Introduction
Neurofibromatosis type 1 (NF1; MIM #162200) is a
commonly inherited autosomal dominant disorder characterized by
variable phenotypic features, including cutaneous manifestations,
such as café au lait spots, neurofibromas and freckling of the
axillary or inguinal regions, as well as extracutaneous
manifestations such as Lisch nodules, optic nerve gliomas,
scoliosis, bone dysplasia, malignant tumors, and cognitive
impairment (1,2). NF1 is caused by neurofibromin 1
(NF1) gene mutations, which encode neurofibromin, a
GTPase-activating protein (GAP) (3). The majority of patients with NF1
develop benign dermal neurofibromas (DNs) and/or plexiform
neurofibromas (PNs) (4,5).
Neurofibromas are composed of a mixture of cell
types including Schwann cells (SCs), fibroblast cells, mast cells,
and perineural cells (6). SCs are
believed to be the primary pathogenic cell source in neurofibromas
(7). As the complete loss of the
NF1 gene has been identified exclusively in neurofibroma SCs
(8–10) and the loss of NF1 in the SC
lineage is sufficient to generate tumors in mice (11), the bi-allelic inactivation of both
NF1 alleles (NF1−/−) in SCs by germline
NF1 mutation at one allele and the additional somatic loss
of heterozygosity (LOH) at the remaining functional NF1
locus, has been suggested to be a major cause of NF1 tumorigenesis.
In addition, haploinsuffiency in other types of cells
(NF1+/−) in neural crest-derived tissues,
including fibroblast cells, mast cells and perineurial cells also
plays an important role in the pathoetiology of NF1 (6,12,13).
Malignant peripheral nerve sheath tumors (MPNSTs)
are a type of aggressive sarcoma and are a major cause of mortality
in patients with NF1 (5,14–16).
The lifetime risk of developing MPNSTs in patients with NF1 is
8–13% (17) or 5.9–10.3% (18). The majority of NF1-associated
MPNSTs (approximately 85% of cases) are high-grade malignant
tumors. The malignant transformation of benign PNs to MPNSTs in
patients with NF1 is notable (19)
and is of far greater concern to patients with NF1 (20); however, the pathogenesis is poorly
understood. The bi-allelic inactivation of the NF1 gene
caused by a germline first-hit mutation and a somatic second-hit
LOH in SCs has been identified in DNs (21,22),
PNs (23,24), and MPNSTs (24,25)
in patients with NF1, indicating that the complete loss of the
NF1 gene (NF1−/−) in SCs contributes to
benign neurofibroma formation and progression to MPNSTs. Since
bi-allelic inactivation of the NF1 gene is insufficient to
explain the pathogenesis of tumor progression in NF1, cooperating
genetic or epigenetic changes have been suggested to be involved in
MPNST pathogenesis. Hence, robust histological and molecular
analyses have been conducted to compare neurofibromas and MPNSTs
(15,26,27),
and recently developed genome-wide DNA copy number change profiling
using array comparative genomic hybridization has identified causal
genes in MPNST development (28,29).
To date, genes involved in regulating the cell cycle and growth
signal transduction have been reported mainly to be dysregulated in
MPNSTs (6,30,31).
A number of studies have focused on genetic alterations in SCs, as
most MPNSTs are thought to arise from SCs (7,32).
We unexpectedly found that the anti-apoptotic
protein, Bcl-xL, is upregulated in primary-cultures and established
NF1-associated MPNST cells. Bcl-xL is responsible for the acquired
anticancer drug resistance of MPNST cells (33). In this study, we compared Bcl-xL
expression levels between normal and MPNST-derived SCs, as well as
between PNs and MPNST tissues from patients with NF1 to determine
whether Bcl-xL upregulation in SCs is involved in MPNST
pathogenesis. Furthermore, we also examined changes in Bcl-xL
expression levels and sensitivity to apoptosis induced by
anti-cancer drugs in NF1+/+ and
NF1−/− SCs when NF1 expression was
manipulated to determine whether Bcl-xL upregulation is associated
with NF1 deficiency in SCs.
Materials and methods
Antibodies and reagents
Anti-Bcl-xL, anti-Bcl2, anti-Bax, anti-caspase 3,
anti-extracellular signal-regulated kinase (Erk)1/2 and
anti-phosphorylated Erk1/2 antibodies were purchased from Cell
Signaling Technology (Danvers, MA, USA). Anti-neurofibromin,
anti-α-tubulin, anti-p53, anti-Mcl-1, HRP-conjugated goat
anti-rabbit IgG and HRP-conjugated goat anti-mouse IgG antibodies
were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA),
and anti-S100 and anti-GFP antibodies were purchased from Thermo
Scientific Pierce (Rockford, IL, USA) and Invitrogen (Carlsbad, CA,
USA), respectively. Doxorubicin and PD98059 were obtained from
Sigma-Aldrich (St. Louis, MO, USA). ABT-737 was purchased from
Santa Cruz Biotechnology.
Cell culture
Human normal Schwann cells (HSCs) and Schwann
cell-like MPNST cells (sNF96.2) were purchased from ScienCell
Research Laboratories (Carlsbad, CA, USA) and the American Type
Culture Collection (Manassas, VA, USA), respectively. Cells were
cultured in Dulbecco’s modified Eagle medium (HyClone Laboratories,
Logan, UT, USA) containing 10% fetal bovine serum supplemented with
penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C
under a humidified atmosphere containing 5% CO2.
Hematoxylin and eosin (H&E)
staining
Tumor tissues were obtained from patients with NF1
by surgical resection. The specimens were formalin-fixed and
embedded in paraffin wax for pathological evaluation by routine
light microscopy. Serial 3-μm sections were prepared on
glass using a cryostat, and the slides were stained with
H&E.
Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded (FFPE) blocks from
six patients with NF1 were cut at 10 μm, and the sections
were dewaxed, rehydrated, followed by antigen retrieval in boiling
citrate buffer. Immunostaining was carried out using an Ultravision
LP-HRP Polymer DAB kit (Thermo Fisher Scientific, Kalamazoo, MI,
USA), according to the manufacturer’s instructions. Briefly, the
sections were incubated with Ultra V Block (Lab Vision, Kalamazoo,
MI, USA) for 5 min at room temperature to reduce the non-specific
background, and were then treated with hydrogen peroxide to block
endogenous peroxidase activity. The sections were incubated with
primary antibody for 1 h and then incubated with HRP polymer for 20
min. The reaction product was visualized with DAB chromogen.
Pathological evaluation was performed under light microscopy. The
present study using human FFPE samples was approved by the
Institutional Review Board of the Ajou University School of
Medicine, Suwon, Korea.
Plasmid constructs and small interfering
RNAs (siRNAs)
Plasmid constructs encoding wild-type Bcl-xL were
generated as described previously (34). The cDNA of the GAP-related domain
(GRD) region (1,181 bp) was amplified by reverse
transcription-polymerase chain reaction (RT-PCR) using the primers
to generate the plasmid construct expressing the human NFl-GRD:
5′-ATAGATCTACCATGGATCTCCAGACAAGAGCTACATTTATG-3′ and
5′-GTAAGCTTAACCAGTGTGTATCTGCCACAGGT-3′, from total RNAs of human
skin tissue cultured fibroblast cells. The cDNAs were subcloned
into the pEYFP-C1 vector (Clontech, Palo Alto, CA, USA) using the
BglII and HindIII restriction enzyme sites. The
siRNAs were synthesized by Genolution Pharmaceuticals, Inc. (Seoul,
South Korea). The target sequences for the siRNAs were as follows:
5′-CAGTGAACGTAAGGGTTCT-3′ for the NF1 gene,
5′-CAGGGACAGCATATCAGAG-3′ for the BCL2L1 (Bcl-xL) gene and
5′-CCTACGCCACCAATTTCGT-3′ for the non-specific negative control.
Cell transfection of the siRNAs and plasmid constructs was
conducted using Lipofectamine RNAiMAX (Invitrogen) and
Lipofectamine 2000 (Invitrogen), respectively, according to the
manufacturer’s instructions.
Cell viability assay
Cell viability was assessed using the EZ-Cytox Cell
Viability Assay kit (Daeil Labservice, Seoul, Korea). Cells were
seeded in a 96-well tissue-culture plate (7×103
cells/well), cultured overnight, and then treated with various
concentrations of doxorubicin and/or ABT-737. After 24 h of
incubation, 10 μl of Ez-Cytox reagent was added to each
well, and the cells were incubated for a further 2 h. The plate was
read with an enzyme-linked immunosorbent assay microplate reader
(Bio-Rad Model 680; Hercules, CA, USA) at 450 nm.
Real-time RT-PCR
Total RNAs were isolated from the cultured cells
using TRIzol reagent (Invitrogen), treated with RNase-free DNase I
(Invitrogen) to avoid amplification of genomic DNA, and were
subsequently reverse-transcribed using the RevertAid™ H Minus
First-Strand cDNA Synthesis kit (Fermentas, Burlington, ON, Canada)
with the oligo(dT)15–18 primer. Real-time RT-PCR was
performed using the SYBR-Green I qPCR kit (Takara, Shiga, Japan).
The specific primers used were as follows:
5′-GTCGGATCGCAGCTTGGATGGCCAC-3′ and 5′-CGTCAGGAACCAGCGGTTGAAGCGT-3′
for BCL2L1. The P238284 primer set (Bioneer, Seoul, Korea)
was used for NF1 and 5′-TGTTGCCATCAATGACCCCTT-3′ and
5′-CTCCACGACGTACTCAGCG-3′ for the GAPDH gene (a relative
quantification standard). All real-time RT-PCR measurements were
performed using the ABI Prism 7500 Fast Real-time PCR System
(Applied Biosystems, Foster City, CA, USA).
Western blot analysis
Cultured cells were lysed in RIPA buffer [150 mM
NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium
dodecyl sulfate (SDS), and 50 mM Tris buffer, pH 8.0]. Proteins
were heated at 100°C for 10 min and analyzed by SDS-polyacrylamide
gel electrophoresis on 8–12% polyacrylamide gels. The proteins were
electroblotted onto PVDF membranes (Millipore, Milford, MA, USA).
The membrane blots were blocked with 5% (w/v) non-fat dried milk,
incubated with primary and secondary antibodies, and then
visualized with the Enhanced Chemiluminescence Western Blotting
Detection System (WEST-ZOL plus; iNtRON Biotechnology, Daejeon,
Korea).
Statistical analysis
In this study, the results are expressed as the
means ± standard deviation. All experiments were repeated at least
three times. Statistical significance was determined by the
two-tailed Student’s t-test, and P-values <0.05 were considered
to indicate statistically significant differences.
Results
Higher Bcl-xL expression observed in
MPNSTs compared to PNs in patients with NF1
Since Bcl-xL hyperexpression has been observed in
NF1-associated MPNST cells (33),
we confirmed this finding in the tumor tissues of patients with
NF1. Tumor speci mens were obtained by surgical resection from six
patients with NF1. The patients were diagnosed with NF1 at the Ajou
University Hospital according to NF1 diagnostic criteria (35). The clinical features of the
patients are summarized in Table
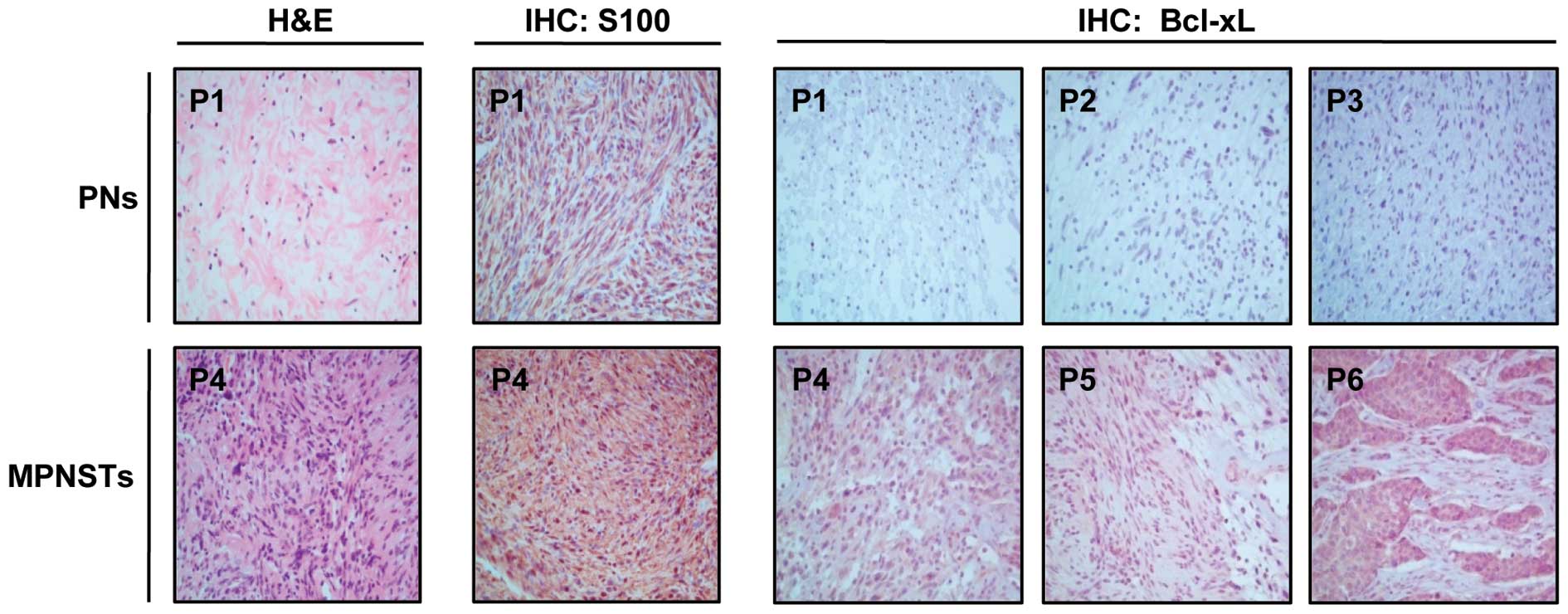
I. Histopathological analysis of the tumor specimens by H&E
staining revealed that three patients [patient (P)1–P3] had benign
PNs and three patients (P4–P6) had MPNSTs (Table I). The H&E results of P1 and P4
are shown in Fig. 1. The PNs were
composed of loosely spaced tumor cells in a myxoid matrix or
collagenous strands, whereas MPNSTs showed densely cellular
atypical spindle cells forming intersecting fascicles. We then
compared the Bcl-xL expression levels between the PN and MPNST
tumor tissues by IHC analysis. The IHC evaluation revealed a higher
Bcl-xL expression in MPNSTs (P4–P6) compared to PNs (P1–P3) in all
patients with NF1 tested.
 | Table I.Clinical and histological
characteristics of the six patients with neurofibromatosis type
1. |
Table I.
Clinical and histological
characteristics of the six patients with neurofibromatosis type
1.
Patient
| Histological
findings
| Clinical
characteristics
| Genotype
|
|---|
| ID | Gender | Age at
diagnosis | H&E | S100 | Bcl-xL | Café au lait
spots | Neurofibromas | Freckling | Optic glioma | Lisch nodule | Skeletal
dysplasia | Family history | NF1 gene
mutation |
|---|
| P1 | Male | 59 | Benign | + | + | Y | Y | Y | N | N | N | Y | N/A |
| P2 | Male | 17 | Benign | + | + | Y | Y | Y | N | Y | Y | N | c.4537C>T |
| P3 | Female | 5 | Benign | + | + | Y | Y | Y | N | N | N | Y | N/A |
| P4 | Male | 39 | Malignant | + | ++ | Y | Y | N | N | N | N | N | N/A |
| P5 | Male | 32 | Malignant | + | ++ | Y | Y | Y | N | N | N | N | c.4861_4862
GT>AG |
| P6 | Female | 41 | Malignant | + | ++ | Y | Y | N | N | N | N | N | N/A |
SCs are generally thought to be the major
progenitors of neurofibromas and MPNSTs and characteristically
express the S100 protein (36).
IHC staining using an antibody against the SC lineage marker, S100,
showed that all tumors contained S100-positive cells (Table I) and more S100-positive cells were
present in MPNSTs than in PNs, as shown in P1 and P4 (Fig. 1). These results suggest that tumors
from patients with NF1 mainly originate from SCs and that most
Bcl-xL-overexpressing cells are SC lineage cells.
Upregulation of Bcl-xL causes an increase
in resistance to doxorubicin in NF1-deficient MPNST SCs
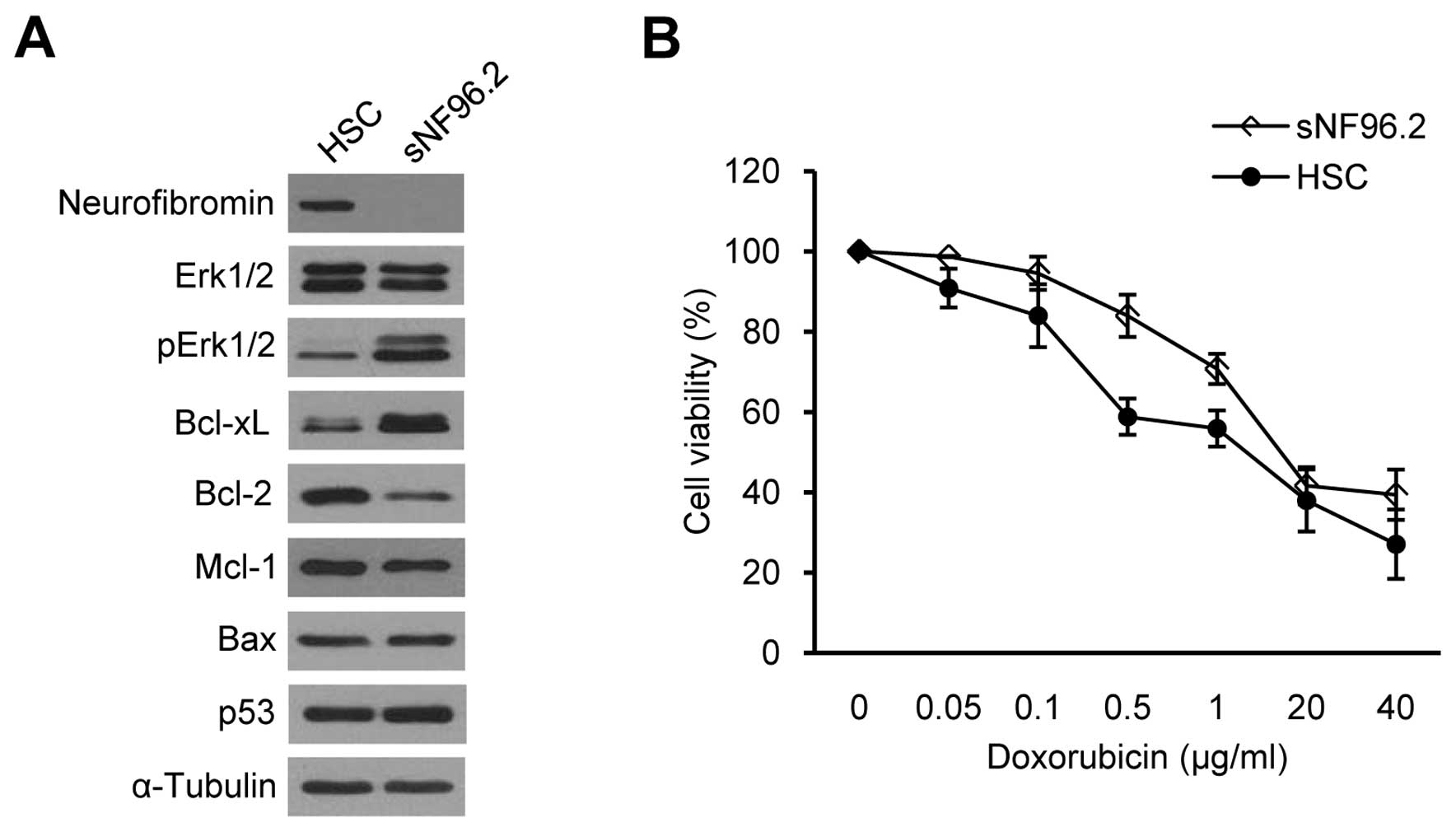
We examined whether the basal Bcl-xL expression
levels were different between SCs derived from normal tissues and
those derived from MPNSTs. We compared the normal human SC line
(HSC) and the sNF96.2 SC line, which was derived from a MPNST in a
patient with NF1. HSCs have both normal NF1 alleles
(NF1+/+), whereas the sNF96.2 cells have a
complete LOH and no remaining NF1 allele
(NF1−/−) (37).
Western blot analysis for neurofibromin confirmed normal
neurofibromin expression in the HSCs and null neurofibromin
expression in the sNF96.2 cells (Fig.
2A). The increased pErk1/2 protein level involved in the
Ras/Raf/Mek/Erk signaling pathway (38), demonstrated that the sNF96.2 cells
were MPNST-derived SCs (Fig. 2A).
Basal Bcl-xL expression was significantly upregulated in the
sNF96.2 cells compared to the HSCs, whereas the lower expression of
Bcl-2, another anti-apoptotic protein, was observed in the sNF96.2
cells (Fig. 2A). No changes in
Mcl-1 and p53 expression levels were observed between the two cell
lines.
Chemoresistance in NF1-associated MPNST cells has
been recently reported (33). We
examined sensitivity to apoptosis induced by anticancer drugs in
HSCs and sNF96.2 cells to determine whether SCs are responsible for
chemoresistance in MPNSTs. We used doxorubicin as doxorubicin has
been suggested to be a good candidate for MPNST chemotherapy
(33,39). The cell viability assay results
demonstrated that the sNF96.2 cells were more resistant to
doxorubicin than the HSCs (Fig.
2B), suggesting that the upregulation of Bcl-xL may decrease
apoptosis sensitivity in sNF96.2 cells.
Bcl-xL expression level is closely
related to sensitivity to doxorubicin-induced apoptosis in normal
SCs and NF1-deficient MPNST-derived SCs
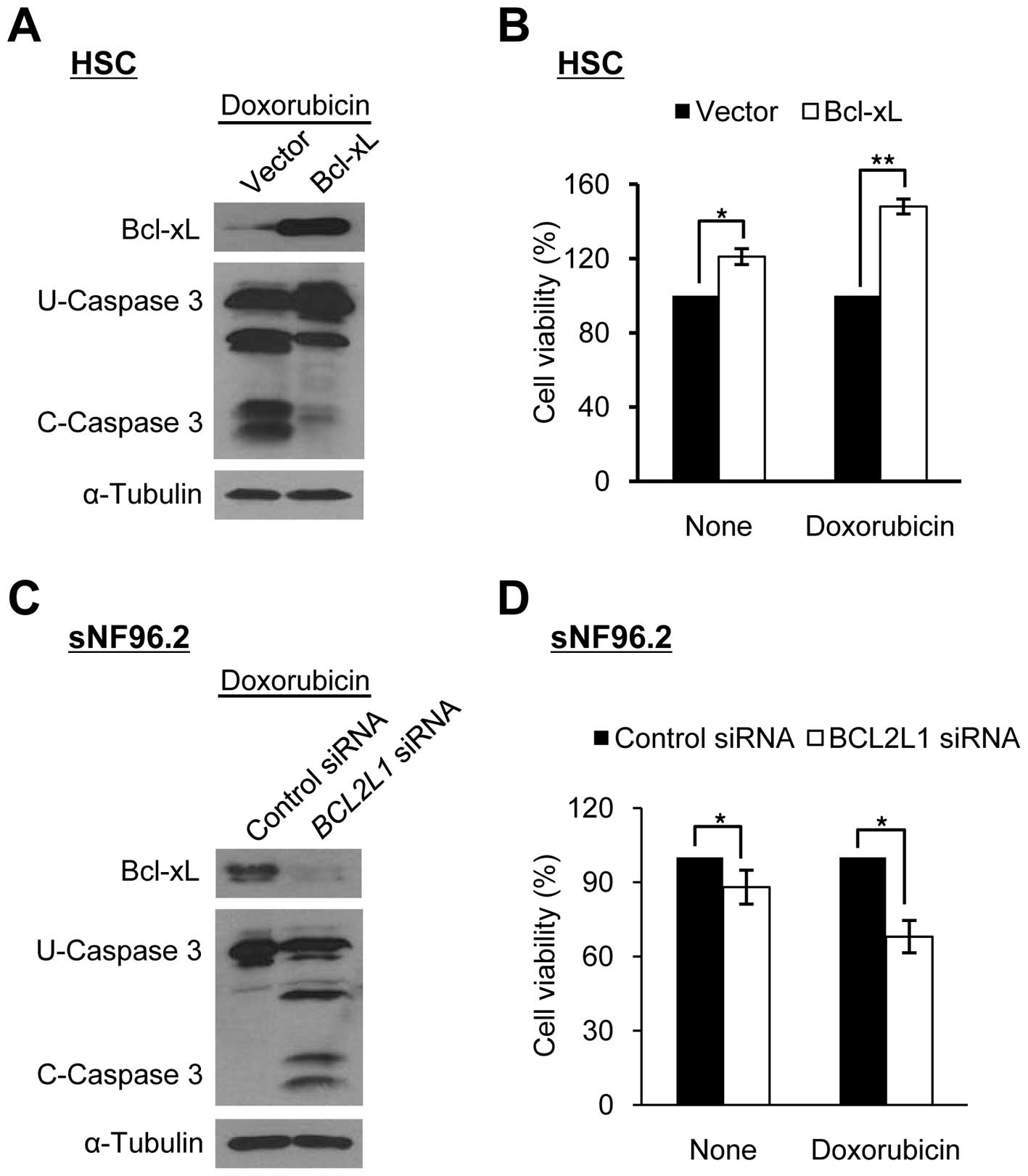
We manipulated Bcl-xL expression levels in HSCs and
sNF96.2 cells to determine whether the sensitivity to
doxorubicin-induced apoptosis in SCs was dependent on the Bcl-xL
expression level. The overexpression of Bcl-xL in HSCs decreased
caspase 3 cleavage activity significantly and increased cell
viability following doxorubicin treatment (Fig. 3A and B). By contrast, the
downregulation of Bcl-xL in the sNF96.2 cells following treatment
with siRNAs targeting the BCL2L1 gene significantly
increased caspase 3 cleavage activity and reduced cell viability
following doxorubicin treatment (Fig.
3C and D). These results indicate that the Bcl-xL level is
closely related to sensitivity to doxorubicin-induced apoptosis in
both types of SC (HSCs and MPNST-derived SCs).
Bcl-xL expression level is mediated by
NF1 gene level in normal SCs and NF1-deficient MPNST-derived
SCs
The genetically noticeable difference between HSCs
and sNF96.2 cells is determined by whether the NF1 gene is
intact (NF1+/+) or inactivated
(NF1−/−), suggesting that the hyperexpression of
Bcl-xL in sNF96.2 cells may be mediated by decreased NF1
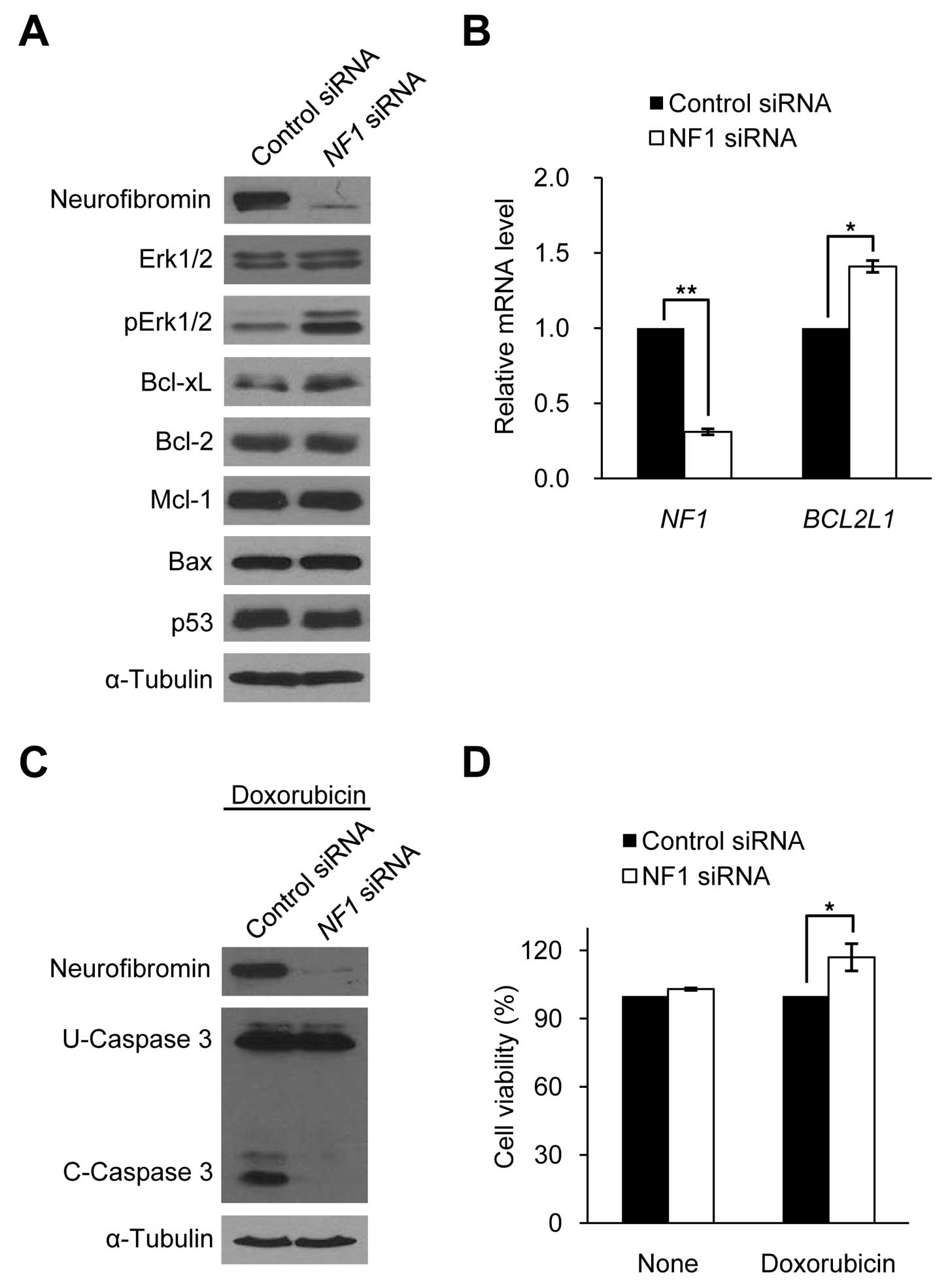
expression. We manipulated neurofibromin expression levels in HSCs
and sNF96.2 cells to determine whether Bcl-xL expression is
dependent on neurofibromin expression in SCs. The downregulation of
neurofibromin expression by siRNA targeting the NF1 gene in
HSCs caused an increase in Bcl-xL expression and pErk1/2 protein
levels, but had no effect on other apoptosis-related proteins such
as Bcl-2, Mcl-1, Bax and p53 (Fig.
4A). Real-time PCR results of BCL2L1 demonstrated that
the increased Bcl-xL expression level in the neurofibromin-depleted
HSCs was caused by the increased BCL2L1 mRNA expression
(Fig. 4B). The
neurofibromin-depleted HSCs showed decreased caspase 3 cleavage
activity and increased cell viability following doxorubicin
treatment for 24 h after 72 h of NF1 siRNA treatment
(Fig. 4C and D), similar to the
Bcl-xL-overexpressing HSCs (Fig. 3A
and B).
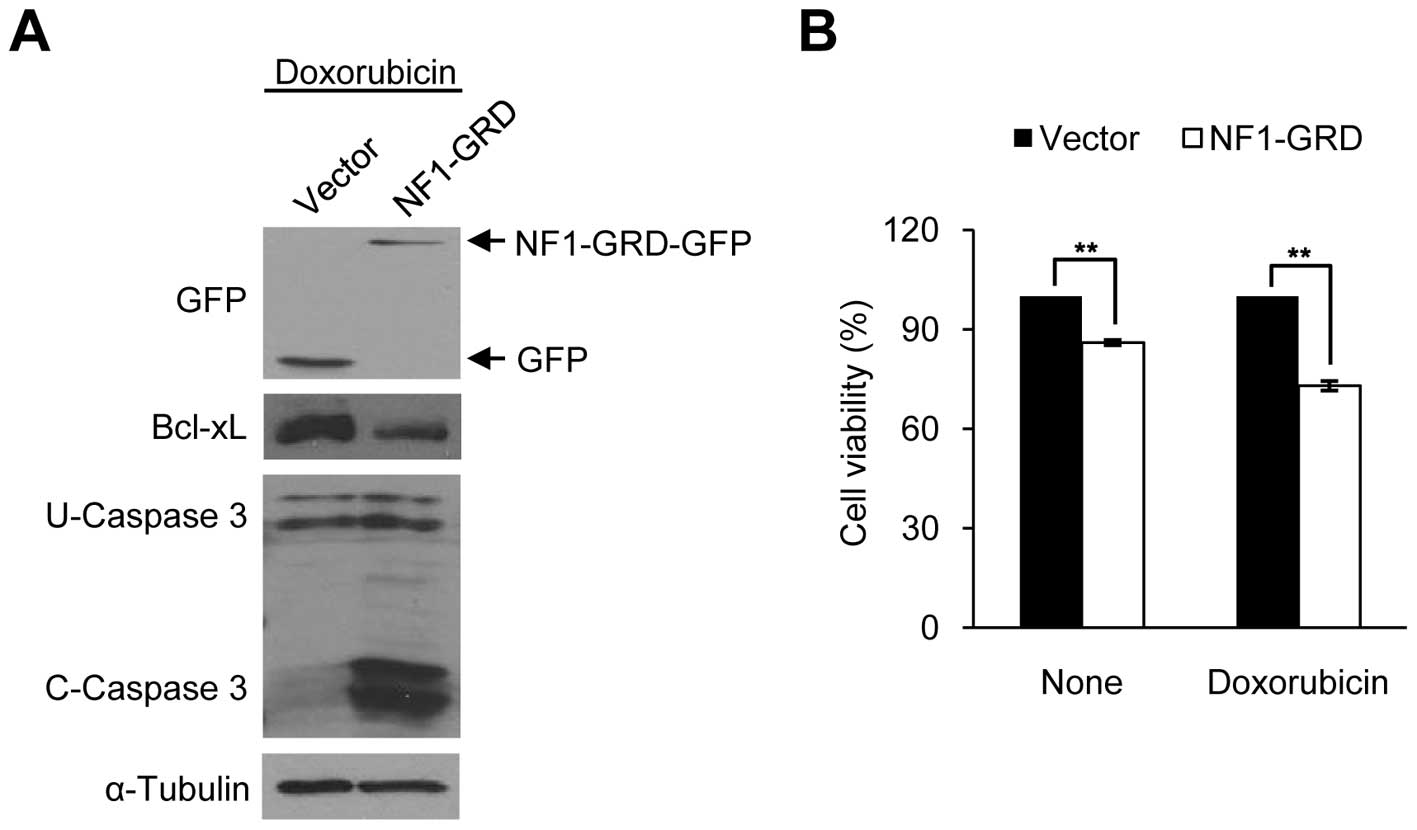
We then overexpressed NF1 in sNF96.2 cells. Since
the NF1 gene is very large, and the NF1-GRD is sufficient to
restore normal growth in mouse NF1−/− cells
(40), we constructed a human
NF1-GRD fused to GFP. NF1-GRD-GFP overexpression in the sNF96.2
cells significantly increased caspase 3 cleavage activity and
reduced cell viability following doxorubicin treatment (Fig. 5A and B), similar to the
Bcl-xL-depleted sNF96.2 cells (Fig. 3C
and D). These results indicate that Bcl-xL expression level is
mediated by the NF1 gene level in both types of SC (HSCs and
MPNST-derived SCs).
NF1 deficiency induces Bcl-xL expression
by activating Erk1/2 in the Ras/mitogen-activated protein kinase
(MAPK) signaling pathway
We then wished to clarify the molecular mechanisms
by which alterations in neurofibromin expression in SCs modulate
the Bcl-xL expression level. As shown in Fig. 2A, Erk1/2 was highly activated in
the sNF96.2 cells when Bcl-xL was highly expressed, suggesting that
the activation of the Erk1/2 downstream effector in the Ras/MAPK
signaling pathway may be involved in NF1 deficiency-mediated
Bcl-xL upregulation in MPNST-derived SCs. We first examined whether
the inhibition of Erk1/2 could influence the Bcl-xL expression
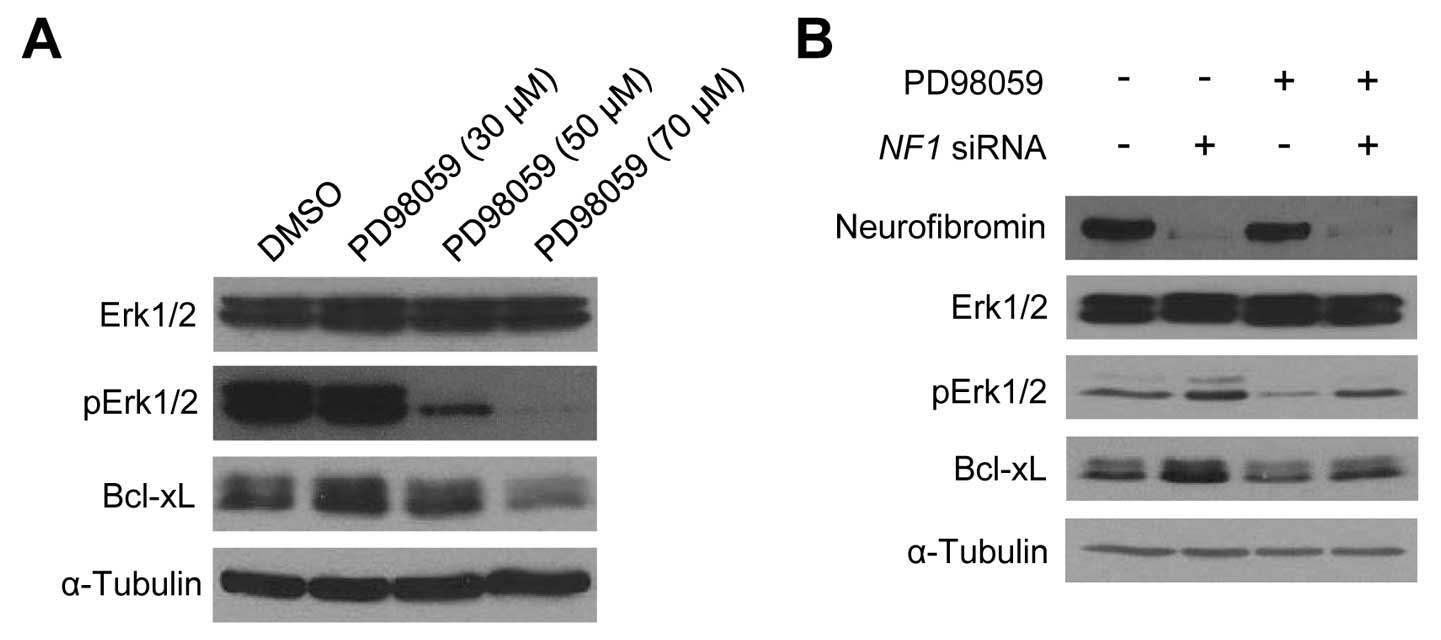
level in sNF96.2 cells. When the sNF96.2 cells were treated with
the Erk1/2 inhibitor, PD98059, for 24 h, the Bcl-xL protein level
decreased in a dose-dependent manner (Fig. 6A). Subsequently, the
neurofibromin-depleted HSCs following transfection with NF1
siRNAs exhibited a significant increase in pErk1/2 and Bcl-xL
levels in the absence of PD98059; however, the
neurofibromin-depleted HSCs showed a slight increase in pErk1/2 and
Bcl-xL levels in the presence of PD98059 (Fig. 6B). These results suggest that the
Erk1/2 activation level may play a crucial role in the NF1
dose-dependent Bcl-xL expression changes in both SCs.
The Bcl-xL inhibitor, ABT-737,
synergistically enhances sensitivity to doxorubicin-induced
apoptosis in NF1-deficient MPNST-derived SCs
Chemotherapy for NF1-associated MPNSTs has not been
extensively investigated. ABT-737, a mimetic of the BH3-only
protein Bad and which binds selectively to Bcl-2, Bcl-xL and Bcl-w
(41), induces synergistic
cytotoxicity in MPNST cells when combined with doxorubicin
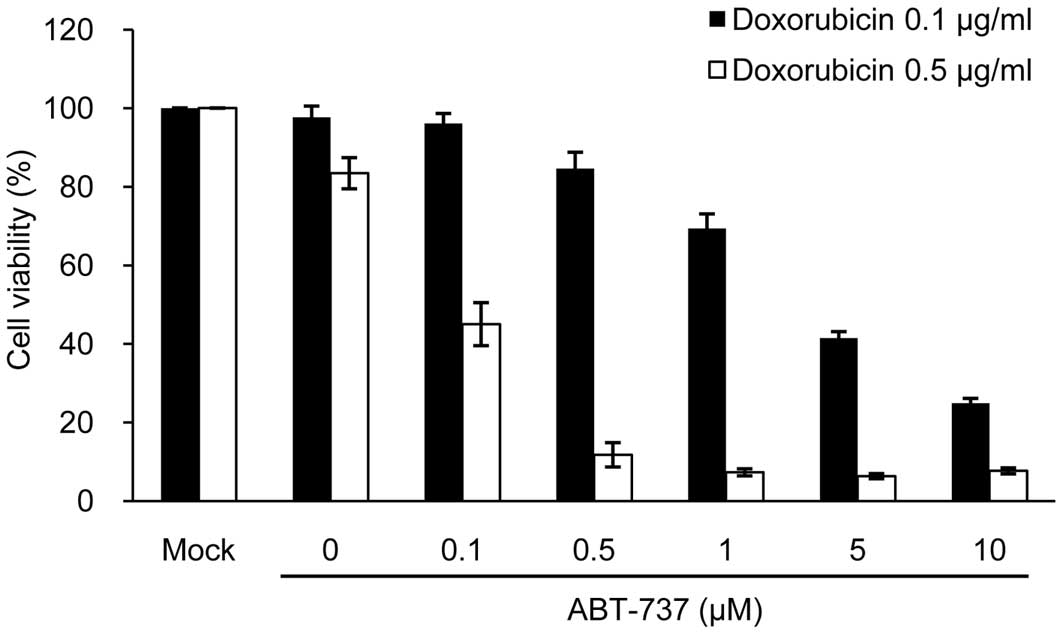
(33). We then investigated
whether ABT-737 would exert a synergistic cytotoxic effect in
sNF96.2 cells when used in combination with doxorubicin. As a
result, ABT-737 effectively enhanced apoptotic cell death in a
dose-dependent manner, compared to the cells treated with
doxorubicin alone at both concentrations of doxorubicin (0.1 and
0.5 μg/ml) (Fig. 7). The
concentrations of ABT-737 and doxorubicin required for 50%
cytotoxicity in the sNF96.2 and MPNST-derived SCs were calculated
to be 0.1 μM ABT-737 plus 0.5 μg/ml doxorubicin.
Discussion
Mouse model studies have reported that the
double-hit inactivation of the NF1 gene
(NF1−/−) in SCs leads benign DNs and/or PNs to
form tumors (42). Other genetic
alterations in SCs may be required for tumors to progress from PNs
to MPNSTs. As loss-of-function mutations in tumor suppressor genes,
such as TP53, RB1 and CDKN2A are particularly
common in MPNSTs (31), the
accumulation of additional loss-of-function mutations in these
tumor suppressor genes in NF1−/− SCs may be
required for MPNST pathogenesis. In addition, the dysregulation of
many genes in MPNSTs has been reported (7,31).
In particular, a large-scale comparison between human MPNST-derived
SCs and normal SCs revealed a relative downregulation of the SC
differentiation markers, SOX10, CNP and PMP22, and nerve growth
factor receptor, as well as the relative upregulation of the neural
crest stem cell markers, SOX9 and TWIST1, in MPNST-derived SCs
(27). Post-transcriptional
modification by microRNAs has also been studied in NF1 and the
results showed the upregulation of miR-10b (43) and the downregulation of miR34a
whose expression is mediated by p53 (44) in MPNST-derived SCs or tissues.
We recently reported the hyperexpression of the
anti-apoptotic protein, Bcl-xL, in primary MPNST cells and a MPNST
cell line (33). Hence, in this
study, we aimed to confirm this result in patient samples as a
first step and demonstrated the upregulation of Bcl-xL in MPNSTs
from patients with NF1 by Bcl-xL immunohistological staining
(Fig. 1). As most Bcl-xL
expressing cells were S100-positive, we investigated Bcl-xL
upregulation in the SCs. As expected, we found a higher Bcl-xL
expression in the sNF96.2 MPNST-derived SCs than in normal HSCs
(Fig. 2). The anti-apoptotic Bcl-2
family member proteinsm Bcl-2, Bcl-xL, Bcl-w, Mcl-1, Bfl1/A-1 and
Bcl-B, bind to and inactivate BH3-domain pro-apoptotic proteins
(45). High expression levels of
these proteins have been found in various types of cancer and have
been related to the development of chemoresistance in malignant
tumor cells (45–47). In our study, we found that
increased Bcl-xL expression, but not that of Bcl-2 or Mcl-1, caused
an increase in resistance to doxorubicin in MPNST-derived SCs
(Figs. 2 and 3). Manipulating Bcl-xL expression levels
demonstrated that the reduced apoptosis sensitivity of
MPNST-derived SCs was caused by Bcl-xL overexpression (Fig. 3).
Although NF1 LOH has been identified in
benign neurofibromas (21–24), a much higher frequency of
NF1 LOH (>4-fold) has been observed in MPNSTs compared to
neurofibromas in patients with NF1 (25). The interaction between
NF1−/− SCs and other types of
NF1+/− cells, including fibroblasts, mast cells
and perineurial cells and the elevated expression of stem cell
factors in NF1−/− SCs in the tumor
microenvironment has been implicated in the tumor progression of
PNs to MPNSTs (12,48). These results strongly indicate that
the bi-allelic inactivation of the NF1 gene in SCs plays a
crucial role in MPNST pathogenesis and NF1 tumorigenesis; however,
little is known about the molecular mechanisms involved. Notably,
NF1 deficiency promotes carcinogenesis by inducing heat
shock factor 1 (HSF1), which is mediated by aberrant Ras/MAPK
signaling (49). HSF1
overexpression and activation has been observed in
NF1-deficient MPNST cells and tumor resections from patients
with NF1 (49). Intriguingly, as
shown in a previous study, NF1 deficiency contributes to the
epithelialmesenchymal transition (EMT) in NF1 (37). The expression levels of the
EMT-related transcription factors, Snail, Twist and ZEB1, were
significantly upregulated in the sNF96.2 MPNST-derived SCs compared
with the normal HSCs. EMT is involved in cancer metastasis via the
Ras/MAPK signaling pathway (50).
Therefore, we investigated whether NF1 deficiency is
directly involved in Bcl-xL upregulation in MPNST SCs. The results
following manipulation of NF1 expression levels demonstrated
a close correlation between neurofibromin and Bcl-xL levels and
sensitivity to doxorubicin-induced apoptosis in sNF96.2 SCs and
HSCs (Figs. 4 and 5). Taken together, these results indicate
that the high Bcl-xL expression in MPNST-derived SCs is caused by
NF1 deficiency.
Neurofibromin depletion by NF1 siRNAs in HSCs
resulted in Erk1/2 activation (Fig.
4A) and an increase in BCL2L1 mRNA levels (Fig. 4B). Treatment with the Erk1/2
inhibitor, PD98059, resulted in a slight increase in Bcl-xL levels
in the neurofibromin-depleted HSCs (Fig. 6B). These results demonstrate that
neurofibromin-mediated Bcl-xL expression is controlled at the
transcriptional level via the Ras/MAPK signaling pathway.
BCL2L1 gene transcription is regulated by a number of
transcription factors, including Ets-1, Ets-2, Rel/nuclear
factor-κB, signal transducers and activators of transcription,
activator protein 1 and Sp1 (51–53).
Of note, these proteins are all downstream of the Ras-signaling
pathway (54). NF1
deficiency-mediated Ras activation has been identified in a
subpopulation of SCs (NF1−/−) but not fibroblasts
(NF1−/−) in mice with neurofibromas (55). Our results suggest that a
neurofibromin deficiency in SCs caused by the bi-allelic
inactivation at the NF1 locus enhanced Ras signaling, which
consequently led to the expression of BCL2L1 transcription
factors.
Bcl-xL overexpression contributes to the inhibtion
of the effects of many chemotherapeutic drugs (46,47).
Reducing Bcl-xL expression restored apoptosis sensitivity to
doxorubicin in sNF96.2 cells (Fig.
3), leading to a reasonable therapeutic strategy for patients
with NF1 and MPNSTs through increased chemosensitization of
malignant SCs by modulating the Bcl-xL expression level. ABT-737
selectively inhibits Bcl-2, Bcl-xL and Bcl-w (41). ABT-737 has been demonstrated to
enhance synergistic chemosensitivity when used in combination with
doxorubicin in other MPNST cells (33), chondrosarcoma cells (56), and hepatoblastoma cells (57). We thus examined apoptosis
sensitivity of sNF96.2 MPNST-derived SCs by the combined treatment
of ABT-737 and doxorubicin. As a result, ABT-737 synergistically
enhanced sensitivity to doxorubicin-induced apoptosis in sNF96.2
cells (Fig. 7). A very low dose of
ABT-737 enhanced the cytotoxic effect of doxorubicin, and the
concentrations required for approximately 50% cytotoxicity in
sNF96.2 cells were 0.5 μg/ml doxorubicin and 0.1 μM
ABT-737. Notably, the concentrations of ABT-737 and doxorubicin
required for effective apoptotic cell death were much lower in
sNF96.2 cells than those in sNF02.2 cells, another MPNST SC line
(33). sNF96.2 SCs are a
NF1 LOH strain (NF1−/−), whereas sNF02.2
SCs harbor one intact NF1 allele (NF1+/−),
suggesting that combining ABT-737 and doxorubicin may increase the
additive effects of the combined treatment in
NF1−/− SCs. Considering that
NF1−/− SCs play a major role in MPNST
pathogenesis in NF1 (31,48) and that a high frequency of
NF1−/− SCs is detected in NF1-associated MPNST
tissues (25), ABT-737 and
doxorubicin seems to be a good combination to effectively treat
NF1-associated MPNSTs with minimal side-effects. Although surgical
resection is the primary treatment for MPNSTs, its limitation due
to tumor location and tumor multiplicity has led to the development
of a drug treatment approach. The proteins involved in the EGFR/Ras
signaling and mTOR pathways have been the main chemotherapeutic
targets for MPNSTs (6,58). In our study, increased cell
survival caused by the prevention of apoptosis was closely related
to the chemoresistance in NF1-associated MPNSTs, suggesting that
Bcl-xL may be good candidate for MPNST-targeted drug treatment.
In conclusion, we found the overexpression of the
anti-apoptotic protein, Bcl-xL, in MPNST tissues from patients with
NF1 and in SCs derived from patients with NF1-associated MPNSTs.
Our results demonstrate that the upregulation of Bcl-xL in
MPNST-derived SCs was caused by NF1 deficiency-mediated
elevation in Ras/MAPK signaling. Our findings may provide an
opportunity for the development of novel chemotherapeutic
strategies for patients with NF1 and MPNSTs.
Acknowledgements
This study was supported by the Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Ministry of Education, Science and
Technology (2009-0093189).
References
|
1.
|
Boyd KP, Korf BR and Theos A:
Neurofibromatosis type 1. J Am Acad Dermatol. 61:1–14. 2009.
View Article : Google Scholar
|
|
2.
|
Ferner RE: Neurofibromatosis 1. Eur J Hum
Genet. 15:131–138. 2007. View Article : Google Scholar
|
|
3.
|
Cawthon RM, Weiss R, Xu GF, et al: A major
segment of the neurofibromatosis type 1 gene: cDNA sequence,
genomic structure, and point mutations. Cell. 62:193–201. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Jett K and Friedman JM: Clinical and
genetic aspects of neurofibromatosis 1. Genet Med. 12:1–11. 2010.
View Article : Google Scholar
|
|
5.
|
Grobmyer SR, Reith JD, Shahlaee A, Bush CH
and Hochwald SN: Malignant peripheral nerve sheath tumor: Molecular
pathogenesis and current management considerations. J Surg Oncol.
97:340–349. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Gottfried ON, Viskochil DH, Fults DW and
Couldwell WT: Molecular, genetic, and cellular pathogenesis of
neurofibromas and surgical implications. Neurosurgery. 58:1–16.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Carroll SL and Stonecypher MS: Tumor
suppressor mutations and growth factor signaling in the
pathogenesis of NF1-associated peripheral nerve sheath tumors: II.
The role of dysregulated growth factor signaling. J Neuropathol Exp
Neurol. 64:1–9. 2005.PubMed/NCBI
|
|
8.
|
Kluwe L, Friedrich R and Mautner VF: Loss
of NF1 allele in Schwann cells but not in fibroblasts derived from
an NF1-associated neurofibroma. Genes Chromosomes Cancer.
24:283–285. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Rutkowski JL, Wu K, Gutmann DH, Boyer PJ
and Legius E: Genetic and cellular defects contributing to benign
tumor formation in neurofibromatosis type 1. Hum Mol Genet.
9:1059–1066. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Serra E, Rosenbaum T, Winner U, et al:
Schwann cells harbor the somatic NF1 mutation in neurofibromas:
evidence of two different Schwann cell subpopulations. Hum Mol
Genet. 9:3055–3064. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zhu Y, Ghosh P, Charnay P, Burns DK and
Parada LF: Neurofibromas in NF1: Schwann cell origin and role of
tumor environment. Science. 296:920–922. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Jouhilahti EM, Peltonen S, Heape AM and
Peltonen J: The pathoetiology of neurofibromatosis 1. Am J Pathol.
178:1932–1939. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Staser K, Yang FC and Clapp DW: Mast cells
and the neurofibroma microenvironment. Blood. 116:157–164. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Katz D, Lazar A and Lev D: Malignant
peripheral nerve sheath tumour (MPNST): the clinical implications
of cellular signalling pathways. Expert Rev Mol Med. 11:e302009.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Brems H, Beert E, de Ravel T and Legius E:
Mechanisms in the pathogenesis of malignant tumours in
neurofibromatosis type 1. Lancet Oncol. 10:508–515. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Spurlock G, Knight SJ, Thomas N, Kiehl TR,
Guha A and Upadhyaya M: Molecular evolution of a neurofibroma to
malignant peripheral nerve sheath tumor (MPNST) in an NF1 patient:
correlation between histopathological, clinical and molecular
findings. J Cancer Res Clin Oncol. 136:1869–1880. 2010. View Article : Google Scholar
|
|
17.
|
Evans DG, Baser ME, McGaughran J, Sharif
S, Howard E and Moran A: Malignant peripheral nerve sheath tumours
in neurofibromatosis 1. J Med Genet. 39:311–314. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
McCaughan JA, Holloway SM, Davidson R and
Lam WW: Further evidence of the increased risk for malignant
peripheral nerve sheath tumour from a Scottish cohort of patients
with neurofibromatosis type 1. J Med Genet. 44:463–466. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Tucker T, Wolkenstein P, Revuz J, Zeller J
and Friedman JM: Association between benign and malignant
peripheral nerve sheath tumors in NF1. Neurology. 65:205–211. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
McQueen M, MacCollin M, Gusella J and
Plotkin SR: Patient and physician attitudes regarding clinical
trials in neurofibromatosis 1. J Neurosci Nurs. 40:341–345. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Sawada S, Florell S, Purandare SM, Ota M,
Stephens K and Viskochil D: Identification of NF1 mutations in both
alleles of a dermal neurofibroma. Nat Genet. 14:110–112. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Wiest V, Eisenbarth I, Schmegner C, Krone
W and Assum G: Somatic NF1 mutation spectra in a family with
neurofibromatosis type 1: toward a theory of genetic modifiers. Hum
Mutat. 22:423–427. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Kluwe L, Friedrich RE and Mautner VF:
Allelic loss of the NF1 gene in NF1-associated plexiform
neurofibromas. Cancer Genet Cytogenet. 113:65–69. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Rasmussen SA, Overman J, Thomson SA, et
al: Chromosome 17 loss-of-heterozygosity studies in benign and
malignant tumors in neurofibromatosis type 1. Genes Chromosomes
Cancer. 28:425–431. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Upadhyaya M, Kluwe L, Spurlock G, et al:
Germline and somatic NF1 gene mutation spectrum in NF1-associated
malignant peripheral nerve sheath tumors (MPNSTs). Hum Mutat.
29:74–82. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Watanabe T, Oda Y, Tamiya S, Masuda K and
Tsuneyoshi M: Malignant peripheral nerve sheath tumour arising
within neurofibroma. An immunohistochemical analysis in the
comparison between benign and malignant components. J Clin Pathol.
54:631–636. 2001. View Article : Google Scholar
|
|
27.
|
Miller SJ, Rangwala F, Williams J, et al:
Large-scale molecular comparison of human schwann cells to
malignant peripheral nerve sheath tumor cell lines and tissues.
Cancer Res. 66:2584–2591. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Mantripragada KK, Spurlock G, Kluwe L, et
al: High-resolution DNA copy number profiling of malignant
peripheral nerve sheath tumors using targeted microarray-based
comparative genomic hybridization. Clin Cancer Res. 14:1015–1024.
2008. View Article : Google Scholar
|
|
29.
|
Upadhyaya M, Spurlock G, Thomas L, et al:
Microarray-based copy number analysis of neurofibromatosis type-1
(NF1)-associated malignant peripheral nerve sheath tumors reveals a
role for Rho-GTPase pathway genes in NF1 tumorigenesis. Hum Mutat.
33:763–776. 2012. View Article : Google Scholar
|
|
30.
|
Upadhyaya M: Genetic basis of
tumorigenesis in NF1 malignant peripheral nerve sheath tumors.
Front Biosci. 16:937–951. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Carroll SL: Molecular mechanisms promoting
the pathogenesis of Schwann cell neoplasms. Acta Neuropathol.
123:321–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Woodruff JM: Pathology of tumors of the
peripheral nerve sheath in type 1 neurofibromatosis. Am J Med
Genet. 89:23–30. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Lee SJ, Park HJ, Kim YH, et al: Inhibition
of Bcl-xL by ABT-737 enhances chemotherapy sensitivity in
neurofibromatosis type 1-associated malignant peripheral nerve
sheath tumor cells. Int J Mol Med. 30:443–450. 2012.PubMed/NCBI
|
|
34.
|
Jeong SY, Gaume B, Lee YJ, et al: Bcl-x(L)
sequesters its C-terminal membrane anchor in soluble, cytosolic
homodimers. EMBO J. 23:2146–2155. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Ferner RE, Huson SM, Thomas N, et al:
Guidelines for the diagnosis and management of individuals with
neurofibromatosis 1. J Med Genet. 44:81–88. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Weiss SW, Langloss JM and Enzinger FM:
Value of S-100 protein in the diagnosis of soft tissue tumors with
particular reference to benign and malignant Schwann cell tumors.
Lab Invest. 49:299–308. 1983.PubMed/NCBI
|
|
37.
|
Arima Y, Hayashi H, Kamata K, et al:
Decreased expression of neurofibromin contributes to
epithelial-mesenchymal transition in neurofibromatosis type 1. Exp
Dermatol. 19:e136–e141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Moretti VM, Crawford EA, Staddon AP,
Lackman RD and Ogilvie CM: Early outcomes for malignant peripheral
nerve sheath tumor treated with chemotherapy. Am J Clin Oncol.
34:417–421. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Hiatt KK, Ingram DA, Zhang Y, Bollag G and
Clapp DW: Neurofibromin GTPase-activating protein-related domains
restore normal growth in Nf1−/− cells. J Biol Chem.
276:7240–7245. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Ni Chonghaile T and Letai A: Mimicking the
BH3 domain to kill cancer cells. Oncogene. 27(Suppl 1): S149–S157.
2008.PubMed/NCBI
|
|
42.
|
Brossier NM and Carroll SL: Genetically
engineered mouse models shed new light on the pathogenesis of
neurofibromatosis type I-related neoplasms of the peripheral
nervous system. Brain Res Bull. 88:58–71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Chai G, Liu N, Ma J, et al: MicroRNA-10b
regulates tumorigenesis in neurofibromatosis type 1. Cancer Sci.
101:1997–2004. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Subramanian S, Thayanithy V, West RB, et
al: Genome-wide transcriptome analyses reveal p53 inactivation
mediated loss of miR-34a expression in malignant peripheral nerve
sheath tumours. J Pathol. 220:58–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Kang MH and Reynolds CP: Bcl-2 inhibitors:
targeting mitochondrial apoptotic pathways in cancer therapy. Clin
Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Kostanova-Poliakova D and Sabova L:
Anti-apoptotic proteins-targets for chemosensitization of tumor
cells and cancer treatment. Neoplasma. 52:441–449. 2005.PubMed/NCBI
|
|
47.
|
Karnak D and Xu L: Chemosensitization of
prostate cancer by modulating Bcl-2 family proteins. Curr Drug
Targets. 11:699–707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Le LQ and Parada LF: Tumor
microenvironment and neurofibromatosis type I: connecting the GAPs.
Oncogene. 26:4609–4616. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Dai C, Santagata S, Tang Z, et al: Loss of
tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J
Clin Invest. 122:3742–3754. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Edme N, Downward J, Thiery JP and Boyer B:
Ras induces NBT-II epithelial cell scattering through the
coordinate activities of Rac and MAPK pathways. J Cell Sci.
115:2591–2601. 2002.PubMed/NCBI
|
|
51.
|
Sevilla L, Zaldumbide A, Pognonec P and
Boulukos KE: Transcriptional regulation of the bcl-x gene encoding
the anti-apoptotic Bcl-xL protein by Ets, Rel/NFkappaB, STAT and
AP1 transcription factor families. Histol Histopathol. 16:595–601.
2001.PubMed/NCBI
|
|
52.
|
Lee J, Kannagi M, Ferrante RJ, Kowall NW
and Ryu H: Activation of Ets-2 by oxidative stress induces Bcl-xL
expression and accounts for glial survival in amyotrophic lateral
sclerosis. FASEB J. 23:1739–1749. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Grad JM, Zeng XR and Boise LH: Regulation
of Bcl-xL: a little bit of this and a little bit of STAT. Curr Opin
Oncol. 12:543–549. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
McCubrey JA, Steelman LS, Abrams SL, et
al: Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in
malignant transformation and drug resistance. Adv Enzyme Regul.
46:249–279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Sherman LS, Atit R, Rosenbaum T, Cox AD
and Ratner N: Single cell Ras-GTP analysis reveals altered Ras
activity in a subpopulation of neurofibroma Schwann cells but not
fibroblasts. J Biol Chem. 275:30740–30745. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
van Oosterwijk JG, Herpers B, Meijer D, et
al: Restoration of chemosensitivity for doxorubicin and cisplatin
in chondrosarcoma in vitro: BCL-2 family members cause
chemoresistance. Ann Oncol. 23:1617–1626. 2012.PubMed/NCBI
|
|
57.
|
Lieber J, Kirchner B, Eicher C, et al:
Inhibition of Bcl-2 and Bcl-X enhances chemotherapy sensitivity in
hepatoblastoma cells. Pediatr Blood Cancer. 55:1089–1095. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Gottfried ON, Viskochil DH and Couldwell
WT: Neurofibromatosis type 1 and tumorigenesis: molecular
mechanisms and therapeutic implications. Neurosurg Focus.
28:E82010. View Article : Google Scholar : PubMed/NCBI
|