Introduction
Hepatocellular carcinoma (HCC) is one of the most
prevalent and lethal types of cancer in Asia and its incidence is
increasing. In Southeastern Asia, the majority of HCCs develops in
individuals with chronic HBV or HCV infection (1). Chronic liver disease and liver
cirrhosis is considered to be a premalignant state because HCC
occurs frequently in the background of liver cirrhosis (2). Currently the treatment modalities in
advance HCC is very limited, and therefore development of new
strategy for HCC therapy based on molecular pathogenesis is
necessary (3).
Adenine nucleotide translocase (ANT), abundantly
located in the inner mitochondrial membrane, participates in the
formation of mitochondrial permeability transition pore complex,
and also plays an important role in the cellular energy metabolism
by catalyzing the exchange of mitochondrial ATP for cytosolic ADP
and thereby influencing mitochondrial oxidative phosphorylation
(4,5). Human ANT has four isoforms (ANT1,
ANT2, ANT3 and ANT4) and the relative expressions of these isoforms
are dependent on developmental stages, proliferation status as well
as tissue types or cell types. ANT2 is specifically expressed in
undifferentiated cells or tissues that are able to proliferate and
regenerate; for example, the lymphocytes, kidney and liver
(6,7). The expression of ANT2 was recently
found to be upregulated in several hormone-dependent cancers
(8) and the induction of ANT2
expression in cancer cells was directly associated with glycolytic
metabolisms, raising a question regarding the role of ANT2 during
carcinogenesis (9–12). ANT2 suppression by a DNA
vector-based RNA interference approach expressing short hairpin RNA
(shRNA) resulted in ATP depletion and induced cell death in breast
cancer cells (13). We also
confirmed that the knockdown of ANT2 by shRNA induced cell death in
HCC cell lines and screened for the identification of ANT2 shRNA
regulated genes in the hepatocellular carcinoma cell line Hep3B. As
a result, we obtained significant candidate genes and reaffirmed by
selecting a specific gene, suppressor of cytokine signaling 1
(SOCS1), among the genes.
Involvement of SOCS1 in carcinogenesis revealed that
SOCS1 is frequently silenced by hypermethylation in HCC (14). Restoration of SOCS1 in HCC cell
lines by transient transfection resulted in growth suppression and
induction of apoptosis (14). The
silencing of gene transcription associated with aberrant DNA
methylation of CpG dinucleotides in normally unmethylated gene
promoter regions is the most widely studied epigenetic abnormality
in tumorigenesis (15–17). SOCS1 is a negative regulator of
Janus kinase and signal transducer and activation of transcription
(Jak-STAT) pathway (18–21). Several studies have indicated that
dysregulation of the Jak-STAT pathway is involved in the malignant
transformation for commonly-encountered human cancers, such as HCC
(14) non-small cell lung cancer,
and head and neck squamous cell carcinoma (22–24).
The high prevalence of the aberrant SOCS1 methylation demonstrated
the importance of the constitutive activation of the Jak-STAT
pathway in the development of HCC (25).
Constitutive activated STAT3 controls the expression
of a number of anti-apoptotic proteins on the transcriptional
level, including survivin, Bcl-2 and Mcl-1, thereby providing an
important fast-track survival signal for tumor cells (26–29).
According to recent report, miR-21 was also identified as a target
of STAT3. siRNA against STAT3 has much reduced levels of miR-21
(30). Moreover, STAT3-induced
miR-21 was important for transformation and tumorigenicity in human
cancer and inhibition of miR-21 dramatically reduced tumorigenicity
(30). In HCC tissues, miR-21 was
markedly increased compared with normal tissues and increased
miR-21 expression was a frequent event in human HCC and may be
involved in hepatocarcinogenesis. More importantly, the relevance
of miR-21 for the oncogenic potential of STAT3 and thereby for its
involvement in the pathogenesis of malignancies. Our results show
that ANT2 shRNA treatment led to restoration of SOCS1 expression
along with its promoter de-methylation in Hep3B cells.
We hypothesized that suppression of Ras/PI3K/Akt
signaling by ANT2 shRNA induced SOCS1 restoration. It is well known
that Ras/PI3K/Akt pathway elevates the cellular DNMT1 protein
level. Upregulation of DNMT1 by Ras/PI3K/Akt signal pathway
involves transcription activation and mRNA stabilization, and that
DNMT1 protein stability was significantly decreased by Ras/PI3K/Akt
inhibition. Eventually, epigenetic silencing was decreased through
the suppression of Ras/PI3K/Akt signaling by ANT2 shRNA. Therefore,
inhibition of DNMT1 activity by suppression of Ras/PI3K/Akt pathway
decreases hyperme-thylated SOCS1 levels.
We assumed that SOCS1 restoration by ANT2 shRNA
might inactivate Jak-STAT signaling pathway, thereby
down-regulating onco-miR-21 expression and subsequent inhibition of
proliferation, in HCC cells. Thus, the purpose of our study was to
determine whether or not ANT2 shRNA-mediated SOCS1 restoration
suppressed HCC cells proliferation, and to investigate the
potential of ANT2 shRNA for future use as a therapeutic agent
targeting HCC cells.
Materials and methods
Cell lines and culture
The human hepatoma cell line Hep3B was purchased
from the American Type Culture Collection (ATCC, Manassas, VA, USA)
and cultured in Dulbecco’s modified Eagle’s medium supplemented
with 10% fetal bovine serum, 100 U/ml penicillin and 100
μg/ml streptomycin (Gibco, Grand Island, NY, USA) in a
humidified 5% CO2/95% air atmosphere at 37°C.
Antibodies and reagents
Anti-phospho-STAT3, anti-STAT3, anti-SOCS1,
anti-phospho-Akt, anti-Akt and anti-β-actin antibodies were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Construction of ANT2 shRNA expression
vectors
The ANT2 shRNA expression vector used to achieve
specific downregulation of ANT2 was described previously. ANT2
siRNA (5′-GCAG AUCACUGCAGAUAAGTT-3′) designed to be complementary
to exon 2 of ANT2 (GenBank accession no. NM001152) was synthesized,
and DNA vectors expressing the shRNA forms of the siRNAs were
generated using pSilencer 3.1-H1 puro plasmids with a TTCAAGAGA
linker sequence that forms looped structures (Ambion, Austin, TX,
USA). The vector expressing ANT2 shRNA was used throughout this
study. A scramble shRNA (Ambion) with no significant homology to
human gene sequences was used as a control to detect nonspecific
effects.
Transfection
For transfection, cells were plated on either 6-well
plates (2×105 cells/well) or 100-mm dishes
(2×106 cells) and were allowed to adhere for 24 h.
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was used for the
transfection. Cells were transfected with either pSilencer™ 3.1-H1
puro ANT2 shRNA or pSilencer™ 3.1-H1 puro scramble shRNA vectors.
Transfected cells were then cultured for 6 h; culture media were
replaced with fresh media supplemented with 10% FBS. The cells were
harvested 24 or 48 h after transfection. pcDNA, pcDNA-ANT2, daRas,
dnRas plasmid or SOCS1 shRNA were transfected into the cells using
the same method.
Microarray analysis and statistical
analysis
Isolated RNA (500–1,000 ng) was converted to cDNA
with reverse transcriptase and an oligo (dT) primer bearing a T7
promoter followed by in vitro transcription with T7 RNA
polymerase to create amplified antisense RNA. The amplified RNA was
labeled with Cy3 or Cy5. As a reference probe universal human
reference RNA (UHR, Stratagene) was amplified and labeled as above.
Amplification and labeling efficiency was controlled using
NanoDrop. Labeled cRNA was hybridized to Agilent Whole Human Genome
Oligo Microarrays per the manufacturer’s protocol. After
hybridization for 17 h the arrays were washed and scanned using an
Agilent scanner and microarray data extracted with Agilent Feature
Extraction software.
Quantitative real-time RT-PCR
(RT-qPCR)
For the RT-qPCR, total-RNA from the same samples
used in microarray analysis was tested by using Bio-Rad iCycler IQ
real-time PCR system. PCR primers were designed with Primer Express
2.0 software (Invitrogen). Results are shown as fold change.
RT-qPCR was carried out with the PrimerScript RT reagent Kit
(Takara, Otsu, Japan) and SYBR-Green real-time PCR Master mix
(Takara) according to manufacturer’s instruction. The housekeeping
gene GAPDH was used for standardization of the initial RNA content
of a sample. Relative changes of gene expression were calculated by
the following formula, and the data are represented as fold
upregulation/downregulation, fold change = 2−ΔΔCt, where
ΔΔCt = (Ct of gene of interest, treated −Ct of HK gene, treated) −
(Ct of gene of interest, control−Ct of HK gene, control), Ct was
the threshold cycle number and HK was the house-keeping gene.
Western blot analysis
For western blot analyses, cells were harvested
after 24 or 48 h of transfection and lysed with lysis buffer (5
mM/l ethylenediamine tetra acetic acid; 300mM/l NaCl; 0.1% NP-40;
0.5 mM/l NaF; 0.5 mM/l Na3VO4; 0.5 mM/l
phenylmethylsulfonyl fluoride; and 10 μg/ml each of
aprotinin, pepstatin and leupeptin; Sigma, St. Louis, MO, USA).
Following centrifugation at 15,000 × g for 30 min, the
concentrations of supernatant proteins were analyzed using the
Bradford reagent (Bio-Rad, Hercules, CA, USA). For analysis of
protein contents, 50 μg of total proteins was
electrophoresed in 10% SDS-PAGE gel and transferred to
polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA),
which were then incubated with the respective antibodies indicated
above. Immunoblots were visualized using an enhanced
chemiluminescence detection system (Amersham Pharmacia Biotech,
Uppsala, Sweden).
Methylation-specific PCR (MSP)
Genomic DNA was obtained and purified using QIAamp
DNA mini Kits (Qiagen, Hilden, Germany). The unmethylated cytosines
in aliquots containing 500 ng DNA were converted to uracil using
MethylCode Bisulfite Conversion Kits (Invitrogen). We modified the
manufacturer’s instructions by extending the desulfonation step to
30 min to ensure complete conversion of the genomic DNA and by
pre-warming the elution buffer to 50°C to increase the DNA yield.
Methylation-specific PCR for SOCS1 was performed using the primer
pair 5′-TTC GCG TGT ATT TTT AGG TCG GTC-3′ (forward) and 5′-CGA CAC
AAC TCC TAC AAC GAC CG-3′ (reverse) for methylated DNA and the pair
5′-TTA TGA GTA TTT GTG TGT ATT TTT AGG TTG GTT-3′ (forward) and
5′-CGA CAC AAC TCC TAC AAC GAC CG-3′ (reverse) for unmethylated
DNA. PCR mixtures contained 1X PCR buffer, 2 mM MgCl2,
200 mM dNTPs, 200 nM of each primer, 0.05 U/ml Taq polymerase and 6
ng/ml converted DNA as the template. PCR products were resolved in
2% agarose gels in 1X Tris/borate/EDTA buffer and stained with
ethidium bromide.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Cells were collected after 24 h of transfection.
Total-RNA was extracted using TRIzol (Invitrogen), according to the
manufacturer’s instructions. For RT-PCR analysis, 5 μg
total-RNA was reverse-transcribed using RT-PCR kits (Promega,
Madison, WI, USA). PCR was used for amplification of target cDNA
under the following conditions: 35 cycles of 94°C for 1 min, 55°C
for 1 min and 72°C for 2 min. PCR products were analyzed using
standard agarose gel electrophoresis. The primer sequences were:
for SOCS1, forward 5′-TTGCCTGGAACCATGTGG-3′ and reverse
5′-GGTCCTGGCCTCCAGATACAG-3′; for β-actin, forward 5′-GCTCCG
GCATGTGCAA-3′ and reverse 5′-GCTC CGGCATGTGCAA-3′.
DNA methyltranserase 1 (DNMT1) activity
assay
Nuclear extracts from Hep3B cells transfected for 24
h with ANT2 or scramble shRNA (control) were evaluated for DNMT1
activity using an EpiQuik Dnmt1 Assay Kit (Epigentek Inc.,
Brooklyn, NY, USA).
Electrophoretic mobility shift assay
(EMSA)
Nuclear extracts of Hep3B transfected with scramble
or ANT2 shRNA, were prepared and EMSA performed. In brief, for
EMSA, 3.4 μg of total nuclear protein were used for each
lane. The oligonucleotide sequence used to assess the STAT3
consensus-binding motif 5′-GATCCTTCTGGGAATTCCTAGATC-3′. All oligos
were synthetized by Bioneer (Dajeon, Korea). The synthetic
oligonucleotides were labeled using a T4 polynucleotide kinase (DNA
5′-End Labeling Kit; Promega, Mannheim, Germany) and
[γ32P]-CTP (Perkin Elmer, Rodgau-Jügesheim, Germany).
Following incubation of the radiolabeled probes with nuclear
extracts, protein-DNA complexes were resolved on a NOVEX 6%
retardation gel (Invitrogen) and detected using a Typhoon 9410
PhosphoImager (Amersham Biosciences, Freiburg, Germany).
Chromatin immunoprecipitation (ChIP)
assays
ChIP assay was carried out using the ChIP-IT Express
Enzymatic kit (Active Motif) according to the manufacturer’s
instructions. In brief, chromatin from cells was cross-linked with
1% formaldehyde (10 min at 22°C), sheared to an average size of
∼500 bp and then immunoprecipitated with anti-STAT3 antibody (Santa
Cruz Biotechnology). The ChIP-PCR primers were designed to amplify
a proximal promoter region containing putative binding sites in the
miR-21 promoter identified by TFSEARCH.
MTT cell proliferation assay
Cell proliferation was measured using the MTT assay
(Sigma). The reduction of tetrazolium salts is now widely accepted
as a reliable way to examine cell proliferation. The yellow
tetrazolium MTT (3-(4, 5-dimethylthiazolyl-2)-2,
5-diphenyltetrazolium bromide) is reduced by metabolically active
cells, in part by the action of dehydrogenase enzymes, to generate
reducing equivalents such as NADH and NADPH. The resulting
intracellular purple formazan can be solubilized and quantified by
spectrophotometric means. Results of cell proliferation assays are
presented as the mean values of three replicate experiments
performed in triplicate.
Results
ANT2 shRNA induces specific restoration
of SOCS1 through the demethylation of SOCS1 promoter in the
hepatocellular carcinoma cell line Hep3B
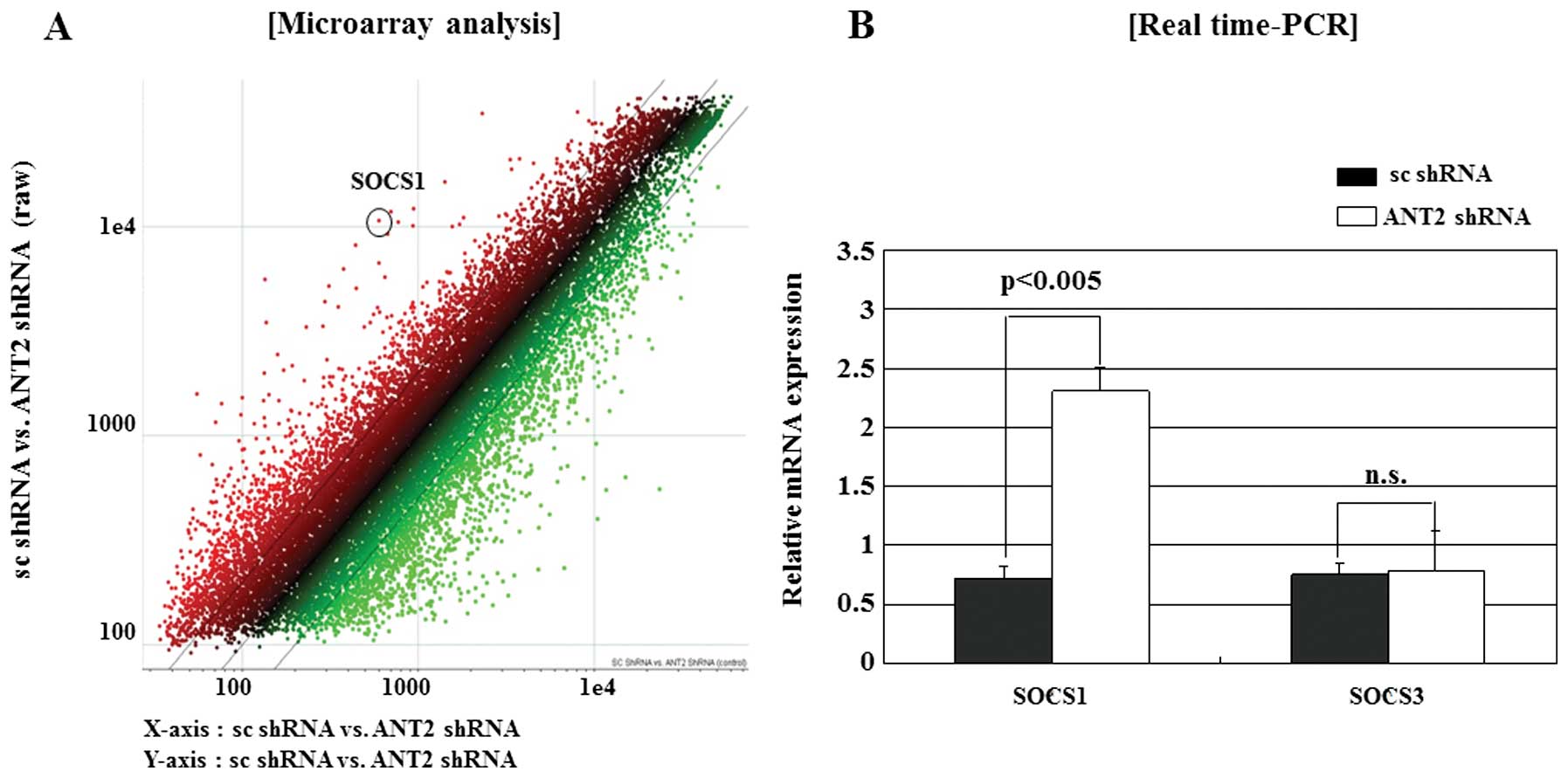
We first microarray screened for the identification
of ANT2 shRNA regulated genes in the hepatocellular carcinoma cell
line Hep3B (Fig. 1A). We obtained
significant candidate genes and reaffirmed by selecting a specific
gene, SOCS1, among the genes. The SOCS1 gene has been demonstrated
to be frequently silenced by methylation of the CpG islands in
human HCC. As shown in Fig. 1B,
SOCS1 was upregulated by ANT2 shRNA. However, ANT2 shRNA had little
influence on SOCS3 expression. These data suggested that ANT2 shRNA
upregulated the SOCS1 expression specifically in Hep3B.
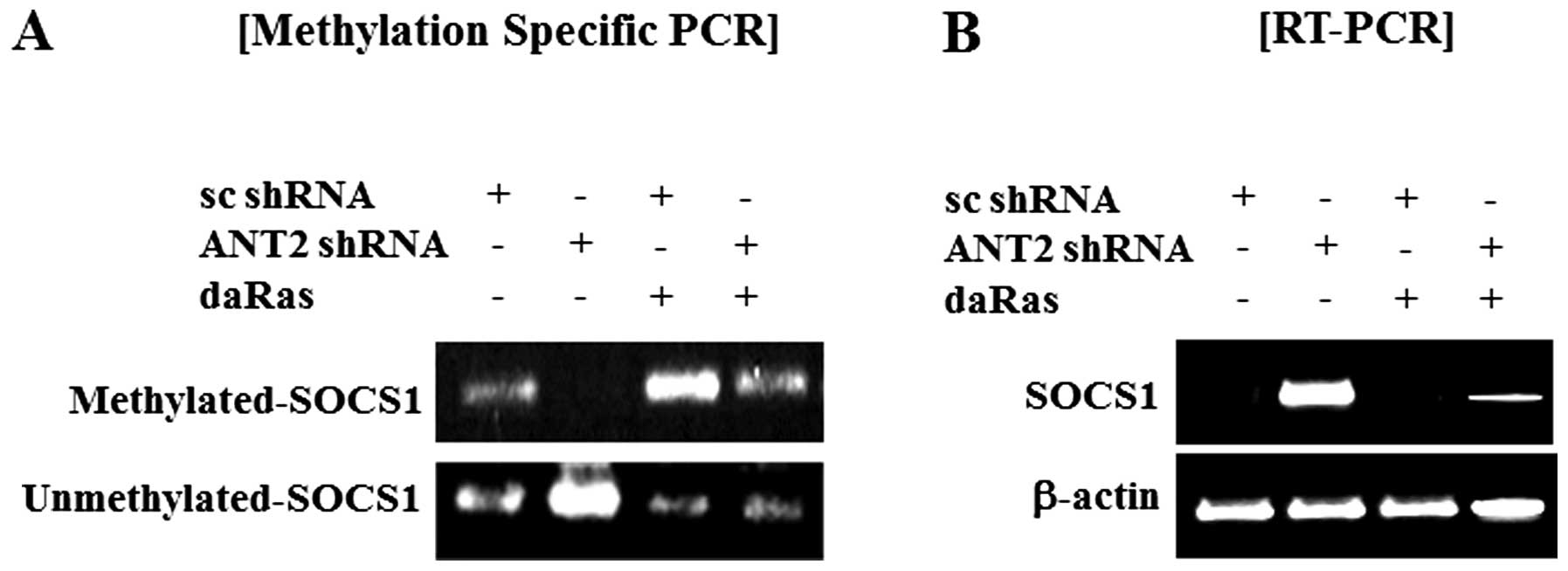
Furthermore, we observed that ANT2 shRNA decreased hyper-methylated
levels and increased unmethylated levels of SOCS1 and that dominant
active Ras suppressed unmethylated levels of SOCS1 and recovered
reduction of hyper-methylated levels of SOCS1 in Hep3B cells
(Fig. 2A). In SOCS1 expression,
the result matched those in Fig.
2B. Thus, SOCS1 upregulation in response to ANT2 knockdown is
mediated by SOCS1 promoter demethylation.
ANT2 shRNA reduces activity of DNA
methyl-transferase 1 (DNMT1), by inhibiting Ras/PI3K/Akt signaling
pathway in the hepatocellular carcinoma cell line Hep3B
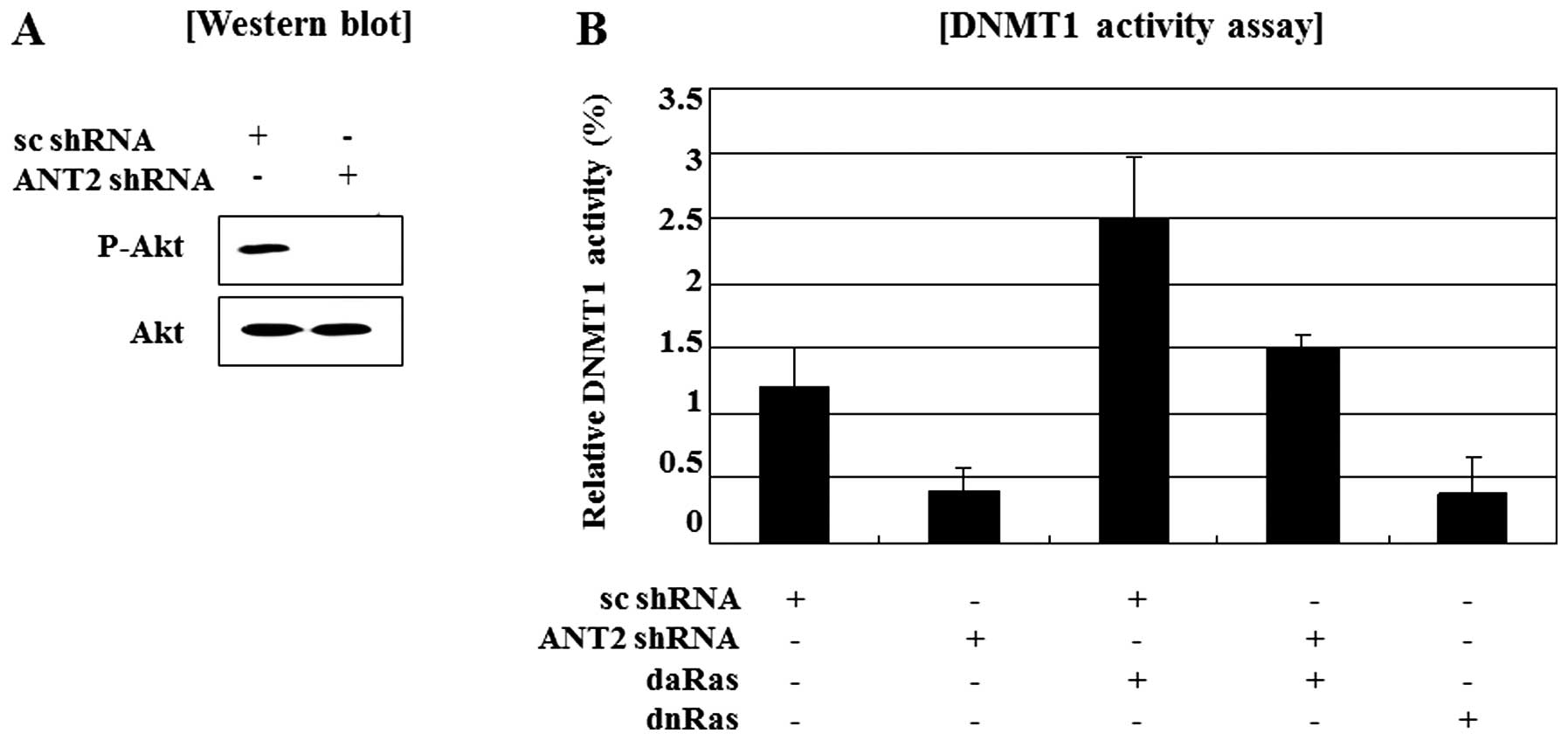
Here, we examined whether ANT2 knockdown by shRNA
affected activity of DNMT1 and whether this might be regulated by
the Ras/PI3K/Akt signaling pathway. Ras protein is the most
well-characterized proto-oncogene and Ras signaling is known to
regulate diverse cellular functions including cell growth, survival
and migration. Indeed, the level of DNA methyltransferase and DNA
methylation are controlled by the RAS signaling pathway. Akt (also
named protein kinase B) is a downstream target of Ras signaling
pathway and is a critical mediator of survival signals for
protection of cells from apoptosis. Especially, relationship
between Ras/PI3K/Akt signaling and epigenetic silencing have been
reported. Activated Ras/PI3K/Akt signaling is a trigger of
epigenetic silencing. To detect the regulation of Ras/PI3K/Akt
activity by ANT2 shRNA, we examined phosphorylated Akt levels was
decreased by knockdown of ANT2 by shRNA (Fig. 3A). A report that DNMT1 activation
is involved in Ras/PI3K/Akt signaling and our observations that
ANT2 knockdown inactivates Ras/PI3K/Akt signaling and causes
demethylation of the SOCS1 promoter in Hep3B cells, led us to
investigate the role of DNMT1 in ANT2 shRNA-induced restoration of
SOCS1. Besides, DNMT1 activity assays showed transfection with
dominant active Ras restored the reduction of DNMT1 activity in
Hep3B cells transfected with ANT2 shRNA and dominant negative Ras
also suppressed DNMT1 activity (Fig.
3B). Taken together, these findings suggested that ANT2 shRNA
treatment led to restoration of SOCS1 expression along with its
promoter demethylation in Hep3B cells, which was accompanied by
decreased DNMT1 activity through suppression of Ras/PI3K/Akt
signaling.
Restoration of SOCS1 by ANT2 shRNA
inhibits STAT3 activity
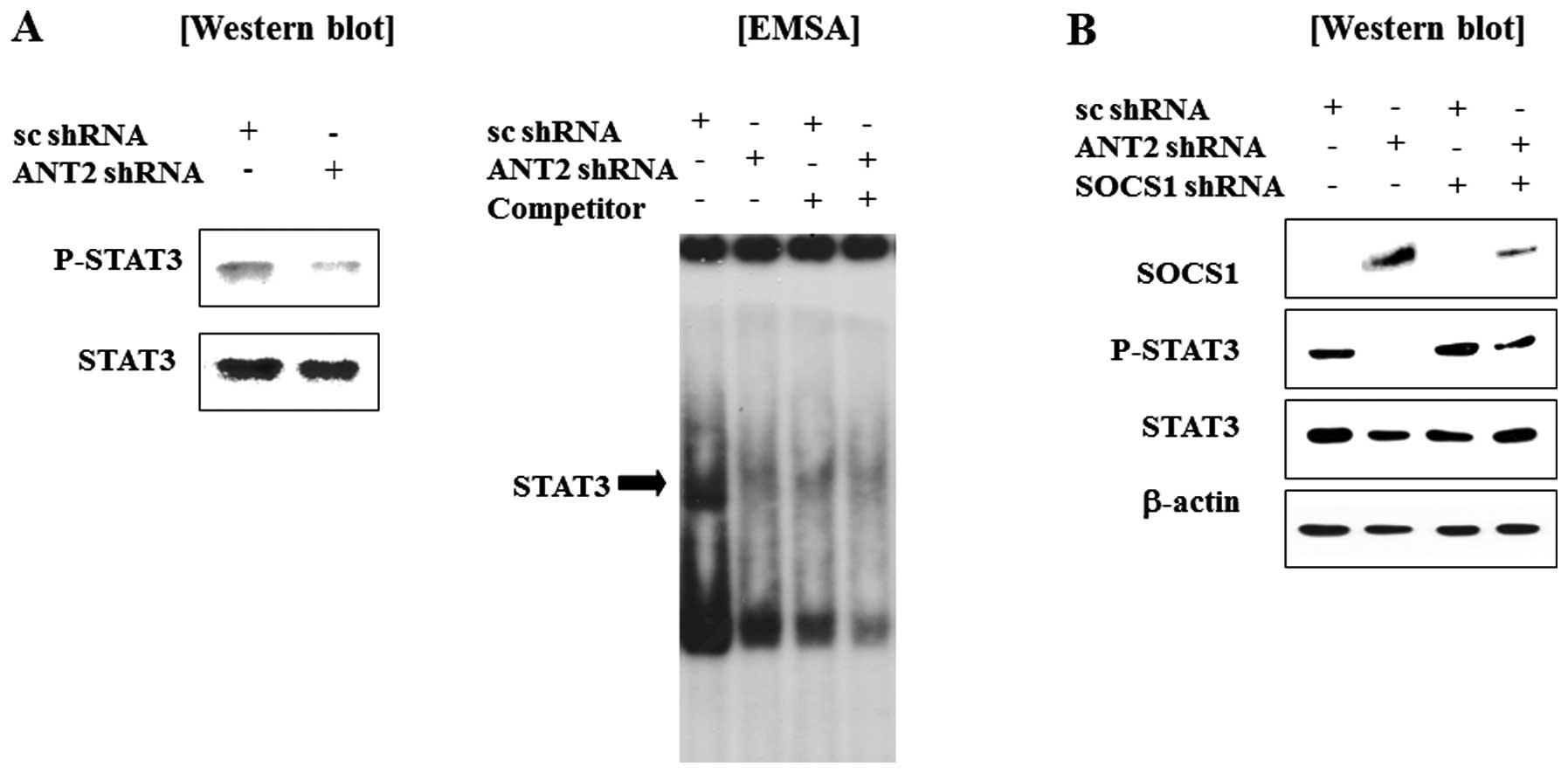
We observed that ANT2 shRNA transfection recovered
SOCS1 expression levels in Hep3B cells. Because SOCS1 is known to a
negative regulator of Janus kinase and signal transducer and
activation of transcription (Jak-STAT) pathway, we anticipated that
STAT3 might be inactivated by restored SOCS1 by ANT2 knockdown. As
expected, ANT2 shRNA treatment inhibits phosphorylation and DNA
binding activity of STAT3 (Fig.
4A). Furthermore, the phosphorylation of STAT3 in ANT2
shRNA-transfected cells was inhibited by the SOCS1 shRNA (Fig. 4B). Taken together, these findings
suggest that ANT2 knockdown reduces hypermethylation of SOCS1
promoter to restore SOCS1 expression, which then inactivates the
Jak-STAT signaling pathway.
Inactivation of STAT3 by ANT2 shRNA
downregulates onco-miR-21 expression
Because silencing of SOCS1 in hepatocellular
carcinoma is attributable to enhanced activation of STAT3, as a
result, induced proliferation and tumorigenesis. A STAT3 target
gene that was described previously is the gene encoding the primary
transcript of miR-21. To assay the suppression of the miR-21 by
ANT2 shRNA, quantitative reverse transcriptase PCR (RT-qPCR)
analysis of RNA isolated from Hep3B cells transfected with scramble
or ANT2 shRNA showed that the expression of miR-21 is suppressed by
ANT2 shRNA (Fig. 5A). We
investigated whether miR-21 is also a direct STAT3 target. Cells
were transfected with ANT2 shRNA to inactivate STAT3, and analyzed
by chromatin immunoprecipitation (ChIP). Chromatin that was
precipitated with STAT3 antibodies was significantly enriched for
the miR-21 promoter sequence when compared with chromatin
precipitated with normal rabbit IgG. As a control, PCR was
performed on the precipitated chromatin for the promoter of
glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a promoter that
lacks STAT3 binding sites. No precipitation of the GAPDH promoter
was observed with or without ANT2 shRNA, demonstrating the
specificity of the ChIP (Fig. 5B).
Collectively, restoration of SOCS1 by ANT2 knockdown, subsequently
inhibited STAT3 activity and downregulated the expression of
miR-21, which has been reported to be an important onco-miR in HCC
and other carcinomas. Furthermore, this suppression of miR-21 by
ANT2 shRNA was restored by ANT2 or SOCS1 overexpression (Fig. 5A and B) and reduction of
phospho-STAT3 levels was restored by ANT2 overexpression (Fig. 5C). Moreover, reduction of miR-21
was recovered by ANT2 or SOCS1 over-expression in ANT2 shRNA
transected Hep3B cells (Fig. 5D and
E). These results suggested that inhibition of Jak-STAT
signaling by ANT2 shRNA might play an important role in the
downregulation of miR-21.
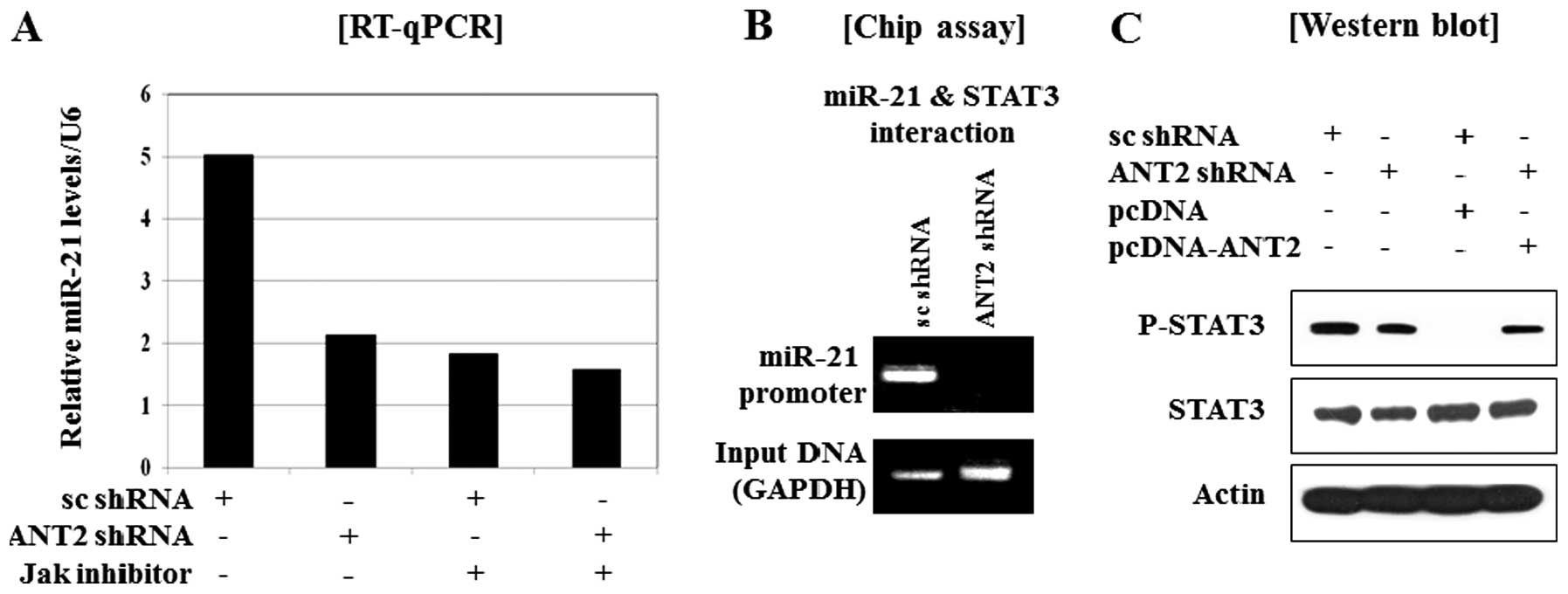 | Figure 5.Restoration of SOCS1 by ANT2 shRNA,
subsequently down-regulated the expression of onco-miR-21 through
the inactivation of STAT3. (A) RT-qPCR analysis of the effect of
ANT2 shRNA on the downregulation of miR-21 expression. Cells were
transfected with scramble or ANT2 shRNA. Total-RNA was extracted 24
h after transfection and subjected to RT-qPCR using specific
primers for human miR-21 or U6 (internal control). (B) After 24 h
of transfection with scramble shRNA or ANT2 shRNA, the cellular
extracts were subjected to chip assay. (C) After 24 h of
transfection with scramble shRNA, ANT2 shRNA, pcDNA and/or
pcDNA-ANT2, the cellular extracts were subjected to western blot
analysis with anti-phospho-STAT3 and anti-STAT3 antibodies. (D)
After 24 h of transfection with scramble shRNA, ANT2 shRNA, pcDNA
and/or pcDNA-ANT2, total-RNA was extracted 24 h after transfection
and subjected to RT-qPCR using specific primers for human miR-21 or
U6 (internal control). (E) After 24 h of transfection with scramble
shRNA, ANT2 shRNA, pcDNA and/or pcDNA-SOCS1, total-RNA was
extracted 24 h after transfection and subjected to RT-qPCR using
specific primers for human miR-21 or U6 (internal control). |
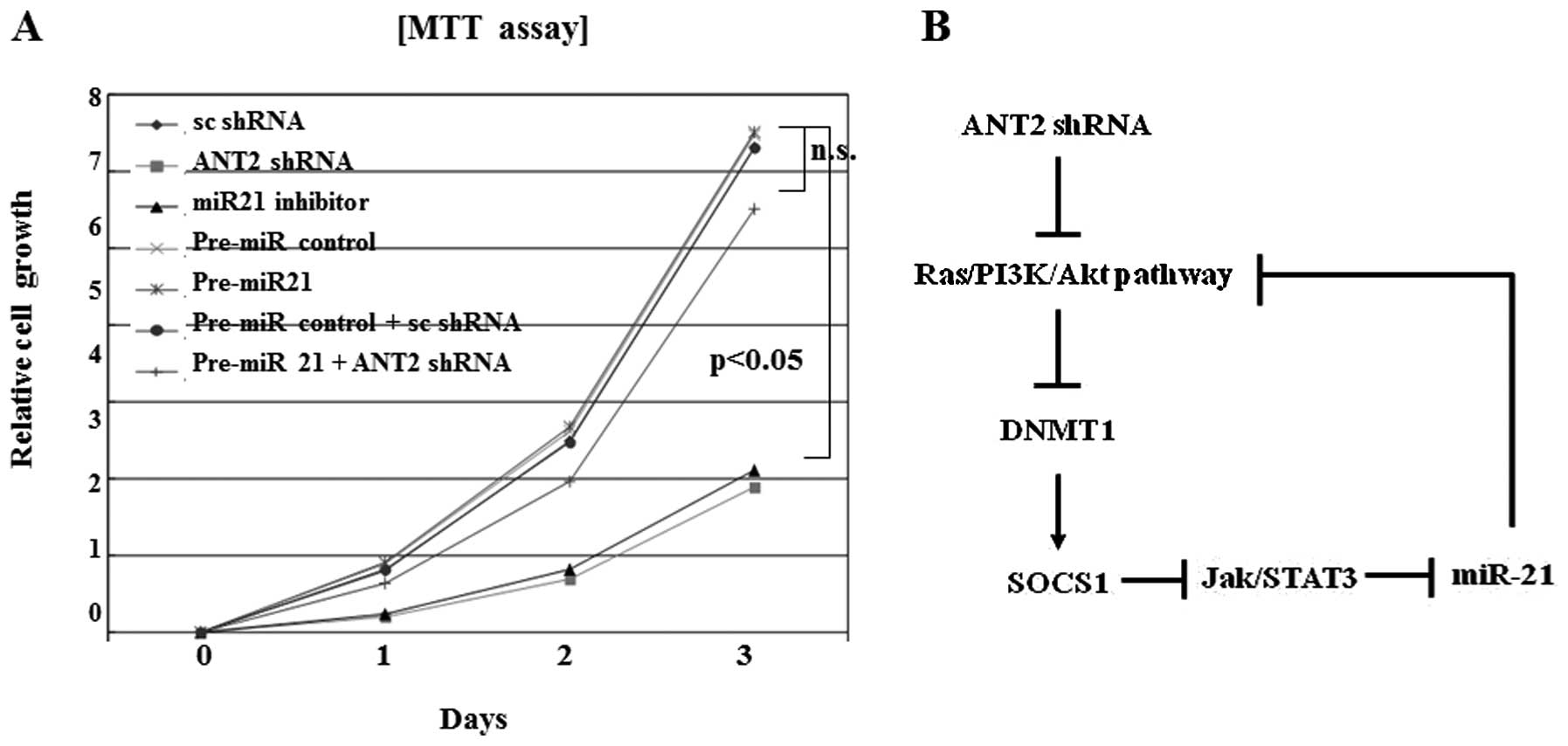
Downregulation of miR-21 by ANT2 shRNA
inhibits cell proliferation
Reduction of miR-21 levels by miR-21 inhibitor
inhibited proliferation. Similar inhibition of cell growth and
proliferation effects was also observed in ANT2 shRNA transfected
cells (Fig. 6A). Downregulation of
miR-21 efficiently suppressed Hep3B cell proliferation in
vitro with a comparable level to ANT2 shRNA treatment. Overall,
the possible pathogenesis of miR-21 suppression by ANT2
shRNA-induced SOCS1 restoration is illustrated in Fig. 6B.
Discussion
Recently, the potential role of the Jak/STAT pathway
in oncogenesis has been proposed in many kinds of tumors (31,32).
The SOCS family has been identified as a negative feedback protein
of Jak/STAT signaling pathway. These proteins are activated by
STATs and negatively regulate the Jak/STAT pathway by inhibiting
the Jaks directly or blocking the access of the STATs (33). Among the SOCS family molecules, the
SOCS1 expression was suppressed through aberrant methylation of the
CpG islands in several HCC studies. Promoter methylation in the
SOCS1 CpG islands was identified in primary HCC. Moreover, SOCS1
methylation was observed more frequently in HCCs derived from
cirrhosis than in those not derived from cirrhosis. This report
suggests that inactivation of SOCS1 might be an important factor in
carcinogenesis of HCC (25,34).
The overexpression of ANT2 in cancer cells was found
to be associated with glycolytic metabolism of cancer cells, which
raised a possible role of ANT2 during carcinogenesis (10). In fact, we and others have shown
that ANT2 knockdown decreased cell viability and induced apoptotic
death in cancer cells and also can sensitize cancer cells to a kind
of chemotherapeutic agents (8,13).
In the work shown here, knockdown of ANT2 by shRNA
restored SOCS1 expression and suppressed HCC cell proliferation. We
also observed that ANT2 shRNA treatment led to restoration of SOCS1
expression along with its promoter demethylation in Hep3B cells,
which was accompanied by decreased DNA methyl-transferase 1 (DNMT1)
activity through suppression of Ras/PI3K/Akt signaling. Generally,
DNMT1 is overexpressed in several cancers and upregulated by
oncogenic pathways and downregulated by tumor suppressive pathways
(35–37). Recently, prolonged half-life of
DNMT1 protein in cancer has also been reported, the mechanism of
its regulation is found that Akt activity positively correlates
with DNMT1 protein level but not mRNA level. Akt is a
serine/threonine protein kinase activated downstream of
phosphatidylinositol 3-kinase (PI3K). The mechanism attributed to
epigenetic role of PI3K/Akt in regulating protein stability of a
pivotal epigenetic molecule-DNMT1. Activated PI3K/Akt signaling
greatly stabilizes DNA methyltransferase 1 (DNMT1) by attenuating
the ubiquitin proteosome reaction in cancer cells (38). In this study, we observed that the
suppression of Ras/PI3K/Akt signaling by ANT2 shRNA induced SOCS1
restoration through the inhibition of DNMT1.
Restoration of SOCS1 by ANT2 knockdown,
subsequently, inhibited STAT3 activity and downregulated the
expression of miR-21, which has been reported to be an important
onco-miR in HCC. Downregulation of miR-21 efficiently suppressed
Hep3B cell proliferation in vitro with a comparable level to
ANT2 shRNA treatment. We demonstrated here for the first time that
ANT2 shRNA induces suppression of proliferation, accompanied by
inactivation of STAT3 through restoration of SOCS1 in HCC cell
lines.
According to another report, the restoration of
SOCS1 suppressed both growth rate and anchorage-independent growth
of cells in which SOCS1 was methylation-silenced and Jak2 was
constitutively activated (14).
This growth suppression was caused by apoptosis and was reproduced
by AG490, a specific, chemical Jak2 inhibitor that reversed
constitutive phosphorylation of STAT3 in SOCS1 inactivated cells.
The high prevalence of the aberrant SOCS1 methylation and its
growth suppression activity demonstrated the importance of the
constitutive activation of the Jak/STAT pathway in the development
of HCC. It also indicates therapeutic strategies for the treatment
of HCC including use of SOCS1 in gene therapy and inhibition of
Jak2 by small molecules, such as AG490 (14). In the present study, ANT2 shRNA
suppressed onco-miR-21 expression and STAT3 activition and preceded
restoration of SOCS1. We thus hypo thesized that ANT2 shRNA may
also inhibit the Jak/STAT signaling pathway through the restoration
of SOCS1, which may explain why ANT2 shRNA can suppress the growth
rate of the HCC cell line. This study provides strong evidence that
inactivation of Jak/STAT signaling pathway by ANT2 shRNA
downregulates onco-miR-21 expression as well as inhibits
proliferation. ANT2 suppression by shRNA might be able to exert
anticancer effect in HCC further by restoring the SOCS1
expression.
miRNAs play important roles in multiple biological
processes such as development, differentiation, and cellular stress
response (39). Recent studies
have linked deregulation of miRNAs to various diseases including
cancer (40). According to recent
reports, dramatic upregulation of miR-21 is observed in HCC and
dysregulation of miR-21 is associated with HCC formation and
progression. Moreover, uncontrolled proliferation, lack of
apoptosis, and invasiveness, all modulated by miR-21, were also
found to be present in tumorigenesis (41). Several questions remain unanswered.
For example, miR-21 targets multiple negative regulators of Ras
signaling, but the molecular alteration caused by miR-21-mediated
Ras regulation remains unidentified (42). miR-21 is a potential onco-miR that
plays important roles in HCC formation and progression. Moreover, a
regulatory loop between Ras/PI3K/Akt pathway and miR-21 is mediated
by STAT3. In conclusion, the ANT2 suppression by shRNA might be
able to exert anticancer effect in HCC further by restoring the
SOCS1 expression.
Abbreviations:
|
ANT2
|
adenine nucleotide translocase-2;
|
|
RNAi
|
RNA interference;
|
|
shRNA
|
short-hairpin RNA;
|
|
siRNA
|
small-interfering RNA;
|
|
SOCS1
|
suppressor of cytokine
signaling-1;
|
|
Jak-STAT
|
janus kinase and signal transducer and
activation of transcription;
|
|
HCC
|
human hepatocellular carcinoma
|
Acknowledgements
This work was supported by the Global
Core Research Center (GCRC) grant (no. 2012-0001190) from the
National Research Foundation (NRF), Ministry of Education, Science
and Technology (MEST), Republic of Korea. This work was supported
by grant (no. SS100003) from the Seoul R&BD Program.
References
|
1.
|
Yang JD and Roberts LR: Hepatocellular
carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 7:448–458.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar
|
|
3.
|
Blum HE: Hepatocellular carcinoma: therapy
and prevention. World J Gastroenterol. 11:7391–7400.
2005.PubMed/NCBI
|
|
4.
|
Marzo I, Brenner C, Zamzami N, Susin SA,
Beutner G, Brdiczka D, Remy R, Xie ZH, Reed JC and Kroemer G: The
permeability transition pore complex: a target for apoptosis
regulation by caspases and bcl-2-related proteins. J Exp Med.
187:1261–1271. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Marzo I, Brenner C, Zamzami N,
Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama
S, Reed JC and Kroemer G: Bax and adenine nucleotide translocator
cooperate in the mitochondrial control of apoptosis. Science.
281:2027–2031. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Dolce V, Scarcia P, Iacopetta D and
Palmieri F: A fourth ADP/ATP carrier isoform in man:
identification, bacterial expression, functional characterization
and tissue. FEBS Lett. 579:633–637. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lunardi J and Attardi G: Differential
regulation of expression of the multiple ADP/ATP translocase genes
in human cells. J Biol Chem. 266:16534–16540. 1991.PubMed/NCBI
|
|
8.
|
Le Bras M, Borgne-Sanchez A, Touat Z, El
Dein OS, Deniaud A, Maillier E, Lecellier G, Rebouillat D, Lemaire
C, Kroemer G, Jacotot E and Brenner C: Chemosensitization by
knockdown of adenine nucleotide translocase-2. Cancer Res.
66:9143–9152. 2006.PubMed/NCBI
|
|
9.
|
Faure Vigny H, Heddi A, Giraud S, Chautard
D and Stepien G: Expression of oxidative phosphorylation genens in
renal tumors and tumoral cell lines. Mol Carcinog. 16:165–172.
1996.PubMed/NCBI
|
|
10.
|
Chevrollier A, Loiseau D, Chabi B, Renier
G, Donay O, Malthiery Y and Stepien G: ANT2 isoform required for
cancer cell glycolysis. J Bioenerg Biomembr. 37:307–316. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Chevrollier A, Loiseau D, Gautier F,
Malthlery Y and Stepien G: ANT2 expression under hypoxic conditions
produces opposite cell-cycle behavior in 143B and HepG2 cancer
cells. Mol Carcinog. 42:1–8. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Chevrollier A, Loiseau D and Stepien G:
What is the specific role of ANT2 in cancer cells? Med Sci (Paris).
21:156–161. 2005.(In French).
|
|
13.
|
Jang JY, Choi Y, Jeon YK and Kim CW:
Suppression of adenine nucleotide translocase-2 by vector-based
siRNA in human breast cancer cells induces apoptosis and inhibits
tumor growth in vitro and in vivo. Breast Cancer Res. 10:R112008.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Yoshikawa H, Matsubara K, Qian GS, Jackson
P, Groopman JD, Manning JE, Harris CC and Herman JG: SOCS-1, a
negative regulator of the JAK/STAT pathway, is silenced by
methylation in human hepatocellular carcinoma and shows
growth-suppression activity. Nat Genet. 28:29–35. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Singal R and Ginder GD: DNA methylation.
Blood. 93:4059–4070. 1999.PubMed/NCBI
|
|
16.
|
Robertson KD and Jones PA: DNA
methylation: past, present and future directions. Carcinogenesis.
21:461–467. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Baylin SB, Esteller M, Rountree MR,
Bachman KE, Schuebel K and Herman JG: Aberrant patterns of DNA
methylation, chromatin formation and gene expression in cancer. Hum
Mol Genet. 10:687–692. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Heinrich PC, Behrmann I, Muller-Newen G,
Schaper F and Graeve L: Interleukin-6-type cytokine signaling
through the gp130/Jak/STAT pathway. Biochem J. 334:297–314.
1998.PubMed/NCBI
|
|
19.
|
Imada K and Leonard WJ: The Jak-STAT
pathway. Mol Immunol. 37:1–11. 2000. View Article : Google Scholar
|
|
20.
|
Alexander WS, Starr R, Metcalf D,
Nicholson SE, Farley A, Elefanty AG, Brysha M, Kile BT, Richardson
R, Baca M, Zhang JG, Willson TA, Viney EM, Sprigg NS, Rakar S,
Corbin J, Mifsud S, DiRago L, Cary D, Nicola NA and Hilton DJ:
Suppressors of cytokine signaling (SOCS): negative regulators of
signal transduction. J Leukoc Biol. 66:588–592. 1999.PubMed/NCBI
|
|
21.
|
Nicola NA and Greenhalgh CJ: The
suppressors of cytokine signaling (SOCS) proteins: important
feedback inhibitors of cytokine action. Exp Hematol. 28:1105–1112.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
He B, You L, Uematsu K, Zang K, Xu Z, Lee
AY, Costello JF, McCormick F and Jablons DM: SOCS-3 is frequently
silenced by hypermethylation and suppresses cell growth in human
lung cancer. Proc Natl Acad Sci USA. 100:14133–14138. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
He B, You L, Xu Z, Mezieres J, Lee AY and
Jablons DM: Activity of the suppressor of cytokine signaling-3
promoter in human non-small-cell lung cancer. Clin Lung Cancer.
5:366–370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Weber A, Hengge UR, Bardenheuer W,
Tischoff I, Sommerer F, Markwarth A, Dietz A, Wittekind C and
Tannapfel A: SOCS-3 is frequently methylated in head and neck
squamous cell carcinoma and its precursor lesions and causes growth
inhibition. Oncogene. 24:6699–6708. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Chu PY, Yeh CM, Hsu NC, Chang YS, Chang JG
and Yeh KT: Epigenetic alteration of the SOCS1 gene in
hepatocellular carcinoma. Swiss Med Wkly. 140:w130652010.PubMed/NCBI
|
|
26.
|
Isomoto H, Kobayashi S, Werneburg NW,
Bronk SF, Guicciardi ME, Frank DA and Gores GJ: Interleukin 6
upregulates myeloid cell leukemia-1 expression through a STAT3
pathway in cholangiocarcinoma cells. Hepatology. 42:1329–1338.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Konnikova L, Kotecki M, Kruger MM and
Cochran BH: Knockdown of STAT3 expression by RNAi induces apoptosis
in astrocytoma cells. BMC Cancer. 3:1–9. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Aoki Y, Feldman GM and Tosato G:
Inhibition of STAT3 signaling induces apoptosis and decreases
survivin expression in primary effusion lymphoma. Blood.
101:1535–1542. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Jourdan M, Veyrune JL, Vos JD, Redal N,
Couderc G and Klein B: A major role for Mcl-1 antiapoptotic protein
in the IL-6-induced survival of human myeloma cells. Oncogene.
22:2950–2959. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk
ML and Struhl K: STAT3 activation of miR-21 and miR-181b-1 via PTEN
and CYLD are part of the epigenetic switch linking inflammation to
cancer. Mol Cell. 39:493–506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Lacronique V, Boureux A, Valle VD, Poirel
H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael
J and Bernard OA: A TEL-JAK2 fusion protein with constitutive
kinase activity in human leukemia. Science. 278:1309–1312. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Bromberg JF, Wrzeszczynska MH, Devgan G,
Zhao Y, Pestell RG, Albanese C and Darnell JE: Stat3 as an
oncogene. Cell. 98:295–303. 1999. View Article : Google Scholar
|
|
33.
|
Cooney RN: Suppressors of cytokine
signaling (SOCS): inhibitors of the Jak/STAT pathway. Shock.
17:83–90. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Okochi O, Hibi K, Sakai M, Inoue S, Takeda
S, Kaneko T and Nakao A: Methylation-mediated silencing of SOCS-1
gene in hepatocellular carcinoma derived from cirrhosis. Clinical
Cancer Res. 9:5295–5298. 2003.PubMed/NCBI
|
|
35.
|
Sun L, Hui AM, Kanai Y, Sakamoto M and
Hirohashi S: Increased DNA methyltransferase expression is
associated with an early stage of human hepatocarcinogenesis. Jpn J
Cancer Res. 88:1165–1170. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Saito Y, Kanai Y, Sakamoto M, Saito H,
Ishii H and Hirohashi S: Expression of mRNA for DNA
methyltransferases and methyl-CpG-binding proteins and DNA
methylation status on CpG islands and pericentromeric
pericentromeric satellite regions during human
hepatocarcinogenesis. Hepatology. 33:561–568. 2001. View Article : Google Scholar
|
|
37.
|
Kanai K, Ushijima S, Kondo Y, Nakanishi Y
and Hirohashi S: DNA methyltransferase expression and DNA
methylation of CpG islands and peri-centromeric satellite regions
in human colorectal and stomach cancers. Int J Cancer. 91:205–212.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Zuo T, Liu TM, Lan X, Weng YI, Shen R, Gu
F, Huang YW, Liyanarachchi S, Deatherage DE, Hsu PY, Taslim C,
Ramaswamy B, Shapiro CL, Lin HJL, Cheng ASL, Jin VX and Huang TH:
Epigenetic silencing mediated through activated PI3K/AKT signaling
in breast cancer. Cancer Res. 71:1752–1762. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Zhang B, Wang Q and Pan X: MicroRNAs and
their regulatory roles in animals and plants. J Cell Physiol.
210:279–289. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Jiang Q, Wang Y, Hao Y, Juan L, Teng M,
Zhang X, Li M, Wang G and Liu Y: miR2 Disease: a manually curated
database for microRNA deregulation in human disease. Nucleic Acids
Res. 37:D98–D104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Meng F, Enson R, Wehbe-Janek H, Ghoshal K,
Jacob ST and Patel T: MicroRNA-21 regulates expression of the PTEN
tumor suppressor gene in human hepatocellular cancer.
Gastroenterology. 133:647–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Hatley ME, Patrick DM, Garcia MR,
Richardson JA, Duby RB, van Rooij E and Olson EN: Modulation of
K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell.
18:282–293. 2010. View Article : Google Scholar : PubMed/NCBI
|