Introduction
With an estimated 1.38 million new cancer cases
worldwide in 2008, breast cancer is the most frequent cancer and
the leading cause of cancer mortality in women (1). If breast cancer is detected and
treated at an early stage, the majority of the patients survive.
These facts highlight an urgent need to develop novel molecular
biomarkers that can detect early stage of breast cancer so that
treatment is initiated promptly.
Carcinogenesis has been demonstrated to involve not
only genetic but also epigenetic alterations that may suppress or
activate multiple genes. The most common epigenetic alteration in
cancer is DNA methylation (2),
which is an early event in carcinogenesis, and may therefore serve
as a biomarker for the early detection of cancer. Promoter
methylation, with a methyl group to the 5-carbon position of
cytosine in CpG islands, is associated with transcriptional
silencing. DNA methylation also occurs in the intragenic region. To
date, little attention has been paid to intragenic methylation.
Recently, aberrant methylations of specific genes
that involve breast carcinogenesis have been widely explored and
the list of aberrant methylated genes is increasing (3,4). Due
to the limitation of techniques for analysis of DNA methylation,
the majority of these efforts have focused only on individual
candidate genes, which have already been identified to play a role
in other cancer models. These genes are mostly tumor suppressor
genes. Thus far, most methylated genes that contribute to breast
cancer, such as methylated p16, CCDN2, MGMT
and hMTLH1 are identified by the candidate approach,
confining to promoter methylation of tumor suppressor genes
(5). This approach is not only
time-consuming and labor-intensive, but also limited by candidate
methylated genes and candidate methylated sites available.
Gene expression microarray based strategy has been
developed for high-throughput screening of DNA methylation
biomarker on a genomic scale in breast cancer. This approach uses
microarray to interrogate upregulated genes after treatment of
breast cancer line with a demethylating agent
5-aza-2′-deoxycytidine (6), and
then uses bisulfite sequencing, restriction analysis, and/or
methylation specific PCR to confirm DNA methylation. This approach
might leave out non-expressed genes in the target tissue, thereby
missing candidate genes, which may actually be potential
methylation biomarkers in cancer diagnosis. For example, the
significantly methylated vimentin gene has been used as a
diagnostic biomarker in colorectal cancer, while colon cancer cells
do not express vimentin (7,8).
Using the gene expression microarray based strategy, methylation of
vimentin could not be identified. However, it is unclear
whether there are that many such non-expressed methylated genes in
cancerous and non-cancerous tissue that critical methylation
biomarkers could not be identified by gene expression microarray
based strategy.
Recently, methyl-DNA immunoprecipitation combined
with high-throughput sequencing (9) and methylated-CpG island recovery
assay (MIRA) combined with CpG island array (10,11)
were also used to screen cancer-associated methylated genes in
clinical samples of breast cancer tissue. To screen novel
biomarkers for the early detection of breast cancer, in the current
study, we utilized a high-throughput genome-wide approach, MIRA
microarray, that is based on the high affinity of the MBD protein
for methylated DNA to identify novel tumor-specific genes with
promoter methylation or intragenic methylation. We also integrated
the corresponding gene expression profile to explore the role of
methylation in regulating gene expression.
Materials and methods
Patient samples
Samples of breast cancer tissue were obtained from
the surgical specimens of 15 patients with early stage invasive
breast cancer. The tissue sections were stained with H&E and
were reviewed by a pathologist to confirm the presence of the
cancer lesions. Breast cancer was staged according to the American
Joint Committee on Cancer staging system protocol. ER, PR and HER2
status were determined by immunohistochemistry. Non-cancerous
breast tissues were collected from the biopsy specimens of 15
patients for the suspicion of malignant lesion but without any
histological findings. Clinicopathological characteristics of the
patient samples are given in Table
I. Ten cancerous and 10 non-cancerous breast tissues were
selected for microarray analysis and the other 5 cancerous and 5
non-cancerous breast tissues were used for validation of the genes.
All samples were snap-frozen in liquid nitrogen and stored at −80°C
until nucleic acid extraction. The study was approved by the Ethics
Committee of our hospital, and all patients provided written
informed consent.
 | Table IClinicopathological characteristics
of the patient samples. |
Table I
Clinicopathological characteristics
of the patient samples.
|
Characteristics | Breast cancer
tissues (n=15) | Breast non-cancer
tissues (n=15) |
|---|
| Mean age (y)
(range) | 41 (34–50) | 38 (32–47) |
| Histological
type | Invasive ductal
carcinoma | No histological
findings |
| Grade (%) | | ND |
| I | 5 (33.3%) | |
| II | 10 (66.7%) | |
| Cancer size
(%) | | ND |
| pT1 | 8 (53.3%) | |
| pT2 | 7 (46.7%) | |
| Lymph node status
(%) | | ND |
| Negative | 15 (100.0%) | |
| Positive | 0 (0.0%) | |
| Estrogen receptor
(%) | | ND |
| Positive | 8 (53.3%) | |
| Negative | 7 (46.7%) | |
| Progesterone
receptor (%) | | ND |
| Positive | 9 (53.3%) | |
| Negative | 6 (46.7%) | |
| HER2 status
(%) | | ND |
| (−)/ (+) | 10 (66.7%) | |
| (++) | 3 (20.0%) | |
| (+++) | 2 (13.3%) | |
MIRA microarray analysis of DNA
methylation
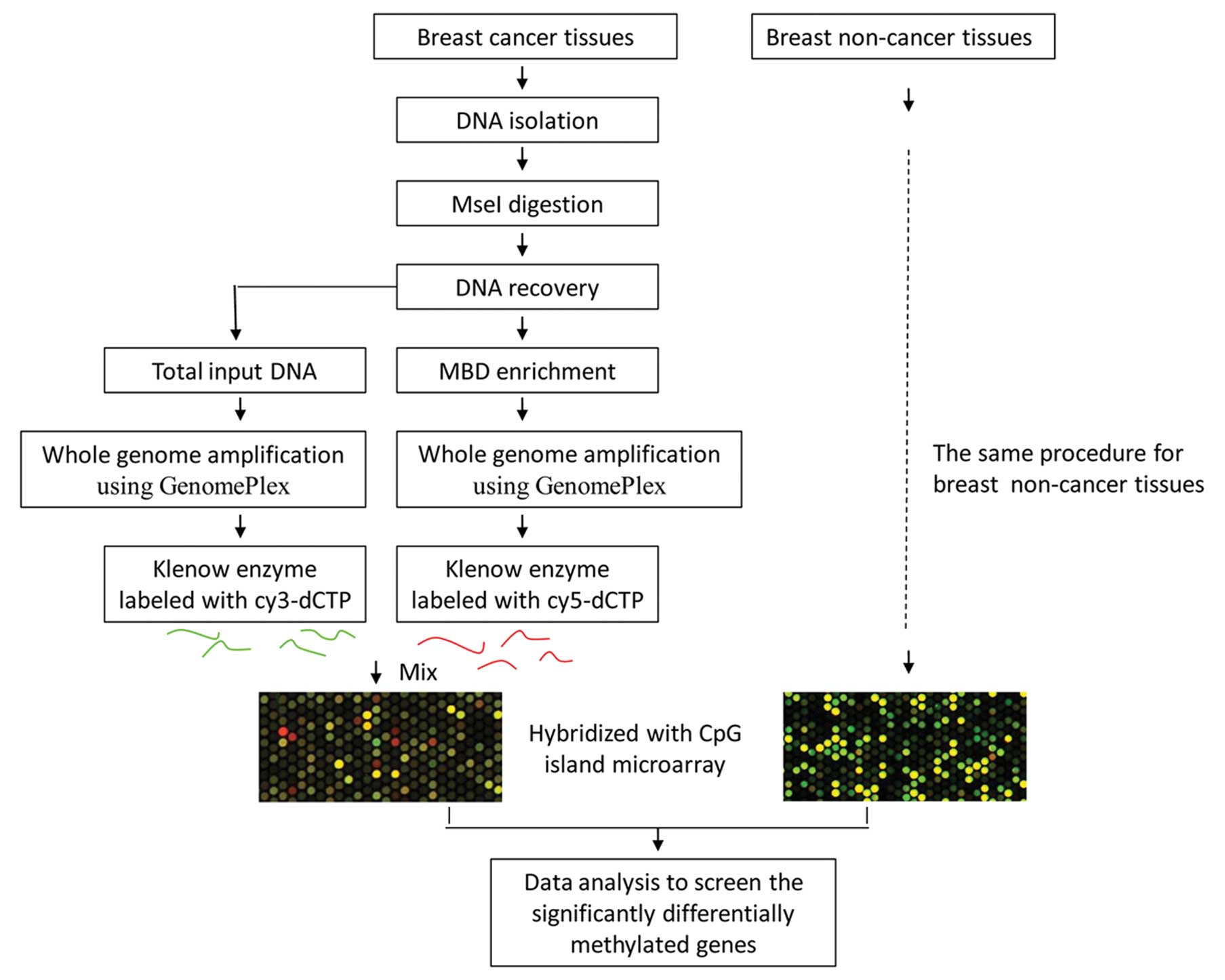
The MIRA microarray analysis was carried out as
described by Rauch et al with minor modifications (12). The schematic diagram for our
MIRA-assisted CpG island microarray analysis is outlined in
Fig. 1. Briefly, DNA was firstly
separated from 10 fresh frozen cancerous and 10 non-cancerous
breast tissues using standard techniques and DNA quality was
verified using agarose gel electrophoresis. Then, 2 μg of genomic
DNA samples were digested by Mse I (5′-TTAA), which generally cuts
outside of the CpG islands and produces 200–1000 bp fragments.
Fragment sizes were verified by gel electrophoresis in 1.5% agarose
gels. To remove fragments smaller than 100 bp, the fragmented DNA
was purified using the MicroDNA Purification Kit (Beijing CoWin
Biotech Co. Ltd, Beijing, China) following the manufacturer’s
instructions. Subsequently, the purified DNA fragments were used to
enrich methylated DNA using the BioChain MBD kit (BioChain,
Hayward, CA, USA) according to the manufacturer’s protocol. This
kit uses the high affinity of the MBD protein for double-stranded
CpG-methylation DNA. This MBD protein can bind methylated DNA
fragments more sensitively and specifically than antibody, and thus
has a low false positive rate in the enrichment of methylated DNA
fragments (12). In addition, its
procedure is simple since MBDs can bind dsDNA (12), natural status of genomic DNA, while
antibody can bind only ssDNA and therefore has to denature the
dsDNA before DNA enrichment in antibody based methods.
The MIRA-captured DNA segments were purified using
the MicroDNA Purification Kit (CoWin Biotech Co.). After
purification, the MIRA-captured DNA segments were amplified using
the GenomePlex Whole Genome Amplification Kit (Sigma, St. Louis,
MO, USA), according to the manufacturer’s instructions. Compared
with the previously reported MIRA method, in which MIRA-captured
methylated DNA was added with oligonucleotide linkers for
subsequent PCR-based amplification (12), the method we emloyed adopted a
whole genome amplification strategy and had a few advantages,
including better liner amplification, less bias in DNA
amplification and did not require adding linker in amplifying
MIRA-captured DNA segments.
The products of whole genome amplification from the
total input DNA without MBD enrichment and methylation-enriched DNA
from each sample were labeled with cy3-dCTP and cy5-dCTP using
Klenow enzyme (Takara, Dalian, China), respectively. Prior to
labeling with fluorescent dye, DNA amplification products were
purified using the MicroDNA Purification Kit. The fluorescent dye
labeled DNA segments were then pooled and hybridized to Agilent
human CpG island microarrays, which were designed to interrogate
61982 CpG dinucleotides, covering 4162 genes. Following
hybridization, washing and scanning were carried out on Agilent’s
microarray platform according to Agilent’s standard protocols.
Agilent Feature Extraction software was used to extract the
hybridization signals from microarrays. Following global mean
normalization, probes with an intensity <400 were discarded from
further analysis.
It is critical to define a proper cut-off value in
fold change in MIRA/input signaling ratios between cancer versus
non-cancer to efficiently screen candidate hypermethylated genes.
However there are still no gold standards on setting up fold change
cutoff for defining candidate methylated genes. In fact,
methylation difference in the MIRA approach is represented by copy
number difference of DNA enriched by MBD protein. It is similar to
DNA copy number difference in array-based comparative genomic
hybridization (aCGH) and quite different from gene expression
difference in gene expression microarray analysis, for one copy of
DNA expresses various copies of mRNA. In well-established aCGH,
cutoff for detecting DNA copy difference is usually defined as
probe ratio value <1.25 (13,14),
which is smaller than that for mRNA detection. Therefore, probes
with more than 1.2 fold changes (p<0.01) in MIRA/input signaling
ratios between cancer versus non-cancer were selected here as
positive probes using SAM software and genes with at least two
positive probes were defined to be significantly hypermethylated
genes. Hypermethylated genes were further analyzed based on a
significant enrichment of GO terms using hyper-geometric
distribution in the R language software package.
Gene expression microarray analysis
RNA from 8 of the 10 cancerous and 8 of the 10
non-cancerous breast tissues subjected to MIRA microarray analysis
were qualified and interrogated for gene expression profiling.
Briefly, total RNA was extracted using TRIzol reagent (Invitrogen
Life Technolgies, Carlsbad, CA, USA). mRNA expression levels were
measured using Agilent Human Whole Genome 8*60K array following the
manufacturer’s protocol. Arrays were washed and scanned on
Agilent’s microarray platform according to Agilent’s standard
protocols. SAM software was used to analyze differentially
expressed genes between cancerous and non-cancerous breast
tissues.
Validation of hypermethylated genes
To validate the reliability of MIRA microarray data
and to explore the methylation extent of hypermethylated genes
identified by methylation microarray analysis, 4 significantly
hypermethylated genes identified by methylation microarray analysis
were selected for further validation using direct bisulfite
sequencing. To validate the reliability of MIRA microarray data and
explore the relationship between methylation and gene expression, 4
significantly hypermethylated genes were selected based on
methylation and gene expression microarray data for further
validation in 5 independent cancerous and 5 independent
non-cancerous samples using combined bisulfite restriction analysis
(COBRA) (15). For PCR
amplification, direct bisulfite sequencing and COBRA assay used the
same PCR reaction system, including the same primers that are
specific for the bisulfite modified DNA and contain minimum CpG
sites in their sequence (Table
II). First, genomic DNA was subjected to bisulfite treatment
using the DNA methylation detection kit (BioChain). Then, 125 ng of
sodium bisulfite-treated DNA was used for a program of 45 cycles of
melting (30 sec at 94°C), annealing (30 sec at 55°C) and extension
(30 sec at 72°C) in a 50 μl reaction mixture containing 1X PCR
buffer (Mg2+ Plus), 200 μM of each dNTP, 0.5 μM of
forward primer, 0.5 μM of reverse primer (primer sequences are
provided in Table I) and 1.25 unit
of Takara Taq HS. PCR products were then purified using the
MicroDNA Purification Kit. For bisulfite sequencing, the purified
PCR products were subjected to sequencing directly using reverse or
forward primers of PCR amplification. For the COBRA assay, the
purified PCR products were subjected for digestion with BstU I
(CG↓CG) restriction enzyme (New England Biolabs, Ipswich, MA, USA)
and then separated on 2% agarose gels supplemented with ethidium
bromide. DNA was visualized under a UV light.
| Table IIPrimer pairs for COBRA validation of
hypermethylated genes. |
Table II
Primer pairs for COBRA validation of
hypermethylated genes.
| Gene | GenBank no. | Primer pairs
(5′-3′) |
|---|
| WT1 | NM_024426.4 | F:
AAAATTATTCGAGGGTTTAGGG
R: TCTACTTACGATTCCTCCACAA |
| OTX2 | NM_172337.1 | F:
AGTTGTGTTAGGTTGAGGGAG
R: AATCCCAAAAACCTTTTTAAA |
| PAX6 | NM_000280.3 | F:
AAAAGGAAGTTAGGAGAAATYGGAA
R: AAATCACTCAAACGAAATTAAACTACAC |
| PITX2 | NM_000325.5 | F:
TAGTGATAGGCGTTTCGGGTT
R: CCACTACATACTAACAAACACTCAAAT |
Gene expression validation of the
hypermethylated genes
To explore the relationship between methylation and
gene expression, the above 4 significantly hypermethylated genes
that were subjected to COBRA validation were selected to validate
their expression levels using reverse transcription PCR. Briefly, 1
μg of DNase-treated total RNAs were reverse transcribed with
oligo(dT)15 using M-MLV reverse transcriptase (Life Technologies)
in a total volume of 20 μl reaction volume. Following reverse
transcription reaction, 1 μl of this mixture was employed for an
RT-PCR program of 45 cycles of melting (30 sec at 94°C), annealing
(3 sec at 58°C) and extension (30 sec at 72°C). The 20 μl reaction
mixture contained 1X PCR buffer (Mg2+ Plus), 200 μM of
each dNTP, 0.5 μM of forward primer, 0.5 μM of reverse primer
(Table III). Total RNA used for
positive control of WT1 and PITX2 was isolated from
uterine tissue and breast cancer tissue, respectively. Total RNA
used for positive control of OTX2 and PAX6 was from
cerebellum tissue. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
was used as internal control. RT-PCR product was separated on 2%
agarose gels supplemented with ethidium bromide. DNA was visualized
under a UV light.
| Table IIIPrimer pairs for RT-PCR validation of
expression levels of hypermethylated genes. |
Table III
Primer pairs for RT-PCR validation of
expression levels of hypermethylated genes.
| Gene | Genbank No | Primer pairs
(5′-3′) |
|---|
| WT1 | NM_024426.4 | F:
CATACCAGTGTGACTTCAAGGA
R: ATGTTGTGATGGCGGACTAAT |
| OTX2 | NM_172337.1 | F:
CCAGACATCTTCATGCGAG
R: GTTCCACTCTCTGAACTCACT |
| PAX6 | NM_000280.3 | F:
CCCAGCCAGACCTCCTCATA
R: CCGGGAACTTGAACTGGAAC |
| PITX2 | NM_000325.5 | F:
ACCTTACGGAAGCCCGAGTC
R: CGGCCCAGTTGTTGTAGGAA |
Results
MIRA microarray analysis of DNA
methylation
To screen novel methylation biomarkers for the early
detection of breast cancer, we utilized the MIRA approach to
identify significantly hypermethylated genes in breast cancer.
Probes in which fold changes in MIRA/input signaling ratios between
cancer versus non-cancer were above 1.2 (p<0.01) were defined as
positive probes, and genes with at least two positive probes were
defined as significantly hypermethylated genes. As a result, a
total of 70 hypermethylated genes were identified (Table IV). Among them, many genes, such as
ITGA4, NFIX and FGF12, were first identified
to be hypermethylated in the breast cancer model.
| Table IVHypermethylated genes identified by
MIRA based microarray. |
Table IV
Hypermethylated genes identified by
MIRA based microarray.
| Gene | Changed probes | Average ratio | Position | Gene
description |
|---|
| CCND2 | 8 | 1.70 | P and I | Cyclin D2 |
| PAX6 | 8 | 2.09 | P and I | Paired box 6 |
| MNX1 | 6 | 2.15 | I and D | Motor neuron and
pancreas homeobox 1 |
| OTX2 | 6 | 1.63 | P and D | Orthodenticle
homeobox 2 |
| PITX2 | 6 | 1.49 | P and I | Paired-like
homeodomain 2 |
| NFIX | 5 | 1.66 | I | Nuclear factor
I/X |
| PDGFRA | 5 | 1.33 | P and I | Platelet-derived
growth factor receptor |
| WT1 | 5 | 1.63 | I | Wilms tumor 1 |
| FGF12 | 4 | 2.06 | I | Fibroblast growth
factor 12 |
| OSR1 | 4 | 1.82 | I | Odd-skipped related
1 |
| SOX9 | 4 | 1.35 | P and I | Sex determining
region Y-box 9 |
| VSX1 | 4 | 1.76 | P | Visual system
homeobox 1 |
| ATOH1 | 3 | 1.68 | P, I and D | Atonal homolog
1 |
| FOXF2 | 3 | 1.33 | P | Forkhead box
F2 |
| GAD1 | 3 | 1.48 | P and I | Glutamate
decarboxylase 1 |
| GATA6 | 3 | 1.62 | P | GATA binding
protein 6 |
| GBX2 | 3 | 1.55 | P and I | Gastrulation brain
homeobox 2 |
| HAND1 | 3 | 1.52 | P, I and D | Heart and neural
crest derivatives expressed 1 |
| HLX | 3 | 1.48 | D | H2.0-like
homeobox |
| HOXD3 | 3 | 1.42 | P and I | Homeobox D3 |
| LHX2 | 3 | 1.63 | I | LIM homeobox 2 |
| MMP9 | 3 | 1.40 | I | Matrix
metallopeptidase 9 |
| NEUROG3 | 3 | 1.53 | P and D | Neurogenin 3 |
| NID2 | 3 | 2.39 | I | Nidogen 2 |
| PCDHGC5 | 3 | 2.21 | D | Protocadherin gamma
subfamily C |
| PDX1 | 3 | 1.85 | P | Pancreatic and
duodenal homeobox 1 |
| SIX1 | 3 | 1.37 | P and D | SIX homeobox 1 |
| TSC2 | 3 | 1.26 | I | Tuberous sclerosis
2 |
| ACSL6 | 2 | 1.39 | D | Acyl-coa synthetase
long-chain family member 6 |
| BDNF | 2 | 1.45 | P and I | Brain-derived
neurotrophic factor |
| CACNA1E | 2 | 1.42 | I | Calcium channel, R
type |
| CACNA1G | 2 | 1.64 | P and I | Calcium channel, T
type |
| CACNA1H | 2 | 1.56 | I | Calcium channel, T
type, |
| CD8A | 2 | 1.57 | I | CD8a molecule |
| CDK6 | 2 | 1.52 | P and I | Cyclin-dependent
kinase 6 |
| CDKN2A | 2 | 1.57 | I | Cyclin-dependent
kinase inhibitor 2A |
| CXCL12 | 2 | 1.40 | P | Chemokine (C-X-C
motif) ligand 12 |
| DAD1 | 2 | 1.63 | I | Defender against
cell death 1 |
| DAPK3 | 2 | 1.44 | P | Death-associated
protein kinase 3 |
| DLL4 | 2 | 1.26 | P and I | Delta-like 4
(Drosophila) |
| EAF1 | 2 | 1.41 | I | ELL associated
factor 1 |
| HOXA6 | 2 | 1.36 | P | Homeobox A6 |
| HOXB13 | 2 | 2.38 | P and I | Homeobox B13 |
| HOXD9 | 2 | 1.37 | P and I | Homeobox D9 |
| ITGA4 | 2 | 1.39 | I | Integrin, alpha
4 |
| MEIS1 | 2 | 1.42 | P and I | Meis homeobox
1 |
| NEDD4L | 2 | 1.40 | P | Neural precursor
cell expressed, developmentally downregulated 4-like |
| NF1 | 2 | 1.42 | I | Neurofibromin
1 |
| NKX2-1 | 2 | 1.36 | P and I | NK2 homeobox 1 |
| NKX2-2 | 2 | 1.55 | P and I | NK2 homeobox 2 |
| NOTCH1 | 2 | 1.40 | I | Notch homolog 1,
translocation-associated |
| NPY | 2 | 1.33 | P and I | Neuropeptide Y |
| NR2F2 | 2 | 1.47 | P | Nuclear receptor
subfamily 2 |
| NR4A3 | 2 | 1.66 | P | Nuclear receptor
subfamily 4, |
| NRP1 | 2 | 1.28 | P and I | Neuropilin 1 |
| P4HB | 2 | 1.52 | P | Prolyl
4-hydroxylase |
| PAX9 | 2 | 1.95 | P | Paired box 9 |
| PHOX2B | 2 | 1.68 | I | Paired-like
homeobox 2b |
| POMC | 2 | 1.41 | P |
Proopiomelanocortin |
| POU4F1 | 2 | 1.42 | P | POU class 4
homeobox 1 |
| POU4F3 | 2 | 1.60 | P and D | POU class 4
homeobox 3 |
| PTGS2 | 2 | 1.56 | P |
Prostaglandin-endoperoxide synthase 2 |
| SALL1 | 2 | 1.46 | P | Sal-like 1 |
| SIM1 | 2 | 1.33 | P and I | Single-minded
homolog 1 |
| SSTR1 | 2 | 1.79 | I | Somatostatin
receptor 1 |
| TCF3 | 2 | 1.28 | I | Transcription
factor 3 |
| TXNRD1 | 2 | 1.54 | I | Thioredoxin
reductase 1 |
| WIT1 | 2 | 1.79 | I | Wilms tumor
upstream neighbor 1 |
| WNT1 | 2 | 1.30 | I | Wingless-type MMTV
integration site family |
| WNT7A | 2 | 1.67 | P and I | Wingless-type MMTV
integration site family |
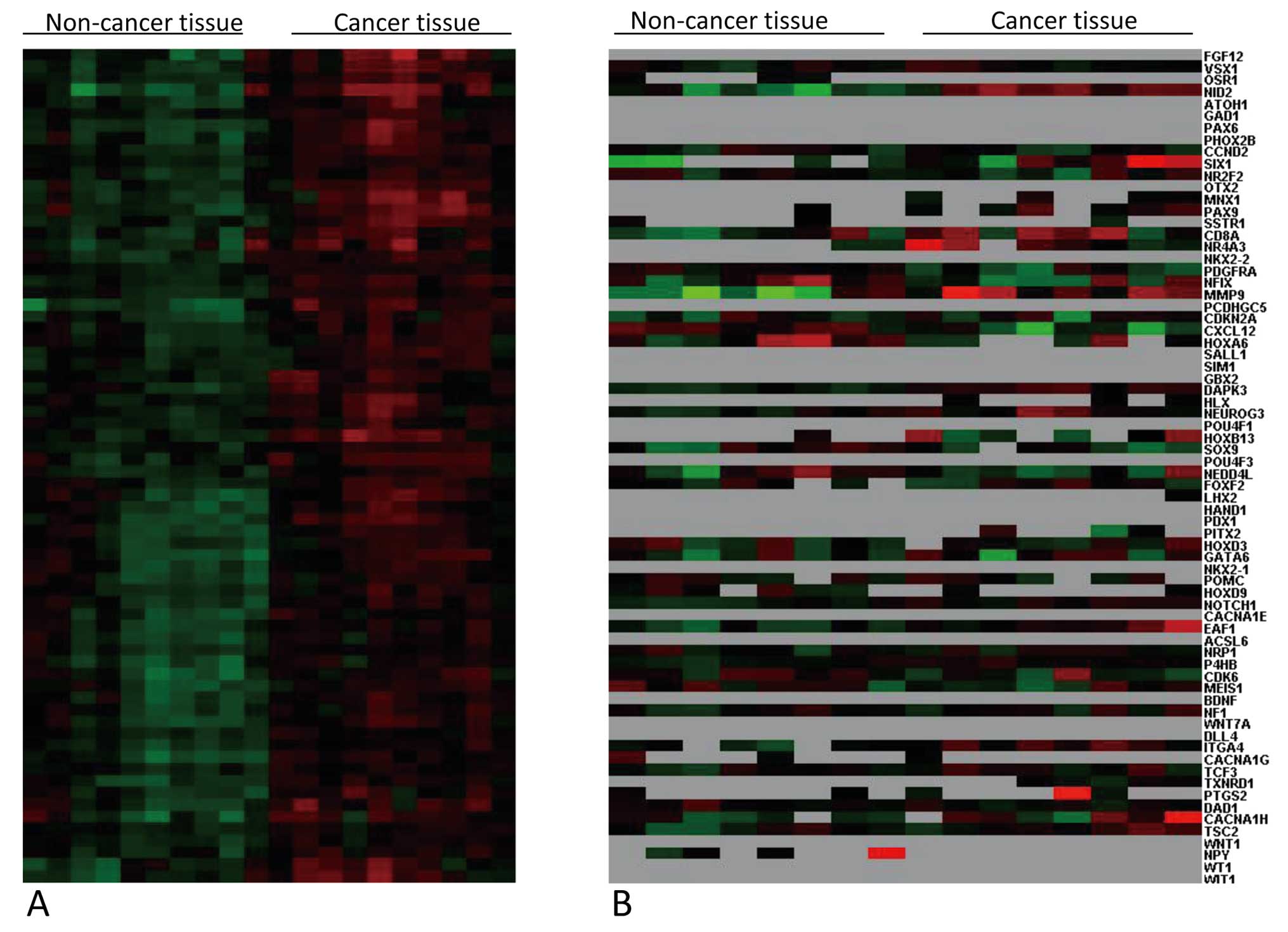
Hierarchical cluster display of these
hypermethylated genes in the 10 cancerous and 10 non-cancerous
breast samples showed 2 separate clusters; one contained all the
breast cancer samples and the other contained all the non-cancerous
breast samples (Fig. 2A).
Enrichment analysis of the GO terms of the 70 hypermethylated genes
showed that these hypermethylated genes were involved in
development, such as the homeobox genes, including the HOX and PAX
gene clusters, and regulation, such as NOTCH1,
NEUROG3, SOX9 and TCF3. The microarray raw
data have been deposited in NCBI’s Gene Expression Omnibus
(http://www.ncbi.nlm.nih.gov/geo) and are
accessible through GEO series accession number GSE33450.
Validation of the hypermethylated
genes
In contrast to traditional bisulfite-modified
sequencing, in which the PCR product is firstly transformed into
single clones, direct bisulfite-modified sequencing directly uses
the PCR product for Sanger sequencing. Even if there are high
background signals, such as heterozygote double peaks at site C of
some CpG islands, originating from PCR products containing
methylated C and T converted from unmethylated C, directing
bisulfite-modified sequencing is time-effective and easily
operated, and therefore has been widely used for rapidly screening
methylated sites and finding methylation levels of target sequences
(16). To confirm the reliability
of our MIRA microarray analysis results, 4 genes were selected for
validation in breast cancer and non-cancerous samples using direct
bisulfite sequencing. Through direct bisulfite sequencing,
widespread methylations were observed in intragenic regions of the
WT1, PAX6 and ITGA4 genes, and in the promoter
region of OTX2 in breast cancer tissue. Methylations were
also observed in non-cancerous tissue for the OTX2 and
WT1 but not for the PAX6 and ITGA4 genes
(Fig. 3).
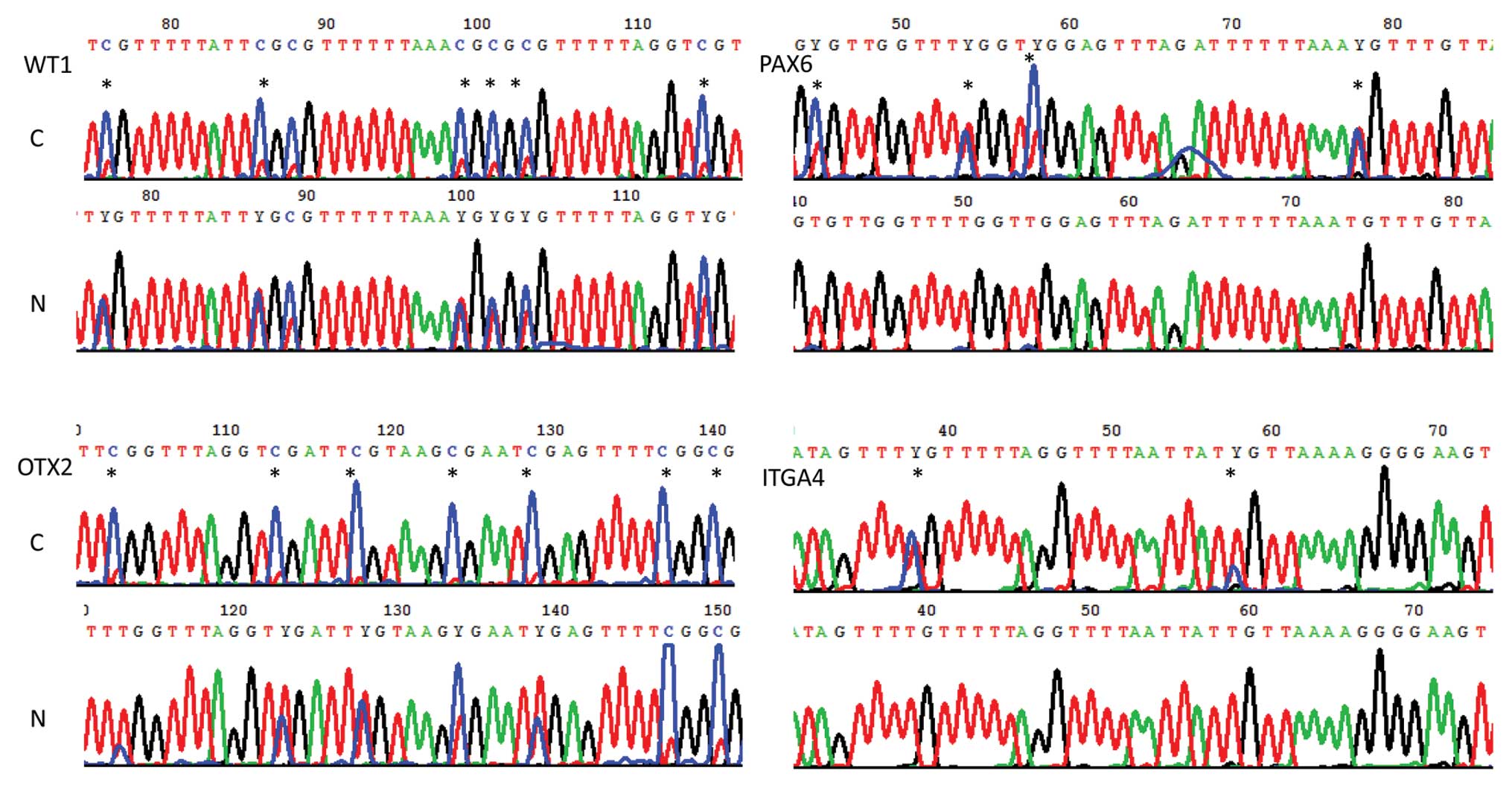 | Figure 3CpG methylations of WT1,
OTX2, PAX6 and ITGA4 were measured by direct
bisulfite sequencing. Hypermethylated CpGs (*) were observed at
nucleotide +262(38) and +282(58) of the ITGA4 gene, at nucleotide
+2094(76), +2104(86), +2117(99), +2119(101), +2121(103) and
+2132(114) of the WT1 gene, at nucleotide +6798(45), +6807(54),
+6811(58) and +6832(79) of the PAX6 gene, and at nucleotide −3992
(103), −4002 (113), −4007 (118), −4018 (129), −4026 (137) and −1029
(140) of positive chain of the OTX2 gene in breast cancer
tissue (C), relative to non-cancerous breast tissue (N). Y
indicates T or C. |
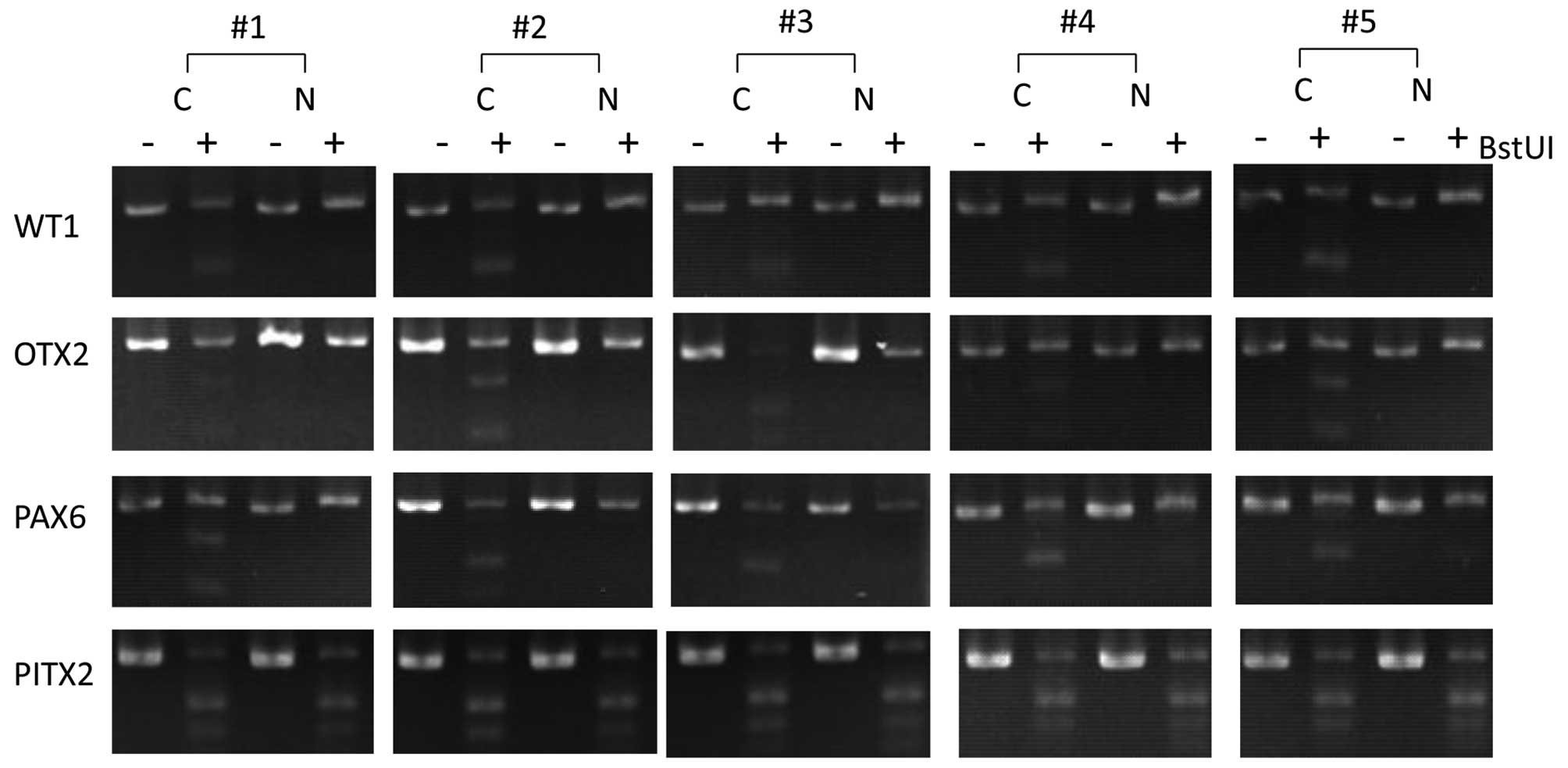
Based on direct sequencing results, we designed
primers that amplify DNA fragments with the richest methylated CpG
and further validated methylation states of the WT1,
OTX2, PAX6 and ITGA4 genes using COBRA assay
in 5 independent cancerous and 5 non-cancerous samples. As shown in
Fig. 4, the WT1,
OTX2 and PAX6 genes were confirmed to be
hypermethylated in breast cancer tissues, compared to non-cancerous
breast tissues. Both direct bisulfite sequencing and COBRA
validation results demonstrate reliability of our MIRA microarray
analysis of DNA methylation.
Relationship between methylation and gene
expression
To further explore the relationship between DNA
methylation and gene expression, we also analyzed gene expression
profiling, with the same batch of cancerous and non-cancerous
samples used in the MIRA microarray analysis, by using gene
expression microarray, which is designed to detect mRNA of 22981
genes (RefSeqAccession). In our study, 62.3% of these genes were
found to be expressed in breast tissues. A total of 1308
transcripts were identified as being significantly differentially
expressed in breast cancer tissues relative to non-cancerous
tissues (GSE33450). Cluster analysis of gene expression data of the
70 hypermethylated genes identified by MIRA microarray revealed
that the relationship between methylation and gene expression was
complex. As shown in Fig. 2B,
expression of some hypermethylated genes (such as MMP9 and NID2) in
cancer tissues were upregulated while expression of some other
genes (such as CXCL12 and NEDD4L) were downregulated. It is worthy
of note that there were many highly methylated genes with
expression levels that were too low to be detected using the gene
expression microarray in both cancerous and non-cancerous breast
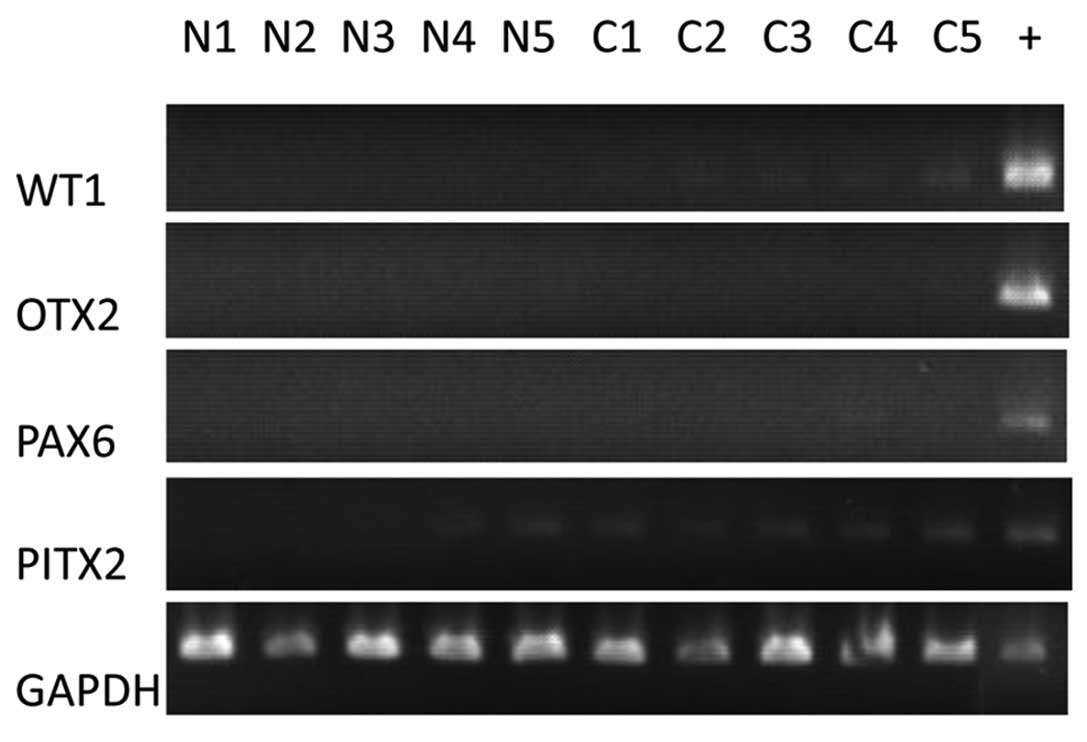
tissues, for examples, PAX6 and WT1. To confirm the gene expression
microarray results, we selected the 4 hypermethylated genes
WT1, PAX6, OTX2 and ITGA4, whose
hypermethylation had been verified by COBRA, for gene expression
detection by RT-PCR assay. The online gene expression database
(www.genecards.org) showed these 4 genes are not
expressed in most breast tissues. For example, SAGE data show that
WT1, OTX2, PAX6 and PITX2 are weakly
expressed in breast cancer tissues and are not expressed in
non-cancerous tissues. Therefore, we used tissues other than breast
tissue as positive controls of RT-PCR for WT1, OTX2
and PAX6. Our RT-PCR results showed that WT1 and
PITX2 were only weakly expressed in the breast cancer
tissues and were not expressed in most non-cancerous tissues.
OTX2 and PAX6 were not expressed in either cancerous
or non-cancerous breast tissues (Fig.
5).
Discussion
In this study, we performed MIRA microarray analysis
in 10 cancerous and 10 non-cancerous breast samples and validated
methylation microarray results in another 5 cancerous and 5
non-cancerous tissues. As the screening was taken at genome-scale
level, the sample size in this study was similar with the sample
size in previous studies which were also performed at a
genome-scale level (10,17). We screened 70 hypermethylated genes
in breast cancer tissues. Numerous hypermethylated genes screened
in this study are homeobox genes. For example, the 4 HOX, 2
PAX and 2 POU genes were found to be hypermethylated
in breast cancer. Our results strengthen the hypothesis that
multiple CpG islands within the HOX clusters and near other
homeobox genes are frequent methylation targets in cancer (11,17).
PITX2 methylation has been reported be
strongly correlated with increased risk of recurrence in breast
cancer (18,19). In this study, we found that
PITX2 was hypermethylated in breast cancer patients.
However, COBRA assay showed that PITX2 was also
hypermethylated in non-cancer patients, suggesting that
PITX2 might not be an ideal methylation biomarker for breast
cancer recurrence in Chinese populations. However, further studies
are required to confirm this hypothesis.
From our MIRA-based methylation screening, a panel
of novel hypermethylated genes that might hold promise as
biomarkers for the early detection of breast cancer was screened,
for example, WT1, OTX2, FGF12, SOX9 and
MNX1. Using COBRA assay, we confirmed that WT1 and
OTX2 were hypermethylated in independent cancerous and
non-cancerous samples. WT1 has been shown to be expressed in
primary breast tumors despite tumor-specific promoter methylation
(20) and its promoter methylation
might not silence the mRNA expression of WT1 during the
development of breast cancer (21). In agreement with previous studies,
WT1 is weakly expressed in breast cancer tissues and is not
expressed in non-cancerous tissues in this study. Contrary to
previous studies, all detected methylated sites of the WT1
gene in our study were located at gene body. Several isoforms of
WT1 have been reported to be expressed in breast tumors (22,23).
Perhaps, intragenic methylation of WT1 is associated with
alternative splicing.
We also explored the relationship between DNA
methylation and gene expression. Gene expression is functionally
more important than DNA methylation and it is generally recognized
that gene promoter hypermethylation results in transcriptional
silence. However, exceptional cases, in which gene expression
levels have nothing to do with their promoter methylation status,
have also been reported. For an example, the promoter of the
vimentin gene has recently been found hypermethylated in
colorectal cancer, while it is not expressed in colorectal tissues
(8). Methylation is also common in
gene body (10). However, the
function of intragenic DNA methylation still remains to be defined.
It has recently been shown that DNA methylation in downstream exons
was only weakly linked to transcription level (24). In our results, the gene body of
WT1, PAX6 and ITGA4 and the promoter of
OTX2 were found hypermethylated in breast cancer tissue and
their expression levels were very low in both cancerous and
non-cancerous breast tissue, suggesting that intragenic DNA
methylation of WT1, PAX6 and ITGA4 and the
promoter methylation of OTX2 may have, if any, only a very
weak function in regulating gene expression.
To display the relationship between methylation and
gene expression from a large perspective, 70 hypermethylated genes
in breast cancer tissues screened by MIRA microarray analysis were
clustered based on their gene expression levels. Our data showed
most of the hypermethylated genes in cancer are not expressed in
either cancerous or non-cancerous tissues. Few studies have paid
attention to this phenomenon before. To validate it, we selected 4
hypermethylated genes that are not expressed in either cancerous
and non-cancerous tissues during tumorigenesis and verified their
methylation status and expression levels using COBRA and RT-PCR,
respectively.
Gene expression microarray based strategy has been
widely used for screening DNA methylation biomarkers on a genomic
scale. This strategy is based on the recognition that gene promoter
hypermethylation results in transcriptional silence. Based on
upregulated genes after demethylating agent treatment,
hypermethylated genes could be detected. In this study, intragenic
DNA methylation of WT1, PAX6 and ITGA4 and the
promoter methylation of OTX2 did not show tight association
with gene expressions. Their methylations would not have been
detected, if gene expression based strategy had been used.
Different from gene expression microarray-based strategy, the MIRA
approach directly detects methylated genes at the DNA level. It can
effectively screen methylated genes irrespective of whether it
expresses mRNA or not. Therefore, we can screen more candidate
methylated genes for further clinical validation using this tool
than the gene expression microarray-based strategy.
To date, no single gene has been found to be
methylated in all types of breast cancer. Methylation signature,
consisting of a panel of genes, has been considered a research
strategy of biomarker for the early detection of breast cancer.
Currently, most studies select genes based on the methylation state
in other cancer modes or known gene function, including regulating
function of methylation on gene expressions. These genes are mostly
tumor suppressor genes, which are hypermethylated in promoter and
highly expressed in non-cancerous tissue. Our results expand our
knowledge of hypermethylated genes and methylation sites in breast
cancer and provide further options other than promoter methylation
of few tumor suppressor genes to study methylation biomarkers of
breast cancer detection, including both single gene methylation and
methylation signature biomarkers. Next, we will further assess the
potential of these hypermethylated genes for their use in the early
detection of breast cancer in well-defined populations, using
accessible samples, for example, nipple aspirate fluid.
In conclusion, we detected 70 genes that were
significantly hypermethylated in promoter and/or gene body in
breast cancer tissues, and most of them were lowly expressed at the
RNA level in both cancerous and non-cancerous breast tissue. These
results expand our knowledge of hypermethylated genes and
methylation sites, deepen our understanding of the relationship
between methylation and gene expression and provide a fundamental
basis to further validate the feasibility of discovered methylation
biomarkers used for the early detection of breast cancer in larger
samples in the future. In addition, we can select more candidate
methylated genes for clinical validation using the MIRA approach
than using the gene expression microarray-based strategy.
Abbreviations:
|
COBRA
|
combined bisulfite restriction
analysis
|
|
RT-PCR
|
reverse transcription PCR
|
|
MIRA
|
methylated-CpG island recovery
assay
|
Acknowledgements
This work was supported by the
Guangdong Natural Science Foundation Grant No. S2011010003747. We
thank Dr Liang Zhang and BioChain (Beijing) Science &
Technology Inc. for their technical assistance.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar
|
|
3
|
Martens JW, Margossian AL, Schmitt M,
Foekens J and Harbeck N: DNA methylation as a biomarker in breast
cancer. Future Oncol. 5:1245–1256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jovanovic J, Ronneberg JA, Tost J and
Kristensen V: The epigenetics of breast cancer. Mol Oncol.
4:242–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brooks J, Cairns P and Zeleniuch-Jacquotte
A: Promoter methylation and the detection of breast cancer. Cancer
Causes Control. 20:1539–1550. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sproul D, Nestor C, Culley J, et al:
Transcriptionally repressed genes become aberrantly methylated and
distinguish tumors of different lineages in breast cancer. Proc
Natl Acad Sci USA. 108:4364–4369. 2011. View Article : Google Scholar
|
|
7
|
Ngan CY, Yamamoto H, Seshimo I, et al:
Quantitative evaluation of vimentin expression in tumour stroma of
colorectal cancer. Br J Cancer. 96:986–992. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen WD, Han ZJ, Skoletsky J, et al:
Detection in fecal DNA of colon cancer-specific methylation of the
nonexpressed vimentin gene. J Natl Cancer Inst. 97:1124–1132. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ruike Y, Imanaka Y, Sato F, Shimizu K and
Tsujimoto G: Genome-wide analysis of aberrant methylation in human
breast cancer cells using methyl-DNA immunoprecipitation combined
with high-throughput sequencing. BMC Genomics. 11:1372010.
View Article : Google Scholar
|
|
10
|
Wu X, Rauch TA, Zhong X, et al: CpG island
hypermethylation in human astrocytomas. Cancer Res. 70:2718–2727.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tommasi S, Karm DL, Wu X, Yen Y and
Pfeifer GP: Methylation of homeobox genes is a frequent and early
epigenetic event in breast cancer. Breast Cancer Res. 11:R142009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rauch TA and Pfeifer GP: The MIRA method
for DNA methylation analysis. Methods Mol Biol. 507:65–75. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Richards AA, Santos LJ, Nichols HA, et al:
Cryptic chromosomal abnormalities identified in children with
congenital heart disease. Pediatr Res. 64:358–363. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Leung TY, Vogel I, Lau TK, et al:
Identification of submicroscopic chromosomal aberrations in fetuses
with increased nuchal translucency and apparently normal karyotype.
Ultrasound Obstet Gynecol. 38:314–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiong Z and Laird PW: COBRA: a sensitive
and quantitative DNA methylation assay. Nucleic Acids Res.
25:2532–2534. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ibanez de Caceres I, Cortes-Sempere M,
Moratilla C, et al: IGFBP-3 hypermethylation-derived deficiency
mediates cisplatin resistance in non-small-cell lung cancer.
Oncogene. 29:1681–1690. 2010.PubMed/NCBI
|
|
17
|
Rauch T, Wang Z, Zhang X, et al: Homeobox
gene methylation in lung cancer studied by genome-wide analysis
with a microarray-based methylated CpG island recovery assay. Proc
Natl Acad Sci USA. 104:5527–5532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maier S, Nimmrich I, Koenig T, et al:
DNA-methylation of the homeodomain transcription factor PITX2
reliably predicts risk of distant disease recurrence in
tamoxifen-treated, node-negative breast cancer patients – Technical
and clinical validation in a multi-centre setting in collaboration
with the European Organisation for Research and Treatment of Cancer
(EORTC) PathoBiology group. Eur J Cancer. 43:1679–1686.
2007.PubMed/NCBI
|
|
19
|
Harbeck N, Nimmrich I, Hartmann A, et al:
Multicenter study using paraffin-embedded tumor tissue testing
PITX2 DNA methylation as a marker for outcome prediction in
tamoxifen-treated, node-negative breast cancer patients. J Clin
Oncol. 26:5036–5042. 2008. View Article : Google Scholar
|
|
20
|
Loeb DM, Evron E, Patel CB, et al: Wilms’
tumor suppressor gene (WT1) is expressed in primary breast tumors
despite tumor-specific promoter methylation. Cancer Res.
61:921–925. 2001.
|
|
21
|
Yang JL, Klinkebiel D, Boland MJ, Tang L
and Christman JK: [Promoter methylation and mRNA expression of WT1
gene in MCF10 breast cancer model]. Zhonghua Bing Li Xue Za Zhi.
36:253–258. 2007.(In Chinese).
|
|
22
|
Silberstein GB, Van Horn K, Strickland P,
Roberts CT Jr and Daniel CW: Altered expression of the WT1 Wilms
tumor suppressor gene in human breast cancer. Proc Natl Acad Sci
USA. 94:8132–8137. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Laux DE, Curran EM, Welshons WV, Lubahn DB
and Huang TH: Hypermethylation of the Wilms’ tumor suppressor gene
CpG island in human breast carcinomas. Breast Cancer Res Treat.
56:35–43. 1999.
|
|
24
|
Brenet F, Moh M, Funk P, et al: DNA
methylation of the first exon is tightly linked to transcriptional
silencing. PLoS One. 6:e145242011. View Article : Google Scholar : PubMed/NCBI
|