacol Toxicol. 45:89–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee MH, Yee Cho S and Han YM: Simvastatin
suppresses self-renewal of mouse embryonic stem cells by inhibiting
RhoA geranylgeranylation. Stem Cell. 25:1654–1663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chan KK, Oza AM and Siu LL: The statins as
anticancer agents. Clin Cancer Res. 9:10–19. 2003.
|
|
6
|
Demierre MF, Higgins PD, Gruber SB, Hawk E
and Lippman SM: Statins and cancer prevention. Nat Rev Cancer.
5:930–942. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mantha AJ, Hanson JE, Goss G, et al:
Targeting the mevalonate pathway inhibits the function of the
epidermal growth factor receptor. Clin Cancer Res. 11:2398–2407.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wong WW, Dimitroulakos J, Minden MD and
Penn LZ: HMBCoA reductase inhibitors and the malignant cell: the
statin family of drugs as triggers of tumor-specific apoptosis.
Leukemia. 16:508–519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu J, Wong WW, Khosravi F, Minden MD and
Penn LZ: Blocking the Raf-MEK-ERK pathway sensitizes acute
myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer
Res. 64:6461–6468. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cafforio P, Dammacco F, Gernone A and
Silvestris F: Statins activate the mitochondrial pathway of
apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis.
26:883–891. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee SK, Kim YC, Song SB, Young BS and Kim
S: Stabilization and translocation of p53 to mitochondria is linked
to Bax translocation to mitochondria in simvastatin-induced
apoptosis. Biochem Biophys Res Commun. 391:1592–1597. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Blanco-Colio LM, Villa A, Ortego M, et al:
3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors,
atorvastatin and simvastatin, induce apoptosis of vascular smooth
muscle cells by down-regulation of Bcl-2 expression and Rho A
prenylation. Atherosclerosis. 161:17–26. 2002. View Article : Google Scholar
|
|
13
|
Herrero-Martin G and Lopez-Rivas A:
Statins activate a mitochondrial-operated pathway of apoptosis in
breast tumor cells by a mechanism regulated by Erb B2 and dependent
on the prenylation of proteins. FEBS Lett. 582:2589–2594. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vousden KH and Lu X: Live or let die: the
cell’s response to p53. Nat Rev Cancer. 2:594–604. 2002.
|
|
15
|
Chipuk JE and Green DR: Dissecting
p53-dependent apoptosis. Cell Death Differ. 13:994–1002. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vaseva AV and Moll UM: The mitochondrial
p53 pathway. Biochim Biophys Acta. 1787:414–420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hwang KE, Na KS, Park DS, et al: Apoptotic
induction by simvastatin in human lung cancer A549 cells via Akt
signaling dependent down-regulation of surviving. Invest New Drugs.
29:945–952. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sambrook J, Fritsch EF and Maniatis T:
Isolation of high molecular weight DNA from mammalian cells.
Molecular Cloning: A Laboratory Manual. Cold Spring Harbor
Laboratory Press; New York: 1989
|
|
19
|
Campbell MJ, Esserman LJ, Zhou Y, et al:
Breast cancer growth prevention by statins. Cancer Res.
66:8707–8714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sassano A, Katsoulidis E, Antico G, et al:
Suppressive effects of statins on acute promyelocytic leukemia
cells. Cancer Res. 67:4524–4532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Xu Z and Zhang M: Down-regulation
of surviving expression and elevation of caspase-3 activity
involved in pitavastatin-induced HepG 2 cell apoptosis. Oncol Rep.
18:383–387. 2007.PubMed/NCBI
|
|
22
|
Adams JM and Cory S: The apoptotic switch
in cancer development and therapy. Oncogene. 26:1324–1337. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shibata MA, Kavanaugh C, Shibata E, et al:
Comparative effects of lovastatin on mammary and prostate
oncogenesis in transgenic mouse models. Carcinogenesis. 24:453–459.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Butterick TA, Igbavboa U, Eckert GP, et
al: Simvastatin stimulates production of the antiapoptotic protein
Bcl-2 via endothelin-1 and NFATc3 in SH-SY5Y. Cells Mol Neurobiol.
41:384–391. 2010. View Article : Google Scholar : PubMed/NCBI
|