Introduction
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) is a member of the tumor necrosis factor cytokine
family and has a homotrimeric structure. TRAIL has been shown to
induce apoptosis in cancer cells with minimal cytotoxicity toward
non-transformed cells. TRAIL exerts its pro-apoptotic effect by
binding to two death domain-containing receptors, TRAIL receptor 1
(TRAIL-R1)/death receptor (DR) 4 and TRAIL-R2/DR5 (1). Binding of TRAIL to TRAIL-R1/2
expressed on the cell surface initiates the extrinsic apoptotic
pathway. TRAIL binding to these DRs induces their oligomerization
and conformational changes in the death domains, resulting in
receptor activation and the formation of a death-inducing signaling
complex. This complex formation allows the binding of an adaptor
molecule, Fas-associated protein with death domain, via death
domain interactions. The adaptor molecule also contains a death
effector domain that binds to caspase-8, resulting in their
oligomerization and autoactivation (2). Activated caspase-8 in turn activates
the effector caspase-3/6/7 that executes the apoptotic process. In
some cell types (type I), the extrinsic pathway is sufficient to
commit the cell to apoptosis, while in other cell types (type II),
activation of caspase-8 is low. In the latter cells, amplification
by the intrinsic mitochondrial pathway is necessary to evoke
substantial apoptosis (3). This
amplification is triggered by activated caspase-8. Activated
caspase-8 can cleave and activate the pro-apoptotic Bcl-2-family
molecule Bid. In turn, truncated Bid activates the other
Bcl-2-family molecules, Bax and Bak, which results in their
oligomerization and the formation of megachannels in the outer
mitochondrial membrane. The release of cytochrome c through the
Bax/Bak megachannels into the cytosol induces the assembly of the
apoptosome, representing the activation-platform for caspase-9
(4,5). Caspase-9 also promotes the activation
of the effector caspase-3/6/7, thereby providing a positive loop of
caspase activation (3).
TRAIL is a promising drug in cancer treatment due to
its selective cytotoxicity toward malignant cells. However, a
growing body of evidence suggests that some cancer cell types,
including malignant melanoma cells, are resistant to TRAIL-induced
apoptosis despite their expression of death-inducing TRAIL-Rs on
the cell surface (6). Moreover,
TRAIL-responsive tumors acquire a resistant phenotype that renders
TRAIL therapy ineffective. Although many possible mechanisms,
including intracellular mechanisms, have been identified, the cause
of the TRAIL-resistance remains under intense scrutiny, as these
mechanisms contribute to TRAIL-resistance to varying extents in
different tumor cells. In any case, overcoming the TRAIL-resistance
of cancer cells is necessary for effective TRAIL therapy, and small
molecular compounds that can amplify TRAIL-induced apoptosis are
urgently required.
Impairment of the intracellular ion homeostasis
leads to depolarization of the plasma membrane potential (7–9).
Depolarization has been shown to be an early event in the apoptosis
induced by divergent agents, including Fas (10), rote-none (11) and arsenic trioxide (12) and is considered to play an
important pro-apoptotic role. By contrast, depolarization has also
been shown to exhibit anti-apoptotic effects. Various
membrane-depolarizing agents, including ouabain, tetraethylammonium
(TEA) and veratridine, protect Purkinje cells against apoptosis
(13). In addition, K+
loading and several K+ channel inhibitors protect
various human tumor cells against staurosporine-induced apoptosis
(14). These observations suggest
that depolarization can act in both a pro-and anti-apoptotic manner
depending on the cell types and the apoptotic stimuli involved.
However, the cellular and molecular mechanisms underlying these
dual functions remains unclear.
The role of depolarization in TRAIL-induced
apoptosis is poorly documented. In the present study, we
investigated whether TRAIL caused depolarization and, if so,
explored its role in TRAIL-induced apoptosis. Results revealed that
TRAIL induced robust depolarization in melanoma cells, but not in
normal melanocytes. Moreover, membrane-depolarizing agents such as
K+ and KATP channel inhibitors sensitized
melanoma cells, but not melanocytes, to TRAIL-induced apoptosis by
promoting endoplasmic reticulum (ER) stress-mediated death pathway,
including caspase-12.
Materials and methods
Reagents
Soluble recombinant human TRAIL and K+
channel inhibitors, glibenclamide (GLB), U37883A (U37),
tetraethylammonium (TEA), 5-hydroxydecanoate (5-HD), α-dendrotoxin
(DTX), charybdotoxin (CTX), bis-oxonol and
DiBAC4(3) were obtained
from Enzo Life Sciences (San Diego, CA, USA). In the present study,
TRAIL was used at concentrations of 6.3–100 ng/ml and K+
channel inhibitors were used at 100 μM except for DTX and
CTX, both of which were used at 100 nM. Thapsigargin (Tg) was
obtained from Sigma-Aldrich (St. Louis, MO, USA). The
cell-permeable general caspase inhibitor z-VAD-fmk (VAD) and
caspase-3/7-specific inhibitor z-DEVD-fmk (DEVD) were obtained from
Calbiochem (La Jolla, CA, USA). The caspase-12-specific inhibitor
z-ATAD-fmk (ATAD) was purchased from BioVision (Mountain View, CA,
USA). The reagents were dissolved in dimethylsulfoxide and diluted
with HBSS to a final concentration of <0.1% before use and used
at 10 or 30 μM. Polyclonal antibodies against X-box-binding
protein-1 (XBP-1) and glucose-related protein 78 (GRP78) were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All
other chemicals were of analytical grade.
Cells
Human melanoma cell lines were obtained from RIKEN
Bioresource Center Cell Bank (Tsukuba, Japan), Health Science
Research Resource Bank (Osaka, Japan) and American Type Culture
Collection (Manassas, VA, USA), and cultured in high
glucose-containing DMEM (Sigma-Aldrich) supplemented with 10% FBS
in a 5% CO2-containing atmosphere. The cells were
harvested by incubation in HBSS containing 1 mM EDTA and 0.25%
trypsin for 5 min at 37°C. Normal human epidermal melanocytes were
obtained from Cascade Biologica (Portland, OR, USA) and cultured
according to the manufacturer’s instructions.
Measurement of depolarization
Depolarization was measured by flow cytometry using
bis-oxonol or DiBAC4(3), an anionic dye that shows an increase
in fluorescence intensity upon membrane depolarization, as
previously described (15). Cells
(4×105 cells/500 μl) suspended in HBSS were
incubated with 100 nM dye for 15 min at 37°C, and then incubated
with the agents to be tested for 2–4 h at 37°C in a 5%
CO2-containing atmosphere. Subsequently,
1×104 cells were counted for their fluorescence using
the FL-2 channel of a FACSCalibur (BD Bioscience, San Jose, CA,
USA) and analyzed using the CellQuest software (BD Bioscience). For
analysis of the depolarization in an earlier time period (0–10
min), the fluorescence in the cells was analyzed using a microplate
fluorometer (Fluoroskan Ascent CF; Labsystems, Helsinki, Finland).
The data were expressed as F/F0, where
F0 is the fluorescence in unstimulated cells and
F is the fluorescence in stimulated cells.
Determination of cell death by
fluorescent microscopy
Cells (1×104) were placed on 8-chamber
coverslips (Asahi Glass Co., Tokyo, Japan) and treated with the
agents to be tested for 24 h at 37°C in a 5%
CO2-containing atmosphere. After removal of the medium,
the cells were stained with 4 μM each of calcein- AM and
ethidium bromide homodimer-1 to label live and dead cells,
respectively, using a commercially available kit
(Live/Dead® Viability/Cytotoxicity Kit; Invitrogen,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
Images were obtained with a fluorescence microscope (IX71 inverted
microscope, Olympus, Tokyo, Japan) and analyzed using the
LuminaVision software (Mitani Corporation, Fukui, Japan).
Determination of apoptotic cell
death
Apoptotic cell death was quantitatively assessed by
staining with propidium iodide (PI) and FITC-conjugated annexin V,
as previously described (16).
Briefly, cells plated in 24-well plates (2×105
cells/well) were treated with TRAIL and the agents to be tested
alone or together for specified times in DMEM containing 10% FBS
(FBS/DMEM). Subsequently, the cells were stained with
FITC-conjugated annexin V and PI using a commercially available kit
(Annexin V FITC Apoptosis Detection Kit I; BD Biosciences) to label
dead or damaged cells. The stained cells were evaluated in the
FACSCalibur and analyzed using the CellQuest software. Four
cellular subpopulations were evaluated: vital cells (annexin
V−/PI−); early apoptotic cells (annexin
V+/PI−); late apoptotic cells (annexin
V+/PI+); and necrotic/damaged cells (annexin
V−/PI+). Annexin V+ cells were
considered to be apoptotic cells.
Measurements of mitochondrial membrane
potential (ΔΨm) depolarization and caspase-3/7
activation
Caspase-3/7 activation and ΔΨm
depolarization were simultaneously measured as previously described
(16). Briefly, cells plated in
24-well plates (2×105 cells/well) were treated with
TRAIL and the agents to be tested alone or in combination in
FBS/DMEM for 24 h. The cells were then stained with the dual sensor
MitoCasp (Cell Technology Inc., Mountain View, CA). Caspase-3/7
activation and ΔΨm depolarization were determined using the
FACSCalibur and the data were analyzed using the CellQuest
software. In some experiments, changes in the ΔΨm were
measured using the lipophilic cation JC-1 as previously described
(17). Briefly, cells
(5×105 cells/500 μl) were loaded with 2 μM
JC-1 at 37°C for 15 min, washed, and resuspended in HBSS. Following
cell stimulation, the green fluorescence (monomeric JC-1) and red
fluorescence (J-aggregates) were measured using the FL-1 and FL-2
channels, respectively, of the FACSCalibur.
Measurement of caspase-12 activation
Activated caspase-12 in living cells was detected
using the caspase-12 inhibitor ATAD-fmk conjugated to FITC as a
marker, since this compound binds to active caspase-12, but not to
inactive caspase-12. Cells (1×106 cells/ml) were stained
with FITCATAD-fmk for 30 min at 37°C using a kit (CaspGlow
Fluorescein Caspase-12 Staining Kit; BioVision) according to the
manufacturer’s protocol. Fluorescence was determined using the FL-1
channel of the FACSCalibur and analyzed using the CellQuest
software.
Western blot analysis
Western blot analysis was carried out as previously
described (18) with minor
modifications. Cells were treated with the agents to be tested for
24 h at 37°C, washed, and lysed with SDS-sample buffer. Whole cell
lysates were subjected to SDS-PAGE and then transferred onto
polyvinylidene difluoride membranes (Nippon Millipore, Tokyo,
Japan). The membranes were blocked with BlockAce (Dainippon
Sumitomo Pharma, Osaka, Japan) at room temperature for 60 min,
washed, incubated with antibodies against GRP78, XBP-1, and
caspase-12 overnight at 4°C and then incubated with horse-radish
peroxidase-conjugated anti-rabbit IgG for 60 min at room
temperature. After extensive washing, the immunoreactive proteins
on the membranes were detected using the ECL Prime Western Blotting
Reagent (GE Healthcare Japan, Tokyo, Japan). To verify equal
loading, the membranes were reprobed with an anti-β-actin antibody.
The signal intensities were quantified relative to the intensity of
the β-actin signal using the NIH image software (NIH, Bethesda,
MD).
Statistical analysis
The statistical significance of differences among
values was analyzed by one-way ANOVA followed by Tukey’s post hoc
test. Values of P<0.05 were considered to indicate a
statistically significant difference.
Results
TRAIL induces depolarization in human
melanoma cells during apoptosis
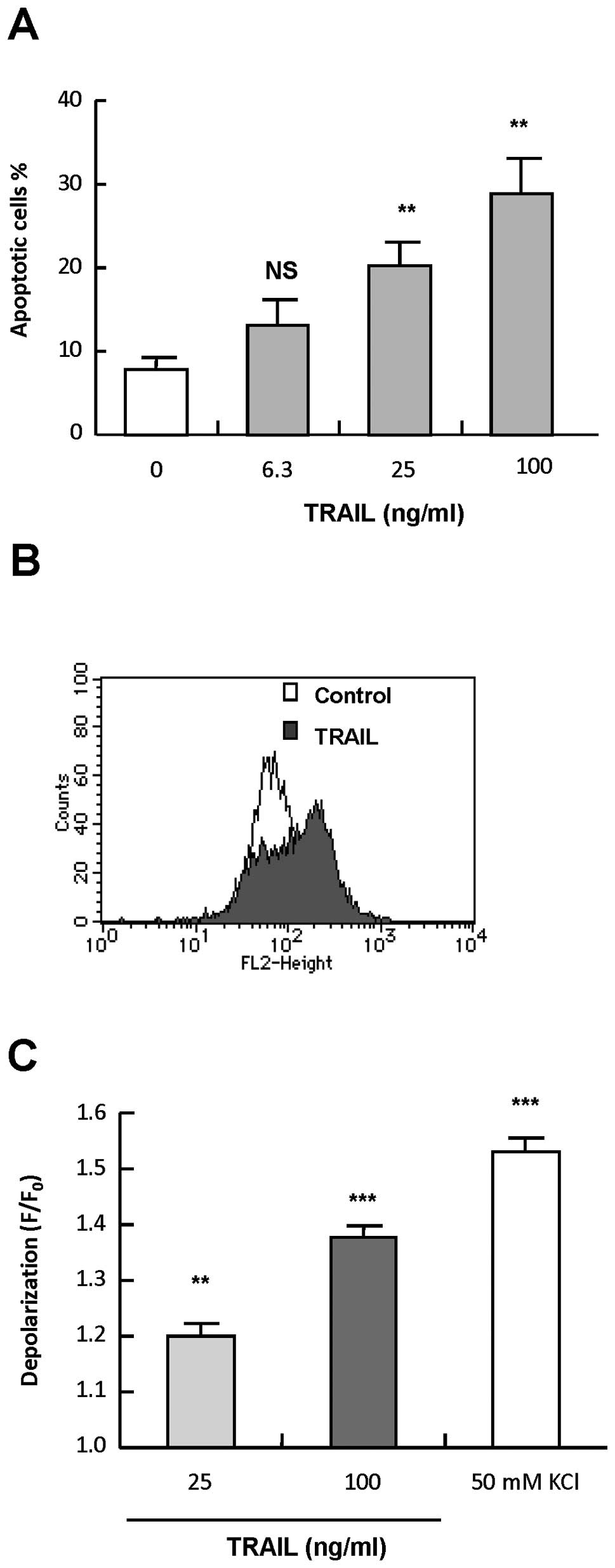
Human melanoma cell line A375 cells were treated
with varying concentrations of recombinant human TRAIL for 24 h,
stained with annexin V-FITC and PI, and analyzed by flow cytometry.
Treatment with TRAIL at concentrations of ≥25 ng/ml resulted in
apoptotic (annexin V+) cells in a dose-dependent manner
(Fig. 1A). Next, we examined
whether TRAIL induced depolarization in the cells. The cells were
treated with 25 and 100 ng/ml TRAIL for various times, and
depolarization of the plasma membrane potential was measured by
flow cytometry using the anionic dye bis-oxonol or
DiBAC4(3). Essentially
similar results were obtained with the two dyes. K+
loading (50 mM) resulted in significant (maximum of 2.2-fold)
increase in the fluorescence, indicating the occurrence of
depolarization. The fluorescence peaked within 5 min, and declined
thereafter, but remained higher than the basal level up to 4 h
(1.5-fold). Although TRAIL induced depolarization in a dose- and
time-dependent manner, unlike K+ loading, it was
observed after 2–4 h of treatment. At 4 h, 25 and 100 ng/ml TRAIL
caused small but significant depolarization (1.2- and 1.4-fold,
respectively) (Fig. 1B and C).
TRAIL also induced robust depolarization within 4 h in SK-MEL-2
melanoma cells and Jurkat leukemia cells at concentrations ranging
from 25–100 ng/ml (data not shown). These data show that TRAIL
induces depolarization in human cancer cells during apoptosis. In
the following experiments, we explored the role of depolarization
in TRAIL-induced apoptosis using melanoma cells as model cell
systems.
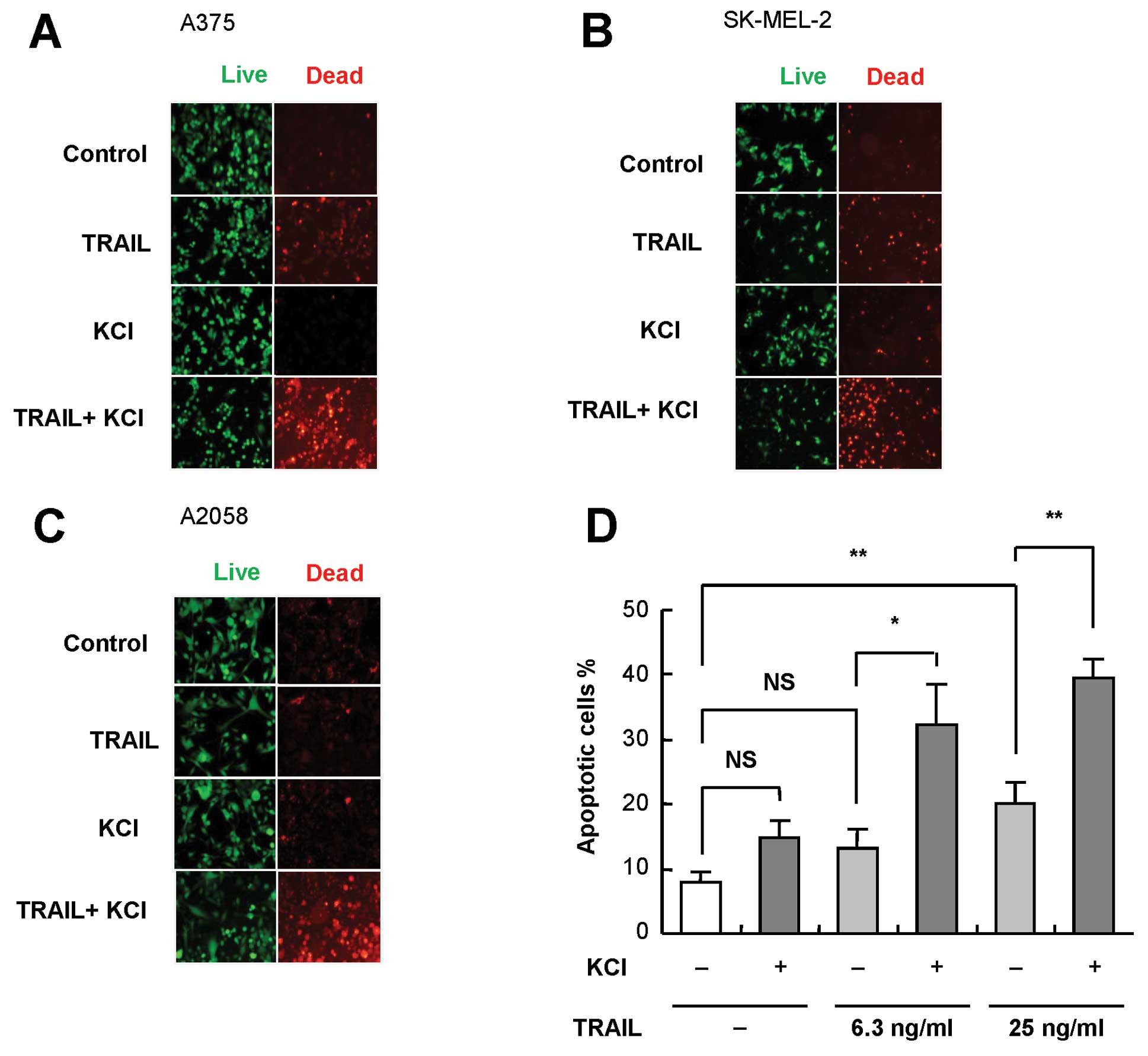
K+ loading sensitizes melanoma
cells to TRAIL-induced apoptosis
Next, we examined whether the modulation of
depolarization affected TRAIL cytotoxicity. To this end, persistent
depolarization was induced by extracellular high K+
loading. Following treatment with TRAIL and K+ alone or
in combination for 24 h, the cells were stained with calcein-AM and
ethidium bromide homodimer, and observed under a fluorescence
microscopy. Live cells were stained green with calcein-AM, whereas
dead cells with compromised cell membranes were stained red with
ethidium bromide homodimer. TRAIL and K+ alone had
minimal or weak cytotoxicity. However, the combined use of the two
agents resulted in considerable cell death (Fig. 2A). Similar synergistic effects were
observed in other TRAIL-sensitive SK-MEL-2 cells as well as in
TRAIL-resistant A2058 cells (Fig. 2B
and C). As shown in Fig. 2D,
K+ alone caused minimal apoptosis but significantly
enhanced TRAIL-induced apoptosis. These data show that
K+ loading sensitizes melanoma cells to TRAIL-induced
apoptosis.
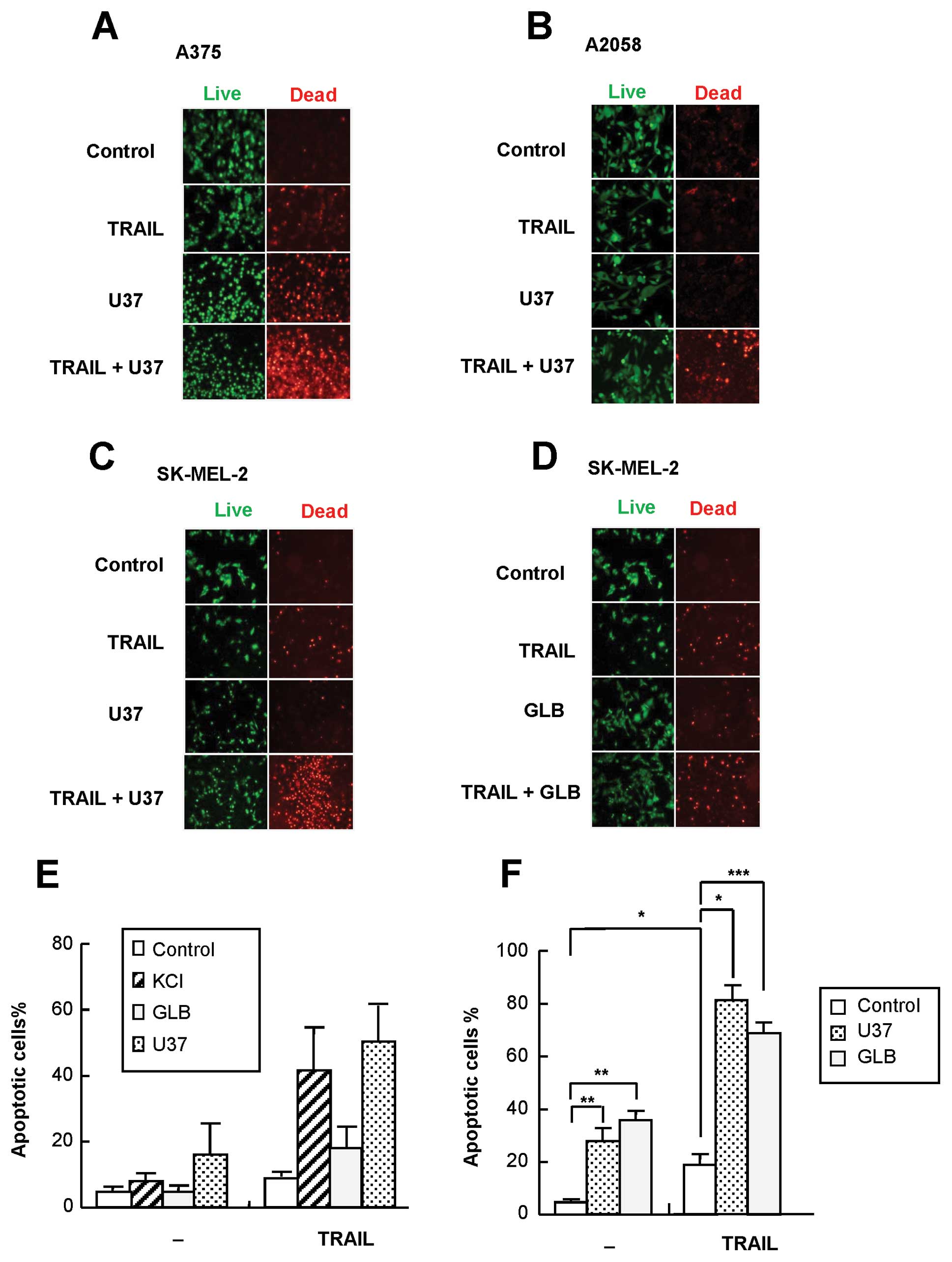
KATP channel inhibitors
specifically sensitize melanoma cells to TRAIL-induced
apoptosis
Since K+ efflux through K+
channels results in repolarization, blockade of the K+
efflux is necessary for persistent depolarization. Such a blockade
can be pharmacologically achieved by inhibition of channels such as
KATP(9). Therefore, we
next examined the effect of KATP channel-specific
inhibitors U37 and GLB on TRAIL cytotoxicity by fluorescence
microscopy. After 24 h of treatment, U37 alone showed substantial
cytotoxicity toward several melanoma cell lines and significantly
enhanced TRAIL cytotoxicity toward all cell lines tested, including
A375, A2058 and SK-MEL-2 (Fig.
3A–C). On the contrary, GLB had a marginal effect on TRAIL
cytotoxicity in SK-MEL-2 cells (Fig.
3D) and A375 cells (data not shown). In agreement with these
observations, U37, but not GLB, caused TRAIL-induced apoptosis at
that time (Fig. 3E). On the other
hand, after 72 h of treatment, U37 and GLB alone enhanced robust
apoptosis. Necrotic/damaged (annexin V−/PI+)
cells often increased significantly. In addition, both agents
significantly enhanced TRAIL-induced apoptosis (Fig. 3F). To determine whether such effect
was specific for KATP channel inhibitors, inhibitors of
other types of K+ channels were examined for their
effects on TRAIL-induced apoptosis. The Kv-specific
inhibitor DTX and KCa-specific inhibitor CTX had minimal
effects on the apoptosis up to 72 h (Fig. 4). The mitochondrial KATP
inhibitor 5-HD had no effect either (Fig. 4), suggesting that plasma membrane
KATP is specifically involved in the sensitization.
Moreover, TEA, which mainly inhibits Kv and
KCa, was also ineffective (data not shown). U37 was as
effective as 100 ng/ml TRAIL in causing depolarization, whereas GLB
had a smaller effect (maximum of 1.3-fold). On the other hand, TEA
caused no significant depolarization. Taken together, these data
show that KATP channel inhibitors specifically sensitize
melanoma cells to TRAIL-induced apoptosis.
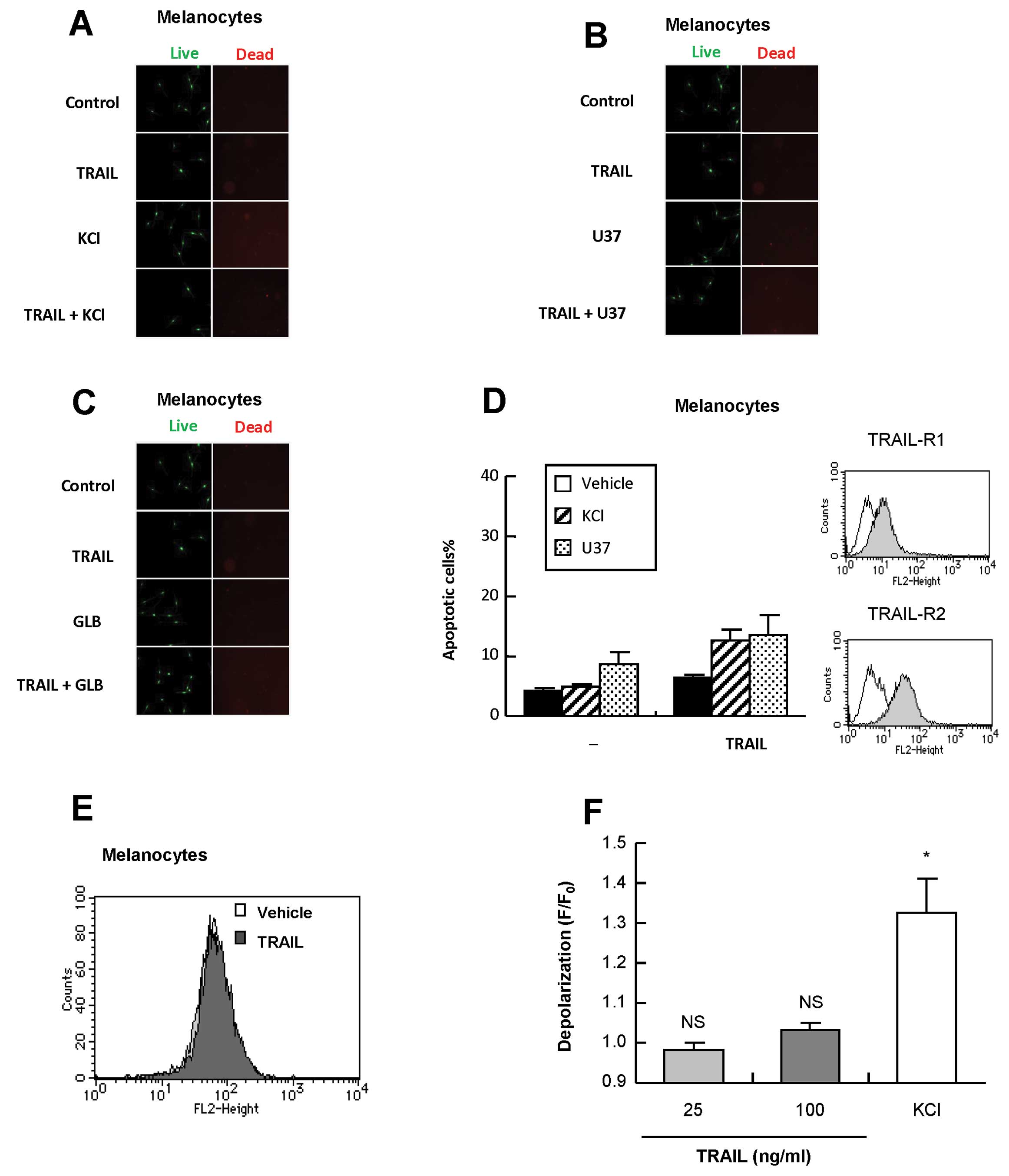
Melanocytes are insensitive to
TRAIL-induced depolarization and apoptosis and to sensitization by
membrane-depolarizing agents
TRAIL has been shown to induce apoptosis in cancer
cells with minimal cytotoxicity in non-transformed cells.
Therefore, we determined whether the membrane-depolarizing agents
affected the tumor-selectivity. Treatment with melanocytes with
TRAIL, K+, U37 and GLB alone or in combination resulted
in minimal apoptosis and cytotoxicity (Fig. 5A–D), although they expressed
substantial levels of DR4 and DR5 on the cell surface (Fig. 5D). TRAIL induced minimal
depolarization in melanocytes (Fig. 5E
and F). On the other hand, K+ loading resulted in
substantial depolarization in the cells, which was comparable to
that observed in A375 cells. Taken together, these data show that
despite their substantial expressions of DRs, melanocytes are
insensitive to TRAIL-induced depolarization and apoptosis as well
as to sensitization by the membrane-depolarizing agents.
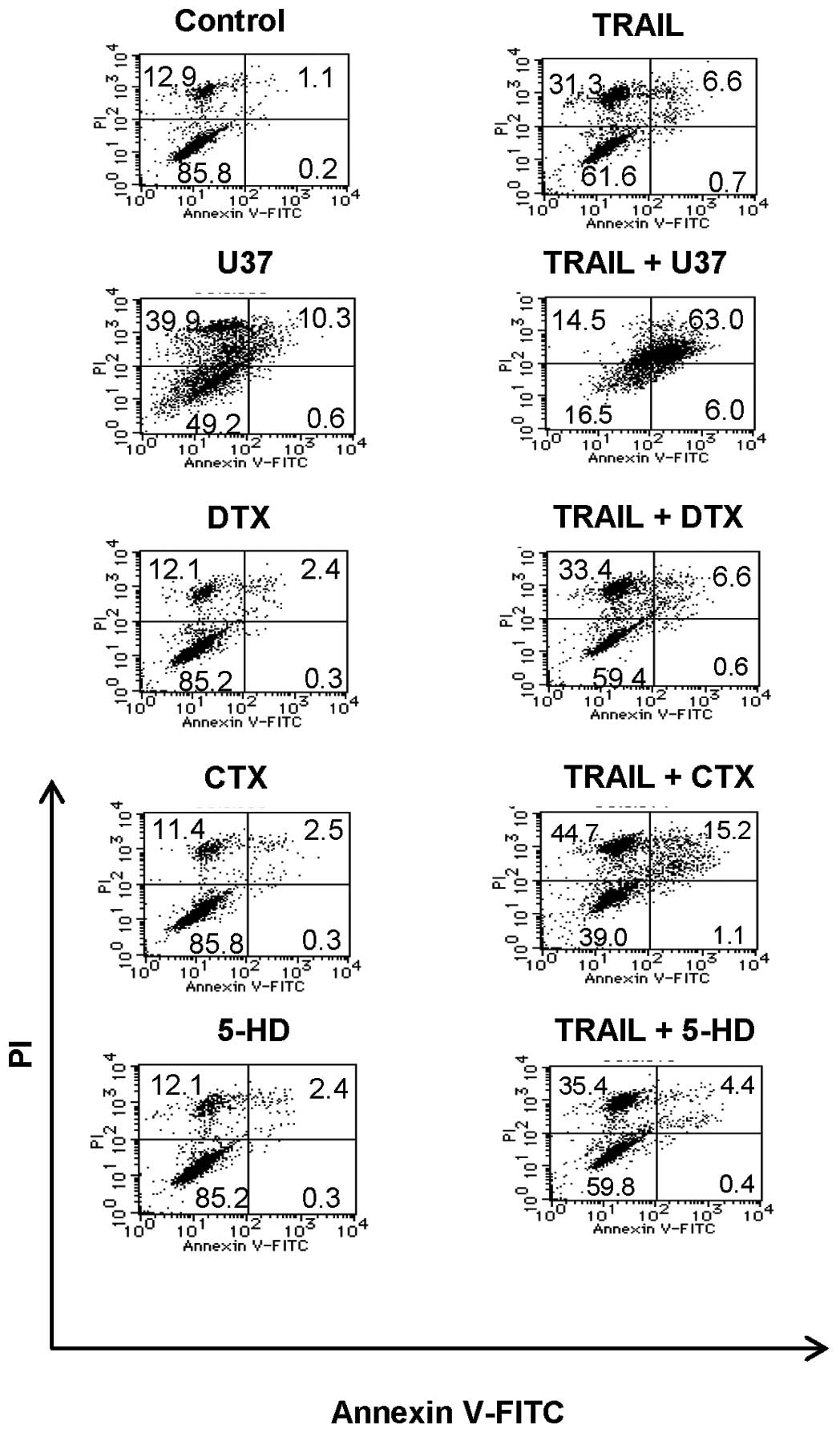
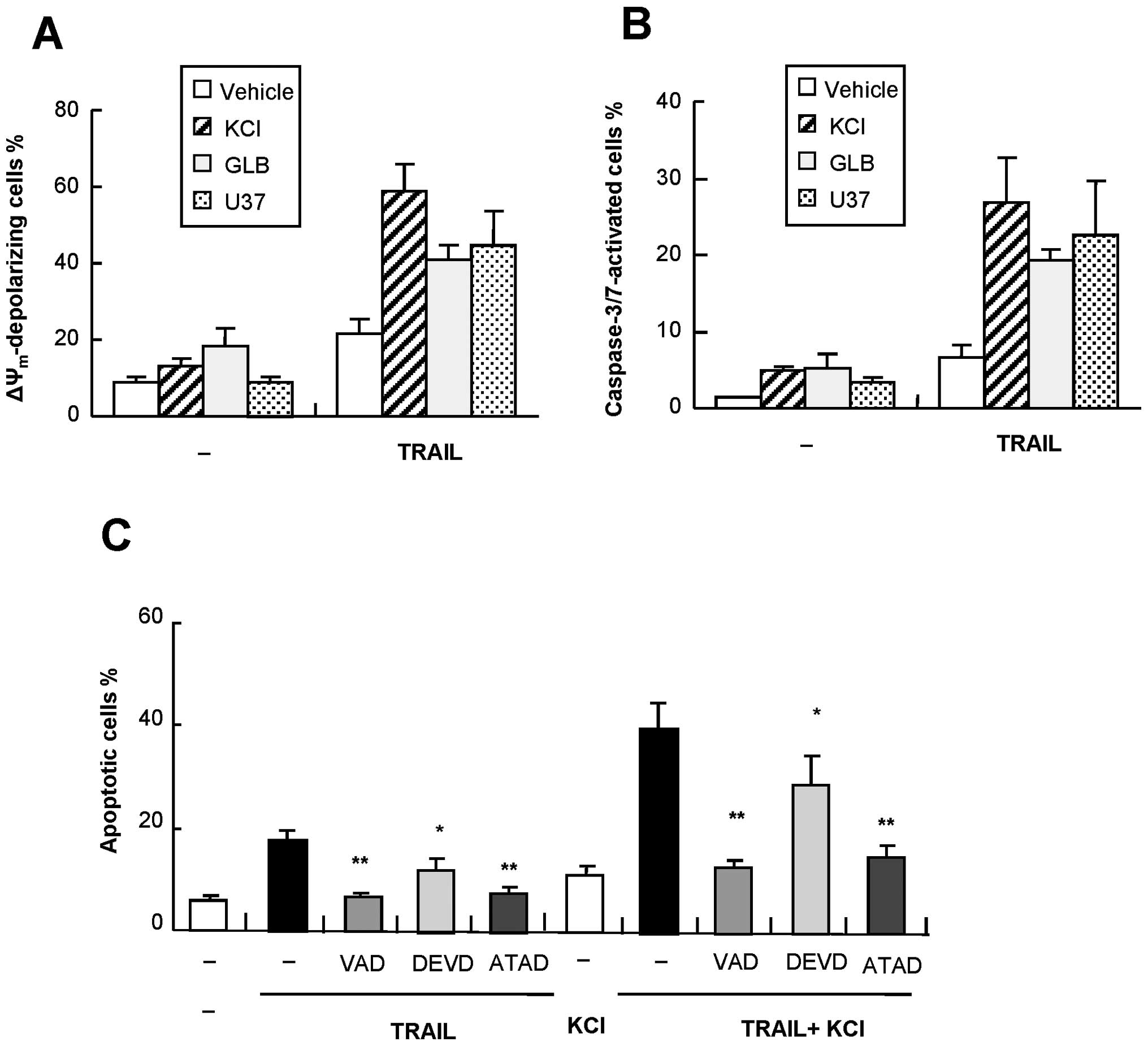
The amplification of apoptosis is
associated with increased ΔΨm collapse and caspase-3/7
activation
Flow cytometric analysis using specific fluorescent
probes revealed that TRAIL-induced apoptosis was accompanied by a
loss of ΔΨm and activation of caspase-3/7, both of which
were augmented by K+, U37, and GLB (Fig. 6A and B). The ΔΨm
depolarization was confirmed by another probe JC-1 (data not
shown). The data showed that despite its weak effect on
TRAIL-induced apoptosis during the initial 24 h, GLB can enhance
the ΔΨm collapse and caspase-3/7 activation, suggesting
that the intrinsic apoptotic pathway is involved in, but is not
sufficient for, the amplification of apoptosis. To explore the role
of caspase-3/7 further, we examined the effect of the
caspase-3/7-specific inhibitor z-DEVD-fmk on apoptosis. As shown in
Fig. 6C, the general caspase
inhibitor VAD completely blocked TRAIL-induced apoptosis. The
caspase-12-specific inhibitor ATAD exhibited similar effects,
whereas DEVD inhibited the apoptosis marginally. On the other hand,
ATAD and to a lesser extent DEVD reduced the K+-mediated
amplification of apoptosis (Fig.
6C), suggesting that caspase-12 as well as caspase-3 play a
role in the amplification of apoptosis.
Role of ER stress and caspase-12 in the
amplification of apoptosis
Caspase-12 is ubiquitously expressed and localized
to the ER membrane, and is specifically activated by ER stress to
play a key role in stress-induced apoptosis (23). Therefore, we hypothesized that the
ER stress-mediated apoptotic pathway, including caspase-12, was
involved in the amplification of TRAIL-induced apoptosis. To test
this hypothesis, we analyzed the unfolded protein response (UPR), a
cellular response to ER stress. A375 cells were treated with 25 and
100 ng/ml TRAIL for 24 h and their expression of two UPR proteins,
GRP78 and XBP1 were analysed by western blotting. Tg, an inhibitor
of sarco/endoplasmic reticulum Ca2+-ATPase, was shown to
be a potent inducer of GRP78 expression in human melanoma cells
(22) and served as a positive
control. Tg treatment caused a robust increase in the expression of
GRP78, while TRAIL treatment had a minimal effect on or often
decreased this expression (Fig.
7A). On the other hand, TRAIL dose-dependently increased the
expression of both the inactive unspliced form of XBP-1 (XBP-1u)
and the active spliced form of XBP-1 (XBP-1s) (maximum of 1.8- and
2.1-fold, respectively), indicating the activation of XBP-1. Then,
we analyzed the effect of membrane-depolarizing agents on
TRAIL-induced UPR response. K+ and U37 synergized with
TRAIL to increase the expression of both forms of XBP-1, whereas
GLB and TEA had minimal effects (Fig.
7B). Next, we examined whether TRAIL induced activation of
caspase-12 and whether membrane-depolarizing agents impacted the
effect. The activation of caspase-12 was initially evaluated by
detecting its processing by western blotting. As shown in Fig. 7B, K+ and U37, but not
GLB or TEA, synergistically induced the processing of caspase-12, a
hallmark of its activation. The functional activation of caspase-12
was assessed by measuring the conversion of a cell-permeable
substrate, FITC-ATAD-fmk. TRAIL induced the activation of
caspase-12 in a dose-dependent manner at concentrations that
induced apoptosis (Fig. 7C). In
addition, K+ and U37 significantly enhanced the effect
(Fig. 7D), whereas other
K+ channel inhibitors had minimal effects even after 72
h of treatment (Fig. 7E). Taken
together, these data show that the ER stress-mediated death pathway
involving caspase-12 plays an important role in the amplification
of TRAIL-induced apoptosis.
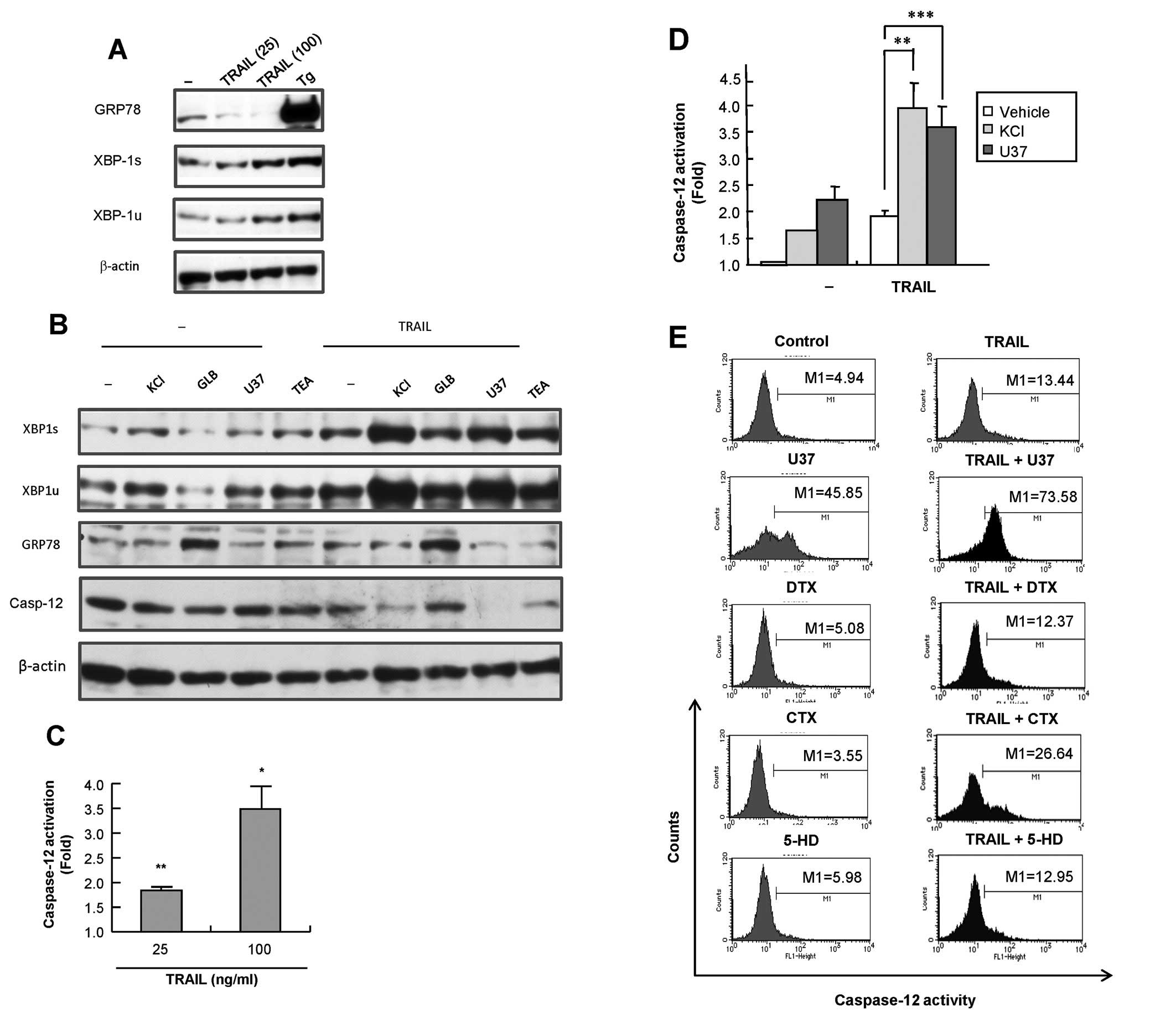 | Figure 7TRAIL induces ER stress and
caspase-12 activation and the membrane-depolarizing agents
potentiate these effects. (A) A375 cells were treated with 25 and
100 ng/ml TRAIL or 1 μM Tg for 24 h. (B) A375 cells were
treated with TRAIL and KCl, U37883A (U37), glibenclamide (GLB) and
TEA alone or in combination for 24 h. The cells were then washed,
lysed with SDS-sample buffer and analyzed for their contents of
GRP78, XBP-1 and/or full-length caspase-12 by western blot analysis
with specific antibodies. To verify equal loading, the blots were
re-probed with an anti-β-actin antibody. The data are
representative of three independent experiments. (C,D) A375 cells
were treated with TRAIL (C) or TRAIL, KCl and U37883A (U37) alone
or in combination for 24 h and caspase-12 activity was measured
using FITC-ATAD-fmk. The data are shown as ratios to the basal
activity and represent the means ± SE from three independent
experiments. *P<0.05; **P<0.01;
***P<0.001. (E) A375 cells were treated with TRAIL
and U37883A (U37), α-dendrotoxin (DTX), charybdotoxin (CTX) and
5-hydroxydecanoate (5-HD) alone or in combination for 72 h, and
caspase-12 activity was measured by flow cytometry using
FITC-ATAD-fmk. The data are representative of two independent
experiments. |
Discussion
The data presented in this paper show that TRAIL
induces robust depolarization of the plasma membrane potential in
human melanoma cells and that membrane-depolarizing agents such as
K+ and KATP channel-selective inhibitors
potentiate TRAIL-induced apoptosis. KATP channels by
themselves caused apoptosis under a long-time treatment, suggesting
that persistent depolarization alone trigger apoptosis.
KATP channels appeared to play a specific role in the
regulation of apoptosis, since inhibitors of other types of
K+ channels, including mitochondrial KATP,
Kv and KCa channels exhibited no effects on
apoptosis. Of note, TRAIL caused minimal membrane depolarization
and apoptosis in melanocytes despite their substantial expression
of DR4 and DR5, and the cells showed unresponsiveness to
KATP channel inhibitors. K+ caused robust
depolarization in melanocytes, indicating depolarization
machineries are intact. Thus, our data show that human melanoma
cells are far more susceptible than melanocytes to the
KATP channel-regulated apoptotic pathway.
The intrinsic mitochondrial pathway plays a crucial
role in amplifying TRAIL-induced apoptosis, and collapse of the
ΔΨm is considered to be a hallmark of this pathway,
although it is still a matter of debate whether this event is a
cause or a result of permeabilization of the outer mitochondrial
membrane (23–25). TRAIL induced both the
ΔΨm collapse and the caspase-3/7 activation in melanoma
cells, and the sensitization of TRAIL-induced apoptosis was
associated with their enhancement, indicating the involvement of
the intrinsic pathway. However, this pathway appeared not to be
sufficient for the sensitization, due to the fact that GLB
upregulated both events but had minimal effects on TRAIL-induced
apoptosis during the initial 24 h. On the other hand, K+
and U37 rapidly sensitized melanoma cells to TRAIL-induced
apoptosis within 24 h. Therefore, it is possible to speculate that
another amplification pathway, which is activated by K+
and U37, but not GLB, may be required for the rapid sensitization.
Consistent with this view, we found that TRAIL-induced apoptosis
involved activation of caspase-12, which was enhanced by
K+ and U37, but not GLB. Caspase-12 has been shown to be
predominantly located at the outer membrane of the ER, to be
specifically activated by ER stress-inducing agents and to play an
important role in the apoptosis induced by ER stress (19,20,26,27).
The emerging view is that, besides mitochondria, the ER is another
key player in the regulation of apoptosis induced by a variety of
death stimuli, including TRAIL. Disparate perturbations in its
normal functions, such as the accumulation of unfolded or misfolded
proteins, ER lipid imbalances or changes in the redox balance or
Ca2+ conditions in the ER lumen, trigger ER stress
(20,26–28).
The cells then activate ER stress responses, including the UPR, to
alleviate the stress, but an excessive and prolonged UPR leads to
apoptosis (27–29). The UPR involves
transcription-dependent upregulation of ER-resident chaperones, and
the ER chaperone GRP78 plays a central role in this response. Upon
ER stress, GRP78 dissociates from ER transmembrane proteins, such
as inositol requiring 1 and activating transcription factor 6, to
bind to unfolded or misfolded proteins, resulting in aggregation of
the transmembrane proteins and their activation. Activated inositol
requiring 1 splices the mRNA for XBP-1 to allow translation of the
mature XBP-1 protein, which acts as a transcription factor and
mediates the transcriptional upregulation of numerous genes
involved in ER function (30).
Consistent with the role of ER stress in TRAIL-induced apoptosis
and the sensitization, we found that TRAIL and U37 induced
activation of XBP-1. Collectively, our data strongly suggest that
ER stress-induced activation of caspase-12 is involved in
TRAIL-induced apoptosis and sensitization. The human caspase-12
homologue exists on chromosome 11 and is 68% homology with murine
caspase-12 and shows 57% homology with human caspase-4 (31). Owing to multiple mutations, this
gene does not seem to be expressed in a functional form in humans,
and therefore the role of caspase-12 in the induction of apoptosis
in human cells is controversial (27,31).
However, there are multiple possibilities that human cells express
caspase-12-like activity, such as (i) a different locus in the
human genome carries a functional caspase-12 gene, (ii) its
function is carried out by other caspases; and (iii) mRNA editing
or other mechanisms can override frame-shift mutations (32). Hence further studies are needed to
clarify the role of caspase-12 in ER stress-induced apoptosis. In
support of this view, there is an increasing body of evidence
suggesting that a caspase-12-like protein exists and is activated
in human cells following the induction of ER stress by divergent
causes, including cisplatin, tetrocarcin A and hyperthermia
(32–36). In the present study, we have
demonstrated the existence of full-length caspase-12 in human
melanoma cells and its processing coincided with the activation of
caspase-12 activity. These findings provide further evidence for a
role of caspase-12 in ER stress-induced apoptosis. It should be
noted that caspase-12 is directly activated by the
Ca2+-dependent protease calpain. Upon apoptotic
stimulation, calpain cleaves the regulatory prodomain, thereby
activating caspase-12, and the activated caspase-12 directly
cleaves caspase-9 for its activation without the need for release
of cytochrome c and apoptotic protease-activating factor-1, which
in turn activates caspase-3 (26,37,38).
Accordingly, caspase-12 can be activated before caspase-9 and
caspase-3/7.
Similar to caspase-12 in rodents, caspase-4 in
humans has been shown to be predominantly located on the outer
membrane of the ER and to play an important role in ER
stress-induced apoptosis (20,30).
Caspase-4 was recently shown to be involved in TRAIL-induced
apoptosis of human melanoma cells. However, inhibition of caspase-4
partially, but not completely, blocked apoptosis (39), suggesting that its role is limited.
Since caspase-4 has been shown to be activated later than
caspase-8, caspase-9 and caspase-3 (39), it is possible that caspase-4 and
caspase-12 may play roles in different processes/stages in ER
stress-induced apoptosis. Our preliminary attempt to establish
caspase-12 gene knockdown cells was hampered by considerable cell
death in the resting state. On the other hand, the caspase-12
inhibitor had a minimal effect on their survival. These
observations suggest that caspase-12 may have an important function
in human melanoma survival that is independent of its enzyme
activity. Further studies are needed to fully elucidate the role of
caspase-12 in the TRAIL-induced apoptosis of human tumor cells.
In conclusion, we have demonstrated for the first
time the pro-apoptotic role of depolarization in TRAIL-induced
apoptosis via ER stress. Since melanoma cells are far more
susceptible to the depolarization-mediated apoptosis than
melanocytes, membrane-depolarizing drugs such as KATP
channel inhibitors may exhibit cytotoxicity in a tumor-selective
manner and have therapeutic potential in the treatment of
TRAIL-resistant melanoma cells.
Acknowledgements
The authors thank Mr. S. Okumura and
Dr H. Nagase for technical assistance and providing human melanoma
cell lines, respectively. This work was supported in part by a
Grant-in-Aid from the Ministry of Education, Culture, Sports,
Science and Technology (KAKENHI 23591631; to Y.S.) and
Grants-in-Aid from Nihon University (to Y.S.).
References
|
1
|
LeBlanc HN and Ashkenazi A: Apo2L/TRAIL
and its death and decoy receptors. Cell Death Differ. 10:66–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kischkel FC, Lawrence DA, Chuntharapai A,
et al: Apo2L/TRAIL-dependent recruitment of endogenous FADD and
caspase-8 to death receptors 4 and 5. Immunity. 12:612–620. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lavrik IN, Golks A and Krammer PH:
Caspases: pharmacological manipulation of cell death. J Clin
Invest. 115:2665–2672. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yan N and Shi Y: Mechanisms of apoptosis
through structural biology. Annu Rev Cell Dev Biol. 21:35–56. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dyer MJ and MacFarlane M: Cohen GM.
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol. 25:4505–4506. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McCarthy JV and Cotter TG: Cell shrinkage
and apoptosis: a role for potassium and sodium ion efflux. Cell
Death Differ. 4:756–770. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lang F, Föller M, Lang K, et al: Cell
volume regulatory ion channels in cell proliferation and cell
death. Methods Enzymol. 428:209–225. 2007.PubMed/NCBI
|
|
9
|
Burg ED, Remillard CV and Yuan JX:
Potassium channels in the regulation of pulmonary artery smooth
muscle cell proliferation and apoptosis: pharmacotherapeutic
implications. Br J Pharmacol. 153:S99–S111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bortner CD, Gomez-Angelats M and Cidlowski
JA: Plasma membrane depolarization without repolarization is an
early molecular event in anti-Fas-induced apoptosis. J Biol Chem.
276:4304–4314. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yin W, Li X, Feng S, et al: Plasma
membrane depolarization and Na, K-ATPase impairment induced by
mitochondrial toxins augment leukemia cell apoptosis via a novel
mitochondrial amplification mechanism. Biochem Pharmacol.
78:191–202. 2009. View Article : Google Scholar
|
|
12
|
Nolte F, Friedrich O, Rojewski M, Fink RH,
Schrezenmeier H and Körper S: Depolarisation of the plasma membrane
in the arsenic trioxide (As2O3)-and
anti-CD95-induced apoptosis in myeloid cells. FEBS Lett. 578:85–89.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ghoumari AM, Piochon C, Tomkiewicz C, et
al: Neuroprotective effect of mifepristone involves neuron
depolarization. FASEB J. 20:1377–1386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Karki P, Seong C, Kim JE, et al:
Intracellular K+ inhibits apoptosis by suppressing the
Apaf-1 apoptosome formation and subsequent downstream pathways but
not cytochrome c release. Cell Death Differ. 4:2068–2075. 2007.
|
|
15
|
Yoshimaru T, Suzuki Y, Inoue T and Ra C:
L-type Ca2+ channels in mast cells: activation by
membrane depolarization and distinct roles in regulating mediator
release from store-operated Ca2+ channels. Mol Immunol.
46:1267–1277. 2009.
|
|
16
|
Suzuki Y, Yoshimaru T, Inoue T and Ra C:
Cav 1.2 L-type Ca2+ channel protects mast cells against
activation-induced cell death by preventing mitochondrial integrity
disruption. Mol Immunol. 46:2370–2380. 2009.
|
|
17
|
Suzuki Y, Yoshimaru T, Inoue T and Ra C:
Mitochondrial Ca2+ flux is a critical determinant of the
Ca2+ dependence of mast cell degranulation. J Leukoc
Biol. 79:508–518. 2006.
|
|
18
|
Inoue T, Suzuki Y, Yoshimaru T and Ra C:
Nitric oxide protects mast cells from activation-induced cell
death: the role of the phosphatidylinositol-3
kinase-Akt-endothelial nitric oxide synthase pathway. J Leukoc
Biol. 83:1218–1229. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boyce M and Yuan J: Cellular response to
endoplasmic reticulum stress: a matter of life or death. Cell Death
Differ. 13:363–373. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Breckenridge DG, Germain M, Mathai JP,
Nguyen M and Shore GC: Regulation of apoptosis by endoplasmic
reticulum pathways. Oncogene. 22:8608–8618. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen LH, Jiang CC, Kiejda KA, et al:
Thapsigargin sensitizes human melanoma cells to TRAIL-induced
apoptosis by up-regulation of TRAIL-R2 through the unfolded protein
response. Carcinogenesis. 28:2328–2336. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai J, Yang J and Jones DP: Mitochondrial
control of apoptosis: the role of cytochrome c. Biochim Biophys
Acta. 1783:924–934. 1998.
|
|
24
|
Düssmann H, Rehm M, Kögel D and Prehn JH:
Outer mitochondrial membrane permeabilization during apoptosis
triggers caspase-independent mitochondrial and caspase-dependent
plasma membrane potential depolarization: a single-cell analysis. J
Cell Sci. 116:525–536. 2003.
|
|
25
|
Adachi S, Cross AR, Babior BM and Gottlieb
RA: Bcl-2 and the outer mitochondrial membrane in the inactivation
of cytochrome c during Fas-mediated apoptosis. J Biol Chem.
272:21878–21882. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Groenendyk J and Michalak M: Endoplasmic
reticulum quality control and apoptosis. Acta Biochim Pol.
52:381–395. 2005.PubMed/NCBI
|
|
27
|
Szegezdi E, Fitzgerald U and Samali A:
Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann
NY Acad Sci. 1010:186–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rutkowski DT and Kaufman RJ: A trip to the
ER: coping with stress. Trends Cell Biol. 14:20–28. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ferri KF and Kroemer G: Organelle-specific
initiation of cell death pathways. Nat Cell Biol. 3:E255–E263.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiang CC, Chen LH, Gillespie S, et al:
Tunicamycin sensitizes human melanoma cells to tumor necrosis
factor-related apoptosis-inducing ligand-induced apoptosis by
up-regulation of TRAIL-R2 via the unfolded protein response. Cancer
Res. 67:5880–5888. 2007. View Article : Google Scholar
|
|
31
|
Fischer H, Koenig U, Eckhart L and
Tschachler E: Human caspase 12 has acquired deleterious mutations.
Biochem Biophys Res Commun. 293:722–726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mandic A, Hansson J, Linder S and Shoshan
MC: Cisplatin induces endoplasmic reticulum stress and
nucleus-independent apoptotic signaling. J Biol Chem.
278:9100–9106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tinhofer I, Anether G, Senfter M, et al:
Stressful death of T-ALL tumor cells after treatment with the
anti-tumor agent Tetrocarcin-A. FASEB J. 16:1295–1297.
2002.PubMed/NCBI
|
|
34
|
Xie Q, Khaoustov VI, Chung CC, et al:
Effect of tauroursodeoxycholic acid on endoplasmic reticulum
stress-induced caspase-12 activation. Hepatology. 36:592–601. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Trisciuoglio D, Uranchimeg B, Cardellina
JH, et al: Induction of apoptosis in human cancer cells by
candidaspongiolide, a novel sponge polyketide. J Natl Cancer Inst.
100:1233–1246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shellman Y, Howe WR, Miller LA, et al:
Hyperthermia induces endoplasmic reticulum-mediated apoptosis in
melanoma and non-melanoma skin cancer cells. J Invest Dermatol.
128:949–956. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rao RV, Castro-Obregon S, Frankowski H, et
al: Coupling endoplasmic reticulum stress to the cell death
program. An Apaf-1-independent intrinsic pathway. J Biol Chem.
277:21836–21842. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Morishima N, Nakanishi K, Takenouchi H,
Shibata T and Yasuhiko Y: An endoplasmic reticulum stress-specific
caspase cascade in apoptosis. Cytochrome c-independent activation
of caspase-9 by caspase-12. J Biol Chem. 277:34287–34294. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mao ZG, Jiang CC, Yang F, Thorne RF,
Hersey P and Zhang XD: TRAIL-induced apoptosis of human melanoma
cells involves activation of caspase-4. Apoptosis. 15:1211–1222.
2010. View Article : Google Scholar : PubMed/NCBI
|