Introduction
Recent progress in the development of chemotherapy
for advanced cancer has improved long-term overall survival rates.
However, acquired drug-resistance during chemotherapy is now a
significant clinical problem in advanced cancer patients because it
inhibits complete remission. The prognosis of patients with
recurrent cancers or advanced malignant tumors would be remarkably
improved, if the mechanisms involved in the acquisition of
anticancer drug-resistance could be clarified and therapies to
overcome drug-resistance or restore drug-sensitivity could be
developed.
Paclitaxel (PTX) is an antineoplastic compound
extracted from the Pacific yew tree, Taxus brevifolia. It
binds to tubulin and inhibits the disassembly of microtubules,
thereby resulting in the inhibition of cell division. PTX is now
widely used to treat patients with various types of cancers such as
lung, ovarian, breast, gastric, endometrial and cervical cancers.
Combination chemotherapy with PTX and carboplatin is now the first
line chemotherapy for ovarian and endometrial cancer patients.
Unfortunately, acquired resistance to PTX in cancer patients has
become a major clinical problem in cancer chemotherapy.
There have been many studies that have investigated
the molecular mechanisms involved in PTX-resistance in cancer
cells. Increased β-1 integrin expression or enhanced cancer cell
adhesion to extracellular matrices has been reported to induce
PTX-resistance (1,2). Morphological changes in proliferating
cancer cells may affect cellular PTX-sensitivity (3). Cancer cells with higher Fas antigen,
an apoptosis-inducing cytokine receptor, are more sensitive to PTX
than cells with lower Fas expression (4). Stimulation of CD40, a cell survival
cytokine receptor, can inhibit PTX-apoptosis (4). Increased expressions of BCL-2 or
BCL-XL, which are apoptosis-inhibitory regulators, have been
reported to induce PTX-resistance in cancer cells (5). Increased expression of MDR-1, a
multi-drug resistance molecule, has been found to be associated
with PTX-resistance (6).
Chromosome instability may induce PTX-resistance in cancer cells
(7). Previous studies on the
mechanisms of PTX-resistance have been performed using various
types of human cancer cells including ovarian and breast cancer
cells. However, until now there has been no study on PTX-resistance
involving human endometrial carcinoma cells.
The purpose of our study was to investigate
molecular mechanisms that are associated with acquired
PTX-resistance and to explore methods for overcoming PTX-resistance
using novel PTX-resistant cell lines derived from human endometrial
adenocarcinoma cells. As a result, we have found a novel type of
PTX-resistance in cancer cells.
Materials and methods
Anticancer drugs
All anticancer drugs were kind gifts from
pharmaceutical companies. PTX and etoposide (VP16) were provided by
Bristol-Myers Squibb Japan Co., Ltd. (Tokyo, Japan). Mitomycin C
(MMC) and 5-fluorouracil (5FU) were provided by Kirin-Kyowa-Hakko
Co., Ltd. (Tokyo, Japan). Pirarubicin-HCl (THP) was provided by
Meiji-Seika Kaisha Ltd. (Osaka, Japan). Cisplatin (CDDP) was
provided by Nihon-Kayaku Co., Ltd. (Tokyo, Japan). 4-Hydroxy-
cyclophosphamide (4OH-CPM), a major active metabolite of
cyclophosphamide, was obtained from Shionogi & Co., Ltd.
(Osaka, Japan).
Cell line and culture
The moderately differentiated human endometrial
endometrioid adenocarcinoma cell line, HEC-1 (8), was purchased from the JCRB Cell Bank
(Japan Collection of Research Bioresources Cell Bank, Tokyo,
Japan). All cells were cultured in OPTI-MEM (Invitrogen Corp.,
Carlsbad, CA, USA) supplemented with 5% fetal calf serum (FCS:
Equitech Bio Inc., Ingram, TX, USA), 100 U/ml penicillin (PC), 100
μg/ml streptomycin (SM) and 0.25 μg/ml fungizone (Invitrogen) in 5%
CO2/95% air at 37°C.
Establishment of PTX-resistant subclones
from HEC-1 cells
To establish PTX-resistant subclones, HEC-1 cells
were cultured with various concentrations of PTX for 3–5 weeks, and
the surviving cells were collected. This collection procedure after
PTX exposure was repeated six times. Finally, five single
cell-derived PTX-resistant subclones, designated PTXr3C, PTXr4B,
PTXr6F, PTXr7A and PTXr11B, were established using the limiting
dilution method. The monoclonality of each PTX-resistant subclone
was also confirmed by chromosome analysis (described below). The
establishment of these PTX-resistant subclones took 14 months.
Anticancer drug-sensitivity assays
The effects of anticancer drugs on cell growth were
assayed as follows. Cells in the log phase were detached with 0.25%
trypsin/1 mM EDTA (Invitrogen) and cultured overnight in 96-well
plates (5x103 cells/well). On day 2, various
concentrations of anticancer drugs were added to the cells. On day
4, the numbers of viable cells were evaluated using a cell
proliferation assay kit (Dojin Corp., Tokyo, Japan) and expressed
as the percentages of viable cells relative to the mean number of
viable untreated cells. All experiments were performed four to
seven times. Data are shown as the mean ± SD, and comparative data
were analyzed using Student's t-test (n=6) and ANOVA.
DNA fragmentation assay
HEC-1 parent cells and PTX-resistant subclones in
the log phase were detached with 0.25% trypsin/1 mM EDTA and
cultured overnight in culture dishes (3x106 cells/dish)
containing OPTI-MEM/5% FCS/PC/SM. On day 2, PTX with a final
concentration of 2.5 μg/ml was added to the cells after removal of
the floating dead cells. On day 4, genomic DNA was extracted from
all cells, including the dead ones, using a SepaGene DNA extraction
kit (Sankyo-Junyaku Co., Ltd., Tokyo, Japan) and treated with 100
μg/ml of RNase A (Sigma Chemical Co., St. Louis, MO, USA) in TE
buffer (10 mM Tris, pH 8.0, 2 mM EDTA) for 90 min at 37°C to remove
any contaminating RNA. Next, approximately 20 μg of the genomic DNA
isolated from 5x105 cells was electrophoresed in a 1.4%
agarose gel at 50 V for approximately 2 h, stained with 5 μg/ml of
ethidium bromide and visualized using UV fluorescence.
Semiquantitative flow cytometric analysis
of cell surface antigens
Cells were detached from culture flasks with 3 mM
EDTA in phosphate-buffered saline (PBS), and immunostained as
follows. The cells (3x105) were incubated with an excess
of the primary antibody for 20 min at 4°C and then washed twice
with wash buffer (PBS containing 2% fetal calf serum and 0.1%
NaN3). Next, the cells were incubated with
FITC-conjugated goat anti-mouse IgG (H+L) (Dako Japan, Kyoto,
Japan) for 20 min at 4°C and washed twice with wash buffer.
Finally, the cells were suspended in 200 μl of wash buffer and
analyzed using a FACSCalibur™ (Beckman Coulter Japan, Tokyo,
Japan). The following primary antibodies were used: mouse
anti-human CD29 monoclonal antibody (clone K20: Immunotech,
Hampshire, UK); mouse anti-human CD30 monoclonal antibody (clone
HRS-4: Immunotech, Marseille, France); mouse anti-human CD40
monoclonal antibody (clone mAb89: Immunotech); mouse anti-human
tumor necrosis factor receptor (TNFR) (CD120a) monoclonal antibody
(Genzyme, Cambridge, MA, USA); mouse anti-human Fas (CD95)
monoclonal IgG (clone UB2: MBL, Nagoya, Japan); mouse anti-human
CD49a monoclonal antibody (clone TS2/7: AbD Serotec Ltd., Oxford,
UK); mouse anti-human CD49b monoclonal antibody (clone 31H4:
Serotec Ltd.); mouse anti-human CD49c monoclonal antibody (clone
11G5: Cymbus Biotech Ltd., Chandlers Ford, UK); mouse anti-human
CD49d monoclonal antibody (clone 44H6: Cymbus Biotech Ltd.); mouse
anti-human CD49e monoclonal antibody (clone SAM1: Beckman Coulter
Japan); and mouse anti-human CD49f monoclonal antibody (clone 4F10:
Cymbus Biotech Ltd.).
Semiquantitative flow cytometric analysis
of intracellular molecules and multidrug-resistance-related
molecules
Flow cytometric analysis of intracellular molecules
and multidrug-resistance-related molecules was performed as
follows. Cells were detached from culture flasks with 3 mM EDTA in
PBS, and then washed with wash buffer (PBS containing 2% fetal calf
serum). The cells were fixed with 4% paraformaldehyde in 0.1 M
NaH2PO4 (pH 7.4) for 10 min on ice. After two
washes with wash buffer, the cells were treated with 100 mg/ml
digitonin (Sigma Chemical Co.) for 10 min at room temperature.
After another two washes, the cells were treated with normal mouse
IgG to block non-specific binding sites for 5 min at room
temperature, followed by incubation with the primary antibody for
30 min at room temperature and two further washes. Finally, the
cells were incubated in FITC-conjugated goat anti-mouse IgG (Gibco
BRL, Carlsbad, CA) for 30 min at room temperature, washed twice and
suspended in 200 μl of wash buffer for analyses using a
FACSCalibur™ (Beckman Coulter Japan). The following primary
antibodies were used: mouse anti-human BAX monoclonal antibody
(clone 2D2: AbD Serotec Ltd.); mouse anti-human BCL-2 monoclonal
antibody (clone 100: AbD Serotec Ltd.); mouse anti-human BCL-XL
monoclonal antibody (clone 2H12: Acris Antibodies Inc., San Diego,
CA); mouse anti-human multi-drug resistance protein-1 (MDR-1)
monoclonal antibody (clone UIC2: Acris Antibodies Inc.); mouse
anti-human MDR-related protein (MRP) monoclonal antibody (clone
QCLR-1: Merck Millipore, Darmstadt, Germany); and mouse anti-human
lung-resistance protein (LRP) monoclonal antibody (clone LRP-56:
Merck Millipore).
Karyotyping analysis
Cytogenetic analysis was performed according to
previously reported methods (9,10)
with the following modifications. Briefly, tumor cell cultures were
washed and incubated with 0.1% (v/v) colcemide (Sigma Chemical Co.)
overnight. The cells were then exposed to a hypotonic solution
composed of 3 g/l KCl, 0.2 g/l EGTA and 4.8 g/l Hepes. The cells
were centrifuged into a pellet and fixed in a methanol solution.
G-banding was performed using the method of Yunis (11).
Results
Establishment of monoclonal PTX-resistant
subclones
Five monoclonal PTX-resistant subclones, PTXr3C,
PTXr4B, PTXr6F, PTXr7A and PTXr11B, were independently established
from HEC-1 cells using long-term PTX-exposure cultures and limiting
dilution cultures. Under the microscope, their cellular appearance
and adherence to the culture dishes could not be discriminated from
those of the parent HEC-1 cells (Fig.
1). Although PTX-sensitivity tests of the PTX-resistant
subclones were performed 4–7 times for each subclone, all of the
five PTX-resistant subclones showed no significant change in
PTX-induced growth inhibition as compared with the HEC-1 parent
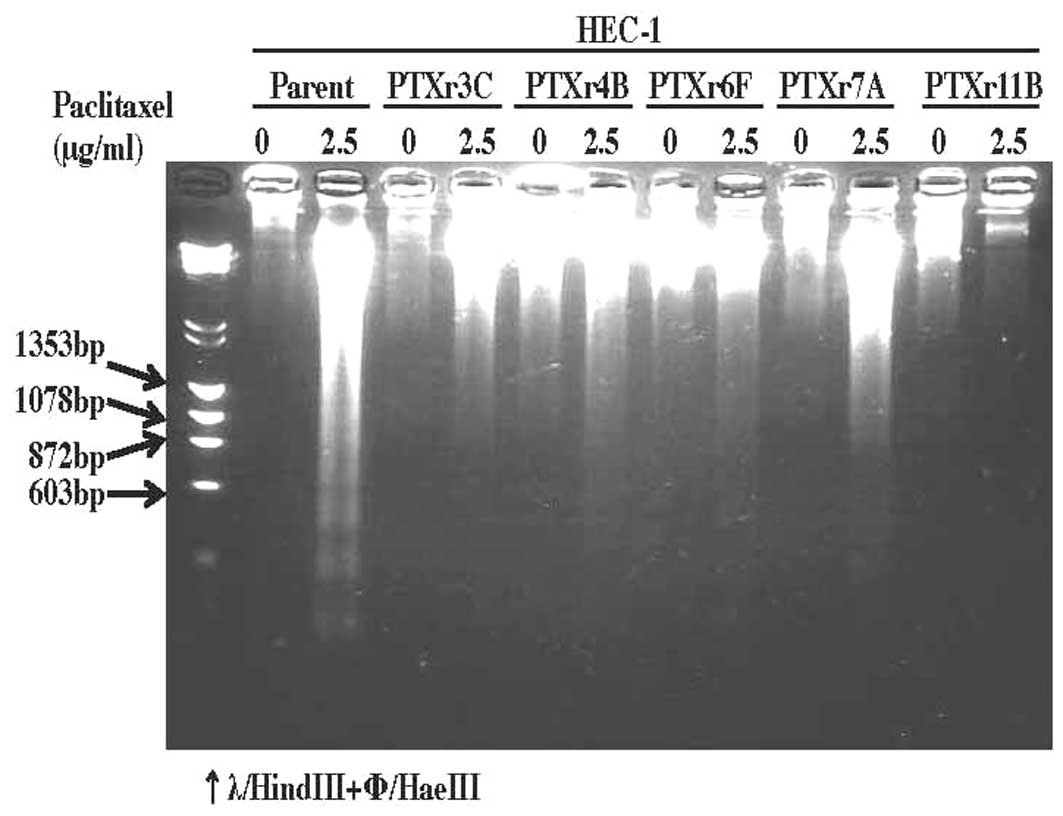
cells (Fig. 2). However, it was
demonstrated using the DNA fragmentation assay that these five
PTX-resistant subclones were apparently more resistant to
PTX-induced DNA fragmentation, or in other words PTX-induced
apoptosis, than the parent HEC-1 cells (Fig. 3).
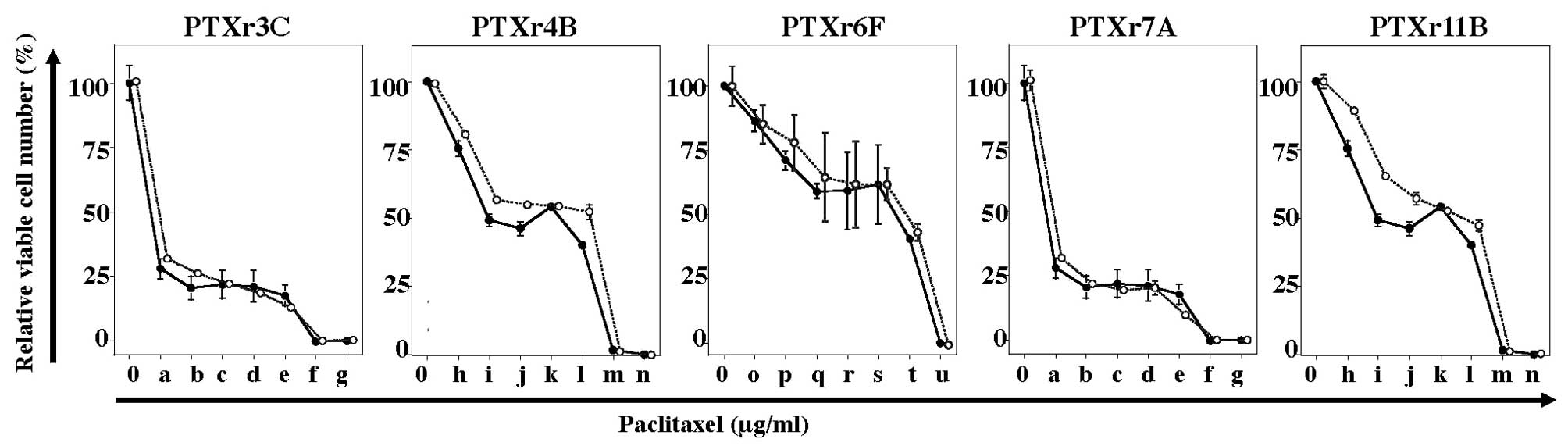 | Figure 2.PTX-sensitivity assays of the five
PTX-resistant subclones. The solid line with closed circles in each
plot is the PTX-sensitivity curve for the parent HEC-1 cells. The
dotted lines with open circles are the PTX-sensitivity curves for
the PTX-resistant subclones. The final concentrations (μg/ml) of
PTX indicated as: 0 and a-g at the bottom of the figures were 0,
0.0064, 0.032, 0.16, 0.8, 4, 20 and 100 μg/ml; 0 and h-n at the
bottom of the figures were 0, 0.0015, 0.0076, 0.0384, 0.192, 0.96,
4.8 and 24 μg/ml; and 0 and o-u at the bottom of the figure were 0,
0.00256, 0.0128, 0.064, 0.32, 1.6, 8 and 40 μg/ml. |
Anticancer drug-sensitivities of the
PTX-resistant subclones
Anticancer drug-sensitivity assays against six
anticancer drugs were performed on the PTX-resistant cell lines. As
shown in Fig. 4, all the
drug-sensitivity curves for the five PTX-resistant subclones showed
very similar results to those for the HEC-1 parent cells. There
were no PTX-resistant subclones that had moderate or high
cross-resistance to any other anticancer drug.
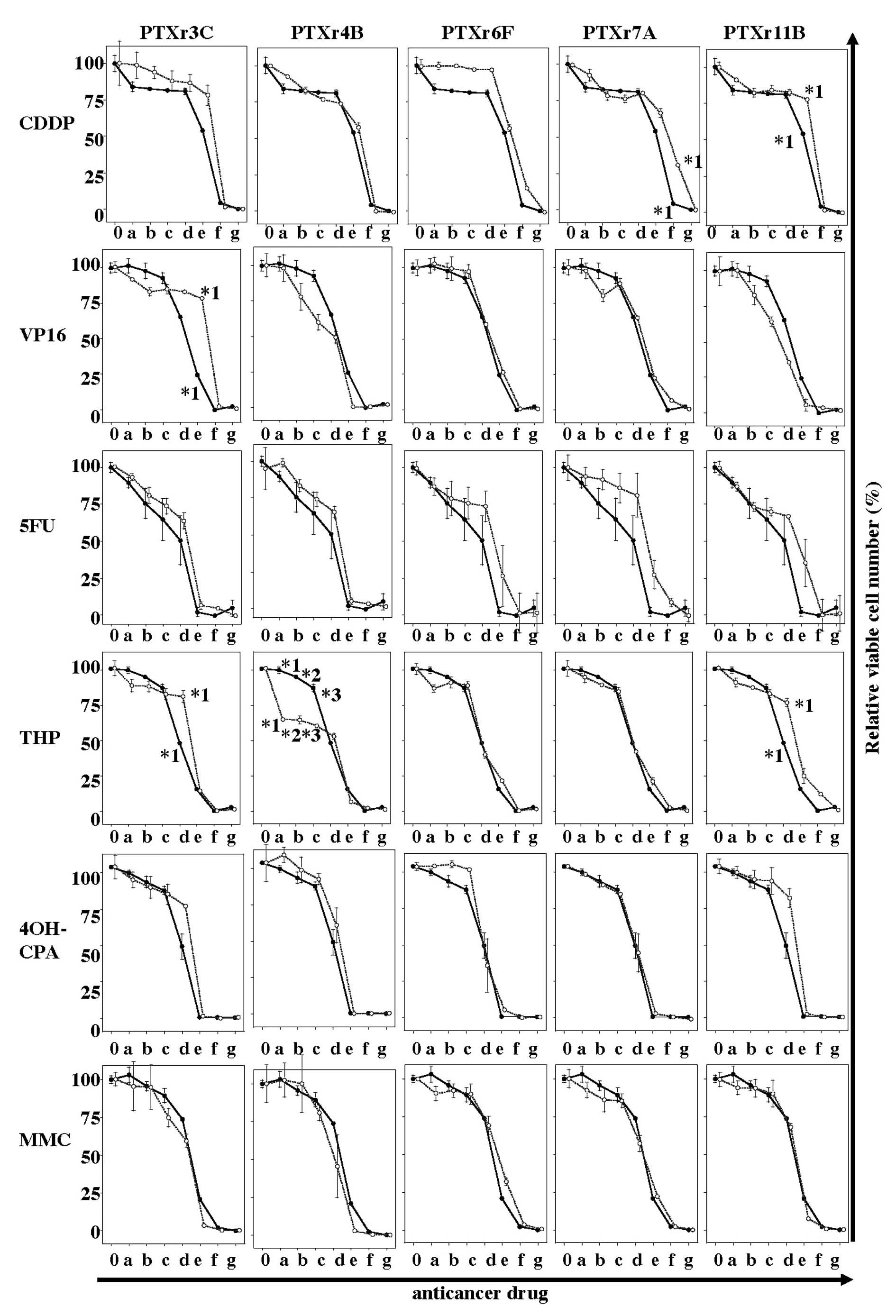 | Figure 4.Anticancer drug-sensitivity assays of
the five PTX-resistant subclones. The solid line with closed
circles in each plot is the drug-sensitivity curve for the parent
HEC-1 cells. The dotted lines with open circles are the
drug-sensitivity curves for the PTX-resistant subclones. The final
concentrations (μg/ml) of each anticancer drug indicated as 0 and
a-g at the bottom of the figures were as follows: CDDP, 0, 0.032,
0.16, 0.8, 4, 20, 100 and 500 μg/ml; VP16, 0, 0.256, 1.28, 6.4, 32,
160, 800 and 4,000 μg/ml; 5FU, 0, 0.32, 1.6, 8, 40, 200, 1,000 and
5,000 μg/ml; THP, 0, 0.001067, 0.00533, 0.0267, 0.133, 0.67, 3.33
and 16.670 μg/ml; 4OH-CPM, 0, 0.32, 1.6, 8, 40, 200, 1,000 and
5,000 μg/ml; and MMC, 0, 0.128, 0.064, 0.32, 1.6, 3.2, 40 and 200
μg/ml. *1–*3, p<0.05. |
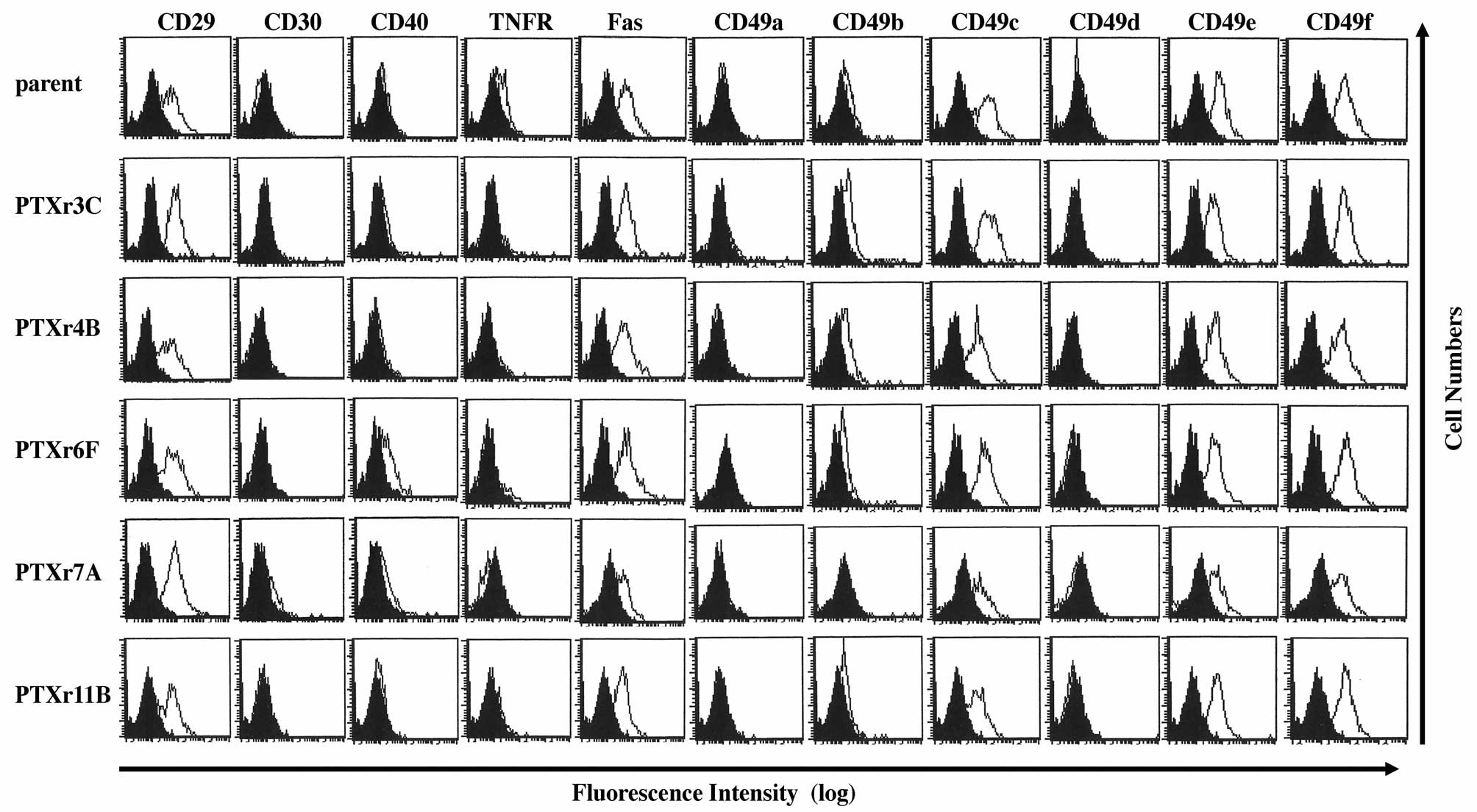
Flow cytometric analysis of cell surface
antigen expressions in the PTX-resistant cells
The cell surface antigen expression profiles of the
PTX-resistant cell lines were investigated using semi-quantitative
flow cytometric analysis. Because all of the PTX-resistant
subclones showed resistance to PTX-induced DNA fragmentation, we
examined the 11 cell surface antigens that were reported to affect
cell apoptosis or cell survival. The flow cytometric data from the
parent HEC-1 cells were compared with those from the PTX-resistant
cells. However, there were no apparent differences among the flow
cytometric profiles of the parent HEC-1 cells and the five
PTX-resistant cell lines (Fig.
5).
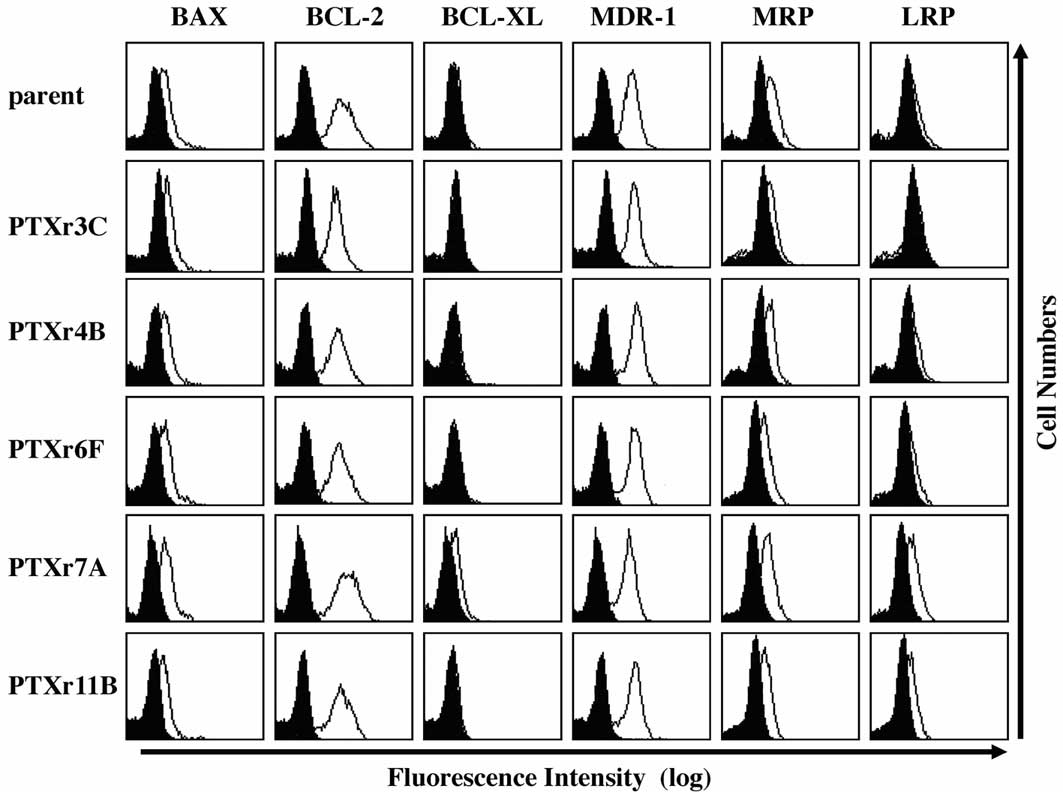
Semiquantitative flow cytometric analysis
of intracellular molecules and multidrug-resistance-related
molecules
To investigate the mechanisms of acquired resistance
to PTX-induced apoptosis in the PTX-resistant subclones,
semiquantitative flow cytometric analysis of three intracellular
apoptosis-regulating molecules and three
multidrug-resistance-related molecules was performed. As shown in
Fig. 6, no apparent differences in
the expression of the six molecules were found among the flow
cytometric profiles of the HEC-1 parent cells and the five
PTX-resistant cell lines.
Karyotyping analysis of the PTX-resistant
cells
Karyotyping analyses of the five in
vitro-cultured PTX-resistant subclones were examined (Fig. 7). The karyotype of the parent HEC-1
cell line was 74, XX, i(X)(p10), +1, add(1)(p36)x2, der(1)t(1;12)
(p22;q11), −2, add(2)(q23)x2, add(3)(p21), −4, +5, −6, add(6)(q21),
+7, −8, +add(9)(p11), i(9)(q10), add(12)(q24), der(12)t(12;14)
(p13;q13), −13, −14, add(15)(p11), +16, +add(17)(p11), add (17)
(p11), −18, −19, −20, +21, add(21)(p11)x2, −22, +8mar (Fig. 7A). The karyotype of the PTXr3C cell
line was 75, XX, i(X)(p10), add(1)(p36), der(1)t(1;12)(p22;q11),
−2, add(2)(q23)x2, add(3) (p21), +5, −6, add(6)(q21), +7, −8,
+add(9)(p11), +11, −12, add(12) (q24), der(12)t(12;14)(p13;q13),
−13, −14, add(15)(p11), +add(17) (p11), add(17)(p11), add(18)(q23),
−19, add(21)(p11)x2, −22, +9 mar (Fig.
7B). The karyotype of the PTXr4B cell line was 75, XX,
i(X)(p10), add(1)(p36), der(1)t(1;12)(p22;q11), −2, add(2) (q23)x2,
add(3)(p21), +5, −6, add(6)(q23), +add(7)(q32), −8, add(9)(p11),
+11, −12, add(12)(q24), der(12)t(12;14)(p13;q13), −13, −14,
add(15)(p11), +add(17)(p11)x2, add(17)(p11), add(18) (q23), −19,
add(21)(p11)x2, −22, +9 mar (Fig.
7C). The karyotype of the PTXr6F cell line was 77, XX,
i(X)(p10), add(1) (p36), der(1)t(1;12)(p22;q11), −2, add(2)(q23)x2,
−3, add(3)(p21), +5, −6, add(6)(q23), +7, +add(9)(p11), −10, +11,
−12, add(12)(q24), der(12)t(12;14)(p13;q13), −13, −14,
add(15)(p11), +add(17)(p11), add(17)(p11), add(18)(q23), −19, +21,
add(21)(p11)x3, −22, +11 mar (Fig.
7D). The karyotype of the PTXr7A cell line was 78, XX,
i(X)(p10), add(1)(p36), der(1)t(1;12)(p22;q11), −2, add(2) (q23)x2,
add(3)(p21), +5, −6, add(6)(q23), +7, −8, +add(9)(p11), +11,
add(12)(q24), der(12)t(12;14)(p13;q13), −13, −14, add(15) (p11),
+add(17)(p11), add(17)(p11), add(18)(q23), −19, −20,
add(21)(p11)x2, −22, +11 mar (Fig.
7E). The karyotype of the PTXr11B cell line was 76, XX,
i(X)(p10), add(1)(p36), der(1) t(1;12)(p22;q11), −2, add(2)(q23)x2,
add(3)(p21), −6, add(6)(q23), +7, −8, +add(9)(p11), add(12)(q24),
der(12)t(12;14)(p13;q13), −14, add(15)(p11), +add(17)(p11),
add(17)(p11)x2, add(18)(q23), −19, add(21)(p11)x2, −22, +9 mar
(Fig. 7F). As a result, common
chromosomal abnormalities were found in chromosome 4 and chromosome
18 among the five PTX-resistant subclones, but not in parent HEC-1
cells.
Discussion
PTX stabilizes microtubules and, as a result,
interferes with the normal breakdown of microtubules during cell
division. Therefore, PTX-treated cells have defects in the mitotic
spindle assembly, chromosome segregation and cell division.
Although PTX is now widely used to treat cancer, acquired or
natural PTX-resistance is a major clinical problem in cancer
chemotherapy. In order to investigate how to overcome acquired
PTX-resistance, we have established five novel monoclonal
PTX-resistant subclones from HEC-1, a human endometrioid
adenocarcinoma cell line. This is probably the first study in which
PTX-resistant cells from human endometrial cancer cells have been
established. Most interestingly, all of the established
PTX-resistant cell lines showed resistance to PTX-induced DNA
fragmentation, but not to PTX-induced growth suppression. The
differential resistance exhibited by the PTX-resistant cell lines
may be a very rare phenomenon. We could not find any reports
concerning anticancer drug-resistant cancer cell lines with such
differential resistance as that shown by the PTX-resistant cell
lines.
The PTX-resistant cell lines showed no significant
changes in sensitivity to drug-induced growth suppression, not only
by PTX but also by the six other anticancer drugs used in our
study. Using the same experimental methods as those used to
establish the present PTX-resistant cell lines, we have previously
established many monoclonal anticancer drug-resistant cell lines
such as CPA-resistant subclones (12), 5FU-resistant subclones (13), CDDP-resistant subclones (14) and VP16-resistant subclones
(15). Almost all of these
drug-resistant subclones demonstrated cross-resistance to several
anticancer drugs including PTX. Both CPA-resistant subclones
(12) and 5FU-resistant subclones
(13) exhibited IC50s
for PTX-sensitivity that were >125 times higher than those of
their parent cell lines, while VP16-resistant subclones (15) had IC50s for
PTX-sensitivity that were up to five times higher than those of
their parent cell lines. The CDDP-resistant subclones had
IC50s that were approximately 25 times higher than their
parent cell line (14). All of the
five PTX-resistant subclones established in the present study
showed no apparent cross-resistance to six other anticancer drugs.
Most interestingly, the PTX-resistant subclones showed almost the
same resistance to 4-OH-CPA and 5FU as the parent HEC-1 cells or
very weak cross-resistance to 4-OH-CPA and 5FU. These results
suggest that after PTX-chemotherapy PTX-resistant cancer cells may
retain high sensitivities to other anticancer drugs. On the other
hand, after chemotherapy with CPA, 5FU or CDDP drug-resistant
cancer cells might have acquired strong cross-resistance to PTX
simultaneously.
All five PTX-resistant subclones showed acquired
resistance to PTX-induced DNA fragmentation (PTX-induced apoptosis)
but not to PTX-induced growth suppression. These results indicate
the possibility that PTX-induced apoptosis is regulated by
different intracellular mechanisms from PTX-induced growth
suppression. For example, strong cross-resistance to PTX-induced
growth suppression was found in CPA-resistant subclones derived
from the cervical carcinoma cell line ME180 (12) and 5FU-resistant subclones derived
from the endometrial carcinoma cell line HHUA (13), while little cross-resistance to
PTX-induced growth suppression was found in VP16-resistant ME180
cells (15). Therefore, the
differential effects between PTX-induced growth suppression signals
and PTX-induced apoptosis signals may depend on the anticancer
drugs that were used to establish the drug-resistant cell
lines.
The PTX-sensitivity of cancer cells can be regulated
by morphological changes in proliferating cancer cells (3) while there was no apparent difference
in microscopic findings between the established PTX-resistant cells
and parent cells. Morphological changes in proliferating cancer
cells are affected by the expression levels and functions of cell
adhesion molecules in the cells. In fact there are several reports
that increased β1-integrin expression or increased cell adhesion to
extracellular matrices induced resistance to PTX (1,2,17).
Another study demonstrated increased integrin expression in
PTX-resistant cells (18). The
VP16-resistant subclones, which have weak cross-resistance to PTX,
showed decreased expression of CD29, CD49a and CD49f (15). However, the five PTX-resistant
subclones established in our study did not show any significant
differences in CD29 and CD49a–f expression profiles from those of
the parent HEC-1 cells. Because there was no difference in integrin
expression and cell proliferation patterns between PTX-resistant
cells and the parent cells, the mechanisms of PTX-resistance in the
established PTX-resistant subclones may not be caused by the
differential cell adhesion ability of the PTX-resistant cells.
Both Fas and TNFR are well-known receptors for
apoptosis-inducing cytokines. There are several reports that PTX
sensitivity can be regulated by apoptosis signals via Fas or TNFR.
The high Fas-expressing cells are reported to be more sensitive to
PTX (4). PTX treatment can
increase production of TNFα (19)
and Fas ligand (20) to induce
apoptosis. On the other hand, PTX can inhibit TNFα functions in
corneal endothelium (21). CD40 is
a well-known receptor for the cell survival cytokine, CD40L.
Increased CD40 expression has been reported in VP16-resistant cells
(15). CD40-stimulation may induce
anticancer multidrug-resistance in cancer cells (22) and inhibit PTX-induced apoptosis
(4). However, in the PTX-resistant
subclones in the present study, there was no significant change in
the expression of Fas, TNFR and CD40 between PTX-resistant cells
and the parent cells.
BCL-2 family products are well-known
apoptosis-regulators. There are several reports that increased
BCL-2 expression and/or BCL-XL expression can induce PTX-resistance
(5,23,24).
Decreased BAX expression has also been reported to be related to
PTX-resistance (25,26). However, in the PTX-resistant
subclones in our study there was no significant change in the
expression of BCL-2, BCL-XL and BAX between PTX-resistant cells and
the parent cells.
MDR-1, MRP and LRP have been identified as
multidrug-resistance-related molecules. Several studies have found
that PTX-resistant cells had increased MDR-1 expression (6) or LRP expression (27), although MRP expression may not have
any relationship to PTX-resistance (5,6). In
the PTX-resistant subclones in the present study, there was no
significant change in the expression of MDR-1, MRP and LRP between
PTX-resistant cells and the parent cells.
Because chromosome instability has been reported to
affect PTX-resistance (7), we
performed karyotype analyses of the established PTX-resistant
subclones. All of the five PTX-resistant subclones had common
changes in chromosomes 4 and 18 that were different from the parent
cells. This finding contrasts with a study involving 5FU-resistant
cells, which reported that there were no apparent changes in
karyotypes relative to the parent cells (13). Chromosomes 4 or 8 might have key
genes that affect sensitivity to PTX-induced apoptosis.
Although we have established novel monoclonal
PTX-resistant subclones from human endometrial cancer cells, the
mechanisms involved in PTX-resistance remain unclear. Most notably,
these subclones showed resistance to PTX-induced apoptosis but not
to PTX-induced growth suppression. During the acquisition process
of PTX-resistance, large differences occurred in the
PTX-concentrations required for the induction of growth suppression
and apoptosis. In the present study, a much higher dose of PTX was
necessary to induce apoptosis in PTX-resistant cells than to induce
growth suppression. As shown in Fig.
2, the PTX growth-inhibition curves are stair-like (12,13,14),
while those for the other anticancer drugs are sigmoid. Since the
docetaxel growth-inhibition curves are also stair-like (15,28),
differential sensitivity to drug-induced apoptosis and drug-induced
growth suppression might be a phenomenon specific to taxane
compounds. In conclusion, the novel PTX-resistant subclones from
HEC-1 established in our study can be used as experimental models
for recurrent cancers occurring after complete remission as a
result of PTX-chemotherapy. They may prove to be very useful tools
for the investigation of methods to prevent or treat recurrent
cancers after PTX-chemotherapy.
References
|
1.
|
Chen YX, Wang Y, Fu CC, Diao F, Song LN,
Li ZB, Yang R and Lu J: Dexamethasone enhances cell resistance to
chemotherapy by increasing adhesion to extracellular matrix in
human ovarian cancer cells. Endocr Relat Cancer. 17:39–50. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Dong Y, Tan OL, Loessner D, Stephens C,
Walpole C, Boyle GM, Parsons PG and Clements JA: Kallikrein-related
peptidase 7 promotes multicellular aggregation via the alpha(5)
beta(1) integrin pathway and paclitaxel chemoresistance in serous
epithelial ovarian carcinoma. Cancer Res. 70:2624–2633. 2010.
View Article : Google Scholar
|
|
3.
|
Loessner D, Stok KS, Lutolf MP, Hutmacher
DW, Clements JA and Rizzi SC: Bioengineered 3D platform to explore
cell-ECM interactions and drug resistance of epithelial ovarian
cancer cells. Biomaterials. 31:8494–8506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Stumm S, Meyer A, Lindner M, Bastert G,
Wallwiener D and Gückel B: Paclitaxel treatment of breast cancer
cell lines modulates Fas/Fas ligand expression and induces
apoptosis which can be inhibited through the CD40 receptor.
Oncology. 66:101–111. 2004. View Article : Google Scholar
|
|
5.
|
Huang Y, Ibrado AM, Reed JC, Bullock G,
Ray S, Tang C and Bhalla K: Co-expression of several molecular
mechanisms of multidrug resistance and their significance for
paclitaxel cytotoxicity in human AML HL-60 cells. Leukemia.
11:253–257. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kamazawa S, Kigawa J, Kanamori Y, Itamochi
H, Sato S, Iba T and Terakawa N: Multidrug resistance gene-1 is a
useful predictor of Paclitaxel-based chemotherapy for patients with
ovarian cancer. Gynecol Oncol. 86:171–176. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Swanton C, Nicke B, Schuett M, Eklund AC,
Ng C, Li Q, Hardcastle T, Lee A, Roy R, East P, Kschischo M,
Endesfelder D, Wylie P, Kim SN, Chen JG, Howell M, Ried T,
Habermann JK, Auer G, Brenton JD, Szallasi Z and Downward J:
Chromosomal instability determines taxane response. Proc Natl Acad
Sci USA. 106:8671–8676. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Satyaswaroop PG, Frost A and Gurpide E:
Metabolism and effects of progesterone in the human endometrial
adenocarcinoma cell line HEC-1. Steroids. 35:21–37. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Gibas Z, Prout GR, Conolly JG, Pontes JE
and Sandberg AA: Nonrandom chromosome changes in transitional cell
carcinoma of the bladder. Cancer Res. 44:1257–1264. 1984.PubMed/NCBI
|
|
10.
|
Yoshida MA, Ohyashiki K, Ochi H, Gibas Z,
Pontes JE, Prout GR Jr, Huben R and Sandberg AA: Cytogenetic
studies of tumor tissue from patients with nonfamilial renal cell
carcinoma. Cancer Res. 46:2139–2147. 1986.PubMed/NCBI
|
|
11.
|
Yunis JJ: New chromosome techniques in the
study of human neoplasia. Hum Pathol. 12:540–549. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Tanaka T, Bai T, Yukawa K, Utsunomiya T
and Umesaki N: Reduced radiosensitivity and increased CD40
expression in cyclophosphamide-resistant subclones established from
human cervical squamous cell carcinoma cells. Oncol Rep.
14:941–948. 2005.
|
|
13.
|
Tanaka T, Bai T and Toujima S:
Establishment and characterization of monoclonal
5-fluorouracil-resistant cell lines derived from human endometrial
adenocarcinoma. Int J Oncol. 37:731–736. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Tanaka T, Toujima S and Umesaki N:
Growth-inhibitory signals by activin A do not affect anticancer
drug-sensitivity and acquired multi-drig-resistance in human
ovarian endometrioid adenocarcinoma OVK-18 cells. Oncol Rep.
11:667–671. 2004.
|
|
15.
|
Tanaka T, Bai T, Yukawa K and Umesaki N:
Optimal combination chemotherapy and chemoradiotherapy with
etoposide for advanced cervical squamous cancer cells in vitro.
Oncol Rep. 15:939–947. 2006.PubMed/NCBI
|
|
16.
|
Ohbayashi M, Yasuda M, Kawakami I, Kohyama
N, Kobayashi Y and Yamamoto T: Effect of interleukins response to
ECM-induced acquisition of drug resistance in MCF-7 cells. Exp
Oncol. 30:276–282. 2008.PubMed/NCBI
|
|
17.
|
To K, Fotovati A, Reipas KM, Law JH, Hu K,
Wang J, Astanehe A, Davies AH, Lee L, Stratford AL, Raouf A,
Johnson P, Berquin IM, Royer HD, Eaves CJ and Dunn SE: Y-box
binding protein-1 induces the expression of CD44 and CD49f leading
to enhanced self-renewal, mammosphere growth, and drug resistance.
Cancer Res. 70:2840–2851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Işeri OD, Kars MD, Arpaci F and Gündüz U:
Gene expression analysis of drug-resistant MCF-7 cells:
implications for relation to extracellular matrix proteins. Cancer
Chemother Pharmacol. 65:447–55. 2010.PubMed/NCBI
|
|
19.
|
Lanni JS, Lowe SW, Licitra EJ, Liu JO and
Jacks T: p53-independent apoptosis induced by paclitaxel through an
indirect mechanism. Proc Natl Acad Sci USA. 94:9679–83. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Biswas RS, Cha HJ, Hardwick JM and
Srivastava RK: Inhibition of drug-induced Fas ligand transcription
and apoptosis by Bcl-XL. Mol Cell Biochem. 225:7–20. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Shivanna M and Srinivas SP: Microtubule
stabilization opposes the (TNF-alpha)-induced loss in the barrier
integrity of corneal endothelium. Exp Eye Res. 89:950–959. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Voorzanger-Rousselot N, Alberti L and Blay
JY: CD40L induces multidrug resistance to apoptosis in breast
carcinoma and lymphoma cells through caspase independent and
dependent pathways. BMC Cancer. 18:752006. View Article : Google Scholar
|
|
23.
|
Wang MY, Chen PS, Prakash E, Hsu HC, Huang
HY, Lin MT, Chang KJ and Kuo ML: Connective tissue growth factor
confers drug resistance in breast cancer through concomitant
up-regulation of Bcl-xL and cIAP1. Cancer Res. 69:3482–3491. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Luo D, Cheng SC, Xie H and Xie Y: Effects
of Bcl-2 and Bcl-XL protein levels on chemoresistance of
hepatoblastoma HepG2 cell line. Biochem Cell Biol. 78:119–126.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Janssen K, Pohlmann S, Jänicke RU,
Schulze-Osthoff K and Fischer U: Apaf-1 and caspase-9 deficiency
prevents apoptosis in a Bax-controlled pathway and promotes
clonogenic survival during paclitaxel treatment. Blood.
110:3662–3672. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Strobel T, Kraeft SK, Chen LB and
Cannistra SA: BAX expression is associated with enhanced
intracellular accumulation of paclitaxel: a novel role for BAX
during chemotherapy-induced cell death. Cancer Res. 58:4776–4781.
1998.PubMed/NCBI
|
|
27.
|
Kitazono M, Sumizawa T, Takebayashi Y,
Chen ZS, Furukawa T, Nagayama S, Tani A, Takao S, Aikou T and
Akiyama S: Multidrug resistance and the lung resistance-related
protein in human colon carcinoma SW-620 cells. J Natl Cancer Inst.
91:1647–1653. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Tanaka T, Toujima S, Toyoda S, Takeuchi S
and Umesaki N: Establishment and characterization of novel human
uterine leiomyosarcoma cell lines. Int J Oncol. 35:125–131.
2010.PubMed/NCBI
|