Contents
Introduction
Papillary thyroid carcinoma
Follicular thyroid carcinoma
Anaplastic thyroid carcinoma
Clinical implications: Potential therapeutic
targets
Conclusion
Introduction
Thyroid carcinoma is the most common type of
malignant endocrine neoplasia, accounting for approximately 1% of
all new malignant diseases with an annual incidence of 5.9 and 17.3
per 100,000 in men and women, respectively (US 2005-2009) (1,2).
Follicular cell-derived thyroid neoplasias include differentiated
thyroid carcinoma (DTC), which represents more than 90% of all
thyroid malignancies and comprise the papillary and follicular
thyroid carcinomas (FTCs). The anaplastic thyroid carcinoma (ATC)
corresponds to 1% of all thyroid tumors and can arise de
novo or by the dedifferentiation of a papillary or follicular
tumor (3). Medullary thyroid
carcinoma (MTC) is a malignancy arising from the parafollicular
C-cells and accounts for approximately 3–8% of all thyroid
carcinomas (4).
The etiology of DTC is not yet fully understood.
External radiation is the only exogenous factor which has been
clearly identified as causing thyroid carcinoma, almost exclusively
the papillary form. Iodine excess has been associated with the
increase in the incidence of papillary thyroid carcinoma (PTC)
(5,6). A number of genetic events have been
described in thyroid carcinoma pathogenesis. Papillary carcinomas
commonly present genetic alterations that lead to the activation of
the mitogen-activated protein kinase (MAPK) pathway (7–9). In
follicular carcinomas, the induction of both the MAPK and
phosphatidylinositol 3-kinase (PI3K) cascades is frequently
observed (10). On the contrary,
anaplastic carcinomas harbor a wide set of additive genetic
alterations, occurring mainly in the gene effectors of the MAPK,
PI3K and β-catenin signaling pathways (11–13).
These distinct signaling pathways have been implicated in
follicular cell-derived thyroid cancer development and progression
(14–16).
In this review, we aimed to present a comprehensive
account of the recent advances in the understanding of the
signaling pathways in follicular cell-derived thyroid carcinomas,
as well as the molecular mechanisms involved in tumor development
and progression.
Papillary thyroid carcinoma
PTC represents ∼80% of all malignant thyroid tumors.
The overall incidence of PTC is 7.7 per 100,000 and is increasing,
in part due to the increase in the detection of small tumors
(16). PTC is often diagnosed at
approximately the 5th decade of life and is known to be a
slow-growing tumor (17,18). Patients usually present with a
palpable nodule and the absence of any other clinical findings is
common (3). The majority of
patients have a favorable outcome; however, ∼10% of the cases have
tumor recurrence and metastatic disease (18,19).
Aberrant activation of the MAPK pathway due to
mutations or gene rearrangements is the most common genetic event
in PTC (7–9). Point mutations in BRAF or
RAS genes and (RET)/PTC or NTRK1 rearrangements are mutually
exclusive and identified in more than 70% of PTCs (7–9). The
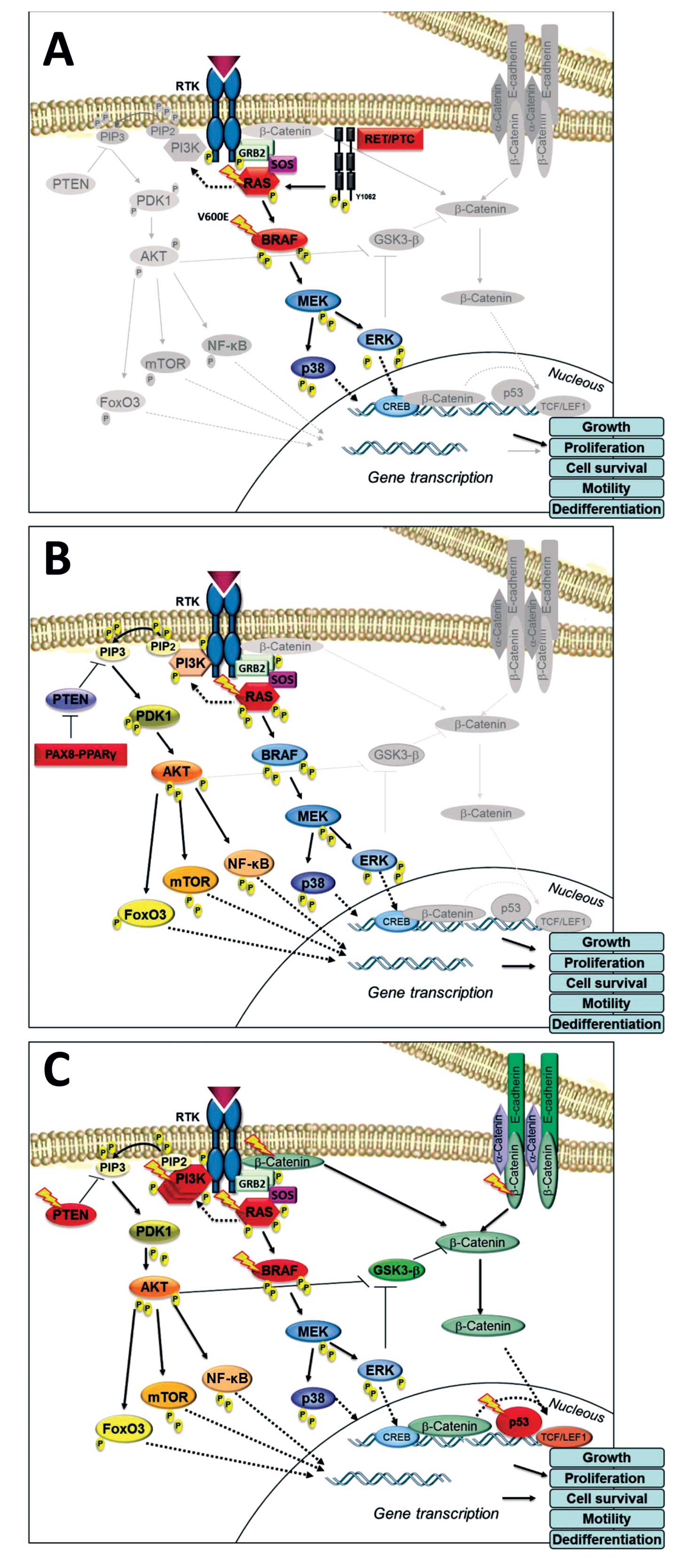
Fig. 1A summarizes the major
signaling pathways involved in PTC.
BRAF oncogene
Mutations in the BRAF gene are the most
common genetic alteration in PTC, occurring in ∼45% of cases
(6). BRAF is a serine-threonine
kinase protein, member of the RAF (v-raf-1 murine leukemia viral
oncogene homolog) family, which comprises the
serine/threonine-specific kinase effectors of the MAPK cascade
(7,20,21).
Briefly, the MAPK cascade effects initiate upon RAS activation,
which recruits BRAF to the plasma membrane initiating its
activation. Once activated, BRAF phosphorylates MEK, which in turn
provides the signal to activate the tyrosine, ERK, in the cytosol
and nucleus, leading to cell proliferation, migration and survival
(22,23) (Fig.
1A). Approximately 95% of all BRAF mutations involve a
T>A transversion at gene position 1799, resulting in valine to
glutamate amino acid substitution at position 600 of the protein
(V600E). Other described alterations in the BRAF gene include the
A>G transversion at gene position 1801 (K601E), fusion with the
A-kinase anchor protein 9 (AKAP9) gene and small in-frame
insertions or deletions around codon 600 (24–26).
The presence of BRAF mutations in micro-PTC (∼40%)
and benign tumors (9,27,28)
suggests a role of this alteration in the early stages of PTC
development. BRAFV600E is an oncogenic protein with
markedly elevated kinase activity that overactivates the MAPK
pathway (34,35). Studies using
BRAFV600E-transgenic mice have shown the development of
PTC with similar properties to those observed in human
BRAF-positive PTCs (29),
whereas mice with the constitutive or doxycycline-inducible
BRAF-mutated gene develop infiltrative PTC with a high rate
of extrathyroidal structures, vascular invasion and a poorly
differentiated aspect (30,31).
The induction of BRAFV600E mutation has been shown to
abolish the expression of several thyroid-specific genes,
radioiodine uptake and cause pronounced hypothyroidism, which may
be partially explained by the down-regulation of the thyroid
hormone activating type 1 and 2 deiodinases and induction of the
thyroid hormone inactivating type 3 deiodinase, as recently
described (31,33).
BRAF mutations are typically identified in
classical and tall cell variant of PTC and are associated with a
more aggressive tumor behavior (9,34,35).
The high growth rates observed in BRAFV600E tumors may
be explained partially by the MAPK-induced hyperphosphorylation
with consequent inhibition of the retinoblastoma (RB) protein,
dependent transcription factors (E2F) and p27 of cyclin-dependent
kinase (CDK) activity (36).
Moreover, the BRAF oncogene induces the expression of matrix
metalloproteinases (MMPs), a large group of enzymes that regulate
cell-matrix composition and are important factors of tumor
invasiveness (37–39). Previous studies have suggested that
MMP proteins are modulated according to the intensity of MAPK
pathway activation and/or signal transducer and activator of
transcription (STAT) expression, which may explain the mechanism of
induction of these proteins in BRAF-mutated PTCs and the increased
propensity of these tumors to invade surrounding tissues (37,40).
The BRAF-mutated protein also induces nuclear factor-κB (NF-κB).
Thyroid cells (WRO) harboring this oncogene display increased
levels of activity in the NF-κB pathway, which results in the
upregulation of anti-apoptotic factors and the induction of cell
invasion (40).
Recently, a novel inhibitory mechanism that may
operate in BRAFV600E-induced PTC was shown. The presence
of BRAFV600E mutation abolished the macrophage
stimulating 1/forkhead box O3 (MST1/FOXO3) pathway transactivation
in a thyroid cell line (FRO), resulting in the suppression of p21
and p27 CDK inhibitors and interrupting the apoptotic process.
Accordingly, the development of BRAFV600E transgenic
mice with the MST1 knockout leads to abundant foci of poorly
differentiated thyroid carcinoma and large areas without follicular
architecture or colloid formation, suggesting that the activity of
the MST1/FOXO3 pathway determines the phenotype of
BRAFV600E tumors (41).
RET/PTC rearrangements
The RET proto-oncogene, located on chromosome
10q11.2, encodes a tyrosine kinase receptor. The RET protein is
usually expressed in cells derived from the neural crest and
gain-of-function mutations are associated with MTC (42). In PTC, genomic rearrangements
juxtapose the RET tyrosine kinase domain to unrelated genes,
thereby creating dominantly transforming oncogenes, denominated
RET/PTC. The RET/PTC rearrangements are the 2nd most common genetic
alteration described in PTC and observed in ∼13–43% of cases,
mostly in pediatric cancers or in individuals exposed to ionizing
radiation from nuclear accidents (12,43–45).
At least 12 types of RET/PTC rearrangements have been reported, all
originating from the RET fusion to different partners (44,46).
RET/PTC1 comprises up to 60% of the rearrangements and is derived
from an intrachromosomal rearrangement (10q), leading to the fusion
of the RET tyrosine kinase domain to the H4 gene
(D10S170). The RET/PTC1 encodes a 585-amino acid protein
with unknown function (47).
RET/PTC3 accounts for 20–30% of the rearrangements and is formed by
the RET gene fusion with the nuclear receptor coactivator 4
(NCOA4) gene (also known as ELE1, RFG or
ARA70) (44,47).
Papillary tumors harboring the RET/PTC1
rearrangement commonly exhibit the classical papillary histology,
whereas RET/PTC3 tumors normally present the solid variant
(48). RET/PTC tumors tend to be
small, with a favorable outcome and usually do not progress to a
more aggressive behavior and/or undifferentiated thyroid carcinoma
(9,49,50).
This alteration has also been associated with a younger age at
diagnosis and a higher rate of lymph node metastasis (9,49).
The high prevalence of RET/PTC in occult (42%) or microscopic PTC
(77%) as well as in follicular adenoma (45%), may indicate a
putative role of this rearrangement during the early stages of PTC
development (51,52). Accordingly, studies performed using
transgenic mice carrying RET/PTC1 and/or RET/PTC3 have shown that
the PTC tumors which develop in these animals are similar to those
occurring in humans (53,54).
The RET/PTC-derived mechanisms of tumor induction
initiate with the fusion of protein partners, resulting in the
ligand-independent autophosphorylation of the RET protein. The RET
intracellular domain contains at least 12 autophosphorylation
sites, and 11 of them are preserved in the RET/PTC protein
(55). The Y1062 and Y1015 RET
residues are constitutively phosphorylated and are required for
cell transformation (56). These
residues are essential binding sites for several proteins, which in
turn, lead to the activation of the MAPK and PI3K/AKT signaling
pathways and play an essential role in RET/PTC signaling with
downstream cellular effects on migration and proliferation
(57–59).
Another dysfunctional signaling pathway identified
in 65–90% of RET/PTC-positive tumors is β-catenin, which is
involved in gene transcription and cell adhesion regulation
(60,61). The β-catenin pathway can be
directly activated by several mechanisms: via RET tyrosine residue,
cAMP response element-binding (CREB), glycogen synthase kinase 3
phosphorylation (GSK3-S) or via effectors of the MAPK and PI3K
pathways (61,62). The increase in the free β-catenin
protein pool promotes proliferation and invasion, possibly due to
the interaction with transcriptional factors, such as the T-cell
factor/lymphoid enhancer factor (TCF/LEF), c-Myc (v-myc
myelocytomatosis viral oncogene homolog), or cyclin D1 (60,61,63).
RAS oncogene
RAS genes (H-RAS, K-RAS, and
N-RAS) encode highly related G-proteins which play a central
role in intracellular signal transduction by the activation of the
MAPK and other signaling pathways, such as PI3K/AKT (see below)
(15). RAS gene mutations
are found in 10–43% of PTCs, particularly in the follicular variant
(64–66). The RAS point mutations generally
occur in codons 12, 13, or 61 of H-RAS, K-RAS, or N-RAS proteins.
RAS-mutated PTC tends to be encapsulated and exhibits a low rate of
lymph node metastasis (9,65). However, previous studies have
reported that this mutation may also be associated with a more
aggressive phenotype and a higher incidence of distant metastasis
(66,67). The molecular mechanism proposed for
RAS-derived tumorigenesis is the constitutive activation of
distinct pathways involved in proliferation, differentiation and
cell survival processes (66).
NTRK1 rearrangements
The neurotrophic tyrosine kinase receptor, type 1
(NTRK1) gene, located on chromosome 1, encodes the
high-affinity nerve growth factor (NGF) receptor and is activated
through the MAPK pathway (68).
NTRK1 rearrangements are usually found in <10% of PTCs and
result from the NTRK1 gene fusion with different partners
(69,70,71).
Experimental evidence suggests that the NTRK1 oncogene
represents an early event in the process of thyroid carcinogenesis.
Transgenic mice carrying NTRK1 oncogene develop thyroid
hyperplasia and PTC (72).
Additionally, crossing NTRK1 mice with p27kip1-deficient mice has
been shown to increase the penetrance of thyroid cancer and shorten
the tumor latency period (73).
NTRK1 rearrangements are associated with a younger age at diagnosis
and a less favorable outcome (69,70).
Follicular thyroid carcinoma
The FTC represents 10–15% of thyroid cancers. These
tumors are generally unifocal and present less lymph node
involvement (<5%) than PTCs. By contrast, distant metastases,
mainly to the lungs and bones, are more frequent at disease
presentation (∼20%) (4). Although
former studies have indicated that FTCs, particularly the invasive
form, have a poorer prognosis than PTCs (74,75),
a recent study that evaluated more than 1,000 patients did not find
differences in tumor-specific survival between PTC and FTC, after
controlling for age, primary tumor size, extrathyroidal invasion or
distant metastasis at diagnosis (76).
The most common genetic events observed in
follicular carcinomas are point mutations in RAS genes and
the rearrangements between the thyroid-specific transcription
factor gene and the peroxisome proliferator-activated receptor gene
[paired box gene 8 (PAX8)-peroxisome proliferator-activated
receptor γ (PPARγ) rearrangements] (80%). Similarly to what is
described in PTC, their oncogenic effects occur through the
activation of the MAPK cascade; however, the induction of the PI3K
pathway is an important event in follicular pathogenesis (15). Fig.
1B summarizes the major signaling pathways involved in FTC.
RAS oncogene
Activating mutations in the RAS gene are
observed in 18–52% of follicular carcinomas and are associated with
tumor dedifferentiation and a less favorable prognosis (77,78).
A number of studies have suggested that RAS mutations are an
early event in follicular thyroid tumorigenesis, since they are
identified in up to 50% of benign follicular tumors (77,79,80,82,83).
Studies using transgenic mice carrying the mutated N-RAS
(Gln61Lys) oncogene demonstrated that these rodents developed
follicular adenomas (11%), invasive follicular carcinomas (∼40%)
and, in certain cases, tumors with a mixed papillary/follicular
morphology. Moreover, 25% of these carcinomas displayed large,
poorly differentiated areas, with vascular invasion and with lung,
bone or liver metastasis (81).
The RAS-mutated protein mediates its effects on
cellular proliferation in part by activation of a cascade of
kinases: RAF (A-RAF B-RAF and C-RAF), dual-specificity
mitogen-activated protein kinases (MEK1/2), extracellular
signal-regulated kinases (ERK1/2) and p38 mitogen-activated protein
kinase. RAS also activates the PI3K pathway, via a direct
interaction with the catalytic subunit of the protein. The PI3K
activation leads to the accumulation of the 2nd messenger,
phosphatidylinositol 3,4,5-trisphosphate (PIP3), resulting in
pyruvate dehydrogenase kinase isozyme 1 (PDK1) and v-akt murine
thymoma viral oncogene homolog (AKT) activation (85,86)
(Fig. 1B). Previous studies using
mice harboring a phosphatase and tensin homolog (PTEN) gene
deletion and a KRASG12D mutation, have shown that the
separate activation of MAPK or PI3K pathways, is unable to
transform thyroid follicular cells; however, their simultaneous
activation is highly oncogenic, leading to locally invasive
follicular carcinomas and distant metastasis (84).
PAX8-PPARγ rearrangements
The thyroid-specific transcription factor
(PAX8) gene is a critical regulator of thyroid
differentiation and growth (87).
PPARγ is a ligand-dependent nuclear transcription factor
highly expressed in adipose tissue, where it plays a critical role
in adipocyte differentiation and fat metabolism regulation
(88). The PAX8-PPARγ
rearrangement arises through a chromosomal translocation, fusing
the 5′ portion of the PAX8 gene with the entire coding
sequence of the PPARγ gene (chromosomes 3p25 and 2q13). It
is detected in ∼35% of FTCs (10,89,90).
The PAX8-PPARγ rearrangement leads to strong
induction of the PPARγ protein and the consequent abrogation of the
normal PPARγ function (95,96).
Under normal conditions, PPARγ inhibits cell proliferation and
induces apoptosis via downstream pathways. The loss of these
functions results in uncontrolled cell growth (14). PPARγ overexpression
abolishes the PTEN-inhibitory effect on immunoactive AKT, which in
turn induces the PI3K signaling pathway (58,97).
The PAX8-PPARγ rearrangement also activates the MAPK, transforming
growth factor β (TGFβ) and Wnt/β-catenin (wingless in
Drosophila) signaling pathways. The increased expression of
the C-terminal binding protein (CTBP2) gene has been
observed in the PAX8-PPARγ-positive-tumors (95). CTBPs are co-repressor proteins
associated with several transcriptional factors involved in Wnt,
TGFβ and MAPK signaling activation, thus explaining their major
role in follicular tumor development (98).
Patients with FTC harboring the PAX8-PPARγ
rearrangement are usually diagnosed at a young age, have a small
tumor size and the majority of tumors are overtly invasive at
presentation (10,89). These findings, however, were not
reproduced in other studies and the impact of PAX8-PPARγ on the
biology and behavior of FTCs remains controversial (10,92).
Follicular adenomas have been shown to have lower
frequency rates of PAX8-PPARγ rearrangements, suggesting that this
chromosomal translocation may be involved in the early phases of
the neoplastic process of FTC, possibly even in premalignant
lesions (90,91,93).
Transfection studies of PAX8-PPARγ in thyroid follicular epithelial
cells have demonstrated accelerated growth rates and a lower number
of cells in the G0/G1 resting state (14,94).
Anaplastic thyroid carcinoma
ATC, also known as undifferentiated thyroid
carcinoma, is the most aggressive form of thyroid neoplasia. It can
originate de novo or represent an advanced stage of
follicular cell-derived thyroid tumors (4,99).
Anaplastic tumors represent <1% of all thyroid tumors and their
annual incidence is ∼1–2 cases per 1,000,000 with a higher overall
incidence in endemic goiter areas (100,101). The ATC typical presentation is
advanced disease at diagnosis. Patients with anaplastic carcinoma
usually have widespread local invasion and distant metastases, most
frequent in the lung, pleura, bone and brain (100). This tumor has poor or no response
to conventional therapeutic modalities. The median survival time
after diagnosis is <1 year (102,103). A younger age (<60 years),
smaller tumor size (<7 cm) and restricted disease have been
associated with a lower mortality rate on multivariate analysis
(104).
ATCs have been described as carrying multiple
distinct genetic alterations with a high prevalence of mutations in
MAPK effectors (13,21). Mutations in the TP53 gene,
β-catenin and PI3K cascade also play a critical role in ATC
development, promoting the dedifferentiation of a previously well
differentiated thyroid tumor (11,105,106). Fig.
1C summarizes the signaling pathways involved in ATC.
Mutations in gene effectors of the MAPK
pathway
MAPK activating genetic alterations have been
described to be involved in the development/progression of ATCs.
ATC tumors present a significant prevalence of RAS (6–55%)
and BRAF mutations (24–50%) (13,14,107). By contrast, RET/PTC, NTRK and
PPARγ-PAX8 rearrangements are rarely observed in these
undifferentiated tumors, supporting the hypothesis that DTCs
associated with these rearrangements do not usually progress to
anaplastic form (108,109).
BRAFV600E mutation is typically found in
ATC tumors which contain areas of well-differentiated PTC, but also
in poorly differentiated and anaplastic tumor areas. These
observations suggest that although this mutation may occur early in
tumorigenesis, it is not sufficient to initiate the
dedifferentiation process. However, it is conceivable that
BRAF mutations may predispose to additional genetic
alterations which in turn activate more aggressive pathways and
lead to dedifferentiation (15,110,111). Of note, BRAFV600E
mutation has also been observed in lymph-node metastasis of ATCs
(111). Of note, patients with
ATCs harboring BRAF mutations have a higher mortality rate
than those patients presenting with RAS or with no
identified mutation, indicating a negative prognosis of these
genetic alterations during all stages of thyroid cancer progression
(13).
RAS mutations are found in a high prevalence
in ATCs (6–55%) (13,14,77).
A previous study suggested that the RAS effect may be due to the
promotion of chromosomal instability, since the expression of
constitutively activated RAS destabilizes the genome of PCCL3
thyroid cells, predisposing to large scale genomic abnormalities
(112).
Genetic alterations in genes involved in
the activation of the PI3K pathway
PIK3CA mutations and copy number
gains
The PIK3CA gene encodes a catalytic subunit
of PI3K and has been described to be mutated in 12–23% of ATC
cases, normally restricted to the undifferentiated thyroid
components. Previous studies have shown a preferential expression
of PIK3CA mutations during the later stages of thyroid
cancer, suggesting that this event may be more important in ATCs
(12–23%) than in DTCs (PTCs, ∼2% and FTCs, <10%) (11,106).
PIK3CA copy number gains are the 2nd most
frequent event in ATC occurring in ∼38–61% of tumors (14,106). Of note, this occurs almost
exclusively in the undifferentiated component of the tumor. The
copy number gain induces the activation of the PI3K cascade through
the enhanced activity of AKT, leading to thyroid cancer
progression. Of note, the PIK3CA mutations and copy number
gain may co-exist with other somatic mutations in ATC, reinforcing
the activation of the distinct signaling pathway in these tumors
(11).
PTEN gene alterations
PTEN is a tumor suppressor gene that
antagonizes signaling through the PI3K pathway. Its action occurs
by removing a phosphate group from the inositol ring of PIP3, which
reduces the downstream activity of the AKT kinase, thereby inducing
cell cycle arrest, apoptosis, or both (113). Several genetic alterations in the
PTEN suppressor gene have been described in ATCs: 12%
present a mutated form (106,108), 28% gene silencing (114) and 69% the hyper-methylated
PTEN gene (115). These
alterations lead to PTEN inactivation by different mechanisms, with
a prominent role in the pathogenesis of follicular
epithelium-derived thyroid carcinomas, particularly in the most
aggressive or undifferentiated forms (114,115). Moreover, PI3K activation produced
by down-regulated PTEN has been shown to correlate with regions of
tumor invasion and metastasis (58,116). Of note, studies using transgenic
mice with a deletion of PTEN or RAS mutations have
shown that the presence of both genetic events is required to
trigger this aggressive form of thyroid cancer (84).
TP53 mutations
The TP53 gene encodes a nuclear protein that
can induce cell cycle arrest, senescence and apoptosis in response
to various stimuli. Alterations in the p53 pathway may contribute
to carcinogenesis, disease progression and resistance to therapy
(117). In thyroid tumors,
TP53 mutations are commonly observed in anaplastic
carcinomas (∼70%) and are rarely described in well-differentiated
thyroid carcinomas (0–9%) (12,105,118). This suggests that TP53
mutations are a late event in tumor progression and that this gene
may play a critical role in the transformation of DTC into the
anaplastic form (105). The
frequent association of p53 inactivation with PI3K activation may
contribute to genomic instability, leading cancer cells to become
resistant to apoptosis and to escape from any growth restriction.
This contributes to a rapidly enlarging neck mass as well as to
chemotherapy and radiotherapy resistance commonly observed in these
tumors (11).
β-catenin genetic alterations
Genetic alterations in the β-catenin
(CTNNB1) gene are observed in ∼65% of thyroid anaplastic
tumors. Gain-of-function mutations can promote β-catenin nuclear
translocation which consequently triggers the transcription process
(119,120). The expression of E-cadherin, a
component of the β-catenin pathway, normally expressed in thyroid
tissue, is usually absent in undifferentiated thyroid carcinomas
(121). These changes appear to
play a pathogenic role in thyroid tumor invasion and regional lymph
node metastasis, due to a decrease in intercellular adhesion and
enhancement of cell motility (122). The lack of E-cadherin expression
is associated with an adverse prognosis for patients with thyroid
carcinoma (123).
Clinical Implications: Potential therapeutic
targets
DTCs demonstrate indolent behavior in the majority
of patients and can be effectively treated by surgery followed by
radioactive iodine and/or thyroid hormone suppressive therapy
(124,125). In patients with metastatic
disease, radioactive iodine therapy can be effective in some cases,
whereas suppressive thyroid hormone therapy can help to delay the
pace of the disease (125,126).
Nevertheless, for those patients with metastatic DTC that
progresses despite radioiodine and thyroid hormone therapy, no
effective treatments are currently available.
Over the last decades, cancer research has been
predominantly focused on the genetic alterations and the advances
in the understanding of the molecular events involved in
differentiated thyroid carcinogenesis have allowed for the
development of new therapies designed for patients with metastatic
disease refractory to radioactive iodine treatment. Specific
tyrosine multikinase inhibitors to target key molecules such as
BRAF, RET/PTC rearrangements, vascular endothelial growth factor
receptors (VEGFRs) and platelet-derived growth factor receptors
(PDGFR) have been evaluated as potential alternatives to DTC
treatment. Table I summarizes the
results obtained to date in several clinical trials. Phase II
studies using BAY 43-9006 (sorafenib) have shown partial response
(15–25%) and stable disease (34–56%) in progressive DTC patients
and the median progression-free survival was significantly longer
in patients harboring BRAF mutations (127–129). A recent study using PLX4032, an
inhibitor of mutant BRAF, in metastatic melanoma patients evaluated
the effect of this drug in 3 PTC patients. The response lasted 8
months in 1 patient (progression-free lasted for 12 months) and
stable disease lasted 11 and 13 months in each of the other 2
patients (130). Although these
compounds have demonstrated the most impressive clinical responses
to date in the treatment of advanced thyroid cancer, the low rate
of partial response, the rare report of complete responses and the
emergence of eventual progression, point out to the need to develop
either more effective single agents or to identify rational
combinations of therapeutic targets.
 | Table I.Clinical trials and follicular
cell-derived thyroid tumors response. |
Table I.
Clinical trials and follicular
cell-derived thyroid tumors response.
| Trade name | Compound | Target | Tumor type | No. of
patients | Partial
responsea [%
(n)] | Stable
diseaseb [% (n)] | Refs. |
|---|
| Sorafenib | BAY 43-9006 | BRAF
(BRAFV600E) | PTC | 41 | 15 (6) | 56 (23) | (127) |
| | VEGFR1-3, PDGFR,
RET, RET/PTC | DTC | 31 | 25 (8) | - | (128) |
| | DTC | 30 | 23 (7) | 34 (10) | (129) |
| Axitinib | AG-013736 | VEGFR1-3,
PDGFR, | PTC | 30 | 26 (8) | 40 (12) | (131) |
| | c-Kit | FTC | 15 | 40 (6) | 46 (7) | |
| | | ATC | 2 | 50 (1) | - | |
| Pazopanib | W786034 | VEGFR1/2,
PDGFR | DTC | 39 | 49 (18) | - | (132) |
| Motesanib | AMG706 | VEGFR1-3, RET,
c-kit | DTC | 93 | 14 (13) | 67 (62) | (133) |
| Gefitinib | ZD1839 | EGFR | DTC | 25 | 0 | 12 (3) | (134) |
| Selumetinib | AZD6244 | MEK1/2 | PTC (IR) | 32 | 3 (1) | 54 (21) | (135) |
| PLX4032 | RG7204 |
BRAFV600E | PTC | 3 | 33 (1) | 66 (2) | (130) |
Conclusion
Thyroid carcinogenesis consists of a complex
process with a large number of molecular alterations among several
thyroid neoplasias. The set of genetic alterations observed in
follicular-cell derived thyroid carcinomas activates specific
pathways, such as the MAPK, PI3K and β-catenin signaling pathways,
which have been shown to play an important role in thyroid cancer
initiation and progression. The screening for follicular
cell-derived specific mutations in association with traditional
diagnosis methods has improved the diagnostic accuracy, impacting
the prognosis of these tumors. Moreover, the advances in the
knowledge of the effects of thyroid oncogenes and related
mechanisms of action have allowed for the development of
multikinase inhibitor targets, promoting new perspectives on
therapy to aggressive thyroid tumors.
Acknowledgements
This study was supported by grants
from Conselho Nacional de Desenvolvimento Científico e Tecnológico
(CNPq), Fundação de Amparo a Pesquisa do Rio Grande do Sul
(FAPERGS), Fundo de Incentivo a Pesquisa do Hospital de Clínicas de
Porto Alegre (FIPE) and Programa de Apoio a Núcleos de Excelência
(PRONEX), Brazil.
References
|
1.
|
Hegedus L: Clinical practice. The thyroid
nodule N Engl J Med. 351:1764–1771. 2004.
|
|
2.
|
Howlader N, Noone AM, Krapcho M, et al:
SEER Cancer Statistics Review, 1975–2009 (Vintage 2009
Populations). National Cancer Institute; Bethesda, MD: http://seer.cancer.gov/csr/1975_2009_pops09/.
Based on November 2011 SEER data submission, posted to the SEER web
site, April 2012.
|
|
3.
|
Wiseman SM, Loree TR, Rigual NR, et al:
Anaplastic transformation of thyroid cancer: review of clinical,
pathologic, and molecular evidence provides new insights into
disease biology and future therapy. Head Neck. 25:662–670. 2003.
View Article : Google Scholar
|
|
4.
|
DeLellis R, Lloyd R, Heitz P and Eng C:
Pathology and genetics of tumours of endocrine origin. World Health
Organization Classification of Tumours. IARC Press; Lyon: pp.
3202004
|
|
5.
|
Harach HR and Ceballos GA: Thyroid cancer,
thyroiditis and dietary iodine: a review based on the Salta,
Argentina model. Endocr Pathol. 19:209–220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Nikiforov YE: Is ionizing radiation
responsible for the increasing incidence of thyroid cancer? Cancer.
116:1626–1628. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Kimura ET, Nikiforova MN, Zhu Z, Knauf JA,
Nikiforov YE and Fagin JA: High prevalence of BRAF mutations in
thyroid cancer: genetic evidence for constitutive activation of the
RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma.
Cancer Res. 63:1454–1457. 2003.
|
|
8.
|
Frattini M, Ferrario C, Bressan P, et al:
Alternative mutations of BRAF, RET and NTRK1 are associated with
similar but distinct gene expression patterns in papillary thyroid
cancer. Oncogene. 23:7436–7440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Adeniran AJ, Zhu Z, Gandhi M, et al:
Correlation between genetic alterations and microscopic features,
clinical manifestations, and prognostic characteristics of thyroid
papillary carcinomas. Am J Surg Pathol. 30:216–222. 2006.
View Article : Google Scholar
|
|
10.
|
Nikiforova MN, Lynch RA, Biddinger PW, et
al: RAS point mutations and PAX8-PPAR gamma rearrangement in
thyroid tumors: evidence for distinct molecular pathways in thyroid
follicular carcinoma. J Clin Endocrinol Metab. 88:2318–2326. 2003.
View Article : Google Scholar
|
|
11.
|
Garcia-Rostan G, Costa AM, Pereira-Castro
I, et al: Mutation of the PIK3CA gene in anaplastic thyroid cancer.
Cancer Res. 65:10199–10207. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kondo T, Ezzat S and Asa SL: Pathogenetic
mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer.
6:292–306. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Ricarte-Filho JC, Ryder M, Chitale DA, et
al: Mutational profile of advanced primary and metastatic
radioactive iodine-refractory thyroid cancers reveals distinct
pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res.
69:4885–4893. 2009. View Article : Google Scholar
|
|
14.
|
Liu Z, Hou P, Ji M, et al: Highly
prevalent genetic alterations in receptor tyrosine kinases and
phosphatidylinositol 3-kinase/akt and mitogen-activated protein
kinase pathways in anaplastic and follicular thyroid cancers. J
Clin Endocrinol Metab. 93:3106–3116. 2008. View Article : Google Scholar
|
|
15.
|
Nikiforov YE: Thyroid carcinoma: molecular
pathways and therapeutic targets. Mod Pathol. 21(Suppl 2): S37–S43.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Paes JE and Ringel MD: Dysregulation of
the phosphatidylinositol 3-kinasepathway in thyroid neoplasia.
Endocrinol Metab Clin North Am. 37:375–387. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Davies L and Welch HG: Increasing
incidence of thyroid cancer in the United States, 1973–2002. JAMA.
295:2164–2167. 2006.
|
|
18.
|
Franceschi S, Boyle P, Maisonneuve P, et
al: The epidemiology of thyroid carcinoma. Crit Rev Oncog. 4:25–52.
1993.
|
|
19.
|
Pacini F, Cetani F, Miccoli P, et al:
Outcome of 309 patients with metastatic differentiated thyroid
carcinoma treated with radioiodine. World J Surg. 18:600–604. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Cohen Y, Rosenbaum E, Clark DP, et al:
Mutational analysis of BRAF in fine needle aspiration biopsies of
the thyroid: a potential application for the preoperative
assessment of thyroid nodules. Clin Cancer Res. 10:2761–2765. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Xing M: BRAF mutation in thyroid cancer.
Endocr Relat Cancer. 12:245–262. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Gutkind JS: Regulation of
mitogen-activated protein kinase signaling networks by G
protein-coupled receptors. Sci STKE. 2000:re12000.PubMed/NCBI
|
|
23.
|
McKay MM and Morrison DK: Integrating
signals from RTKs to ERK/MAPK. Oncogene. 26:3113–3121. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Ciampi R, Knauf JA, Kerler R, et al:
Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway
activation in thyroid cancer. J Clin Invest. 115:94–101. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Carta C, Moretti S, Passeri L, et al:
Genotyping of an Italian papillary thyroid carcinoma cohort
revealed high prevalence of BRAF mutations, absence of RAS
mutations and allowed the detection of a new mutation of BRAF
oncoprotein (BRAF(V599lns)). Clin Endocrinol (Oxf). 64:105–109.
2006. View Article : Google Scholar
|
|
26.
|
Hou P, Liu D and Xing M: Functional
characterization of the T1799-1801del and A1799-1816ins BRAF
mutations in papillary thyroid cancer. Cell Cycle. 6:377–379. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lupi C, Giannini R, Ugolini C, et al:
Association of BRAF V600E mutation with poor clinicopathological
outcomes in 500 consecutive cases of papillary thyroid carcinoma. J
Clin Endocrinol Metab. 92:4085–4090. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Basolo F, Torregrossa L, Giannini R, et
al: Correlation between the BRAF V600E mutation and tumor
invasiveness in papillary thyroid carcinomas smaller than 20
millimeters: analysis of 1060 cases. J Clin Endocrinol Metab.
95:4197–4205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Knauf JA, Ma X, Smith EP, et al: Targeted
expression of BRAFV600E in thyroid cells of transgenic mice results
in papillary thyroid cancers that undergo dedifferentiation. Cancer
Res. 65:4238–4245. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Franco AT, Malaguarnera R, Refetoff S, et
al: Thyrotrophin receptor signaling dependence of Braf-induced
thyroid tumor initiation in mice. Proc Natl Acad Sci USA.
108:1615–1620. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Chakravarty D, Santos E, Ryder M, Knauf
JA, Liao XH, West BL, Bollag G, Kolesnick R, Thin TH, Rosen N,
Zanzonico P, Larson SM, Refetoff S, Ghossein R and Fagin JA:
Small-molecule MAPK inhibitors restore radioiodine incorporation in
mouse thyroid cancers with conditional BRAF activation. J Clin
Invest. 121:4700–4711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Romitti M, Wajner SM, Zennig N, Goemann
IM, Bueno AL, Meyer EL and Maia AL: Increased type 3 deiodinase
expression in papillary thyroid carcinoma. Thyroid. 22:897–904.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Meyer EL, Dora JM, Wagner MS and Maia AL:
Decreased type 1 iodothyronine deiodinase expression might be an
early and discrete event in thyroid cell dedifferentiation towards
papillary carcinoma. Clin Endocrinol (Oxf). 62:672–678. 2005.
View Article : Google Scholar
|
|
34.
|
Xing M, Westra WH, Tufano RP, et al: BRAF
mutation predicts a poorer clinical prognosis for papillary thyroid
cancer. J Clin Endocrinol Metab. 90:6373–6379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Handkiewicz-Junak D, Czarniecka A and
Jarzab B: Molecular prognostic markers in papillary and follicular
thyroid cancer: current status and future directions. Mol Cell
Endocrinol. 322:8–28. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Motti ML, De Marco C, Califano D, et al:
Loss of p27 expression through RAS-->BRAF-->MAP
kinase-dependent pathway in human thyroid carcinomas. Cell Cycle.
6:2817–2825. 2007.
|
|
37.
|
Mesa C Jr, Mirza M, Mitsutake N, et al:
Conditional activation of RET/PTC3 and BRAFV600E in thyroid cells
is associated with gene expression profiles that predict a
preferential role of BRAF in extracellular matrix remodeling.
Cancer Res. 66:6521–6529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Ahmed M, Uddin S, Hussain AR, et al: FoxM1
and its association with matrix metalloproteinases (MMP) signaling
pathway in papillary thyroid carcinoma. J Clin Endocrinol Metab.
97:E1–E13. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Bommarito A, Richiusa P, Carissimi E, et
al: BRAFV600E mutation, TIMP-1 upregulation, and NF-kappaB
activation: closing the loop on the papillary thyroid cancer
trilogy. Endocr Relat Cancer. 18:669–685. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Palona I, Namba H, Mitsutake N, et al:
BRAFV600E promotes invasiveness of thyroid cancer cells through
nuclear factor kappaB activation. Endocrinology. 147:5699–5707.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Lee SJ, Lee MH, Kim DW, et al:
Cross-regulation between oncogenic BRAF(V600E) kinase and the MST1
pathway in papillary thyroid carcinoma. PLoS One. 6:e161802011.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Ceolin L, Siqueira DR, Romitti M, Ferreira
CV and Maia AL: Molecular basis of medullary thyroid carcinoma: the
role of RET polymorphisms. Int J Mol Sci. 13:221–239. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Fugazzola L, Pilotti S, Pinchera A, et al:
Oncogenic rearrangements of the RET proto-oncogene in papillary
thyroid carcinomas from children exposed to the Chernobyl nuclear
accident. Cancer Res. 55:5617–5620. 1995.PubMed/NCBI
|
|
44.
|
Nikiforov YE: RET/PTC rearrangement in
thyroid tumors. Endocr Pathol. 13:3–16. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Zhu Z, Ciampi R, Nikiforova MN, Gandhi M
and Nikiforov YE: Prevalence of RET/PTC rearrangements in thyroid
papillary carcinomas: effects of the detection methods and genetic
heterogeneity. J Clin Endocrinol Metab. 91:3603–3610. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Tallini G and Asa SL: RET oncogene
activation in papillary thyroid carcinoma. Adv Anat Pathol.
8:345–354. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Grieco M, Santoro M, Berlingieri MT, et
al: PTC is a novel rearranged form of the ret proto-oncogene and is
frequently detected in vivo in human thyroid papillary carcinomas.
Cell. 60:557–563. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Nikiforov YE, Rowland JM, Bove KE,
Monforte-Munoz H and Fagin JA: Distinct pattern of ret oncogene
rearrangements in morphological variants of radiation-induced and
sporadic thyroid papillary carcinomas in children. Cancer Res.
57:1690–1694. 1997.
|
|
49.
|
Tallini G, Santoro M, Helie M, et al:
RET/PTC oncogene activation defines a subset of papillary thyroid
carcinomas lacking evidence of progression to poorly differentiated
or undifferentiated tumor phenotypes. Clin Cancer Res. 4:287–294.
1998.
|
|
50.
|
Smyth P, Finn S, Cahill S, et al: ret/PTC
and BRAF act as distinct molecular, time-dependant triggers in a
sporadic Irish cohort of papillary thyroid carcinoma. Int J Surg
Pathol. 13:1–8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Viglietto G, Chiappetta G, Martinez-Tello
FJ, et al: RET/PTC oncogene activation is an early event in thyroid
carcinogenesis. Oncogene. 11:1207–1210. 1995.PubMed/NCBI
|
|
52.
|
Sugg SL, Ezzat S, Rosen IB, Freeman JL and
Asa SL: Distinct multiple RET/PTC gene rearrangements in multifocal
papillary thyroid neoplasia. J Clin Endocrinol Metab. 83:4116–4122.
1998.PubMed/NCBI
|
|
53.
|
Jhiang SM, Sagartz JE, Tong Q, et al:
Targeted expression of the ret/PTC1 oncogene induces papillary
thyroid carcinomas. Endocrinology. 137:375–378. 1996.PubMed/NCBI
|
|
54.
|
Powell DJ Jr, Russell J, Nibu K, et al:
The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas
in murine thyroids. Cancer Res. 58:5523–5528. 1998.PubMed/NCBI
|
|
55.
|
Kawamoto Y, Takeda K, Okuno Y, et al:
Identification of RET autophosphorylation sites by mass
spectrometry. J Biol Chem. 279:14213–14224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Salvatore D, Barone MV, Salvatore G, et
al: Tyrosines 1015 and 1062 are in vivo autophosphorylation sites
in ret and ret-derived oncoproteins. J Clin Endocrinol Metab.
85:3898–3907. 2000.PubMed/NCBI
|
|
57.
|
Knauf JA, Kuroda H, Basu S and Fagin JA:
RET/PTC-induced dedifferentiation of thyroid cells is mediated
through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene.
22:4406–4412. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Vasko V, Saji M, Hardy E, et al: Akt
activation and localisation correlate with tumour invasion and
oncogene expression in thyroid cancer. J Med Genet. 41:161–170.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Melillo RM, Castellone MD, Guarino V, et
al: The RET/PTC-RAS-BRAF linear signaling cascade mediates the
motile and mitogenic phenotype of thyroid cancer cells. J Clin
Invest. 115:1068–1081. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Gujral TS, van Veelen W, Richardson DS, et
al: A novel RET kinase-beta-catenin signaling pathway contributes
to tumorigenesis in thyroid carcinoma. Cancer Res. 68:1338–1346.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Castellone MD, De Falco V, Rao DM, et al:
The beta-catenin axis integrates multiple signals downstream from
RET/papillary thyroid carcinoma leading to cell proliferation.
Cancer Res. 69:1867–1876. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Pradeep A, Sharma C, Sathyanarayana P, et
al: Gastrin-mediated activation of cyclin D1 transcription involves
beta-catenin and CREB pathways in gastric cancer cells. Oncogene.
23:3689–3699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Peifer M and Polakis P: Wnt signaling in
oncogenesis and embryogenesis-a look outside the nucleus. Science.
287:1606–1609. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Esapa CT, Johnson SJ, Kendall-Taylor P,
Lennard TW and Harris PE: Prevalence of Ras mutations in thyroid
neoplasia. Clin Endocrinol (Oxf). 50:529–535. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Zhu Z, Gandhi M, Nikiforova MN, Fischer AH
and Nikiforov YE: Molecular profile and clinical-pathologic
features of the follicular variant of papillary thyroid carcinoma.
An unusually high prevalence of ras mutations. Am J Clin Pathol.
120:71–77. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Santarpia L, Myers JN, Sherman SI,
Trimarchi F, Clayman GL and El-Naggar AK: Genetic alterations in
the RAS/RAF/mitogen-activated protein kinase and
phosphatidylinositol 3-kinase/Akt signaling pathways in the
follicular variant of papillary thyroid carcinoma. Cancer.
116:2974–2983. 2010. View Article : Google Scholar
|
|
67.
|
Hara H, Fulton N, Yashiro T, Ito K,
DeGroot LJ and Kaplan EL: N-ras mutation: an independent prognostic
factor for aggressiveness of papillary thyroid carcinoma. Surgery.
116:1010–1016. 1994.PubMed/NCBI
|
|
68.
|
Djakiew D, Delsite R, Pflug B, Wrathall J,
Lynch JH and Onoda M: Regulation of growth by a nerve growth
factor-like protein which modulates paracrine interactions between
a neoplastic epithelial cell line and stromal cells of the human
prostate. Cancer Res. 51:3304–3310. 1991.
|
|
69.
|
Bongarzone I, Vigneri P, Mariani L,
Collini P, Pilotti S and Pierotti MA: RET/NTRK1 rearrangements in
thyroid gland tumors of the papillary carcinoma family: correlation
with clinicopathological features. Clin Cancer Res. 4:223–228.
1998.PubMed/NCBI
|
|
70.
|
Musholt TJ, Musholt PB, Khaladj N, Schulz
D, Scheumann GF and Klempnauer J: Prognostic significance of RET
and NTRK1 rearrangements in sporadic papillary thyroid carcinoma.
Surgery. 128:984–993. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Martin-Zanca D, Mitra G, Long LK and
Barbacid M: Molecular characterization of the human trk oncogene.
Cold Spring Harb Symp Quant Biol. 51:983–992. 1986. View Article : Google Scholar
|
|
72.
|
Russell JP, Powell DJ, Cunnane M, et al:
The TRK-T1 fusion protein induces neoplastic transformation of
thyroid epithelium. Oncogene. 19:5729–5735. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Fedele M, Palmieri D, Chiappetta G, et al:
Impairment of the p27kip1 function enhances thyroid carcinogenesis
in TRK-T1 transgenic mice. Endocr Relat Cancer. 16:483–490. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Passler C, Scheuba C, Prager G, et al:
Prognostic factors of papillary and follicular thyroid cancer:
differences in an iodine-replete endemic goiter region. Endocr
Relat Cancer. 11:131–139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Gulcelik MA, Gulcelik NE, Kuru B, Camlibel
M and Alagol H: Prognostic factors determining survival in
differentiated thyroid cancer. J Surg Oncol. 96:598–604. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Verburg FA, Mader U, Luster M and Reiners
C: Histology does not influence prognosis in differentiated thyroid
carcinoma when accounting for age, tumour diameter, invasive growth
and metastases. Eur J Endocrinol. 160:619–624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Lemoine NR, Mayall ES, Wyllie FS, et al:
High frequency of ras oncogene activation in all stages of human
thyroid tumorigenesis. Oncogene. 4:159–164. 1989.PubMed/NCBI
|
|
78.
|
Garcia-Rostan G, Zhao H, Camp RL, et al:
ras mutations are associated with aggressive tumor phenotypes and
poor prognosis in thyroid cancer. J Clin Oncol. 21:3226–3235. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Namba H, Rubin SA and Fagin JA: Point
mutations of ras oncogenes are an early event in thyroid
tumorigenesis. Mol Endocrinol. 4:1474–1479. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Bond JA, Wyllie FS, Rowson J, Radulescu A
and Wynford-Thomas D: In vitro reconstruction of tumour initiation
in a human epithelium. Oncogene. 9:281–290. 1994.PubMed/NCBI
|
|
81.
|
Vitagliano D, Portella G, Troncone G, et
al: Thyroid targeting of the N-ras(Gln61Lys) oncogene in transgenic
mice results in follicular tumors that progress to poorly
differentiated carcinomas. Oncogene. 25:5467–5474. 2006. View Article : Google Scholar
|
|
82.
|
Kiaris H and Spandidos DA: Mutations of
ras genes in human tumours. Int J Oncol. 7:413–429.
1995.
|
|
83.
|
Malumbres M and Barbacid M: RAS oncogenes:
the first 30 years. Nat Rev Cancer. 3:459–465. 2003.PubMed/NCBI
|
|
84.
|
Miller KA, Yeager N, Baker K, Liao XH,
Refetoff S and Di Cristofano A: Oncogenic Kras requires
simultaneous PI3K signaling to induce ERK activation and transform
thyroid epithelial cells in vivo. Cancer Res. 69:3689–3694. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
85.
|
Vojtek AB and Der CJ: Increasing
complexity of the Ras signaling pathway. J Biol Chem.
273:19925–19928. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
86.
|
Krasilnikov MA: Phosphatidylinositol-3
kinase dependent pathways: the role in control of cell growth,
survival, and malignant transformation. Biochemistry (Mosc).
65:59–67. 2000.PubMed/NCBI
|
|
87.
|
Damante G, Tell G and Di Lauro R: A unique
combination of transcription factors controls differentiation of
thyroid cells. Prog Nucleic Acid Res Mol Biol. 66:307–356. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
88.
|
Desvergne B and Wahli W: Peroxisome
proliferator-activated receptors: nuclear control of metabolism.
Endocr Rev. 20:649–688. 1999.PubMed/NCBI
|
|
89.
|
Kroll TG, Sarraf P, Pecciarini L, et al:
PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma
[corrected]. Science. 289:1357–1360. 2000.
|
|
90.
|
Cheung L, Messina M, Gill A, et al:
Detection of the PAX8-PPAR gamma fusion oncogene in both follicular
thyroid carcinomas and adenomas. J Clin Endocrinol Metab.
88:354–357. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
91.
|
Marques AR, Espadinha C, Catarino AL, et
al: Expression of PAX8-PPAR gamma 1 rearrangements in both
follicular thyroid carcinomas and adenomas. J Clin Endocrinol
Metab. 87:3947–3952. 2002.PubMed/NCBI
|
|
92.
|
Lacroix L, Mian C, Barrier T, et al: PAX8
and peroxisome proliferator-activated receptor gamma 1 gene
expression status in benign and malignant thyroid tissues. Eur J
Endocrinol. 151:367–374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
93.
|
Klemke M, Drieschner N, Belge G, Burchardt
K, Junker K and Bullerdiek J: Detection of PAX8-PPARG fusion
transcripts in archival thyroid carcinoma samples by conventional
RT-PCR. Genes Chromosomes Cancer. 51:402–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
94.
|
Gregory Powell J, Wang X, Allard BL, et
al: The PAX8/PPARgamma fusion oncoprotein transforms immortalized
human thyrocytes through a mechanism probably involving wild-type
PPARgamma inhibition. Oncogene. 23:3634–3641. 2004.
|
|
95.
|
Lui WO, Foukakis T, Liden J, et al:
Expression profiling reveals a distinct transcription signature in
follicular thyroid carcinomas with a PAX8-PPAR(gamma) fusion
oncogene. Oncogene. 24:1467–1476. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
96.
|
Reddi HV, McIver B, Grebe SK and Eberhardt
NL: The paired box-8/peroxisome proliferator-activated
receptor-gamma oncogene in thyroid tumorigenesis. Endocrinology.
148:932–935. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
97.
|
Farrow B and Evers BM: Activation of
PPARgamma increases PTEN expression in pancreatic cancer cells.
Biochem Biophys Res Commun. 301:50–53. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
98.
|
Chinnadurai G: CtBP, an unconventional
transcriptional core-pressor in development and oncogenesis. Mol
Cell. 9:213–224. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
99.
|
Neff RL, Farrar WB, Kloos RT and Burman
KD: Anaplastic thyroid cancer. Endocrinol Metab Clin North Am.
37:525–538. 2008. View Article : Google Scholar
|
|
100.
|
Ain KB: Anaplastic thyroid carcinoma:
behavior, biology, and therapeutic approaches. Thyroid. 8:715–726.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
101.
|
Giuffrida D and Gharib H: Anaplastic
thyroid carcinoma: current diagnosis and treatment. Ann Oncol.
11:1083–1089. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
102.
|
Kitamura Y, Shimizu K, Nagahama M, et al:
Immediate causes of death in thyroid carcinoma: clinicopathological
analysis of 161 fatal cases. J Clin Endocrinol Metab. 84:4043–4049.
1999. View Article : Google Scholar
|
|
103.
|
Smallridge RC, Marlow LA and Copland JA:
Anaplastic thyroid cancer: molecular pathogenesis and emerging
therapies. Endocr Relat Cancer. 16:17–44. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
104.
|
Kim TY, Kim KW, Jung TS, et al: Prognostic
factors for Korean patients with anaplastic thyroid carcinoma. Head
Neck. 29:765–772. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105.
|
Nikiforov YE: Genetic alterations involved
in the transition from well-differentiated to poorly differentiated
and anaplastic thyroid carcinomas. Endocr Pathol. 15:319–327. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
106.
|
Hou P, Liu D, Shan Y, et al: Genetic
alterations and their relationship in the phosphatidylinositol
3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res.
13:1161–1170. 2007. View Article : Google Scholar
|
|
107.
|
Nikiforova MN, Kimura ET, Gandhi M, et al:
BRAF mutations in thyroid tumors are restricted to papillary
carcinomas and anaplastic or poorly differentiated carcinomas
arising from papillary carcinomas. J Clin Endocrinol Metab.
88:5399–5404. 2003. View Article : Google Scholar
|
|
108.
|
Costa AM, Herrero A, Fresno MF, et al:
BRAF mutation associated with other genetic events identifies a
subset of aggressive papillary thyroid carcinoma. Clin Endocrinol
(Oxf). 68:618–634. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
109.
|
Sobrinho-Simoes M, Maximo V, Rocha AS, et
al: Intragenic mutations in thyroid cancer. Endocrinol Metab Clin
North Am. 37:333–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
110.
|
Begum S, Rosenbaum E, Henrique R, Cohen Y,
Sidransky D and Westra WH: BRAF mutations in anaplastic thyroid
carcinoma: implications for tumor origin, diagnosis and treatment.
Mod Pathol. 17:1359–1363. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
111.
|
Santarpia L, El-Naggar AK, Cote GJ, Myers
JN and Sherman SI: Phosphatidylinositol 3-kinase/akt and
ras/raf-mitogen-activated protein kinase pathway mutations in
anaplastic thyroid cancer. J Clin Endocrinol Metab. 93:278–284.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
112.
|
Saavedra HI, Knauf JA, Shirokawa JM, et
al: The RAS oncogene induces genomic instability in thyroid PCCL3
cells via the MAPK pathway. Oncogene. 19:3948–3954. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
113.
|
Sansal I and Sellers WR: The biology and
clinical relevance of the PTEN tumor suppressor pathway. J Clin
Oncol. 22:2954–2963. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
114.
|
Frisk T, Foukakis T, Dwight T, et al:
Silencing of the PTEN tumor-suppressor gene in anaplastic thyroid
cancer. Genes Chromosomes Cancer. 35:74–80. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
115.
|
Hou P, Ji M and Xing M: Association of
PTEN gene methylation with genetic alterations in the
phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid
tumors. Cancer. 113:2440–2447. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116.
|
Ringel MD, Hayre N, Saito J, et al:
Overexpression and overactivation of Akt in thyroid carcinoma.
Cancer Res. 61:6105–6111. 2001.PubMed/NCBI
|
|
117.
|
Petitjean A, Mathe E, Kato S, et al:
Impact of mutant p53 functional properties on TP53 mutation
patterns and tumor phenotype: lessons from recent developments in
the IARC TP53 database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
118.
|
Ito T, Seyama T, Mizuno T, et al: Unique
association of p53 mutations with undifferentiated but not with
differentiated carcinomas of the thyroid gland. Cancer Res.
52:1369–1371. 1992.PubMed/NCBI
|
|
119.
|
Cerrato A, Fulciniti F, Avallone A,
Benincasa G, Palombini L and Grieco M: Beta- and gamma-catenin
expression in thyroid carcinomas. J Pathol. 185:267–272. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
120.
|
Garcia-Rostan G, Tallini G, Herrero A,
D’Aquila TG, Carcangiu ML and Rimm DL: Frequent mutation and
nuclear localization of beta-catenin in anaplastic thyroid
carcinoma. Cancer Res. 59:1811–1815. 1999.PubMed/NCBI
|
|
121.
|
Motti ML, Califano D, Baldassarre G, et
al: Reduced E-cadherin expression contributes to the loss of
p27kip1-mediated mechanism of contact inhibition in thyroid
anaplastic carcinomas. Carcinogenesis. 26:1021–1034. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
122.
|
Naito A, Iwase H, Kuzushima T, Nakamura T
and Kobayashi S: Clinical significance of E-cadherin expression in
thyroid neoplasms. J Surg Oncol. 76:176–180. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
123.
|
Von Wasielewski R, Rhein A, Werner M, et
al: Immunohistochemical detection of E-cadherin in differentiated
thyroid carcinomas correlates with clinical outcome. Cancer Res.
57:2501–2507. 1997.PubMed/NCBI
|
|
124.
|
Maia AL, Ward LS, Carvalho GA, Graf H,
Maciel RM, Maciel LM, Rosário PW and Vaisman M: Thyroid nodules and
differentiated thyroid cancer: Brazilian consensus. Arq Bras
Endocrinol Metabol. 51:867–893. 2007.PubMed/NCBI
|
|
125.
|
Cooper DS, Doherty GM, Haugen BR, et al:
Revised American thyroid association management guidelines for
patients with thyroid nodules and differentiated thyroid cancer.
Thyroid. 19:1167–1214. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
126.
|
Fernandes JK, Day TA, Richardson MS and
Sharma AK: Overview of the management of differentiated thyroid
cancer. Curr Treat Options Oncol. 6:47–57. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
127.
|
Kloos RT, Ringel MD, Knopp MV, et al:
Phase II trial of soafenib in metastatic thyroid cancer. J Clin
Oncol. 27:1675–1684. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
128.
|
Hoftijzer H, Heemstra KA, Morreau H, et
al: Beneficial effects of sorafenib on tumor progression, but not
on radioiodine uptake, in patients with differentiated thyroid
carcinoma. Eur J Endocrinol. 161:923–931. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
129.
|
Gupta-Abramson V, Troxel AB, Nellore A, et
al: Phase II trial of sorafenib inadvanced thyroid cancer. J Clin
Oncol. 26:4714–4719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
130.
|
Flaherty KT, Puzanov I, Kim KB, et al:
Inhibition of mutated, activated BRAF in metastatic melanoma. N
Engl J Med. 363:809–819. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
131.
|
Cohen EE, Rosen LS, Vokes EE, et al:
Axitinib is an active treatment for all histologic subtypes of
advanced thyroid cancer: results from a phase II study. J Clin
Oncol. 26:4708–4713. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
132.
|
Bible KC, Suman VJ, Molina JR, et al:
Efficacy of pazopanib in progressive, radioiodine-refractory,
metastatic differentiated thyroid cancers: results of a phase 2
consortium study. Lancet Oncol. 11:962–972. 2010. View Article : Google Scholar
|
|
133.
|
Sherman SI, Wirth LJ, Droz JP, et al:
Motesanib diphosphate in progressive differentiated thyroid cancer.
N Engl J Med. 359:31–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
134.
|
Pennell NA, Daniels GH, Haddad RI, et al:
A phase II study of gefitinib in patients with advanced thyroid
cancer. Thyroid. 18:317–323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
135.
|
Hayes DN, Lucas AS, Tanvetyanon T, et al:
Phase II efficacy and pharmacogenomic study of Selumetinib
(AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid
carcinoma with or without follicular elements. Clin Cancer Res.
18:2056–2065. 2012. View Article : Google Scholar : PubMed/NCBI
|