Introduction
The transcriptional inactivation of genes through
CpG islands hypermethylation at their promoters is a widely
reported mechanism in human malignancies, including colorectal
cancer (CRC) (1,2). Reduced expression of genes, in
particular the tumor suppressor genes, due to epigenetic masking
can contribute to cancerous transformation by inducing selective
growth advantages (3).
Microarray analysis can lead to the identification
of changes in gene expression after the treatment of cells with
5-aza-2′-deoxycitidine (5-aza-dC), a methyltransferase inhibitor
that can cause induction of epigenetically silenced genes (4–7).
Profiling of methylated genes is growing rapidly as
a powerful diagnostic tool for the early detection, prognosis and
even prediction of clinical response to treatment of various cancer
types (8). This encourages the
search for new candidate genes using global demethylation and
microarray analysis that could effectively find genes silenced in
association with promoter hypermethylation in several tumor types,
including CRC, with a role in cell proliferation, tumor
progression, apoptosis or angiogenesis (8–10).
In CRC, the path of epigenetic silencing has not yet been fully
explored and further searches for methylation-silenced genes are
required. Despite the fact that hundreds of genes have been
identified to be silenced through promoter CpG island methylation
in CRC (11).
In this study, genome-wide demethylation and
expression microarray screening were carried out to discover
putative genes inactivated by methylation in CRC.
Materials and methods
Cell lines and 5-aza-dC treatment
Cell lines used in this study were obtained from the
Cell Resource Center for Biomedical Research, Tohoku University
(Miyagi, Japan) and the American Type Culture Collection (Manassas,
VA, USA). Fifteen CRC cell lines (CCK81, CoCM1, Colo205, Colo320,
ColoTC, DLD1, HCT116, HCT15, HT29, LoVo, RCM1, RKO, SW48, SW480 and
WiDr) were cultured in appropriate medium and under conditions
described by the providers, with media obtained from Gibco (Grand
Island, NY, USA) or Sigma (St. Louis, MO, USA), supplemented with
10% heat-inactivated fetal bovine serum (FBS; Nichirei, Tokyo,
Japan), 100 μg/ml streptomycin, and 100 U/ml penicillin
(Invitrogen, Carlsbad, CA, USA).
HCT116, RKO, Colo320, SW480 and HT29 CRC cell lines
were treated with 5-aza-dC, each experiment was performed in
triplicate. The five cell lines were cultured at 37°C in 5%
CO2. Cells (5×103 per well) were plated in
their respective culture media on day 1, on day 3 cultured cells
were treated with PBS dissolved and filter-sterilized 0.5 μM
5-aza-dC (Sigma) for 72 h with media changed every 24 h, treatments
were performed in parallel with drug-free phosphate-buffered saline
(PBS) as a control. The dose of 5-aza-dC was administered on the
basis of its pharmacological dose and the results of our
preliminary experiments (12). On
day 6, cells were harvested following incubation with trypsin-EDTA
then stained with trypan blue and counted. For the analysis of mRNA
expression total RNA was extracted using QIAshreder with RNeasy
minikit (Qiagen, Hilden, Germany). Genomic DNA (gDNA) was extracted
by proteinase K (Invitrogen) digestion, phenol/chloroform
extraction and ethanol precipitation method for methylation
analysis.
Clinical samples
Paired samples from 23 patients, who underwent
surgical treatment for CRC in 2007 at Tokyo Medical and Dental
University Hospital were included in this study. All specimens
(macroscopically resected) were stored at −80°C until further use.
Mean age was 66.3±10.9 years (median 67.0 years; range 38–81 years)
and male-to-female ratio was 1.3:1. The number of cases for stages
I, II, III and IV was six, six, five and six, respectively. Written
informed consent was obtained from all patients and the
Institutional Review Board at Tokyo Medical and Dental University
approved the study. All the samples were used in methylation
analysis, of them 14 assigned to gene expression study.
Oligonucleotide microarray analysis
RNA was extracted from cell lines before and after
treatment with 5-aza-dC. Contaminant DNA was removed by digestion
with RNase-free DNase (Qiagen). The integrity of the total RNA
obtained was assessed using an Agilent 2100 BioAnalyzer (Agilent
Technologies, Palo Alto, CA, USA). All samples had an RNA integrity
number of ≥5.0 prior to gene expression analysis. Complementary RNA
(cRNA) was prepared from 2 μg total RNA using one-cycle
target labeling and a control reagents kit (Affymetrix, Santa
Clara, CA, USA). Hybridization and signal detection of the Human
Genome (HG) U133 Plus 2.0 array (Affymetrix) were performed
according to the manufacturer’s protocol.
The gene expression data sets were submitted to Gene
Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, accession no.
GSE32323). To investigate the difference in expression patterns,
the raw gene expression data were derived from each probe signal
intensity (CEL file) using the model-based robust multi-array
average method algorithm as implemented in the Affymetrix
Expression Console™ software (version 1.1). Expression levels were
log2-transformed, and 62 control probe sets were removed
from subsequent analyses. R 2.11.1 statistical software together
with a Bioconductor package (R Foundation for Statistical
Computing, Vienna, Austria) was used to calculate fold-change (FC)
values for each of the 54,613 probes on the HG-U133 Plus 2.0 array.
FC ≥1.2 in all cell lines was selected as the threshold for
up-regulation of gene expression. Genes with an FC difference
greater than threshold in all of the five cell lines with FC
>1.5 in at least one cell line were selected.
DNA extraction and methylation-specific
PCR (MSP)
gDNA was extracted using phenol/chloroform method.
Bisulfite treatment was performed using EpiTect Bisulfite kit
(Qiagen) according to the manufacturer’s instructions. Sodium
bisulfite treatment of gDNA converts unmethylated cytosine residues
(but not methylated cytosine) to uracil, which is then converted to
thymidine during the subsequent PCR step, giving sequence
differences between methylated and unmethylated DNA.
The methylation status of the cyclin-dependent
kinase inhibitor 2A (CDKN2A/p14ARF),
cyclin-dependent kinase inhibitor 2A
(CDKN2A/p16INK4A), growth arrest and
DNA-damage-inducible beta (GADD45B), protein-tyrosine
phosphatase receptor type O gene (PTPRO) and thrombospondin,
type I, domain containing 1 (THSD1) genes was determined by
methylation-specific polymerase chain reaction (MSP) with 1
μl of bisulfite-treated DNA as template and AmpliTaq Gold
360 Master Mix (Applied Biosystems, Foster City, CA, USA) for
amplification, as previously described (13). Bisulfite-converted gDNA, methylated
and unmethylated human control DNA (EpiTect control DNA, Qiagen)
was used as a positive control for the methylated and unmethylated
experiments, respectively.
Methylated and unmethylated primer sequences (Life
Technologies Inc., Rockville, MD, USA), amplicon lengths and PCR
conditions have been previously reported (12,14–16).
None of the primer sets amplified non-bisulfite-treated gDNA (data
not shown), this eliminate the possibility of PCR product
amplification from unconverted DNA. All reactions were performed in
duplicate and PCR products were loaded onto a 2.0% agarose gel,
stained with 0.5 μg/ml ethidium bromide and visualized under
ultraviolet illumination.
PTPRO gene was included as a positive control
for target gene methylation analysis and verified the integrity of
gDNA used in this study (i.e., showed unmethylated bands in both
normal and tumor tissues) (17).
Methylation status of p14ARF and
p16INK4A gene was used to confirm the efficient
demethylation of CpG-dinucleotides in the treated cell lines
(17).
Quantitative real-time RT-PCR
(qRT-PCR)
Total RNA was extracted from primary CRC and
adjacent normal tissues (14 pairs) using RNeasy minikit (Qiagen)
and from 14 colon cancer cell lines. Total RNA (10 μg) was
reverse-transcribed into complementary DNA (cDNA) samples using
High Capacity cDNA Reverse Transcription Kit (Applied Biosystems)
according to the manufacturer’s protocol. TaqMan gene expression
assays (Applied Bioystems: THSD1, Hs00938785_m1, and
β-actin, Hs99999903_m1) were used to determine the expression of
THSD1. β-actin was used as an internal control. The PCR
reaction was carried out with the TaqMan Universal PCR Master Mix
(Applied Biosystems). Thermal cycling conditions (7300 ABI PRISM,
TaqMan; Applied Biosystems) were as follows: 50°C for 2 min, 95°C
for 10 min, 40 cycles of 15 sec denaturation at 95°C and 1 min
annealing at 60°C. All calculated concentrations of target genes
were normalized by the amount of the endogenous reference with the
comparative Ct method for relative quantification (ΔΔCt method)
using Relative Quantification Study software (7300 Sequence
Detection System version 1.2.1, Applied Biosystems). Each assay was
performed in 20 μl including 1 μl of cDNA and all
assays were done in duplicate.
Statistical analysis
Differences between groups were estimated using; the
χ2 test, Fisher’s exact test, Mann-Whitney U-test and
Wilcoxon signed ranks test, where appropriate. A probability level
of 0.05 was used for statistical significance. Statistical analyses
of gene expression were performed with SPSS (version 17.0, SPSS
Inc., Chicago, IL, USA) for Windows software.
Results
Identification of 20 candidate genes
up-regulated in 5-aza-dC-treated CRC cell lines
To find hypermethylated genes associated with CRC,
microarray analysis comparison of the mock and 5-aza-dC treatment
expression patterns in five colon cancer cell lines (HCT116, RKO,
Colo320, SW480 and HT29) was done, this identified the reactivated
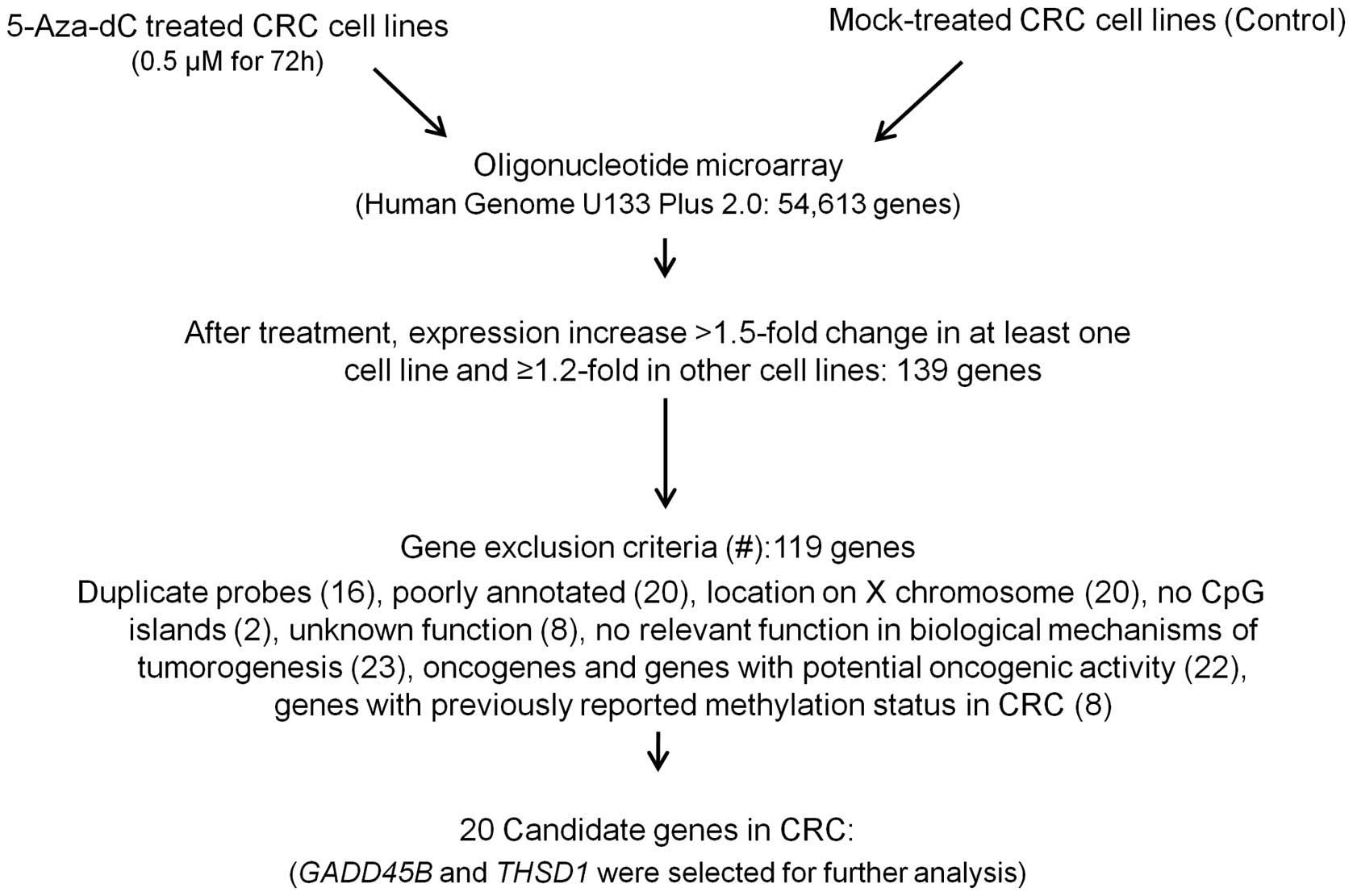
genes expression profiles (method summarized in Fig. 1). Comparison of the resultant gene
expression profiles revealed 139 genes that had FC >1.5 in
signal intensities in at least one cell line with ≥1.2-fold change
in the remaining four 5-aza-dC-treated cell lines collectively. Of
note, this group of up-regulated transcripts contains several genes
of cancer-germline antigen families (i.e., GAGE, MAGE, PAGE
and XAGE), which is consistent with previous reports
(18). Also the functional
analysis by DAVID bioinformatics resource [(19); http://david.niaid.nih.gov] showed the significant
enrichment (FDR <0.05%) of genes located on X chromosome (20
genes). In addition, the reactivation of gene transcripts known to
be hypermethylated in CRC was also detected including DDB1 and CUL4
associated factor 4-like 1 (DCAF4L1), DEAD (Asp-Glu-Ala-Asp)
box polypeptide 43 (DDX43), intercellular adhesion molecule
1 (ICAM1), msh homeobox 1 (MSX1), placental growth
factor (PGF), PTPRO and zinc finger protein 42
homolog (ZFP42) (11,16,20–23).
In order to identify candidate genes potentially
affected by methylation with a putative tumor suppressor activity,
first we removed duplicated probes (16 genes), then we excluded
genes with poor annotation (20 genes) and genes with a chromosomal
location on X chromosome (20 genes). Genes with no CpG islands in
the promoter (two genes), genes with unknown function (8 genes) and
genes with no relevant function in biological mechanisms of
tumorigenesis (23 genes), were also excluded. Oncogenes and genes
with potential oncogenic activity (22 genes) were also removed. And
lastly genes for which methylation status of their promoter CpG
island loci has already been reported for CRC (8 genes) were
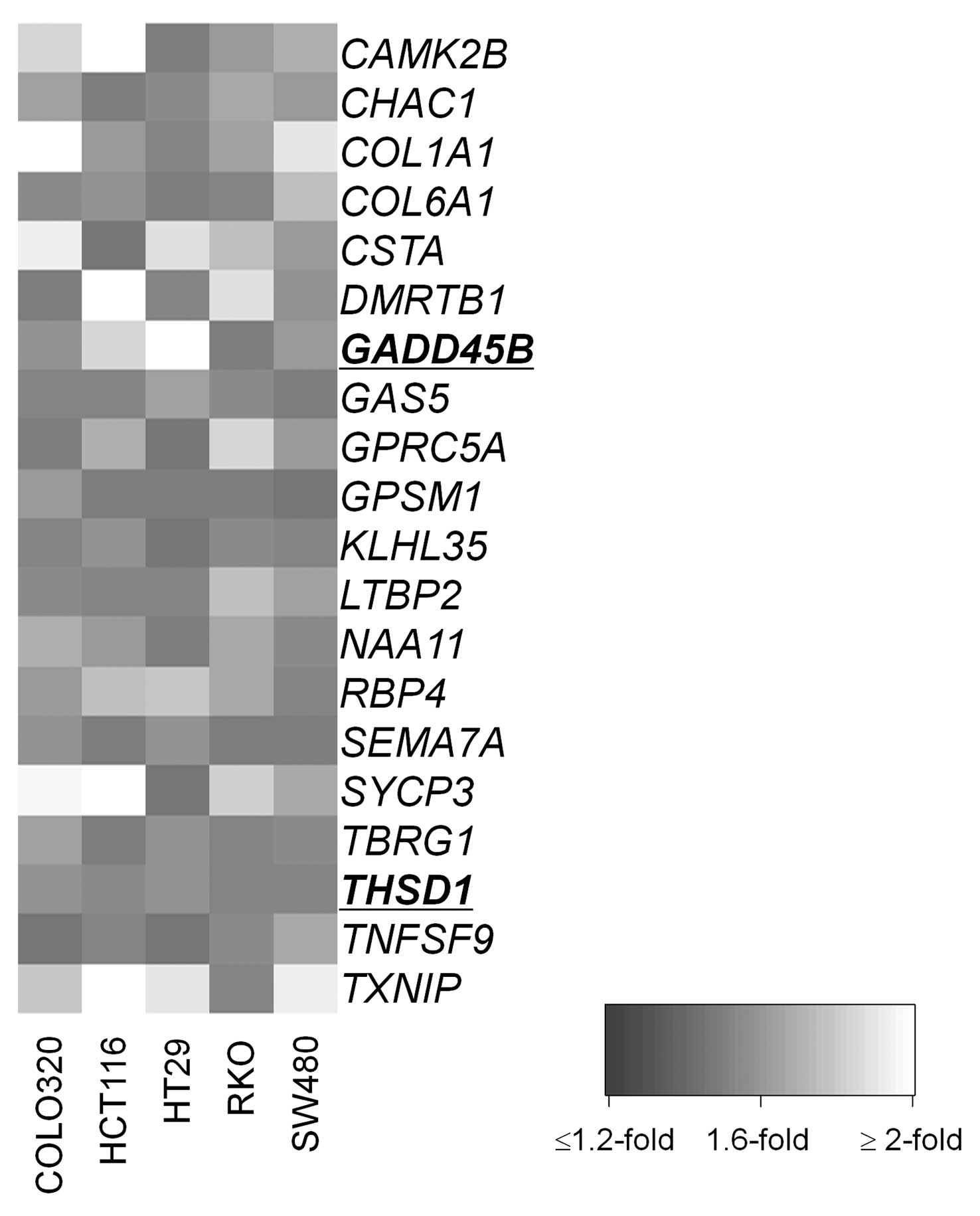
eliminated. We identified 20 candidates (Fig. 2) that have not been reported to be
affected by epigenetic mechanism in CRC (Table I) of which GADD45B and
THSD1 were selected for further analysis. Both genes have
been recently reported to be frequently methylated in cancer types
other than CRC (14,15). Also they were identified as having
CpG-rich sequences fulfilling the criteria of CpG island (GC
content ≥50%, CpG:GpC ratio ≥0.6, and minimum length 200 bp) in
their 5′ regions (24).
 | Table ITwenty putative methylated genes in
colorectal cancer up-regulated in 5-aza-2′-deoxycitidine-treated
colorectal cancer cell lines compared to mock-treated cell lines by
oligonucleotide microarray with a fold change >1.5 in at least
one cell line.a |
Table I
Twenty putative methylated genes in
colorectal cancer up-regulated in 5-aza-2′-deoxycitidine-treated
colorectal cancer cell lines compared to mock-treated cell lines by
oligonucleotide microarray with a fold change >1.5 in at least
one cell line.a
| Gene symbol | Probes | Gene name | Locus | Function | Mean FC |
|---|
| CAMK2B | 209956_s_at |
Calcium/calmodulin-dependent protein
kinase II beta | 7p14.3-p14.1 |
Calmodulin-dependent protein kinase
activity | 2.52 |
| CHAC1 | 219270_at | ChaC, cation
transport regulator homolog 1 (E. coli) | 15q15.1 | Protein binding,
proapoptotic component of the unfolded protein response | 1.65 |
| COL1A1 | 1556499_s_at | Collagen, type I,
alpha 1 | 17q21.33 | Extracellular
matrix structural constituent, platelet-derived growth factor
binding, transcription activator activity | 2.47 |
| COL6A1 | 212091_s_at | Pollagen, type VI,
alpha 1 | 21q22.3 | Platelet-derived
growth factor binding | 1.61 |
| CSTA | 204971_at | Cystatin A (stefin
A) | 3q21 | Cysteine-type
endopeptidase inhibitor activity | 2.38 |
| DMRTB1 | 240313_at | DMRT-like family B
with proline-rich C-terminal 1 | 1p32.3 | Metal ion binding,
sequence-specific DNA binding | 3.24 |
|
GADD45B | 207574_s_at | Growth arrest and
DNA-damage-inducible beta | 19p13.3 | Apoptosis,
regulation of MAPKK activity | 2.28 |
| GAS5 | 224841_x_at | Growth
arrest-specific 5 (non-protein coding) | 1q25.1 | Control of
mammalian apoptosis and cell population growth | 1.43 |
| GPRC5A | 203108_at | G-protein-coupled
receptor, family C, group 5, member A | 12p13-p12.3 | G-protein coupled
receptor activity | 1.83 |
| GPSM1 | 226043_at | G-protein signaling
modulator 1 (AGS3-like, C. elegans) | 9q34.3 | G-protein alpha
subunit binding, GTPase activator activity | 1.33 |
| KLHL35 | 1553611_s_at | Kelch-like 35
(Drosophila) | 11q13.4 | Protein
binding | 1.37 |
| LTBP2 | 204682_at | Latent transforming
growth factor beta binding protein 2 | 14q24 | Calcium ion
binding, growth factor binding | 1.67 |
| NAA11 | 210603_at |
N(alpha)-acetyltransferase 11, NatA
catalytic subunit | 4q21.21 | N-acetyltransferase
activity | 1.69 |
| RBP4 | 219140_s_at | Retinol binding
protein 4, plasma | 10q23-q24 | Retinol binding,
retinol transporter activity | 1.96 |
| SEMA7A | 230345_at | Semaphorin 7A, GPI
membrane anchor (John Milton Hagen blood group) | 15q22.3-q23 | Integrin binding,
receptor activity | 1.4 |
| SYCP3 | 1553599_a_at | Synaptonemal
complex protein 3 | 12q | DNA binding | 5.77 |
| TBRG1 | 226318_at | Transforming growth
factor beta regulator 1 | 11q24.2 | DNA, protein
binding | 1.48 |
|
THSD1 | 219477_s_at | Thrombospondin,
type I, domain containing 1 | 13q14.3 | Extracellular
matrix | 1.46 |
| TNFSF9 | 206907_at | Tumor necrosis
factor (ligand) superfamily, member 9 | 19p13.3 | Cytokine activity,
tumor necrosis factor receptor binding | 1.43 |
| TXNIP | 201009_s_at | Thioredoxin
interacting protein | 1q21.1 | Enzyme inhibitor
activity, ubiquitin protein ligase binding | 3.04 |
CpG island searcher (25) and UCSC Table Browser (26) using the current genome assembly
(GRCh37/hg19), were used for the potential CpG islands screening.
In addition, we searched the published literature for additional
analysis of the up-regulated genes.
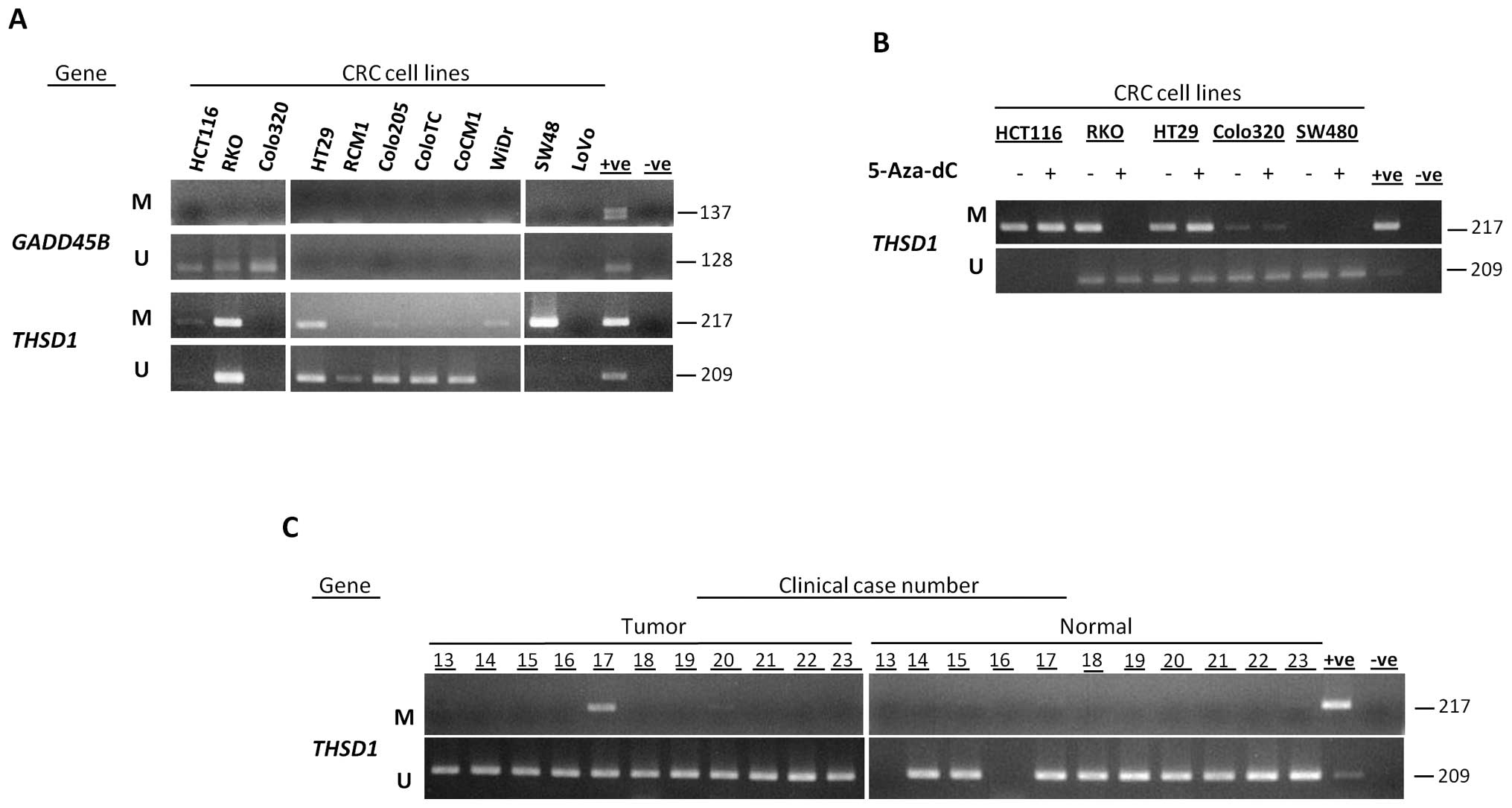
Methylation of THSD1 was observed in CRC
cell lines
To identify novel targets for aberrant methylation
in CRC, we further studied methylation status of two candidate
genes, GADD45B and THSD1, in colon cancer cell lines.
By using MSP, we determined their methylation status in 15 colon
cancer cells. GADD45B showed no methylation in colon cancer
cell lines and was excluded from further analysis, while
THSD1 was methylated in 27% (Table II; Fig. 3A).
| Table IIMethylation frequency of candidate
genes in 15 CRC cell lines and 23 matched clinical CRC tissue
samples.a |
Table II
Methylation frequency of candidate
genes in 15 CRC cell lines and 23 matched clinical CRC tissue
samples.a
| GADD45B | THSD1 |
|---|
| CRC cell lines
(%) | 0 (n=0) | 4 out of 15
(n=27) |
| Primary CRC tissue
(%) | np | 2 out of 23
(n=9) |
| Normal colon tissue
(%) | np | 0 out of 23
(n=0) |
| P-value, tumor vs.
normal (two-tailed Fisher’s exact test) | | 0.49 |
THSD1 promoter region was demethylated in
CRC cell lines after 5-aza-dC treatment
Methylation status of THSD1 in five CRC cell
lines before and after 5-aza-dC treatment was analyzed by MSP.
5-aza-dC treatment caused complete demethylation in the RKO cell
line, partial demethylation in the Colo320 cell line and no changes
in methylation were seen in SW480, HCT116 and HT29 cell lines
(Fig. 3B). This verified that
THSD1 promoter is affected by demethylation effect of
5-aza-dC and confirms the data obtained by oligonucleotide
microarray analysis.
Methylation of THSD1 was observed in
tumor samples of CRC patients
To test whether the aberrant methylation identified
in colon cancer cell lines was also present in primary colon cancer
specimens, we studied the methylation status of THSD1 gene
in 23 matched CRC samples. Aberrant methylation in primary colon
cancers was identified in 9% for THSD1. Distinctively,
THSD1 was completely unmethylated in all the samples from
normal tissues (Table II; Fig. 3C). There was no significant
difference in the methylation of THSD1 between tumor and
normal tissues (P=0.49; Table
II).
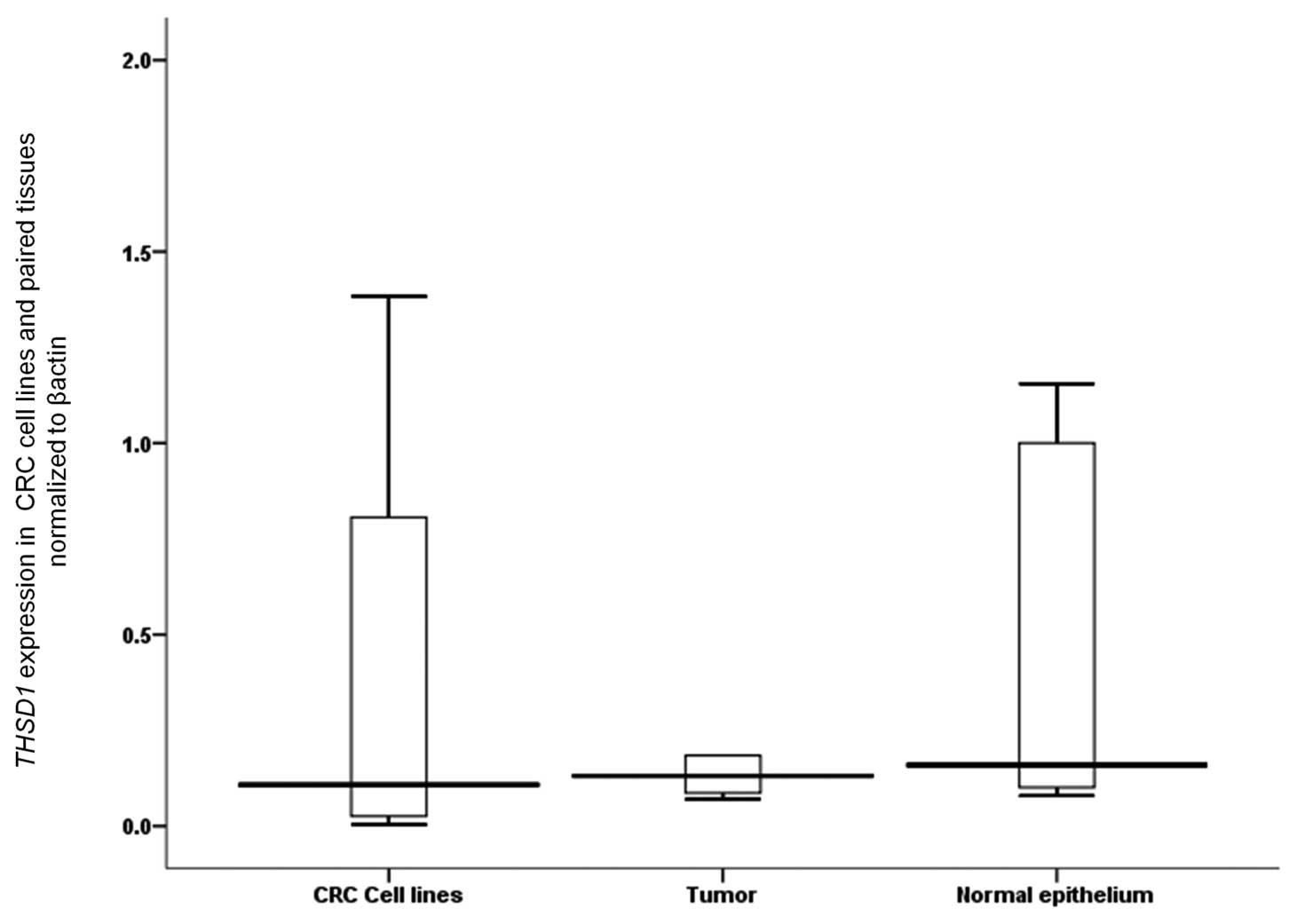
Down-regulation of expression of THSD1
was found in tumor tissues of CRC patients
To investigate the relationship between gene
expression and promoter hypermethylation, we analyzed THSD1
mRNA expression in 14 CRC cell lines and 14 paired CRC tissues. The
level of THSD1 mRNA was reduced (Fig. 4) in all tumor tissues relative to
cancer cell lines and adjacent non-tumor tissues, however it did
not reach statistical significance (P=0.613 and 0.158,
respectively).
Discussion
In this study, we carried out a systematic global
search for aberrantly methylated genes in CRC. 5-aza-dC was used to
demethylate silenced genes in five colon cancer cell lines and then
followed by expression microarray to identify overexpressed
genes.
The exposure of cell lines to demethylating agent
(5-aza-dC) induced expression changes in a group of genes in all
treated cell lines. A total of 139 genes were up-regulated,
including genes that have been previously reported to be
up-regulated by demethylation in CRC (11,16,20–23).
Treatment of the cell lines with 5-aza-dC can induce
genes without a direct consequence of CpG island demethylation
(27). We examined the methylation
status in two putative genes by MSP and one of them,
GADD45B, was not methylated in any of the 15 CRC cell lines
(Table II, Fig. 3A). In addition to the up-regulation
of two genes with no CpG islands, up-regulation of such genes might
be a secondary effect caused by an induced upstream factor (e.g.
genes in the p53 DNA damage pathway) (27).
THSD1 encodes a transmembrane molecule
containing a thrombospondin type 1 repeat, which might be involved
in cell adhesion and angiogenesis (15). Although THSD1 has no clear
function, other proteins possessing the thrombospondin type 1
repeat (TSR) have been shown to inhibit tumor angiogenesis and
growth (28), such as
thrombospondin 1 (TSP1), ADAM metallopeptidase with
thrombospondin type 1 motif 1 (ADAMTS1) and ADAM
metallopeptidase with thrombospondin type 1 motif 12
(ADAMTS12), all were reported as being methylated in CRC
(29,30). High THSD1 expression
positively correlated with a better distant metastasis survival in
breast cancer (15), hence its
loss possibly is associated with metastatic tumor spread.
The chromosomal location of THSD1 at 13q14.3
is similar to retinoblastoma 1 (RB1) gene region which has
been reported with widely differing frequencies of allelic loss
detected by LOH in CRC (31). This
region is strongly associated with the progression of colorectal
adenomas towards carcinomas (32).
THSD1 might have a role in radiation response
as it was one of the consensus radiation response genes in primary
human fibroblasts (33). The
thrombospondins (TSPs) might act to support tumor progression to
metastasis through their effects on the degradation of the
extracellular matrix and the ability of tumor cells to invade the
surrounding tissues, although few studies have addressed the role
of the TSPs in metastasis in vivo (34).
Based on combined genomic and transcriptomic
information from 6 patients, with tumor stage Dukes A, B, C and D,
respectively (n=24), a recent study showed the presence of
THSD1 in 17 genes that have been expressed in Dukes D that
may be relevant to tumor progression (35).
Silencing of THSD1 involved LOH and promoter
hypermethylation in esophageal cancer cell lines and tumor tissues
with gene expression down-regulation (15). We identified THSD1 as
aberrantly methylated in CRC cell lines and CRC positive samples.
THSD1 showed a tumor specific methylation with no abnormal
methylation in non-cancerous tissues (Table II, Fig. 3C). Moreover, low expression in
tumor tissues rather than normal for THSD1 besides no
significant correlation between methylation and mRNA expression
suggests that down-regulation of THSD1 in CRC specimens
could be attributed to allelic deletion, or epigenetic events other
than methylation of the CpG islands.
GADD45B belongs to GADD45 genes family and
proteins encoded by this family and recognized as stress sensors in
signaling responses to various physiological or environmental
stresses. GADD45 genes family mediates their activity via
interactions with other cellular proteins (36) and can inhibit cell proliferation at
different stages and induce cell apoptosis (37). Results from a mouse model showed
that Gadd45b is involved in tumor surveillance by CD8+ T
cells and the Gadd45 signaling pathway is important in containing
cancer (38). GADD45 genes become
repressed in cancer by different mechanisms such as methylation,
NF-κB activation or mutation through which deregulated expression
may lead to tumorigenesis (36).
Altogether, previous reports suggest that GADD45B is
functionally a tumor suppressor gene.
Down-regulation due to promoter hypermethylation of
GADD45B was reported in certain cancer types, such as
hepatocellular carcinoma and non-small cell cancer (14,39).
Na et al (39) showed no
methylation of GADD45B promoter in five CRC cell lines in
their efforts to investigate the GADD45 genes methylation profile
in multiple tumors was consistent with our results.
A substantial number of putatively hypermethylated
genes remain unexamined at present (Table I) and those remaining candidates
may have a role in tumorigenesis of CRC.
In conclusion, this study adds an additional step in
global profiling of gene promoter hypermethylation to identify
novel aberrantly methylated genes in CRC. This may help in further
understanding of CRC pathogenesis and may lead to identification of
diagnostic, prognostic methylation markers or therapeutic targets.
Also our data showed that coherent mining of the microarray data
can further widen the gene detection results. The current study is
the first to identify THSD1 down-regulation and methylation
in primary CRC tissues. Further functional studies are required to
clarify the role of THSD1 in tumorigenesis and to examine
its clinical significance in CRC.
Acknowledgements
This work was funded by the Ministry
of Education, Culture, Sports, Science, and Technology of Japan
(no. 90706001089).
References
|
1
|
Jones PA and Laird PW: Cancer epigenetics
comes of age. Nat Genet. 21:163–167. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baylin SB, Herman JG, Graff JR, Vertino PM
and Issa JP: Alterations in DNA methylation: a fundamental aspect
of neoplasia. Adv Cancer Res. 72:141–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esteller M: Dormant hypermethylated tumour
suppressor genes: questions and answers. J Pathol. 205:172–180.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karpf AR and Jones DA: Reactivating the
expression of methylation silenced genes in human cancer. Oncogene.
21:5496–5503. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamashita K, Upadhyay S, Osada M, et al:
Pharmacologic unmasking of epigenetically silenced tumor suppressor
genes in esophageal squamous cell carcinoma. Cancer Cell.
2:485–495. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Suzuki H, Gabrielson E, Chen W, et al: A
genomic screen for genes upregulated by demethylation and histone
deacetylase inhibition in human colorectal cancer. Nat Genet.
31:141–149. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koul S, Houldsworth J, Mansukhani MM, et
al: Characteristic promoter hypermethylation signatures in male
germ cell tumors. Mol Cancer. 1:82002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Konduri SD, Srivenugopal KS, Yanamandra N,
et al: Promoter methylation and silencing of the tissue factor
pathway inhibitor-2 (TFPI-2), a gene encoding an inhibitor
of matrix metalloproteinases in human glioma cells. Oncogene.
22:4509–4516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Knobbe CB, Reifenberger J, Blaschke B and
Reifenberger G: Hypermethylation and transcriptional downregulation
of the carboxyl-terminal modulator protein gene in glioblastomas. J
Natl Cancer Inst. 96:483–486. 2004. View Article : Google Scholar
|
|
10
|
Sawa H, Murakami H, Ohshima Y, et al:
Histone deacetylase inhibitors such as sodium butyrate and
trichostatin A inhibit vascular endothelial growth factor
(VEGF) secretion from human glioblastoma cells. Brain Tumor
Pathol. 19:77–81. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schuebel KE, Chen W, Cope L, et al:
Comparing the DNA hypermethylome with gene mutations in human
colorectal cancer. PLoS Genet. 3:1709–1723. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ishiguro M, Iida S, Uetake H, et al:
Effect of combined therapy with low-dose 5-aza-2′-deoxycytidine and
irinotecan on colon cancer cell line HCT-15. Ann Surg Oncol.
14:1752–1762. 2007.
|
|
13
|
Herman JG, Graff JR, Myöhänen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qiu W, Zhou B, Zou H, et al:
Hypermethylation of growth arrest DNA damage-inducible gene 45 beta
promoter in human hepatocellular carcinoma. Am J Pathol.
165:1689–1699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ko JM, Chan PL, Yau WL, et al:
Monochromosome transfer and microarray analysis identify a critical
tumor-suppressive region mapping to chromosome 13q14 and
THSD1 in esophageal carcinoma. Mol Cancer Res. 6:592–603.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mori Y, Yin J, Sato F, et al:
Identification of genes uniquely involved in frequent
microsatellite instability colon carcinogenesis by expression
profiling combined with epigenetic scanning. Cancer Res.
64:2434–2438. 2004. View Article : Google Scholar
|
|
17
|
Khamas A, Ishikawa T, Shimokawa K, et al:
Screening for epigenetically masked genes in colorectal cancer
using 5-aza-2′-deoxycytidine, microarray and gene expression
profile. Cancer Genomics Proteomics. 9:67–75. 2012.PubMed/NCBI
|
|
18
|
Liang G, Gonzales FA, Jones PA, Orntoft TF
and Thykjaer T: Analysis of gene induction in human fibroblasts and
bladder cancer cells exposed to the methylation inhibitor
5-aza-2′-deoxycytidine. Cancer Res. 62:961–966. 2002.PubMed/NCBI
|
|
19
|
Huang DW, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
20
|
Ali D: Identification of novel epigenetic
biomarkers in colorectal cancer, GLDC and PPP1R14A.
Department of Molecular Biosciences (IMBV), Faculty of Mathematics
and Natural Sciences, University of Oslo; pp. 972010
|
|
21
|
Easwaran HP, van Neste L, Cope L, et al:
Aberrant silencing of cancer-related genes by CpG hypermethylation
occurs independently of their spatial organization in the nucleus.
Cancer Res. 70:8015–8024. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim MS, Lee J and Sidransky D: DNA
methylation markers in colorectal cancer. Cancer Metastasis Rev.
29:181–206. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu L and Jain RK: Down-regulation of
placenta growth factor by promoter hypermethylation in human lung
and colon carcinoma. Mol Cancer Res. 5:873–880. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gardiner-Garden M and Frommer M: CpG
islands in vertebrate genomes. J Mol Biol. 196:261–282. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takai D and Jones PA: The CpG island
searcher: a new WWW resource. In Silico Biol. 3:235–240.
2003.PubMed/NCBI
|
|
26
|
Karolchik D, Hinrichs AS, Furey TS, et al:
The UCSC table browser data retrieval tool. Nucleic Acids Res.
32:D493–D496. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lind GE, Thorstensen L, Løvig T, et al: A
CpG island hypermethylation profile of primary colorectal
carcinomas and colon cancer cell lines. Mol Cancer. 3:282004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X and Lawler J: Thrombospondin-based
antiangiogenic therapy. Microvasc Res. 74:90–99. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rojas A, Meherem S, Kim YH, et al: The
aberrant methylation of TSP1 suppresses TGF-beta1 activation
in colorectal cancer. Int J Cancer. 123:14–21. 2008.
|
|
30
|
Moncada-Pazos A, Obaya AJ, Fraga MF, et
al: The ADAMTS12 metalloprotease gene is epigenetically
silenced in tumor cells and transcriptionally activated in the
stroma during progression of colon cancer. J Cell Sci.
122:2906–2913. 2009.
|
|
31
|
Lai PS, Cheah PY, Kadam P, et al:
Overexpression of RB1 transcript is significantly correlated
with 13q14 allelic imbalance in colorectal carcinomas. Int J
Cancer. 119:1061–1066. 2006.PubMed/NCBI
|
|
32
|
Derks S, Postma C, Carvalho B, et al:
Integrated analysis of chromosomal, microsatellite and epigenetic
instability in colorectal cancer identifies specific associations
between promoter methylation of pivotal tumour suppressor and DNA
repair genes and specific chromosomal alterations. Carcinogenesis.
29:434–439. 2008. View Article : Google Scholar
|
|
33
|
Kis E, Szatmári T, Keszei M, et al:
Microarray analysis of radiation response genes in primary human
fibroblasts. Int J Radiat Oncol Biol Phys. 66:1506–1514. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kazerounian S, Yee KO and Lawler J:
Thrombospondins in cancer. Cell Mol Life Sci. 65:700–712. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lagerstedt KK, Kristiansson E, Lönnroth C,
et al: Genes with relevance for early to late progression of colon
carcinoma based on combined genomic and transcriptomic information
from the same patients. Cancer Inform. 9:79–91. 2010.PubMed/NCBI
|
|
36
|
Cretu A, Sha X, Tront J, Hoffman B and
Liebermann DA: Stress sensor Gadd45 genes as therapeutic targets in
cancer. Cancer Ther. 7:268–276. 2009.PubMed/NCBI
|
|
37
|
Ying J, Srivastava G, Hsieh WS, et al: The
stress-responsive gene GADD45G is a functional tumor
suppressor, with its response to environmental stresses frequently
disrupted epigenetically in multiple tumors. Clin Cancer Res.
11:6442–6449. 2005.
|
|
38
|
Ju S, Zhu Y, Liu L, et al: Gadd45b and
Gadd45g are important for anti-tumor immune responses. Eur J
Immunol. 39:3010–3018. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Na YK, Lee SM, Hong HS, Kim JB, Park JY
and Kim DS: Hypermethylation of growth arrest DNA-damage-inducible
gene 45 in non-small cell lung cancer and its relationship with
clinicopathologic features. Mol Cells. 30:89–92. 2010. View Article : Google Scholar : PubMed/NCBI
|