Introduction
Cervical cancer is the second most common malignancy
in women worldwide. Approximately 80% of these tumors are squamous
cell carcinomas (SCCs) and 5–20% are adenocarcinomas (AdCAs)
(1,2). Cervical SCCs are often preceded by a
premalignant disease known as cervical intraepithelial neoplasia
(CIN) which is graded 1–3 with increasing atypia. Infection with
high-risk subtypes of human papillomavirus (HPV), namely HPV-16 and
HPV-18, is the major etiological factor and is the primary
initiator of premalignant lesions (3). The viral oncoproteins E6 and E7 play
an important role in the process of CIN, inhibiting a variety of
cellular targets, including the tumor suppressor protein p53 and
retinoblastoma (Rb), which disrupts key cellular processes such as
apoptosis and cell cycle control processes and leads to genomic
instability and neoplastic development. However, only a small
fraction of women infected with oncogenic HPV subtypes develop
cervical cancer, which indicates that HPV infection alone is not
sufficient to cause disease and that there are other host factors
associated with the development of invasive cervical cancer
(4).
The process of increased genomic instability in
addition to HPV infection has been proposed to contribute to the
need for precursor lesions to progress to invasive cancer (4,5).
Progression from CIN1 to CIN3 and SCC is admitted and is consistent
with the concept of lesional continuum (6,7).
However, because of the elevated rate of spontaneous regression of
CIN1, it is probably a lesion of very low potential aggressivity
and its role as a precursor is uncertain (8). Recently, It has been shown that
genomic profiling of p16INK4a immunopositive CIN2/3
lesions can distinguish histologically similar high-grade CIN
lesions into potentially early and more advanced lesions (9). This indicates that the progression of
CIN might be reflected in its chromosomal profile. In addition, the
involvement of clonal selection and expansion process during the
progression of CIN was also suggested (10).
However, the chromosomal aberrations which might
affect the progression of cervical lesion to SCC have not been
systematically examined. The differential and common chromosomal
aberrations among CIN1, CIN2, CIN3 and SCC might be important clues
for understanding of the progression of cervical lesions. In this
study, we applied array-based comparative genomic hybridization
(array CGH) to specimens of 60 patients to identify differential
and common chromosomal aberrations among CIN1, CIN2, CIN3 and
cervical squamous cell carcinoma (SCC). Using an open database, our
study showed driver genes to genetic alterations that provide tumor
cells with a growth advantage during carcinogenesis or tumor
progression.
Materials and methods
Tissue specimens
Sixty cervical tissue samples including 15 cases
each of CIN1, CIN2, CIN3 and SCC were obtained from the Department
of Obstetrics and Gynecology, Seoul St. Mary’s Hospital, The
Catholic University of Korea. This study followed the guidelines of
the Institutional Review Board of The Catholic University of Korea
and written informed consent was obtained from all patients
included.
Microdissection, extraction of nucleic
acids and HPV DNA testing
Paraffin-embedded tissues were first sectioned in 10
μm slices, which were hematoxylineosin stained for selection of the
appropriated tissue area. The corresponding selected areas of each
tissue sample were then collected under microscopic observation
with a 30-gauge needle (Becton-Dickinson, Franklin Lakes, NJ).
Genomic DNA of microdissected tissue was extracted by proteinase K
digestion followed by standard phenol-chloroform extraction
(11). HPV DNA test was performed
with the hybrid capture II (HCII) assay system according to the
manufacturer’s instructions (Digene Diagnostics Inc., Gaithersburg,
MD). Briefly, the isolated DNA from the above microdissected sample
were denatured at 65°C for 45 min and hybridized under high
stringency conditions with a mixture of RNA probes that detect 13
different high-risk HPV types: 16, 18, 31, 33, 35, 39, 45, 51, 52,
56, 58, 59 and 68. The resultant DNA-RNA hybrids were captured on
the surface of the microtiter plate wells coated with anti-DNA-RNA
hybrid antibody. The immobilized hybrids were then reacted with an
alkaline phosphatase-conjugated anti-hybrid monoclonal antibody.
Light intensity was measured with a luminometer. A positive cutoff
value was set at 1 pg HPV DNA per mm in each specimen.
Array CGH
The array CGH experiments were performed with a
MacArray™ Karyo 1440 (Macrogen, Seoul, Korea) according to the
manufacturer’s instructions (http://www.macrogen.co.kr/eng/biochip/karyo_summary.jsp)
where the BAC chip information together with chromosomal location
of each clone was also provided. The array consisted of 1,440 human
BAC clones spotted in triplicate, including 356 cancer-related
genes at an average interval of 2.3 Mb. Sample DNA and 9948 male
reference DNA (Promega Corp., Madison, WI) (500 ng each) were
labeled by the random priming method with fluorescence dyes, Cy3
and Cy5, respectively. The use of a male reference DNA enabled the
determination of whether the hybridization had succeeded based on
the expected gain of chromosome X and loss of chromosome Y in the
female test samples. The labeled DNAs were mixed with Cot-1 DNA (50
μg; Gibco BRL, Gaithersburg, MD) and then hybridized to the array
slides for 2 days at 37°C in a moist chamber. The array slides were
rinsed in a washing buffer and dried well. The array slides were
scanned using a GenePix 4000A scanner (Axon Instruments, Union
City, CA). Spots were quantified using the MAC View™ software
program (Macrogen) with the flagging of poor quality spots.
Array CGH data analysis
After exclusion of clones with one or more flagged
spots, the average of the triplicate spots was calculated for each
BAC clone. Log2 Cy3/Cy5 ratios were normalized using the
locally weighted regression known as lowess smoothing for each
array followed by scale normalization between arrays with R package
limma (www.r-project.org). Selection of abnormal clones
used the averaged log2 ratio for each type of cervical
lesion. Chromosomal aberrations were classified as a gain when the
normalized log2 ratio was ≥0.2 and as a loss when the
ratio was ≤−0.2. This number was determined as a 3-fold difference
of the average standard deviation of normal versus normal array CGH
hybridization. In addition, the log2 ratio >0.6 was
defined as amplification. After selection of clones with
aberrations, resampling-based t-test and multiple testing with R
package multi-test (www.r-project.org) were performed to
identify BAC clones showing significantly differential DNA copy
number aberration (DCNA) between and among CIN1, CIN2, CIN3 and
SCC, respectively. The adjusted P-values based on permutation
method (1000 permutations) <0.05 were considered statistically
significant (12).
Validation of DCNA
Polymerase chain reaction (PCR) was carried out to
test the reliability of chromosomal aberrations estimated by the
array CGH experiments (13). PCR
was conducted against pooled genomic DNA of 15 cases each of CIN1,
CIN2, CIN3 and SCC rather than individual case because the average
log2 ratio for each cervical lesion was used for the
discrimination of DCNA. Two genes were selected including
MTR and HDAC9, which show the same state of DCNA at
CIN1, CIN2, CIN3 and SCC. PCR was performed in a reaction tube
containing a PCR mixture composed of 1 ng sample DNA, 20 μM
deoxynucleotide triphosphate (a mixture of dATP, dCTP, dGTP and
dTTP), 2 μl 10X PCR buffer (50 mM KCl, 4 mM MgCl2, 10 mM
Tris-HCl, pH 8.3), 20 pM of each primer, and 2.5U Taq polymerase
(Takara, Shiga, Japan). The 9947A female reference DNA was obtained
from Promega and β-globin was used as internal control. The mixture
was cycled in a PTC-200 thermal cycler (MJ Research, Waltham, MA)
at 94°C for 30 sec, 48°C for 30 sec and 72°C for 30 sec for 35
cycles. PCR products were analyzed by electrophoresis on an agarose
gel. The amplicons were visualized with an ultraviolet
transilluminator Chemi Imager™ 4400 (Alpha Innotech, San Leandro,
CA).
Cervical squamous cell carcinoma datasets
in public database
To validate the data generated by array CGH, we
directly accessed another independent public cervical cancer gene
expression datasets (GEO accession no. GSE7803) (14). In total, 38 microdissected squamous
epithelial samples from 10 normal cervices, 7 high-grade squamous
intra-epithelial lesions (HSIL) and 21 SCCs were profiled for
differential gene expression discovery.
Results
Chromosomal aberrations in CIN1, CIN2,
CIN3 and SCC
The mean age and HPV status of patients are shown in
Table I. Ninety-three percent of
patients showed infection with high-risk HPV types and the mean
ages of women were 40.4, 37.3, 42.7 and 53.0, respectively. We have
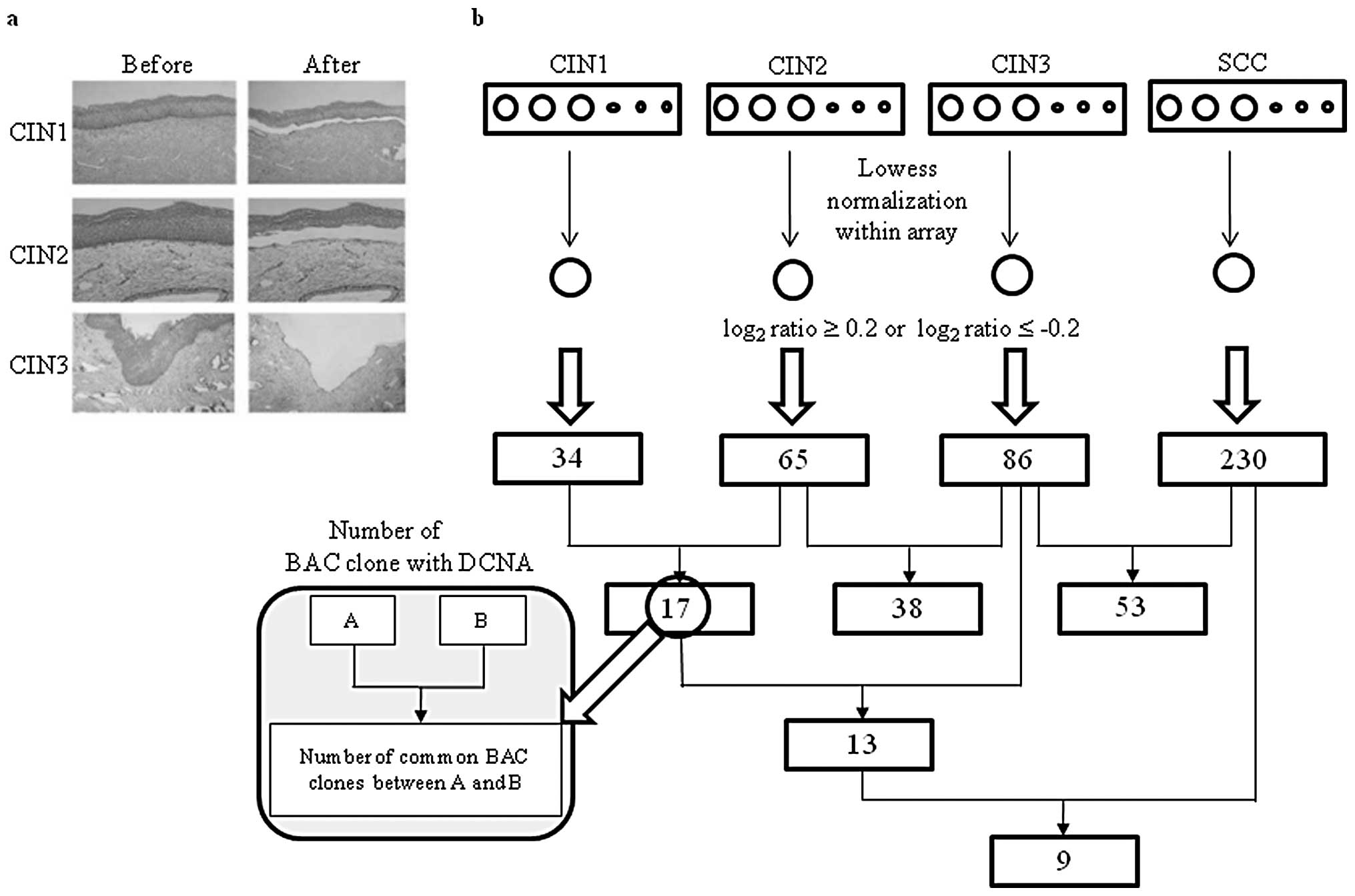
microdissected 60 samples under a microscope to obtain DNA samples
(Fig. 1a). To determine BAC clones
with DCNA, the log2 ratios of 15 samples for each case
were averaged (Fig. 1b). The sex
chromosome and BAC clones with >2 missing values in each type
were excluded from the analysis. A total of 276 BAC clones showed
at least a DCNA among CIN1, CIN2, CIN3 and SCC and the number of
BAC clones with DCNA increased with the progression of CIN.
Approximately half of the BAC clones with DCNA were also retained
at the next grade of cervical lesion. This indicates that the
clonal evolution and selection process might be involved in
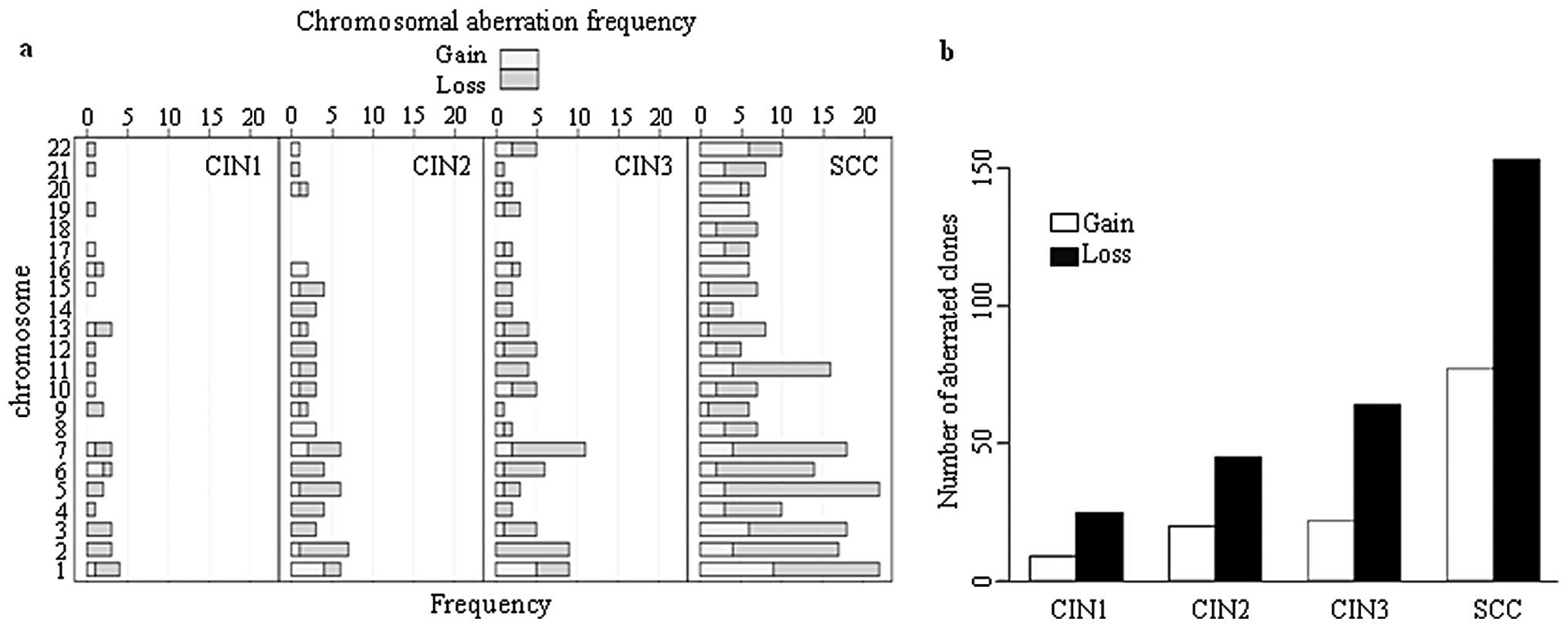
progression of CIN. Distribution of chromosomal aberration at each
type of cervical lesion and SCC is shown in Fig. 2. The number of chromosomes having
>5 BAC clones with DCNA was markedly increased in SCC, which
indicates that severe chromosomal instability might be required
during transition from CIN3 to SCC.
 | Table IPatient characteristics according to
cytopathology. |
Table I
Patient characteristics according to
cytopathology.
| CIN 1 | CIN2 | CIN3 | SCC |
|---|
| No. of
patients | 15 | 15 | 15 | 15 |
| Age (years) | | | | |
| Mean | 40.4±6.0 | 37.3±7.6 | 42.7±13.4 | 53.0±11.3 |
| HPV infection | | | | |
| Positive | 12 | 14 | 15 | 15 |
| Negative | 3 | 1 | 0 | 0 |
The BAC clones showing the same status of DCNA at
CIN1, CIN2, CIN3 and SCC were shown in Table II. These BAC clones included genes
such as 5-methyltetrahydrofo-late-homocysteine methyltransferase
(MTR; loss), histone deacetylase (HDAC9; gain),
calcium/calmodulin-dependent 3′,5′-cyclic nucleotide
phosphodiesterase (PDE1C; loss), component of the exosome
3′→5′ exoribonuclease complex (EXOSC1; gain), zinc finger
DHHC domain-containing protein 16 (ZDHHC16; gain), G
protein-coupled receptor (EDNRB; loss), ethanolamine
phosphate transferase (PIGO; gain), and Ras-related protein
(RAB40C; gain). The DCNAs of these nine BAC clones might
have important role in the progress of cervical lesions because
they were maintained throughout CIN progression to SCC.
| Table IIBAC clones with the same status of
DCNA for CIN1, CIN2, CIN3 and SCC. |
Table II
BAC clones with the same status of
DCNA for CIN1, CIN2, CIN3 and SCC.
| | | Copy no. aberration
|
|---|
| Clone ID | Chromosomal
region | Candidate gene | CIN1 | CIN2 | CIN3 | SCC |
|---|
| 1157 | 1q43 | MTR | − | − | − | − |
| 408 | 2p11.2 | RPIA | − | − | − | − |
| 998 | 6p11.2 | RAB23,
PRIM2A | − | − | − | − |
| 207 | 7p21.1 | HDAC9 | + | + | + | + |
| 1329 | 7p14.3 | PDE1C | − | − | − | − |
| 335 | 10q24.1 | KIAA0690, PGAM1,
EXOSC1, ZDHHC16 | + | + | + | + |
| 472 | 13q22.3 | EDNRB | − | − | − | − |
| 709 | 13q34 | GRTP1,
DKFZp451A211 | + | + | + | + |
| 456 | 16p13.3 | SOLH, LOC146325,
FLJ36208, PIGQ, RAB40C | + | + | + | ++ |
Identification of BAC clones with
significant differential in DCNA among CIN1, CIN2, CIN3 and
SCC
The resampling-based t-test and multiple testing
were performed to detect BAC clones with significantly differential
DCNA between and among CIN1, CIN2, CIN3 and SCC, respectively. The
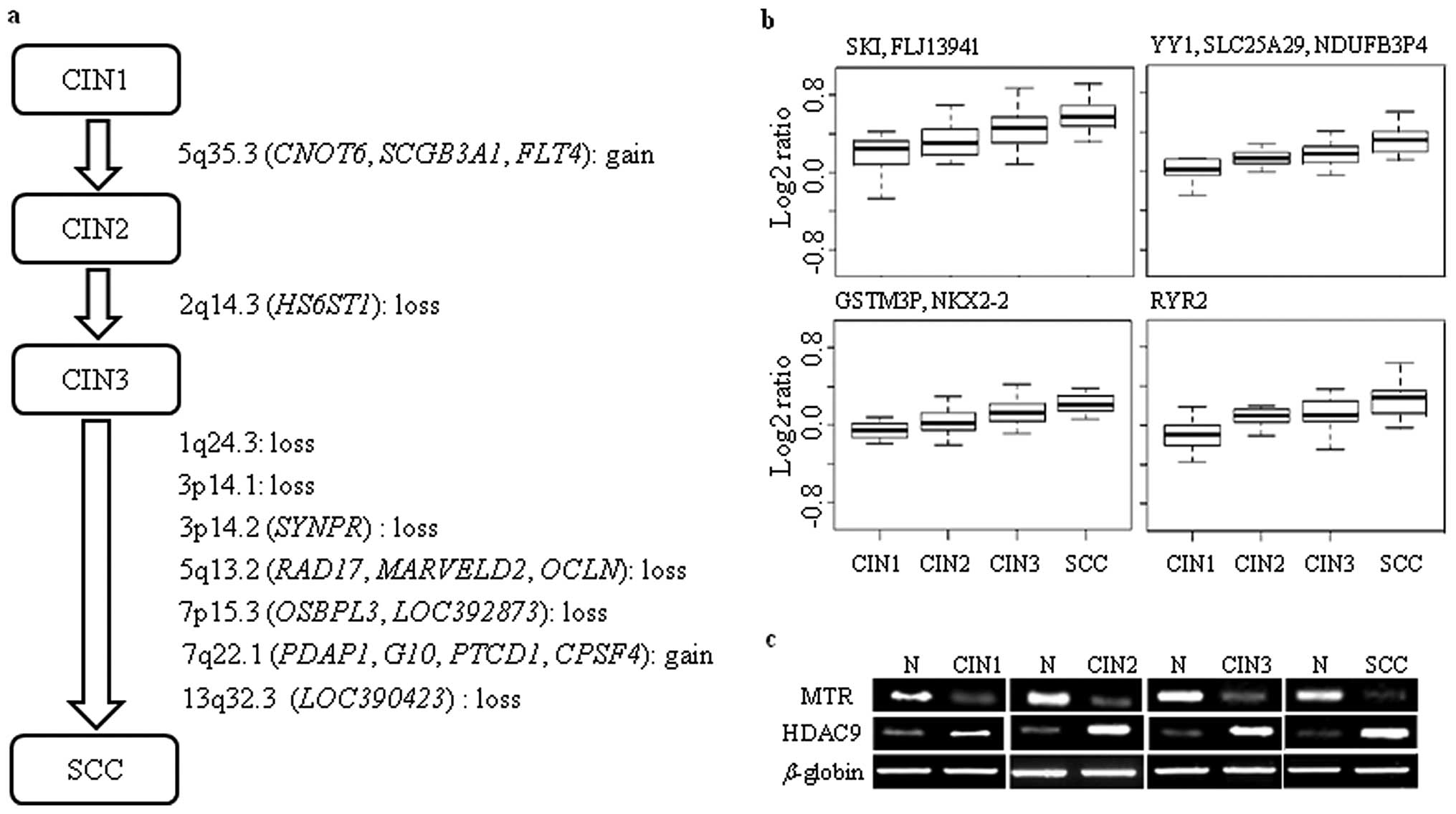
chromosomal regions of 5q35.3 and 2q14.3 showed significantly
differential DCNA between CIN1 and CIN2, and between CIN2 and CIN3,
respectively (Fig. 3a). The
chromosomal region of 5q35.3 included poly(A) nuclease
(CNOT6), potential growth inhibitory cytokine
(SCGB3A1), and tyrosine kinase receptor for vascular
endothelial growth factors C and D (FLT4). The loss of
heparan sulfate biosynthetic enzyme (HS6ST1) located at
2q14.3 during the progression of CIN2 to CIN3 might lead to failure
of heparan sulfate synthesis. In the case of progression of CIN3 to
SCC, seven BAC clones showed significantly different DCNA. Both
PDAP1 and RAD17 showed DCNA only at SCC. This
suggests that the gain of PDAP1 and loss of RAD17
might be potential markers distinguishing SCC from CINs.
The multiple testing among CIN1, CIN2, CIN3 and SCC
identified 57 BAC clones showing significant DCNA (Table III). Among them, 45 BAC clones
showed DCNA only at SCC (Fig. 3b).
This indicates that the marked increase of DCNA might be required
for progression from CIN3 to SCC. The gains of 1p36.33-1p36.32
including SKI (v-ski sarcoma viral oncogene homolog), 5q35.3
including FLT4 and 8q24.3 including ZC3HDC3 (zinc
finger CCCH domain-containing protein 3) were maintained from CIN2
to SCC while the loss of 2q12.1 including SLC9A2
(sodium/hydrogen exchanger) was kept from CIN2 to SCC. These
conserved DCNAs from CIN2 to SCC might play an important role in
the progression of cervical lesions like the BAC clones in Table II. To verify the chromosomal
aberrations estimated by array-CGH experiment, PCR was conducted
against pooled genomic DNA of 15 cases each of CIN1, CIN2, CIN3 and
SCC. The targets of PCR were two genes, the MTR and
HDAC9, which showed the same state of DCNA at CIN1, CIN2,
CIN3 and SCC (Table II). The PCR
amplicons of MTR and HDAC9 generated from test
samples presented a clear contrast with those generated from normal
samples, while there were no significant differences in the
amplicons of β-globin between test and normal samples (Fig. 3c). This result showed a good
agreement with DCNAs evaluated by array-CGH experiments.
| Table IIIFifty-seven BAC clones with
significantly different DCNA among CIN1, CIN2, CIN3 and SCC. |
Table III
Fifty-seven BAC clones with
significantly different DCNA among CIN1, CIN2, CIN3 and SCC.
| | | Copy no. aberration
|
|---|
| Clone ID | Chromosomal
region | Candidate gene | CIN1 | CIN2 | CIN3 | SCC |
|---|
| 99 | 1p34.3 | COL8A2, CSF3R,
RSPO1 | | | | − |
| 1243 | 1p33 |
LOC388630 | | | | − |
| 841 | 1p31.3 |
KIAA1573 | | | | − |
| 196 | 1p13.2 | MRP63P1 | | | | − |
| 1002 | 1q24.3 | MYOC,
TNFSF6 | | − | | − |
| 894 | 2p25.2 | SOX11,
CMPK2 | | | | − |
| 842 | 2p14 |
FLJ16124 | | − | | − |
| 857 | 2q12.1 | SLC9A2,
MGC11332 | | − | − | − |
| 950 | 3p21.33 | ABHD5 | | | | − |
| 1005 | 3p14.1 | ATXN7,
EPM5 | | | | − |
| 1227 | 4p15.33 | BAPX1,
LOC285548, FAM44A | | | | − |
| 1009 | 4p15.32 | MED28,
KIAA1276 | | | | − |
| 816 | 4q31.3 |
KIAA0922 | | − | | − |
| 820 | 5q13.2 | RAD17, MARVELD2,
OCLN | | | | − |
| 295 | 5q34–5q35.1 | SLIT3 | | | | − |
| 1093 | 5q35.1 | KCNIP1 | | | | − |
| 1133 | 6p21.1 | TRERF1 | | | | − |
| 838 | 6q24.3 |
LOC389432 | | | | − |
| 168 | 6q25.2 | MYCT1,
VIP | | | | − |
| 1215 | 7p15.3 | OSBPL3,
LOC392873 | | | | − |
| 886 | 7p15.2 | KIAA0087,
LOC442614 | | | | − |
| 489 | 7p13 | LOC442745,
NUDCD3, LOC442301 | | | − | − |
| 222 | 7q32.3 |
FLJ40288 | | | | − |
| 318 | 8q24.22 | TG,
NDRG1 | | | | − |
| 978 | 9q33.3 | ANGPTL2 | | | | − |
| 1282 | 11q23.3 | APOA5,
APOA1 | | | | − |
| 1000 | 11q24.1 | SCN3B | | | | − |
| 1018 | 13q12.11 | FEOM4,
GJA3 | | | | − |
| 1006 | 13q32.3 |
LOC390423 | | | | − |
| 4 | 17p13.1 | FXR2, SAT2,
SHBG, ATP1B2, TP53 | | | | − |
| 189 | 17q11.2 | HCA66, SUZ12,
LOC114659 | | | | − |
| 1098 | 21q22.2 | C21orf24, ETS2,
FLJ45139 | | | | − |
| 345 | 21q22.2 | DSCAM | | | | − |
| 828 | 22q12.1 | SEZ6L, LOC57168,
HPS4 | | | | − |
| 460 | 22q12.1 |
KIAA1043 | | | | − |
| 1391 | 1p36.33 | PRKCZ,
FLJ31031, LOC440554, SKI | | + | + | + |
| 122 |
1p36.33–1p36.32 | SKI,
FLJ13941 | | + | + | + |
| 1594 | 1q43 |
RYR2 | | | | + |
| 1534 | 2q35 | WNT6, WNT10A,
LOC391485 | | | | + |
| 1562 | 3q26.31 | GHSR,
SPATA16 | | | + | + |
| 1167 | 3q27.3 |
SST | | | | + |
| 1339 | 4p16.1 |
SLC2A9 | | | | + |
| 1387 | 5q31.3 | PCDHB7,
PCDHB8, PCDHB16, PCDHB9, PCDHB10, PCDHB11, PCDHB12, PCDHB13,
PCDHB14, PCDHB18, PCDHB19P, PCDHB15 | | | | + |
| 174 | 5q35.3 | CNOT6,
SCGB3A1, FLT4 | | + | + | + |
| 1272 | 7q22.1 | PDAP1, G10,
PTCD1, CPSF4, ATP5J2 | | | | + |
| 1580 | 7q22.1 | SERPINE1,
AP1S1, VGF, FLJ39237, MOGAT3, PLOD3, ZNHIT1, CLDN15,
TTC11 | | | | + |
| 1284 | 8q24.3 | ZC3HDC3,
GSDMDC1, PP3856, EEF1D, PTK2 | | + | + | + |
| 607 | 8q24.3 | CYHR1, KIFC2,
FOXH1, PPP1R16A, GPT, LOC113655, RECQL4, LRRC14, LOC441381,
MGC70857, KIAA1688 | | + | | + |
| 1203 | 11p15.5 | TOLLIP,
BRSK2 | | | | + |
| 1389 |
12q24.12 |
ATXN2 | | | + | + |
| 363 | 14q32.2 | YY1,
SLC25A29, C14orf68, WARS, NDUFB3P4 | | | | + |
| 760 | 16p11.2 | SBK1,
LOC388237, LOC388229 | | | | + |
| 1083 |
18p11.22 | C18orf30,
C18orf58 | | | | + |
| 312 | 18q23 | ZNF236,
MBP | | | | + |
| 1589 | 20p13 | ATRN, GFRA4,
ADAM33 | | | | + |
| 1581 |
20p11.22 | GSTM3P,
NKX2-2 | | | | + |
| 260 | 21q22.3 | AIRE, PFKL,
C21orf2, TRPM2 | | | | + |
Clustering of array-CGH data including
CIN1, CIN2, CIN3 and SCC
To test the differentiability of chromosomal profile
during CIN progression, hierarchical clustering was carried out for
the 57 BAC clones which showed significantly differential in DCNA
among CIN1, CIN2, CIN3 and SCC (Table
III). The clustering was done on both samples and BAC clones
(Fig. 4a). Group 1 included 3%
CIN1, 19% CIN2, 22% CIN3 and 56% SCC, while group 2 was composed of
43% CIN1, 30% CIN2 and 27% CIN3. This suggests that CIN1 could be
efficiently distinguished from SCC, while classification between
CIN2 and CIN3 might be difficult. The CIN2/3s in group 1 including
all cases of SCC might have a high potential to progress to SCC,
while the CIN2/3s of group 2 including 93% of CIN1s might have a
high potential to regress or persist. Groups 1 and 2 seemed to have
contrast pattern between cluster 1 and 2. This was more apparently
observed when the average log2 ratios were plotted
against each type of cervical lesion (Fig. 4b). The BAC clones which belong to
cluster 1 and 2 showed contrast pattern in terms of the average
log2 ratio. CIN2 and CIN3 appeared to be a kind of
intermediate state, where only minor change was evident, with a few
exceptions. This indicates that severe chromosomal aberrations
might not occur during the progression of CIN2 to CIN3. To validate
the genes located in a region of genomic copy gain/loss in SCC, we
directly accessed another independent public cervical cancer gene
expression datasets (GEO accession no. GSE7803) (14). Differentially expressed genes among
the genes in Table III were
identified by comparison of SCCs to normal cervix samples
(P<0.05) from a one-way ANOVA. This comparison yielded 15 probe
sets with higher expression in SCCs and 19 genes with lower
expression in SCCs. A heat map of expression for 15 genes
(SERPINE1, WARS, RECQL4, MED28, EEF1D, HDAC9, LRRC14, PDAP1,
ZNF236, PTK2, ATXN2, and YY1) that correlated with gain copy number
in SCC is shown in Fig. 4c. This
analysis also showed 19 probe sets (ATXN7, ANGPTL2, EDNRB, SLC9A2,
SOX11, DSCAM, RAB23, TP53, ABHD5, SEZ6L, COL8A2, PFKL, SEZ6L,
SLIT3, and RAD17) that were correlated with loss copy number in SCC
(Fig. 4d). Using 34 probesets as a
signature, the SCCs showed a dominant feature of expression pattern
of the gene sets compared to HSILs and normal cervix samples. The
results suggest that the 34 probes might correlate with CIN
progression to invasive cervical carcinoma.
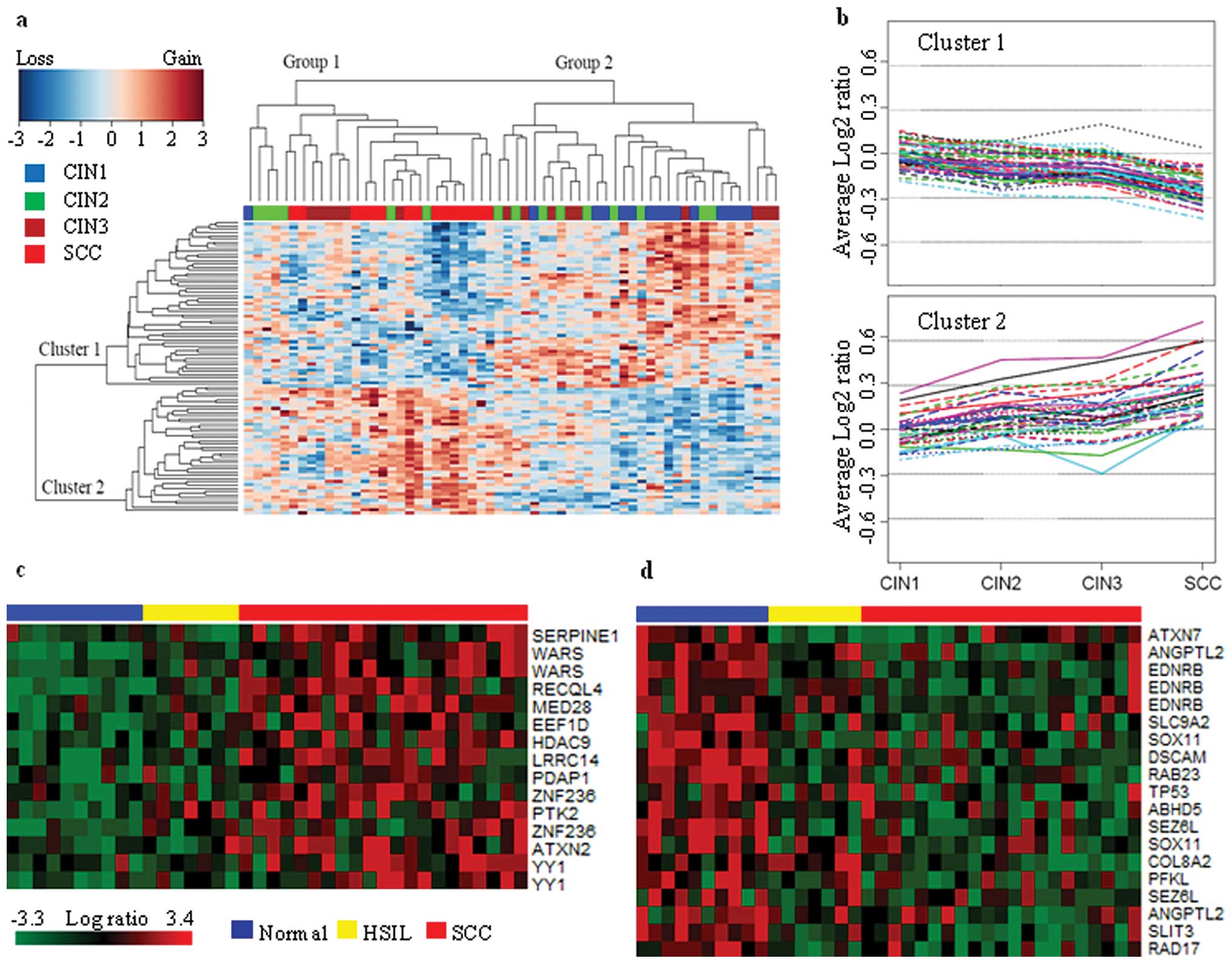 | Figure 4Two-way hierarchical clustering of 60
arrays each including 15 cases of CIN1, CIN2, CIN3 and SCC. (a)
Clustering of 60 samples. Vertical columns correspond to samples,
horizontal lines correspond to BAC clones. Red squares correspond
to loss, green squares to gain, and grey squares are
non-informative. (b) The average log2 ratio profiles of
BAC clones which belong to clusters 1 and 2. Except for few BAC
clones, the DNA copy number of BAC clones in cluster 1 tends to
decrease while that in cluster 2 tends to increase with the
progression of cervical lesion. (c) This analysis identified gene
sets whose expression most strongly correlated with gain copy
number in SCC as shown in Table
III (P<0.05). Then, we performed a supervised clustering
with the sets and samples such as normal, HSIL, and SCC. The genes
are presented in matrix format, where rows represent individual
genes and columns represent each tissue. Samples are ordered
according to progression status of the tumors (blue, normal;
yellow, HSIL; red, SCC). Each cell in the matrix represents the
expression level of a gene in an individual tissue. Red and green
cells reflect high and low expression levels, respectively. (d)
This analysis identified gene sets whose expression most strongly
correlated with loss copy number in SCC (P<0.05). |
Discussion
Integration of high-risk HPV associated with host
genomic alterations has been provided in a study that showed more
numerical chromosomal aberrations were found to progress to
invasive cancer for precursor lesions (15). The present study surveyed the
chromosomal aberrations for CIN1, CIN2, CIN3 and SCC with array-CGH
to determine whether the progression of CIN is reflected in their
genomic signature. The DCNAs of chromosomal locations including
1q43, 2p11.2, 6p11.2, 7p21.1, 7p14.3, 10q24.1, 13q22.3, 13q34 and
16p13.3 might play a crucial role in the progression of cervical
lesion because they were conserved throughout the progression of
CIN to SCC. The chromosomal region of 16p13.3 showed amplification
at SCC (16). HDAC9 located
in 7p21.1 is involved in the alteration of chromosome structure and
affects the transcription factor access to DNA. HDAC9 are
significantly upregulated in high-risk medulloblastoma in
comparison to low-risk medulloblastoma, and their expression is
associated with poor survival (17). The amplification of RAS
oncogene family, RAB40C, might be a potential maker for SCC
(18). The DCNAs of these nine BAC
clones might have important role in the progress of cervical
lesions because they were maintained throughout CIN progression to
SCC.
The chromosomal regions of 5q35.3 and 2q14.3 showed
significant differential in DCNA between CIN1 and CIN2, and between
CIN2 and CIN3, respectively. The gain of 5q35.3 including
FLT4 at CIN2 might induce the activation of
lymphangiogenesis and maintenance of the lymphatic endothelium,
while the loss of 2q14.3 including HS6ST1 at CIN3 might lead
to failure in generating a myriad of distinct heparan sulfate fine
structures that carry out multiple biological activities. The gain
of FLT4 might play important role in the progression of CIN1
to CIN2 because the vascular endothelial growth factor (VEGF)
C/FLT4 autocrine loop in tumor cells is known as a potential
enhancer system to promote cancer progression (19). The heparan sulfate biosynthetic
enzyme family is key components in generating a myriad of distinct
heparan sulfate fine structures that carry out multiple biological
activities (20). The chromosomal
regions with significantly differential DCNA were markedly
increased in the progression of CIN3 to SCC and 55% of newly
appeared clones of DCNA at SCC included at least a cancer
associated gene. Region on chromosome 7q22 was commonly gained,
whereas chromosome 5q frequently showed losses in cervical
malignant lesions (21,22). Among genes included in those seven
BAC clones, PDAP1 located at 7q22.1 is known to enhance
platelet-derived growth factor A (PDGFA)-stimulated cell growth in
fibroblasts, but inhibits the mitogenic effect of PDGFB while
RAD17 located at 5q13.2 encodes a product that is highly
similar to the gene product of S. pombe rad17, a cell cycle
checkpoint gene required for cell cycle arrest and DNA damage
repair in response to DNA damage (23). In addition, both PDAP1 and
RAD17 showed DCNA only at SCC. This implicates that the SCC
could have different chromosome profile from CINs (24). As it is difficult to distinguish
between CIN3 lesion and early invasive SCC on clinical grounds
alone, the gain of PDAP1 and loss of RAD17 might be
potential markers distinguishing SCC from CINs (25).
The multiple testing among CIN1, CIN2, CIN3 and SCC
identified 57 BAC clones showing significant DCNA. The marked
increase of DCNA at SCC only might be required for progression from
CIN3 to SCC. The gains of 1p36.33 including SKI and 5q35.3
including FLT4 were maintained from CIN2 to SCC, which were
not common chromosomal aberrations. There have been many reports on
chromosomal aberrations, including duplication or triplication and
deletions in 1p36.33 and 5q35.3, associated with various genetic
diseases (26,27). It is however likely that deletion
is more common in both aberrations than either duplication or
triplication (28). The 8q24
amplicon has been attributed to PTK2 (which encodes
FAK) in squamous carcinoma cell lines (29). PTK2/FAK encodes a
cytoplasmic tyrosine kinase, and seems to be specific to the
ability of integrins to crosstalk through Ras and PI3K for
oncogenesis (30). Integrin
signaling seems to maintain the cancer stem cell population in
tumors, as ablation of PTK2 decreased the pool of cancer
stem cells in spontaneously forming mouse mammary tumors (31). Thus the 8q24 amplicon has a
plausible role in cancer biology. It is also consistent that the
loss of 2q12.1 including SLC9A2 (sodium/hydrogen exchanger)
was kept from CIN2 to SCC (21).
These conserved DCNAs from CIN2 to SCC might play an important role
in the progression of cervical lesions.
Seventy-nine percent of these BAC clones presented
DCNA only at SCC. This indicates that the progression to SCC
accompanies a dramatic increase in DCNA. The gain of oncogenes such
as RAB40C and SKI was retained from CIN1 and CIN2,
respectively, to SCC. The loss of tight junction membrane proteins
such as MARVELD2 and OCLN at SCC might induce
destruction of the epithelial barriers (32,33).
The severe DCNAs at SCC might be due to the genome instability by
the integration of HPV DNA. The HPV initially exists in an episomal
state after infection (34).
However, the viral DNA often integrates into the host genome as
lesions progress (35). The fact
that most invasive cases contain integrated copies of the virus
genome suggests that this event may be selected during clonal
evolution and, thus, contributes to tumor development (36). The viral DNA integration into host
genome leads to an increase of E6 and E7 transcription at the
interruption of the viral E2 regulatory component, escalating the
ability of the virus to induce neoplastic transformation (37). The viral integration may also be
located within fragile sites or area of the genome that contain
tumor suppressor genes or oncogenes (38,39).
In addition, DCNAs and other complex rearrangements have been
observed near sites of viral integration (39). These reports implicate that HPV
mediated tumorigenesis might act at the direct integration-related
disruption of genomic segmental DNA copy number as well as at viral
oncoproteins.
Gene expression is useful for identifying target
genes within a region of genomic copy gain/loss. Gene expression
data with DCNA could derive correlation of genes in determining the
ranking of genes. Not all genes studied here, however, had a change
in expression level. It was found that 62% of highly amplified
genes in breast cancer exhibit ≥2-fold increased expression
(40). Another study showed that
44% of the highly amplified genes were overexpressed and 10.5% of
the highly overexpressed genes were amplified in breast cancer cell
lines (41). Overall, ≥12% of all
variation in gene expression was directly attributed to variation
in gene copy number. Transcription regulation, such as upstream
genes and feedback regulators, is related to this unparalleled
changes in the expression level. Our study identified 34 probesets
consistently overexpressed when amplified, suggesting an unbiased
identification of candidate drivers in SCC beyond transcription
factors or signaling proteins.
In conclusion, the presence of differential and
common DCNAs among CIN1, CIN2, CIN3 and SCC supports that the CIN
progression might include continual clonal selection and evolution.
In addition, the functional study of genes showing those DCNAs
might give clues to understanding the progression of cervical
lesions.
Acknowledgements
This study was supported by a grant
from the National R&D Program for Cancer Control, Ministry for
Health, Welfare and Family affairs, Republic of Korea (grant no.
0820330).
References
|
1.
|
Green J, Berrington de Gonzalez A,
Sweetland S, et al: Risk factors for adenocarcinoma and squamous
cell carcinoma of the cervix in women aged 20–44 years: the UK
National Case-Control Study of Cervical Cancer. Br J Cancer.
89:2078–2086. 2003.
|
|
2.
|
Pisani P, Bray F and Parkin DM: Estimates
of the world-wide prevalence of cancer for 25 sites in the adult
population. Int J Cancer. 97:72–81. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Termini L, Maciag PC, Soares FA, et al:
Analysis of human kallikrein 7 expression as a potential biomarker
in cervical neoplasia. Int J Cancer. 127:485–490. 2010.PubMed/NCBI
|
|
4.
|
Lockwood WW, Coe BP, Williams AC, MacAulay
C and Lam WL: Whole genome tiling path array CGH analysis of
segmental copy number alterations in cervical cancer cell lines.
Int J Cancer. 120:436–443. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Heselmeyer K, Schrock E, du Manoir S, et
al: Gain of chromosome 3q defines the transition from severe
dysplasia to invasive carcinoma of the uterine cervix. Proc Natl
Acad Sci USA. 93:479–484. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Rapp L and Chen JJ: The papillomavirus E6
proteins. Biochim Biophys Acta. 1378:F1–19. 1998.PubMed/NCBI
|
|
7.
|
Hillemanns P, Wang X, Staehle S, Michels W
and Dannecker C: Evaluation of different treatment modalities for
vulvar intraepithelial neoplasia (VIN): CO(2) laser vaporization,
photodynamic therapy, excision and vulvectomy. Gynecol Oncol.
100:271–275. 2006. View Article : Google Scholar
|
|
8.
|
Tranbaloc P: [Natural history of precursor
lesions of cervical cancer]. Gynecol Obstet Fertil. 36:650–655.
2008.(In French).
|
|
9.
|
Wilting SM, Steenbergen RD, Tijssen M, et
al: Chromosomal signatures of a subset of high-grade premalignant
cervical lesions closely resemble invasive carcinomas. Cancer Res.
69:647–655. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ueda Y, Enomoto T, Miyatake T, et al:
Monoclonal expansion with integration of high-risk type human
papillomaviruses is an initial step for cervical carcinogenesis:
association of clonal status and human papillomavirus infection
with clinical outcome in cervical intraepithelial neoplasia. Lab
Invest. 83:1517–1527. 2003. View Article : Google Scholar
|
|
11.
|
van Zeeburg HJ, Snijders PJ, Pals G, et
al: Generation and molecular characterization of head and neck
squamous cell lines of fanconi anemia patients. Cancer Res.
65:1271–1276. 2005.PubMed/NCBI
|
|
12.
|
van der Laan MJ, Dudoit S and Pollard KS:
Augmentation procedures for control of the generalized family-wise
error rate and tail probabilities for the proportion of false
positives. Stat Appl Genet Mol Biol. 3:Jun 15–2004.(Epub ahead of
print).
|
|
13.
|
Fitzpatrick MA, Funk MC, Gius D, et al:
Identification of chromosomal alterations important in the
development of cervical intraepithelial neoplasia and invasive
carcinoma using alignment of DNA microarray data. Gynecol Oncol.
103:458–462. 2006. View Article : Google Scholar
|
|
14.
|
Zhai Y, Kuick R, Nan B, et al: Gene
expression analysis of preinvasive and invasive cervical squamous
cell carcinomas identifies HOXC10 as a key mediator of invasion.
Cancer Res. 67:10163–10172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Alazawi W, Pett M, Strauss S, et al:
Genomic imbalances in 70 snap-frozen cervical squamous
intraepithelial lesions: associations with lesion grade, state of
the HPV16 E2 gene and clinical outcome. Br J Cancer. 91:2063–2070.
2004. View Article : Google Scholar
|
|
16.
|
Kang JU, Koo SH, Kwon KC, Park JW and Kim
JM: Identification of novel candidate target genes, including
EPHB3, MASP1 and SST at 3q26.2–q29 in squamous cell carcinoma of
the lung. BMC Cancer. 9:2372009.PubMed/NCBI
|
|
17.
|
Milde T, Oehme I, Korshunov A, et al:
HDAC5 and HDAC9 in medulloblastoma: novel markers for risk
stratification and role in tumor cell growth. Clin Cancer Res.
16:3240–3252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Yang Q, Jie Z, Cao H, et al: Low-level
expression of let-7a in gastric cancer and its involvement in
tumorigenesis by targeting RAB40C. Carcinogenesis. 32:713–722.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Matsuura M, Onimaru M, Yonemitsu Y, et al:
Autocrine loop between vascular endothelial growth factor (VEGF)-C
and VEGF receptor-3 positively regulates tumor-associated
lymphangiogenesis in oral squamoid cancer cells. Am J Pathol.
175:1709–1721. 2009. View Article : Google Scholar
|
|
20.
|
Habuchi H, Miyake G, Nogami K, et al:
Biosynthesis of heparan sulphate with diverse structures and
functions: two alternatively spliced forms of human heparan
sulphate 6-O-sulphotransferase-2 having different expression
patterns and properties. Biochem J. 371:131–142. 2003. View Article : Google Scholar
|
|
21.
|
Narayan G, Pulido HA, Koul S, et al:
Genetic analysis identifies putative tumor suppressor sites at
2q35–q36.1 and 2q36.3–q37.1 involved in cervical cancer
progression. Oncogene. 22:3489–3499. 2003.PubMed/NCBI
|
|
22.
|
Rao PH, Arias-Pulido H, Lu XY, et al:
Chromosomal amplifications, 3q gain and deletions of 2q33–q37 are
the frequent genetic changes in cervical carcinoma. BMC Cancer.
4:52004.
|
|
23.
|
Zimmerman ES, Chen J, Andersen JL, et al:
Human immuno-deficiency virus type 1 Vpr-mediated G2 arrest
requires Rad17 and Hus1 and induces nuclear BRCA1 and gamma-H2AX
focus formation. Mol Cell Biol. 24:9286–9294. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Liang M, Ueno M, Oomizu S, et al:
Galectin-9 expression links to malignant potential of cervical
squamous cell carcinoma. J Cancer Res Clin Oncol. 134:899–907.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Tan GC, Sharifah NA, Shiran MS, Salwati S,
Hatta AZ and Paul-Ng HO: Utility of Ki-67 and p53 in distinguishing
cervical intraepithelial neoplasia 3 from squamous cell carcinoma
of the cervix. Asian Pac J Cancer Prev. 9:781–784. 2008.PubMed/NCBI
|
|
26.
|
Tonk VS, Wilson GN, Yatsenko SA, et al:
Molecular cytogenetic characterization of a familial
der(1)del(1)(p36.33)dup(1) (p36.33p36.22) with variable phenotype.
Am J Med Genet A. 139A:136–140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Chen M, Ye Y, Yang H, et al: Genome-wide
profiling of chromosomal alterations in renal cell carcinoma using
high-density single nucleotide polymorphism arrays. Int J Cancer.
125:2342–2348. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Heilstedt HA, Ballif BC, Howard LA,
Kashork CD and Shaffer LG: Population data suggest that deletions
of 1p36 are a relatively common chromosome abnormality. Clin Genet.
64:310–316. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Garnis C, Coe BP, Ishkanian A, Zhang L,
Rosin MP and Lam WL: Novel regions of amplification on 8q distinct
from the MYC locus and frequently altered in oral dysplasia and
cancer. Genes Chromosomes Cancer. 39:93–98. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Schlaepfer DD and Mitra SK: Multiple
connections link FAK to cell motility and invasion. Curr Opin Genet
Dev. 14:92–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Luo M, Fan H, Nagy T, et al: Mammary
epithelial-specific ablation of the focal adhesion kinase
suppresses mammary tumorigenesis by affecting mammary cancer
stem/progenitor cells. Cancer Res. 69:466–474. 2009. View Article : Google Scholar
|
|
32.
|
Ohkuni T, Kojima T, Ogasawara N, et al:
Expression and localization of tricellulin in human nasal
epithelial cells in vivo and in vitro. Med Mol Morphol. 42:204–211.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Benedicto I, Molina-Jimenez F, Bartosch B,
et al: The tight junction-associated protein occludin is required
for a postbinding step in hepatitis C virus entry and infection. J
Virol. 83:8012–8020. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Li W, Wang W, Si M, et al: The physical
state of HPV16 infection and its clinical significance in cancer
precursor lesion and cervical carcinoma. J Cancer Res Clin Oncol.
134:1355–1361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Singh RK, Dasgupta S, Bhattacharya N, et
al: Deletion in chromosome 11 and Bcl-1/Cyclin D1 alterations are
independently associated with the development of uterine cervical
carcinoma. J Cancer Res Clin Oncol. 131:395–406. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Gallego MI, Schoenmakers EF, van de Ven WJ
and Lazo PA: Complex genomic rearrangement within the 12q15
multiple aberration region induced by integrated human
papillomavirus 18 in a cervical carcinoma cell line. Mol Carcinog.
19:114–121. 1997. View Article : Google Scholar
|
|
37.
|
Lazo PA: The molecular genetics of
cervical carcinoma. Br J Cancer. 80:2008–2018. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Reuter S, Bartelmann M, Vogt M, et al:
APM-1, a novel human gene, identified by aberrant co-transcription
with papillomavirus oncogenes in a cervical carcinoma cell line,
encodes a BTB/POZ-zinc finger protein with growth inhibitory
activity. EMBO J. 17:215–222. 1998. View Article : Google Scholar
|
|
39.
|
Thorland EC, Myers SL, Gostout BS and
Smith DI: Common fragile sites are preferential targets for HPV16
integrations in cervical tumors. Oncogene. 22:1225–1237. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Pollack JR, Sorlie T, Perou CM, et al:
Microarray analysis reveals a major direct role of DNA copy number
alteration in the transcriptional program of human breast tumors.
Proc Natl Acad Sci USA. 99:12963–12968. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Hyman E, Kauraniemi P, Hautaniemi S, et
al: Impact of DNA amplification on gene expression patterns in
breast cancer. Cancer Res. 62:6240–6245. 2002.PubMed/NCBI
|