Introduction
Over 200,000 cases of renal cancer are diagnosed
annually, with ∼75% of renal cell carcinomas being of the clear
cell subtype (ccRCC). Loss of function of the von Hippel-Lindau
(VHL) tumour suppressor gene occurs in both sporadic ccRCC and VHL
disease, a familial cancer syndrome predisposing to a variety of
malignant and benign tumours including RCC (1,2). The
germline mutation spectrum in patients with VHL disease includes
exon deletion, as well as nonsense, frameshift, splice site and
missense mutations (3). VHL
is a classical tumour suppressor gene and somatic inactivation of
the second copy by LOH, methylation or mutation is observed in
tumours from VHL patients (2,4). The
VHL gene has also been implicated in the majority of
sporadic ccRCC cases, with mutation frequencies of 75–82% reported
(5,6). Together with loss of heterozygosity
(LOH) and promoter methylation, the evidence implicates biallelic
inactivation of VHL in 86% of ccRCC (6).
The VHL gene on chromosome 3p25, consists of
three exons (1,7). It produces two transcripts, and is
expressed in a wide range of tissues and developmental stages
(1,7). The larger transcript contains all
three exons and encodes two biologically active protein isoforms,
pVHL19 and pVHL30. The longer isoform contains all 213 residues of
the VHL ORF whereas the shorter isoform uses an internal
translation start site (8). The
shorter, less abundant transcript (Δ2) lacks exon 2 (2,9). No
protein product from Δ2 has been identified, although in-frame. A
non-expressed processed pseudogene is present on chromosome 1q12
(10).
The primary amino acid sequence of VHL has
little similarity to other sequences in the human genome (1,11)
but conservation amongst mammals is high. The main exception is the
N-terminus which in humans and other higher primates contains eight
copies of an acidic pentamer repeat but has fewer copies in rats
and mice. Lack of mutation coupled with the relatively low
evolutionary conservation of this repeat motif and its absence from
the pVHL19 isoform suggests it is of lesser importance to pVHL
function than the rest of the gene (11).
The role played by pVHL as the substrate recognition
domain of an E3 ubiquitin ligase complex targetting HIF-α for
ubiquitination and degradation by the proteasome is well understood
and stabilisation of HIF-α (in particular HIF-2α) resulting from
loss of VHL function in RCC has been shown to be central to
tumourigenesis (12). However, the
relative importance of HIF-independent pVHL activity remains to be
determined. Knowledge relating to the consequences of disruption of
VHL with stabilisation of HIF and consequent upregulation of
factors such as vascular endothelial growth factor (VEGF) has been
exploited with drugs targeting these pathways such as sorafenib,
sunitinib and bevacizumab which have improved response rates and
relapse-free survival in RCC (13).
Truncating and missense mutations are rarely
reported in the first half of exon 1 but are otherwise distributed
across the entire coding sequence (3,6). Any
correlations between mutation type and aspects of phenotype as
observed in familial VHL (3) have
not yet been found in sporadic RCC although such information might
be of value prognostically and in treatment response. Following our
previous analysis of the genetic and epigenetic status of
VHL in tumour specimens from a large series of patients with
sporadic RCC (6,14), we have extended the
characterisation of a subset of these samples to examine the
functional consequences of such changes on the VHL
transcript to understand further the potential biological
impact.
Materials and methods
Samples and RNA and DNA extraction
Frozen tissue samples from 84 patients with RCC (80
conventional and 4 other subtypes), previously screened for VHL
mutation, methylation and LOH and covering a spectrum of changes
(6,14) were selected for analysis together
with 11 RCC cell lines (Table
IA–C). For tissue samples, 5–10 sections were cut for RNA
extraction from frozen samples embedded in OCT, with flanking
sections used to determine the percentage of tumour cells. Sections
were immersed in 500 μl of RNAlater (Qiagen: http://www1.qiagen.com) and stored at −20°C for 24–72
h. Total-RNA was extracted using RNeasy (Qiagen) following the
manufacturer’s animal tissue protocol and assessed using a Nanodrop
spectrophotometer. RNA was extracted from cultured cells using
RNeasy, and DNA using QIAamp (Qiagen), following the animal cell
protocol.
 | Table IMutation, methylation and RNA
expression status in renal tumour tissue and cell lines. |
Table I
Mutation, methylation and RNA
expression status in renal tumour tissue and cell lines.
A, Tumours and cell
lines with nonsense or frameshift mutations.
|
|---|
| Sample ID | Mutation
(cDNA) | Mutation (amino
acid) | VHL RNA
present | Mutant present in
VHL RNA | Estimated % tumour
cells in sample | Methylation at VHL
promoter detected [tumour data from (6,14)] | Estimated relative
abundance of mutation in DNA and RNA | Site of predicted
termination (codon no.) | Length of
frameshift (no. aa) |
|---|
| R215T | c.171delG | p.R58fs | + | + | 60 | − | Not tested | 66 | 8 |
| R197T | c.183delC | p.V62fs | + | + | 20–40 | + | Not tested | 66 | 4 |
| R35T | c.194C>A |
p.S65* | + | + | 60 | − | Not tested | 65 | n/a |
| R271T | c.194C>A |
p.S65* | + | + | 75 | + | Not tested | 65 | n/a |
| R19T | c.203C>A |
p.S68* | + | + | 25 | − | Not tested | 68 | n/a |
| R138T | c.208G>T |
p.E70* | + | + | 45–80 | − | Not tested | 70 | n/a |
| R189T | c.226_235del10 | p.F76fs | + | + | 70 | − | DNA≈RNA | 155 | 79 |
| R244T | c.248_255del8 | p.V83fs | + | + | 80 | − | DNA>RNA | 128 | 45 |
| R236T | c.274_278del5 | p.D92fs | + | + | 75 | − | DNA>RNA | 129 | 37 |
| R231Ta | c.315_330del16 | p.G106fs | + | + | 75 | + | DNA≈RNA | 153 | 47 |
| R243T | c.337C>T |
p.R113* | + | + | 50 | − | Not tested | 113 | n/a |
| R248Ta | c.350G>A |
p.W117* | + | + | 60–70 | + | DNA>RNA | 117 | n/a |
| R255T | c.360_367del8 | p.A122fs | + | + | 40 | + | DNA>RNA | 128 | 6 |
| R87T | c.400G>T |
p.E134* | + | − | 60 | − | n/a | 134 | n/a |
| R224T | c.404dupT | p.L135fs | + | + | 70 | + | DNA≈RNA | 143 | 8 |
| R166T | c.422dupA | p.N141fs | + | + | 75 | − | DNA>RNA | 143 | 2 |
| R181T | c.443_444delTT | p.F148fs | + | + | 30–50 | − | RNA>DNA | 172 | 24 |
| R117T | c.464_474del11 | p.V155fs | + | + | 60 | − | DNA>RNA | 169 | 14 |
| R182T | c.467dupA |
p.Y156* | + | + | 75 | − | RNA>DNA | 156 | n/a |
| R222T | c.473_498dup26 |
p.R167* | + | + | 75 | − | RNA>DNA | 167 | n/a |
| R177T | c.477delA | p.E160fs | + | + | 80 | + | DNA≈RNA | 169 | 9 |
| R134T | c.481C>T |
p.R161* | + | + | 10–40 | − | DNA≈RNA | 161 | n/a |
| R164T | c.525C>A |
p.Y175* | + | + | 50 | − | DNA≈RNA | 175 | n/a |
| R202T | c.563dupT | p.E189fs | + | + | 60–70 | − | Not tested | 3′UTR | 66 |
| R252T | c.620_635dup16 | p.D213fs | + | + | 30 | − | Not tested | 3′UTR | 47 |
| Cell lines | | | | | | | | | |
| 786-0b,c | c.311delG | p.G104fs | + | + | n/a | − | n/a | 158 | 54 |
| HTB47b,d | c.529A>T |
p.R177* | + | + | n/a | − | n/a | 177 | n/a |
| HTB44b,e | c.426_429del4 | p.G144fs | + | + | n/a | − | n/a | 158 | 13 |
B, Tumours and cell
lines with missense mutations.
|
|---|
| Sample ID | Mutation
(cDNA) | Mutation (amino
acid) | VHL RNA
present | Mutant present in
VHL RNA | Estimated % tumour
cells in sample | Methylation at VHL
promoter detected [tumour data from (6,14)] |
|---|
| R185Tk | c.25G>A | p.D9N | + | + | 80 | − |
| R187T | c.74C>T | p.P25L | + | + | 40–60 | + |
| R124T | c.232A>G | p.N78D | + | + | 70–85 | − |
| R157T | c.256C>G | p.P86A | + | + | 80 | − |
| R247T | c.262T>A | p.W88R | + | + | 80 | + |
| R261T | c.269A>T | p.N90I | + | + | 80 | − |
| R292T | c.302T>C | p.L101P | + | + | 75 | − |
| R160T |
c.320_321delinsCA | p.R107P | + | + | 85–90 | − |
| R283T | c.340G>C | p.G114R | + | − | 40–60 | + |
| R69T | c.341G>C | p.G114D | + | − | 75 | − |
| R10T | c.343C>G | p.H115D | + | + | 90 | + |
| R17T | c.343C>A | p.H115N | + | + | 50 | − |
| R273T | c.349T>C | p.W117R | + | + | 80 | − |
| R183T | c.361G>T | p.D121Y | + | + | 65–70 | + |
| R267T | c.388G>C | p.V130L | + | + | 95 | + |
| R142T | c.406T>G | p.F136V | + | + | 65–90 | − |
| R295T | c.413C>G | p.P138R | + | + | 80 | − |
| R294T | c.446C>A | p.A149D | + | + | 70 | − |
| R123T | c.452T>A | p.I151N | + | + | 75 (s1
necrotic) | − |
| R118T | c.461C>T | p.P154L | + | − | 40 | − |
| R151T | c.473T>A | p.L158Q | + | + | 60–75 | − |
| R279T | c.485G>A | p.C162Y | + | + | 75 | + |
| R218T | c.497T>G | p.V166G | + | + | 70 | + |
| R200T | c.551T>C | p.L184P | + | + | 60 | + |
| Cell lines | | | | | | |
|
CRL1933b,f | c.539T>A | p.I180N | − | n/a | n/a | + |
| RCC4b,g | c.194C>G | p.S65W | + | + | n/a | − |
| A704b,h | c.539T>A | p.I180N | − | n/a | n/a | + |
C, Tumours with
intronic mutations.
|
|---|
| Sample ID | Mutation
(cDNA) | Mutation (amino
acid) | VHL RNA
present | Mutant present in
VHL RNA | Estimated % tumour
cells in sample | Methylation at VHL
promoter detected [tumour data from (6,14)] |
|---|
| R1T |
c.332_340+1del10 | n/a | + | − | 65 | + |
| R86T | c.340+1G>T | n/a | + | − | 90 | + |
| R167T | c.340+2delT | n/a | + | − | 75 | + |
| R139T | c.341-1G>A | n/a | + | − | 45 | + |
| R281T | c.[ 341-11T>A
(;)463+43A>G] | n/a | + | − | 50–60 | + |
| R179T | c.463+43A>G | n/a | + | − | 80 | + |
| R230T | c.463+43A>G | n/a | + | − | 75 | + |
| R233T | c.463+43A>G | n/a | + | − | 60 | + |
| R276T | c.463+43A>G | n/a | + | − | 80 | − |
D, Tumours and cell
lines without mutations.
|
|---|
| Sample ID | Mutation
(cDNA) | Mutation (amino
acid) | VHL RNA
present | Mutant present in
VHL RNA | Estimated % tumour
cells in sample | Methylation at VHL
promoter detected [tumour data from (6,14)] |
|---|
| Tumours | | | | | | |
| R108Tk | No mutation
detected | n/a | + | n/a | 95 | − |
| R128T | No mutation
detected | n/a | + | n/a | 80 | − |
| R140T | No mutation
detected | n/a | + | n/a | 35–75 | − |
| R143T | No mutation
detected | n/a | + | n/a | 10 | − |
| R150T | No mutation
detected | n/a | + | n/a | 40 | − |
| R153Tk | No mutation
detected | n/a | + | n/a | 85 | − |
| R158Tk | No mutation
detected | n/a | + | n/a | 70 | − |
| R163T | No mutation
detected | n/a | + | n/a | 80–90 | − |
| R165T | No mutation
detected | n/a | + | n/a | 80–85 | − |
| R195T | No mutation
detected | n/a | + | n/a | 95 | − |
| R201T | No mutation
detected | n/a | + | n/a | 20–30 | − |
| R203T | No mutation
detected | n/a | + | n/a | 90 | − |
| R207T | No mutation
detected | n/a | + | n/a | n/d | − |
| Tumours | | | | | | |
| R208T | No mutation
detected | n/a | + | n/a | n/d | − |
| R217T | No mutation
detected | n/a | + | n/a | 80 | − |
| R220T | No mutation
detected | n/a | + | n/a | 70 | − |
| R225Tk | No mutation
detected | n/a | + | n/a | 0 | − |
| R226T | No mutation
detected | n/a | + | n/a | 75–85 | − |
| R228T | No mutation
detected | n/a | + | n/a | 65–75 | − |
| R230T | No mutation
detected | n/a | + | n/a | 80–95 | + |
| R242T | No mutation
detected | n/a | + | n/a | 30–50 | + |
| R253T | No mutation
detected | n/a | + | n/a | 80–90 | − |
| R256T | No mutation
detected | n/a | + | n/a | 60 | + |
| R280T | No mutation
detected | n/a | + | n/a | 70–75 | − |
| R282T | No mutation
detected | n/a | + | n/a | 90 | + |
| R290T | No mutation
detected | n/a | + | n/a | 90 | + |
| Cell lines | | | | | | |
| HTB46b,i | No mutation
detected | n/a | + | n/a | n/a | − |
| SN12Cb,j | No mutation
detected | n/a | + | n/a | n/a | − |
| TK10b,j | No mutation
detected | n/a | + | n/a | n/a | − |
| HTB49b | No mutation
detected | n/a | − | n/a | n/a | + |
| U031b,j | No mutation
detected | n/a | + | n/a | n/a | n/d |
Mutation screening of cell line DNA
Screening for mutations in genomic DNA from cell
lines was carried out by direct DNA sequencing using the primers
and conditions previously described (6).
RT-PCR and PCR
VHL mRNA was amplified using primer set 9
(codons 1–170) and/or primer set 3 (codons 89 to 214) depending on
the location of the mutation. Primer sequences are: set 3 forward
(M3537) cgtcgtgctgcccgtatg and reverse ccatcaaaagctgagatgaaacag
(M3548); set 9 forward cccgggtggtctggatcg (M3529) and reverse
tggcaaaaataggctgtcc (M3540). RT-PCR was specific for fully
processed VHL mRNA and did not amplify unprocessed
VHL pre-mRNA, VHL from co-purified genomic DNA or the
processed VHL pseudogene. To achieve this, the 3′ end at
least one of each pair of primers was positioned over a nucleotide
which discriminates between VHL and the pseudogene and each
primer pair spanned at least one exon-exon junction.
RNA-specificity of RT-PCT products was further verified by showing
the absolute RT-dependence of the reaction by heat-inactivation of
the reverse transcriptase prior to addition of RNA (data not
shown). Sequencing of RT-PCR products confirmed that they were
derived from the gene and not from the pseudogene. The VHL
splice variant lacking exon 2 (Δ) co-amplified with the full length
product when using primer set 3.
One step RT-PCR was carried out using Superscript
III one-step RT-PCR kit (Invitrogen). Reactions contained 10 ng of
total RNA, 10 pmol forward primer, 10 pmol reverse primer, 1X
Superscript III reaction buffer, 0.4 μl of Superscript III enzyme
mix and, for primer set 9 only, 10% DMSO in a total volume of 10
μl. For primer set 3, cycling conditions were: reverse
transcription at 55°C for 30 min, denaturation at 94°C for 5 min,
38 cycles of denaturation at 94°C for 15 sec, annealing at 60°C for
30 sec and extension at 68°C for 1 min. For primer set 9, cycling
conditions were as above except that reverse transcription was
carried out at 60°C and the cycle number was increased to 40.
SDHA (chromosome 5p) and HPRT1 (Xq26) were used as
controls for RT-PCR in samples from which no VHL RT-PCR
product was obtained. Primers were: SDHA forward tgggaaca
agagggcatctg and reverse ccaccactgcatcaaattcatg and HPRT1
forward gacactggcaaaacaatgca and reverse cttcgtggggtccttt tcacc.
RT-PCR conditions were as described as for VHL primer set 3
except annealing was at 55°C and the cycle number was increased to
40. 4 μl of each RT-PCR product were examined using 1.5% agarose
gels in 1X TBE buffer to determine yield and purity.
PCR from trace levels of DNA present in the RNA
preparations was carried out in order to confirm the presence of
mutations absent from RNA or to compare the relative levels of
variants in DNA and RNA. Reactions contained 10 ng of total RNA, 10
pmol forward primer, 10 pmol reverse primer and 5 μl of HotStarTaq
master mix (Qiagen) in a total volume of 10 μl. Cycling conditions
were: denaturation at 95°C for 15 min, 40 cycles of (denaturation
at 95°C for 30 sec, annealing at 60°C for 30 sec and extension at
72°C for 30 sec), final extension at 72°C for 10 min. Primers were
as follows: M3537 VHL1_1F cgtc gtgctgcccgtatg; M3563 VHL 1_2F
gctgcgctcggtgaactcg; M3538 VHL1_1R accgtgctatcgtccctgct; M3539
VHL2_1F ggctctttaacaacctttgctt; M3540 VHL2_1R tggcaaaaataggctgtcc;
M3541 VHL2_2F ccaaactgaattatttgtgccatc; M3542 VHL2_2R
tggtctatcctgtactyaccacaacaa; M3543 VHL3_1F gaccctagtctgcc actgagga;
M3544 VHL3_1R agagcgacctgacgatgtcc.
Sequencing
RT-PCR and PCR products were prepared for sequencing
by treatment of 2.5 μl of product with 1 μl of ExoSAP-IT (USB,
http://www.usbweb.com) and 10 μl sequencing
reactions were carried out using 1 μl of prepared PCR product, 1.6
pmol of primer, BigDye ready reaction mix (Applied Biosystems,
http://www3.appliedbiosystems.com)
version 3.1 diluted 1 in 8 with Half Big Dye reagent (Genetix,
http://www.genetix.com/). Cycle sequencing
conditions were 25 cycles of 96°C for 10 sec, 50°C for 5 sec and
60°C for 4 min. Unincorporated nucleotides and primers were removed
by ethanol precipitation. Sequencing products were resuspended in
20 μl of formamide and run on a 3130 genetic analyser (Applied
Biosystems) using POP7 and 36 cm well-to-read capillaries. Data
analysis was carried out by visual inspection of electropherograms
and using Mutation Surveyor software (SoftGenetics). RT-PCR product
9 and all PCR products were sequenced using the same primers that
had been used for RT-PCR/PCR. RT-PCR product 3 was sequenced using
forward primer tccacagctaccgaggtcac and reverse primer
tctttcagagtatacactgg. These primers bind over the exon 1–2 and exon
2–3 junctions, respectively, and consequently do not bind to the
exon 2-lacking product, which co-amplifies in the primer set 3
reaction.
Methylation analysis
The methylation status of the VHL promoter was
examined by methylation-specific PCR (15). Genomic DNA (1 μg) was
bisulphite-treated using EZ DNA methylation kit (Zymo Research).
Methylated and unmethylated DNA were amplified in separate
reactions with primers as previously described (14). CpGenome universal methylated DNA
(Millipore) was used as a control. Products were examined by
agarose gel electrophoresis.
Prediction of splice sites and exonic
splicing enhancers (ESE)
Splice site prediction by neural network (http://www.fruitfly.org/seq_tools/splice.html) was
used to predict the effect of mutations on splice sites. The
presence of exonic splicing enhancers was predicted using ESEfinder
3.0 (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home)
together with possible effects of mutations.
Results
The tumour samples were selected to give a
distribution of point mutations and premature termination codons
(PTCs) throughout the 3 coding exons of VHL (Table I) with samples from 56 patients
containing 52 unique VHL mutations or polymorphisms (Table IA–C) together with a further 28
tumours (24 ccRCC and 4 other subtypes) in which no variant had
been identified. In the tumour samples, based on the predicted
change to the primary amino acid sequence, 24 of the mutations were
missense, 11 resulted in frameshifted amino acid sequence followed
by a premature truncation codon (PTC), 9 resulted in immediate
truncation at the site of the mutation and 2 were frameshifts which
extended the reading frame beyond the normal stop codon into the
3′UTR. Tumours with intronic variants (Table IC) included three tumours with
intronic substitutions affecting invariant residues at splice
sites, one tumour with a deletion which removed nucleotides
spanning the exonintron junction (R1T), and one tumour with an
intronic variant of unknown function (R281T). An intronic
polymorphism, c.463+43 A>G (rs34661876) was present in 5
tumours. This polymorphism and two of the missense variants (p.D9N
and p.P25L (rs35460768)) were germline in origin, all of the other
variants were somatic (6,14). p.P25L is not associated with VHL
disease (3). p.D9N has not, to our
knowledge, been reported previously, but affects a non-conserved
residue at the extreme N-terminus and is unlikely to be associated
with VHL disease. Additionally 11 RCC cell lines were analysed.
Truncating mutations were identified in three cell lines (Table IA), missense mutations in a further
three (Table IB), while five had
no detectable mutation (Table ID).
All mutations detected were homozygous. Although mutation status of
many of these cell lines has previously been reported, mutation
nomenclature may have used alternative nucleotide numbering
systems, so for consistency we have included these results in
Table I. In the cell lines
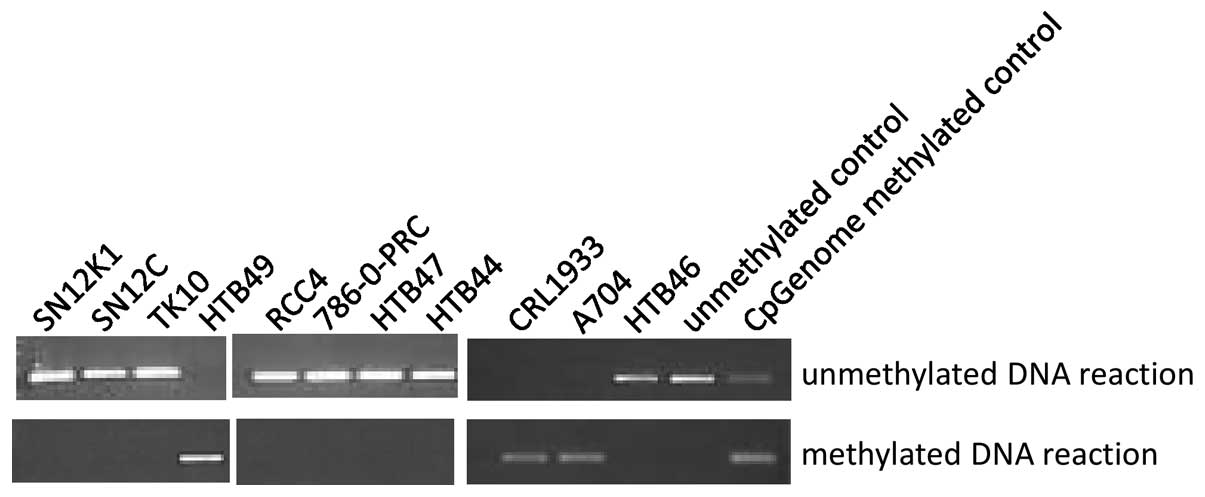
CRL1933, A704 and HTB49 the VHL promoter was methylated (Fig. 1) and no VHL RT-PCR product
was found in these 3 cell lines only although control reactions
confirmed the presence of amplifiable RNA in these samples (data
not shown). CRL1933 and A704 were also homozygously mutated,
showing that mutation and methylation can occur on the same
allele.
VHL produces two mRNA species (2); the full length product consisting of
exons 1–3 and a shorter product lacking exon 2 (Fig. 2A). No aberrantly sized bands
indicating an effect on pre-mRNA splicing were seen in any sample
other than R222T which has 26 bp duplication (data not shown);
smaller insertion or deletion mutations present in other samples
were below the resolving power of the system. An increased
intensity of the lower band when compared to the upper band was
seen in four tumours (Fig. 2B).
R17T and R69T both have exonic mutations, c.343C>A and
c.341G>C respectively, located close to the exon 2 splice
acceptor site. R281T contains two variants, c.463+43A>G and
c.341-11T>A. None of the tumours with c.463+43A>G alone
showed any change to the product ratios, suggesting that the rare
somatic variant c.341-11T>A could be responsible for the change.
One other tumour, R248T, in which we had not previously detected a
variant, showed a more subtle change to the band ratio.
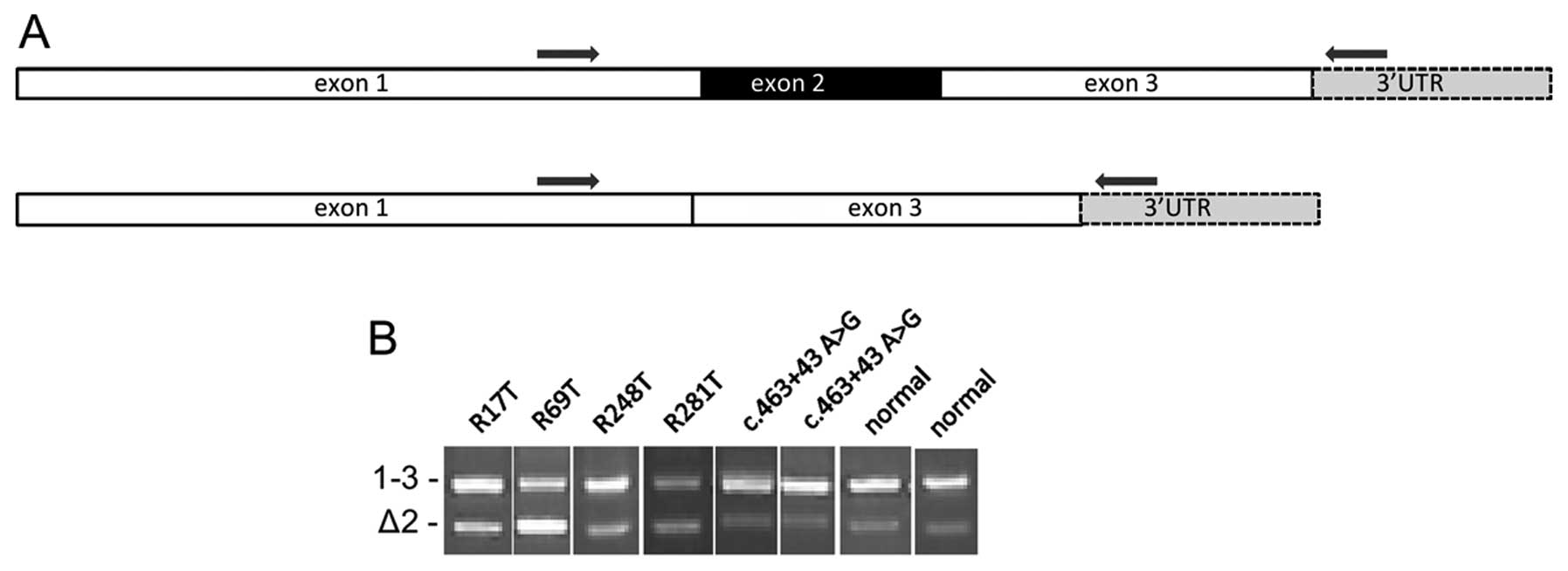 | Figure 2Agarose gel images of RT-PCR
products. (A) Schematic showing the two transcripts produced from
the VHL gene. Amplification using primer set 3, in which the
primers are located in exons 1 and 3, produces two products. The
larger product, 1–3, (442 bp) is produced from full length VHL
mRNA. The smaller product, Δ2, (319 bp) lacks exon 2. (B) Agarose
gel images of RT-PCR products showing 1–3 and Δ2 products. In
normal kidney tissue, the Δ2 product is much weaker than the full
length product. The same pattern was observed in the majority of
the tumour samples (data not shown). We observed four tumours in
which the relative intensity of the bands was notably different.
Tumour R69T had a clearly brighter lower band; in tumour R281T both
bands were of similar intensity and in tumours R17T and R248T,
although the full length band was stronger, there was less
difference in intensity between the two bands than in control
samples. Also shown are two tumours which contain the common
polymorphism c.463+43 A>G. As this image is compiled from
several individual gels, the absolute distance separating the two
bands may differ but in every case the mobility relative to
molecular weight markers and to controls was the same. |
The VHL RT-PCR products were sequenced in
order to confirm use of the correct splice sites and to determine
whether exonic mutations were present in the message. Splice site
usage was exclusively at the wild-type sites, no activation of
local cryptic splice sites was observed. Mutant RNA containing the
expected exonic variant was detected in all 13 tumours containing
frameshift variants, 9/10 tumours containing nonsense variants and
21/24 tumours containing missense variants (Table IA and B). Mutant RNA was also
detected in each of the four cell lines which had mutations and
expressed VHL RNA. There were four tumours in which we were unable
to detect the previously identified mutation. One of these tumours
had a nonsense variant, c.400G>T/p.E134*; the other
three tumours had the missense variants c.340G>C/p.G114R,
c.341G>C/p.G114D and c.461C>T/p.P154L. Previous work showed
methylation of the VHL promoter in the tumour with c.340G>C but
not in the other three tumours (6,14).
The tumour R1T has a 10 bp deletion,
c.332_340+1del10, which removes the final 9 nucleotides of exon 1
and the first nucleotide of the splice donor site in intron 1.
Deletion of a splice site would be expected to prevent splicing,
and splice site prediction software indeed predicts that the splice
site is completely destroyed (Table
II). Sequencing of the RT-PCR product from R1T, as expected,
showed only wild-type sequence, consistent with the mutant being
unable to form a mature VHL mRNA.
| Table IIPredicted effects on splice site
strength using http://www.fruitfly.org/seq_tools/splice.html. |
Table II
Predicted effects on splice site
strength using http://www.fruitfly.org/seq_tools/splice.html.
| Splice site | Tumour | Variant | Predicted
strength |
|---|
| Exon 1 donor | | Wild-type | 0.99 |
| R1T | c.332_340+1 del
10 | No site
predicted |
| R283T | c.340 G>C | 0.75 |
| R86T | c.340+1 G>T | No site
predicted |
| R167T | c.340+2 del T | No site
predicted |
| Exon 2
acceptor | | Wild-type | 0.97 |
| R281T | c.341-11
T>A | 0.88 |
| R139T | c.341-1 G>A | No site
predicted |
| R69T | c.341 G>C | 0.85 |
| R17T | c.343 C>A | 0.97 |
| R10T | c.343 C>G | 0.99 |
| R248T | c.350 G>A | 0.97 |
| Exon 2 donor | | Wild-type | 0.82 |
| R118T | c.461 C>T | 0.46 |
| R179T, R230T,
R233T, | c.463+43
A>G | 0.82 |
| R276T, R281T | | |
| Exon 3
acceptor | | Wild-type | 0.84 |
| R117T | c.464_474
del11 | 0.96 |
Sequencing of the RT-PCR products from the 28
tumours in which no mutation had previously been identified
revealed the presence of mutations in two tumours, c.350G>A and
c.315_330del16 (Table IA). In the
five mutant samples in which no mutation was present in RNA and the
two samples in which a mutation was newly identified in the RNA,
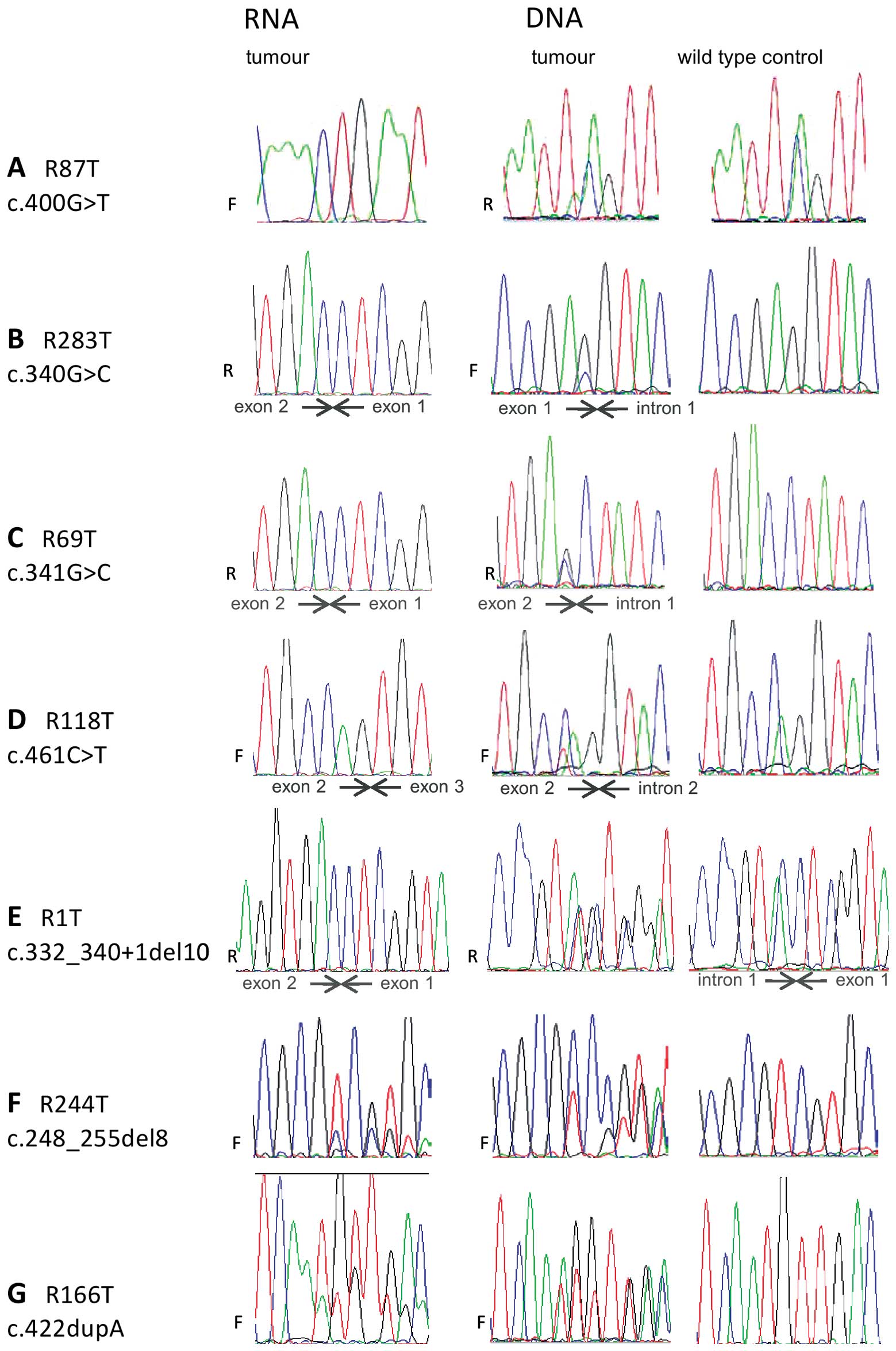
co-purifying DNA was examined. In R1T, from which the absence of
the mutation from RNA was not unexpected, as well as each of the
four samples where the expected mutation was not detected in RNA,
the mutation was clearly present in DNA, confirming that the sample
contained sufficient tumour material for the mutation to be
detectable (Fig. 3A–E). In the two
samples in which mutations were newly detected, the mutations were
clearly present in DNA (data not shown) and our failure to detect
these in our previous study presumably reflects the heterogeneity
of the tissue samples.
Nonsense-mediated decay may reduce the level of
mutant RNA in tumours containing premature termination codons
(PTCs). We compared the level of mutation in RNA and DNA in samples
containing PTCs and expressing mutant RNA except those where the
mutation was located 5′ of nucleotide c.208, as no satisfactory PCR
product could be obtained from these samples. In each case, the
expected mutation was present in co-purifying DNA. However, the
level of mutation observed in the DNA sequencing traces from R117T,
R166T, R244T, R246T, R248T and R255T was markedly different from
the level observed in RNA sequencing traces (Fig. 3 F–G, Table I).
Splice site prediction software (16) was used to examine whether there was
a change to the predicted strength of the splice sites of variants
located in introns, within the few nucleotides adjacent to introns
or in samples which had altered ratios of RT-PCR products (Table II). For the four variants which
affected absolutely conserved residues at splice sites, the
software predicted that the variant eliminated the existing splice
site. However, none of these samples had any evidence for exon
skipping as determined by altered RT-PCR product ratios. Five
variants either produced no change or increased the strength of the
prediction. These were the common variant c.463+43A>G, the
substitutions c.343C>A, c.343C>G and c.350G>A which are
located 3–10 nucleotides into exon 2 and c.464_474 del 11 which
removes the first 11 nucleotides of exon 3. Of these, c.343C>A
and c.350G>A had altered band ratios by RT-PCR suggestive of
exon skipping whereas the others had a normal appearance. Four
variants had reduced scores and of these c.341G>C and
c.341-11T>A had a RT-PCR band ratio suggestive of exon skipping
whereas c.340G>C and c.461C>T had a normal appearance. The
possibility of c.341-11T>A introducing a cryptic splice site was
considered but although the donor splice site consensus sequence
YAG is introduced 9 nucleotides 5′ of the true exon 2 donor site,
the software did not predict the creation of a site and no RT-PCR
products using this cryptic site were observed. Finally, the
variant c.400G>T, in which mutant RNA is absent was examined,
and no creation of a cryptic splice site was predicted (data not
shown).
Exonic splicing enhancers (ESEs) are cis-acting
signals located within exons which act as binding sites for
proteins essential for splicing. Using ESE-finder software
(17) to examine the predicted
effects of the mutations c.340G>C, c.341G>C, c.343C>T,
c.350G>A, c.400G>T and c.461C>T which have experimental
evidence to suggest that they perturb RNA processing, only
c.341G>C and c.461C>T showed any changes in predicted ESEs:
an SF2/ASF binding site was created in c.341G>C and an SF2/ASF
binding site was replaced with a SC35 binding site in
c.461G>C.
Discussion
Examination of VHL mRNA in RCC clearly
complements genomic analyses illustrating the unpredictability of
some findings and the potential for multiple truncated forms of VHL
protein based on the presence of transcript. Four tumours showed an
increase in the Δ2 product relative to the full length RT-PCR
product. R17T and R69T both have exonic mutations, c.343C>A and
c.341G>C respectively, located close to the exon 2 splice
acceptor site (discussed further below). In R281T, c.341-11T>A
is the only rare variant we have detected. Predictions using this
sequence change (Table II) suggest
a small decrease in the strength of the exon 2 splice acceptor,
which may be able to account for the exon skipping observed. This
mutation is within the polypyrimidine tract, a loosely defined
sequence which is involved in 3′ splice site definition (18). Rare reports of polypyrimidine tract
mutations exist (19). R248T
contains the exonic variant c.350G>A. No changes to splice site
strength (Table II) or to ESE
sequences were predicted and the cause of exon skipping is unclear.
PCR bias towards shorter products could contribute toward a bias to
under-reporting of use of distant cryptic sites and intron
retention. However, exon skipping is the most common consequence of
mutation-related aberrant splicing, followed by use of local
cryptic sites, with intron retention accounting for only a minority
of cases (20,21). Other reported examples of aberrant
splicing of VHL also describe mutations which cause skipping
of exon 2 (2,22). As germline deletion of exon 2 alone
is sufficient to cause VHL disease (23), Δ2 is unlikely to retain VHL
function. Four tumours (R1T, R86T, R139T, R167T) have mutations
which affect at least one of the absolutely conserved nucleotides
within consensus splice sites (24) and all are predicted to remove the
affected splice site (Table II).
Interestingly, none of the VHL RNA from of these tumours
showed an increase in the Δ2 isoform. The exon skipping mutations
reported previously (2,22) affect either conserved but not
invariant intronic residues or exonic residues, as do the mutations
that we have observed which cause exon skipping.
Coding exon sequences describe the primary amino
acid sequence of the protein but the same nucleotides may also
contain information required for the correct processing of gene
transcripts (reviewed in ref. 25). Mutations which appear to be silent,
missense, nonsense or frameshift in their effects on the amino acid
sequence, may instead act by disrupting RNA processing by a variety
of mechanisms including creation of cryptic splice sites (26,27),
disruption of ESE sequences (28,29)
and disruption of the exonic portion of consensus splice sites
(30,31). We observed three exonic
substitutions (c.340G>C, c.341G>C and c.461C>T) which
encode missense changes to the primary amino acid sequence and in
which mutant RNA was not detectable (Fig. 3). These mutations are located,
respectively, in the final nucleotide of exon 1, the first
nucleotide of exon 2 and at the −3 position of exon 2. Two further
mutations included in this study were located at the +3 position of
exon 2: c.243C>A and c.243C>G, of which c.243C>A but not
c.243C>G showed evidence for exon skipping. The three tumours
which had no detectable mutant RNA all had mutations which reduced
the predicted strength of the splice site (Table II). Exonic nucleotides immediately
adjacent to the consensus splice site sequences have non-random
nucleotide sequences, with the 3′-most nucleotide of the exon
having the greatest sequence constraint (24,32).
Disease-causing mutations are reported in most of these positions,
most frequently at the 3′-most nucleotide of the exon (21).
As c.340G>C, c.341G>C and c.461C>T do not
produce mutant mRNA, they must therefore function solely as
splicing mutations. c.243C>A appears to be associated with some
degree of exon skipping but is still detectable in VHL mRNA
and may produce its effects through multiple mechanisms including
splicing and missense change to pVHL whereas c.243C>G does not
appear to manifest any deleterious consequences at the RNA
level.
Introduction of a PTC into a gene product accounts
for a substantial proportion of the mutations underlying both
inherited genetic disease and many forms of cancer (33). Of familial VHL mutations, 13% are
frameshift and 11% are nonsense (3) while in sporadic RCC we have found 51%
of detected mutations to be frameshift and 11% nonsense (6). Nonsense-mediated decay (NMD)
(reviewed in refs. 34,35) degrades mRNAs which contain PTCs,
protecting cells from potentially deleterious consequences of
truncated proteins. Numerous studies report severe reduction in the
level of mRNAs containing nonsense codons (36–38).
However, not all PTCs elicit NMD and the level of reduction can be
highly variable (39,40). Location of the PTC in the
transcript can influence susceptibility to NMD (39,40)
with PTCs in the 3′ terminal exon or within 50–54 nucleotides of
the final exon-exon junction not triggering NMD (41). In the case of VHL, only
those mutations which introduce PTCs into exon 1 or into the 5′ end
of exon 2 (before codons 137–138) could trigger NMD. Because
VHL contains fairly long ORFs in both alternative reading
frames, it is possible for the PTC associated with a mutation to be
located some considerable distance downstream of the mutation. Two
of the tumours in this study (R189T and R231T) have sufficiently
long ARFs that the PTC is located after the 50–54 nt boundary,
although the mutation itself is located well before the
boundary.
Previously, two cell lines with frameshift mutations
which terminated after the 50–54 nt boundary were shown not to be
susceptible to NMD (42). Of four
other mutations examined (43),
two of which were of the intronless Drosophila Vhl gene, the
data showed that in both humans and Drosophila, the presence
of a truncating mutation does not necessarily trigger NMD and that
the site of the PTC within the transcript was likely to be the
factor which determined whether NMD occurred (43). In 22 of 23 RCC tumours containing
PTC mutations, mutant mRNA was readily detectable (Table IA). In one tumour, which contained
the nonsense-variant p.E134*, mutant mRNA was
undetectable (Table IA) and as the
PTC associated with this mutation is located just 5′ of the 50–54
nt boundary, NMD is a plausible explanation. In a further 6 tumours
out of 15 in which the relative abundance of mutant RNA and DNA was
estimated, there was evidence to support a detectable reduction in
the amount of mutant RNA. Five of the seven tumours showing
evidence for loss of mutant mRNA had PTCs which were located more
than 50–54 nucleotides upstream of the final exon-exon boundary.
The two tumours (R189T and R231T) in which a mutation located prior
to the 50–54 nt boundary introduces a PTC after the boundary did
not show any evidence for reduction of mutant RNA compared to
mutant DNA. The cell lines 786-0-PRC, HTB47 and HTB44 contain
truncating mutations, all of which result in a PTC located 3′ of
the 50–54 nt boundary and all expressed mutant VHL RNA. Our
study greatly expands the number of truncating VHL mutations
which have been examined at the RNA level. Our data suggest that in
the majority of RCC tumours in which VHL has a truncating
mutation, even if the PTC is located before the 50–54 nt boundary,
mutant mRNA is present, albeit at a reduced level in some tumours.
There is therefore the potential for the production of truncated
VHL protein in these RCCs which may exhibit partial function. This
could ideally be checked by western blotting providing antibodies
but would ideally require a range of antibodies to cover unaffected
epitopes and additionally our studies find that currently available
antibodies are only able to detect VHL protein when grossly
overexpressed in transfectant cell lines and not at endogenous
levels.
In our previous study with 74.6% of RCC cases having
a detectable VHL mutation and 31.3% methylation of the
VHL promoter (6), 32
tumours had both mutation and methylation and of these, 30 also had
LOH. Although other studies have observed methylation and mutation
to be mutually exclusive (5), it
is possible, especially where LOH has also occurred, that mutation
and methylation have occurred on the same allele, although tumour
heterogeneity may also explain the finding. Our finding that
CRL1933 and A704 contain only mutant VHL and have only
methylated DNA show mutation and methylation are not mutually
exclusive in these cell lines. The link between VHL promoter
hypermethylation and silencing of the gene is well recognised
(44) and all cell lines showing
promoter methylation had complete absence of VHL RNA
(Fig. 1). Seven of the 23 tumours
containing truncating mutations (30.4%) and 8 of 24 tumours with
missense mutations (33.3%) also had methylation providing an
alternative explanation for the lack of or reduced level of
detectable mutant RNA. However, 3 of the four tumours in which the
exonic mutation was not detected in RNA were not methylated.
Eight of the 9 tumours with intronic variants also
had methylation (88.9%) which differs markedly not only to the
other two categories and but also to the average for unselected
tumours. It is, perhaps, unsurprising that there should be a high
level of methylation of tumours with the polymorphism
c.463+43A>G. It is more surprising that all of the tumours with
intronic splicing mutations should also have methylation although
possibly a consequence of the small numbers. As germline mutations
of VHL splice sites are sufficient to cause VHL disease
(45), there is no reason to
suppose that splicing mutations are less inactivating than other
types of mutation.
Deep intronic mutations activating the splicing of
cryptic exons (46,47) are more readily detected using RNA
than genomic DNA, as conventional mutation scanning of introns is
expensive and time-consuming. To our knowledge, deep intronic
mutations have not been reported in VHL disease (3) or in cancer (http://www.sanger.ac.uk/genetics/CGP/cosmic/). In
the 28 tumours without VHL mutation in our previous study,
mutations were identified in two using RNA. However, as they were
subsequently confirmed in DNA, we speculate that they were not
detected previously due to inadequate tumour cell numbers. Had we
used RNA-level mutation screening alone, we would have been unable
to detect the presence of the four exonic mutations which prevented
the production of mature, stable VHL mRNA or of the four mutations
affecting invariant consensus splice site sequences that did not
show an altered mRNA isoform ratio. Clearly RNA screening
complements DNA mutation screening. Our data show that the
biological consequences of VHL mutations are not necessarily
predictable from the sequence change of the mutation and that for
the majority of VHL truncating mutations, the potential for the
generation of mutant protein exists. Challenges due to the low
level of endogenous VHL protein and the paucity of good VHL
antibodies recognising the various domains currently limit study at
the protein level but clearly expression studies coupled with
functional studies may shed further light on the consequences of
the genetic and epigenetic changes in VHL and their potential
clinical significance.
Acknowledgements
We are grateful to Cancer Research UK
for financial support, to the patients for their participation, and
the oncology and urology staff at St James’s University hospital
and technical staff in the CR-UK Centre for their assistance.
References
|
1
|
Gnarra JR, Tory K, Weng Y, et al:
Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat
Genet. 7:85–90. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Latif F, Tory K, Gnarra J, et al:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nordstrom-O’Brien M, van der Luijt RB, van
Rooijen E, et al: Genetic analysis of von Hippel-Lindau disease.
Hum Mutat. 31:521–537. 2010.PubMed/NCBI
|
|
4
|
Vortmeyer AO, Huang SC, Pack SD, Koch CA,
Lubensky IA, Oldfield EH and Zhuang X: Somatic point mutation of
the wild-type allele detected in tumours of patients with VHL
germline deletion. Oncogene. 21:1167–1170. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nickerson ML, Jaeger E, Shi Y, et al:
Improved identification of von Hippel-Lindau gene alterations in
clear cell renal tumors. Clin Cancer Res. 14:4726–4734. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Young AC, Craven RA, Cohen D, et al:
Analysis of VHL gene alterations and their relationship to clinical
parameters in sporadic conventional renal cell carcinoma. Clin
Cancer Res. 15:7582–7592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richards FM, Crossey PA, Phipps ME, et al:
Detailed mapping of germline deletions of the von Hippel-Lindau
disease tumour suppressor gene. Hum Mol Genet. 3:595–598. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Richards FM, Schofield PN, Fleming S and
Maher ER: Expression of the von Hippel-Lindau disease tumour
suppressor gene during human embryogenesis. Hum Mol Genet.
5:639–644. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schoenfeld A, Davidowitz EJ and Burk RD: A
second major native von Hippel-Lindau gene product, initiated from
an internal translation start site, functions as a tumor
suppressor. Proc Natl Acad Sci USA. 95:8817–8822. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bradley JF and Rothberg PG: Processed
pseudogene from the von Hippel-Lindau disease gene is located on
human chromosome 1. Diagn Mol Pathol. 8:101–106. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Woodward ER, Buchberger A, Clifford SC,
Hurst LD, Affara NA and Maher ER: Comparative sequence analysis of
the VHL tumour suppressor gene. Genomics. 65:253–265. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stebbins CE, Kaelin WG Jr and Pavletich
NP: Structure of the VHL-ElonginC-ElonginB complex implications for
VHL tumor suppressor function. Science. 284:455–461. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rini BI, Campbell SC and Escudier B: Renal
cell carcinoma. Lancet. 373:1119–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Banks RE, Tirukonda P, Taylor C, et al:
Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene
alterations and relationship with clinical variables in sporadic
renal cancer. Cancer Res. 66:2000–2011. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herman JG, Graff JR, Myohanen S, Nelking
BD and Baylin SB: Methylation-specific PCR a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reese MG, Eeckman FH, Kulp D and Haussler
D: Improved splice site detection in genie. J Comp Biol. 4:311–323.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cartegni L, Wang J, Zhu Z, Zhang MQ and
Krainer AR: ESEfinder: a web resource to identify exonic splicing
enhancers. Nucleic Acids Res. 31:3568–3571. 2003.PubMed/NCBI
|
|
18
|
Valcarcel J, Gaur RK, Singh R and Green
MR: Interaction of U2AF65 RS region with pre-mRNA of
branch point and promotion base pairing with U2 snRNA. Science.
273:1706–1709. 1996.PubMed/NCBI
|
|
19
|
Lefevre SH, Chauveinc L, Stoppa-Lyonnet D,
et al: A T to C mutation in the polypyrimidine tract of the exon 9
splicing site of the RB1 gene responsible for low penetrance
hereditary retinoblastoma. J Med Genet. 39:e212002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakai K and Sakamoto H: Construction of a
novel database containing aberrant splicing mutations of mammalian
genes. Gene. 141:171–177. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krawczak M, Thomas NST, Hundrieser B, Mort
M, Wittig M, Hampe J and Cooper DN: Single base-pair substitutions
in exon-intron junctions of human genes: nature, distribution, and
consequences for mRNA splicing. Hum Mutat. 28:150–158. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martella M, Salviati L, Casarin A, et al:
Molecular analysis of two uncharacterised sequence variants of the
VHL gene. J Hum Genet. 51:964–968. 2006. View Article : Google Scholar
|
|
23
|
McNeill A, Rattenberry E, Barber R,
Killick P, MacDonald F and Maher ER: Genotype-phenotype
correlations in VHL exon deletions. Am J Med Genet Part A.
149A:2147–2151. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang MQ: Statistical features of human
exons and their flanking regions. Hum Mol Genet. 7:919–932. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Z and Burge C: Splicing regulation:
from a parts list of regulatory elements to an integrated splicing
code. RNA. 14:802–813. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pohlenz J, Rosenthal IM, Weiss RE, Jhiang
SM, Burant C and Refetoff S: Congenital hypothyroidism due to
mutations in the sodium/iodide symporter. J Clin Invest.
101:1028–1035. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pymar LS, Platt FM, Askham JM, Morrison EE
and Knowles MA: Bladder tumour-derived somatic TSC1 missense
mutations cause loss of function via distinct mechanisms. Hum Mol
Genet. 17:2006–2017. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lorson CI, Hahnen E, Androphy EJ and Wirth
B: A single nucleotide in the SMN gene regulates splicing and is
responsible for spinal muscular atrophy. Proc Natl Acad Sci USA.
96:6307–6311. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu W, Qian C and Francke U: Silent
mutation induces exon skipping of fibrillin-1 gene in Marfan
syndrome. Nat Genet. 16:328–329. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang C-H, Reid M, Daniels G and
Blumenfeld OO: Alteration of splice site selection by an exon
mutation in the human glycophorin A gene. J Biol Chem.
268:25902–25908. 1993.PubMed/NCBI
|
|
31
|
Teraoka SN, Telatar M, Becker-Catania S,
et al: Splicing defects in the ataxia-telangiectasia gene ATM:
underlying mutations and consequences. Am J Hum Genet.
64:1617–1631. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eskesen ST, Eskesen FN and Ruvinsky A:
Natural selection affects frequencies of AG and GT dinucleotides at
the 5′ and 3′ ends of exons. Genetics. 167:543–550. 2004.PubMed/NCBI
|
|
33
|
Frischmeyer PA and Dietz HC:
Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet.
8:1893–1900. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Holbrook JA, Neu-Yilik G, Hentze MW and
Kulozik AE: Nonsense-mediated decay approaches the clinic. Nat
Genet. 36:801–808. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Silva AL and Romao L: The mammalian
nonsense-mediated mRNA decay pathway: to decay or not to decay!
Which players make the decision? FEBS Lett. 583:499–505. 2009.
|
|
36
|
Hamosh A, Rosenstein BJ and Cutting GR:
CFTR nonsense mutations G542X and W1282X associated with severe
reduction of CFTR mRNA in nasal epithelial cells. Hum Mol Genet.
1:542–544. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gossage DL, Norby-Slycord CJ, Hershfield
MS and Markert ML: A homozygous 5 base-pair deletion in exon 10 of
the adenosine deaminase (ADA) gene in a child with severe combined
immunodeficiency and very low levels of ADA mRNA and protein. Hum
Mol Genet. 2:1493–1494. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rajavel KS and Neufeld EF:
Nonsense-mediated decay of human HEXA mRNA. Mol Cel Biol.
21:5512–5519. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Perrin-Vidoz L, Sinilnikova OM,
Stoppa-Lyonnet D, Lenoir GM and Mazoyer S: The nonsense-mediated
mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing
premature truncation codons. Hum Mol Genet. 11:2805–2814. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Inacio A, Silva AL, Pinto J, et al:
Nonsense mutations in close proximity to the initiation codon fail
to trigger full nonsense-mediated mRNA decay. J Biol Chem.
279:32170–32180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nagy E and Maquat LE: A rule for
termination-codon position within intron-containing genes: when
nonsense affects RNA abundance. Trends Biochem Sci. 23:198–199.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ong KR, Woodward ER, Killick P, Lim C,
Macdonald F and Maher ER: Genotype-phenotype correlations in von
Hippel-Lindau disease. Hum Mutat. 28:143–149. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Micale L, Muscarella LA, Marzulli M, et
al: VHL frameshift mutation as target of nonsense-mediated mRNA
decay in Drosophila melanogaster and human HEK293 cell line.
J Biomed Biotechnol. 2009:8607612009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Herman JG, Latif F, Weng Y, et al:
Silencing of the VHL tumour-suppressor gene by DNA methylation in
renal carcinoma. Proc Natl Acad Sci USA. 91:9700–9704. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zbar B, Kishida T, Chen F, et al: Germline
mutations in the von Hippel-Lindau disease (VHL) gene in families
from North America, Europe and Japan. Hum Mutat. 8:348–357. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Highsmith WE, Burch LH, Zhou Z, et al: A
novel mutation in the cystic fibrosis gene in patients with
pulmonary disease but normal sweat chloride concentrations. New
Engl J Med. 331:974–980. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Christie PT, Harding B, Nesbit MA, Whyte
MP and Thakker RV: X-linked hypophosphatemia attributable to
pseudoexons of the PHEX gene. J Clin Endocrinol Metab.
86:3840–3844. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Whaley JM, Naglich J, Gelbert L, et al:
Germ-line mutations in the von Hippel-Lindau tumor-suppressor gene
are similar to somatic von Hippel-Lindau aberrations in sporadic
renal cell carcinoma. Am J Hum Genet. 55:1092–1102. 1994.PubMed/NCBI
|
|
49
|
Maxwell PH, Wiesener MS, Chang GW, et al:
The tumour suppressor protein VHL targets hypoxia-inducible factors
for oxygen-dependent proteolysis. Nature. 399:271–275. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ikediobi ON, Davies H, Bignell G, et al:
Mutation analysis of 24 known cancer genes in the NCI-60 cell line
set. Mol Cancer Ther. 5:2606–2612. 2006. View Article : Google Scholar : PubMed/NCBI
|