Introduction
Breast cancer remains one of the leading causes of
cancer-related death worldwide. Although chemotherapy has improved
outcomes for patients, the marginal benefits achieved with
cytotoxic agents seem to have reached a plateau (1). Recently, preventive agents and
targeted therapies directed at the estrogen receptor, progesterone
receptor, and human epidermal growth factor 2 receptor have
resulted in improved clinical outcomes for many women with breast
cancer (2). However, further
challenges remain in treating tumors that do not express these
molecular targets or tumor cells that become resistant for these
molecular targets. Hence, the development of new therapeutic agents
for these clinically intractable tumors is highly desirable.
The mammalian target of rapamycin (mTOR), a
serine/threonine kinase, integrates multiple signaling pathways,
including cell growth and survival (3). Activation of phosphatidylinositol
3-kinase (PI3K)/Akt/mTOR signaling contributes to the pathogenesis
of many tumor types including breast cancer (4–6). Two
distinct mTOR complexes, mTORC1 and mTORC2, have been identified
and have differential sensitivity to rapamycin (Rapa). mTORC1 is
downstream of Akt, sensitive to Rapa inhibition, and controls
cap-dependent protein translation (6). One of the best-studied mTOR
substrates is p70 ribosomal S6 kinase (p70S6K). In contrast, mTORC2
is directly upstream of Akt and is resistant to Rapa. Akt can be
activated by phosphorylation at two different sites, S473 by mTORC2
and T308 by phosphoinositide-dependent kinase 1 (PDK1).
Constitutive activation of the PI3K/Akt/mTOR signaling axis leads
to uncontrolled tumor cell proliferation and survival (4,7).
Rapa is an allosteric inhibitor of mTOR. Rapa analogues (rapalogs)
have been approved for the treatment of neuroendocrine tumors,
renal cell carcinoma, and subependymal giant cell astrocytoma
associated with tuberous sclerosis, and have very promising
clinical benefits in other tumor types such as breast and
endometrial cancer. However, single agent rapalogs have only
achieved modest antitumor activity in a clinical setting (8). The limited anticancer efficacy of
rapalogs can be explained by two possible mechanisms. First,
rapalogs have been shown to induce Akt activation. Insulin-like
growth factor-I (IGF-I) and insulin-dependent induction of the
PI3K/Akt pathway leads to feedback inhibition of signaling due to
mTOR/S6K-mediated phosphorylation and degradation of IRS-1.
Rapa-induced Akt activation has been primarily attributed to the
loss of this negative feedback loop. This feedback loop activation
of Akt was not only in vitro but was also observed in a
phase I clinical trial of the Rapa analog everolimus (7,8).
Second, rapalogs incompletely block mTORC1 downstream signaling
(9,10).
We have examined whether inducers of differentiation
in leukemia cells can control the growth of solid tumors. Cotylenin
A (CN-A), which is a fucicoccan-diterpene glycoside with a complex
sugar moiety, was originally isolated as a plant growth regulator
and has been shown to affect several physiological processes in
higher plants (11). We previously
reported that CN-A has a potent differentiation-inducing activity
in several human and murine myeloid leukemia cell lines and in
leukemia cells that were freshly isolated from patients with acute
myeloid leukemia (12–15). We previously found that treatment
with CN-A plus Rapa, which also has a potent
differentiation-inducing activity in myeloid leukemia cells
(16), effectively inhibited the
proliferation of human breast cancer cell line MCF-7 cells in
vitro and in vivo(17).
In the present study we found that CN-A inhibited Rapa-induced
phosphorylation of Akt (Ser473). Our results suggest that the
inhibition of Rapa-induced Akt phosphorylation by CN-A correlates
with their effective growth inhibition of cancer cells.
Materials and methods
Cell culture
Human breast cancer cell line MCF-7 cells,
adriamycin-resistant MCF-7/Adr cells, and human lung cancer A549
cells were cultured in RPMI-1640 supplemented with 10% fetal bovine
serum at 37°C in a humidified atmosphere of 5% carbon dioxide in
air.
Materials
Rapamycin, arsenic trioxide and LY294002 were
purchased from Sigma-Aldrich (St. Louis, MO, USA).
17-Allylamino-17-demethoxygeldanamycin (17-AAG) was obtained from
LC Laboratories (Woburn, MA, USA). CN-A, ISIR-005 and fusicoccin J
(FC-J) were prepared as described previously (11,18).
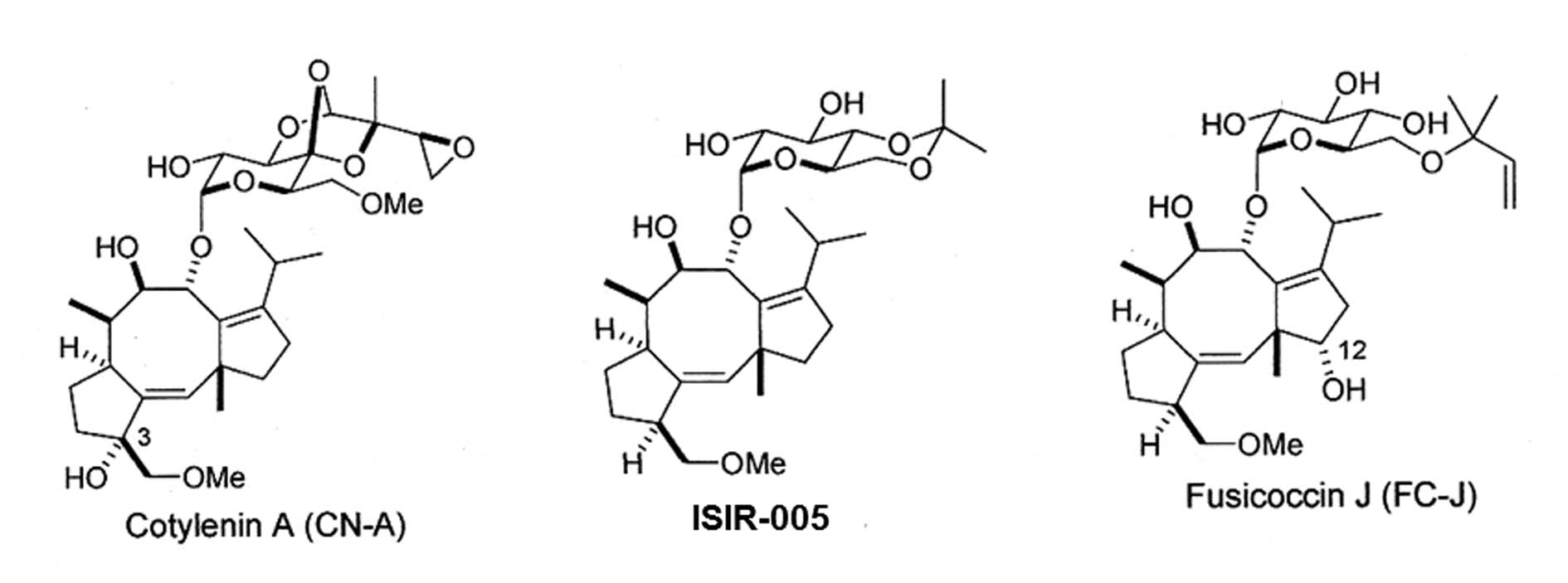
The structures of CN-A, ISIR-005 and FC-J were shown in Fig. 1. Anti-phospho-Akt (Ser473),
anti-Akt and anti-phospho-p70 S6 kinase (Thr389) antibodies were
purchased from Cell Signaling Technology (Danvers, MA, USA).
Anti-α-tubulin antibody, Akt1/2 siRNA and control siRNA were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Assay of cell growth
Cells were seeded at 1–3×104 cells/ml in
a 24-well multidish. After culture with or without test compounds
for the indicated times, viable cells were examined by a modified
MTT (3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide)
assay (17).
Western blot analysis
Cells were packed after washing with cold PBS and
then lysed at a concentration of 1×107 cells/ml in lysis
buffer CelLytic™-M (Sigma-Aldrich Inc.) supplemented with a
proteinase inhibitor cocktail and phosphatase inhibitor cocktail
1/2 (Sigma-Aldrich Inc.). Equal amounts of protein were separated
on 10–20% SDS-polyacrylamide gels (Wako, Osaka, Japan). Proteins
were electrophoresed on gels and transferred to an Immobilon-P
membrane (Millipore, Bedford, MA, USA) using the following
antibodies: rabbit anti-phospho-Akt (Ser473) antibody (1:1,000),
rabbit anti-Akt antibody (1:1,000), rabbit anti-phospho-p70 S6
kinase (Thr389) antibody (1:1,000) or mouse anti-α-tubulin antibody
(1:5,000). An anti-rabbit or anti-mouse IgG HRP-linked antibody
(Cell Signaling Technology,) was used as a secondary antibody
(1:2,000 dilution). Bands were identified by treatment with
Immune-Star™ HRP chemiluminescence (Bio-Rad Laboratories, Hercules,
CA, USA) for 5 min at room temperature and detected using a
FujiLumino Image Analyser LAS-4000 system (Fuji Film Co. Ltd,
Tokyo, Japan) (19). All western
blots shown are representative of at least 3 independent
experiments.
siRNA transfection
A total of 0.75×104 cells were seeded
into the 24-well multidish and transfected with siRNA (20 nM) by
siLentFect™ Lipid Reagent (Bio-Rad Laboratories). The next day,
Rapa was added to cells and cultured for a further 4 days. Viable
cells were then examined by the MTT assay. To determine the
knockdown efficiency, western blot analysis was done using cell
lysates from siAkt- or control siRNA-transfected cells.
Results
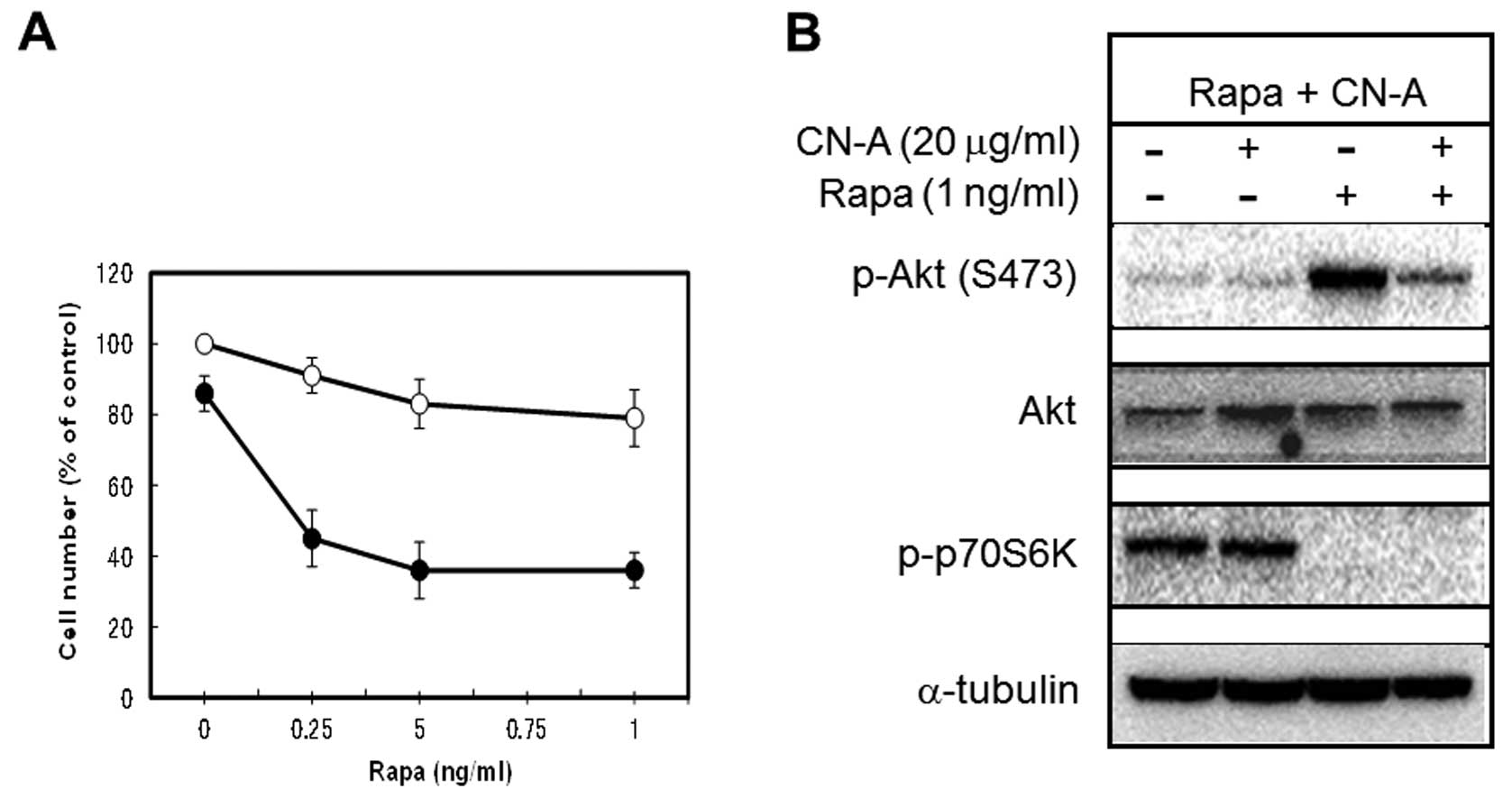
The suppression of Rapa-induced
phosphorylation of Akt (Ser473) by CN-A correlates with their
synergistic growth inhibition of MCF-7 cells
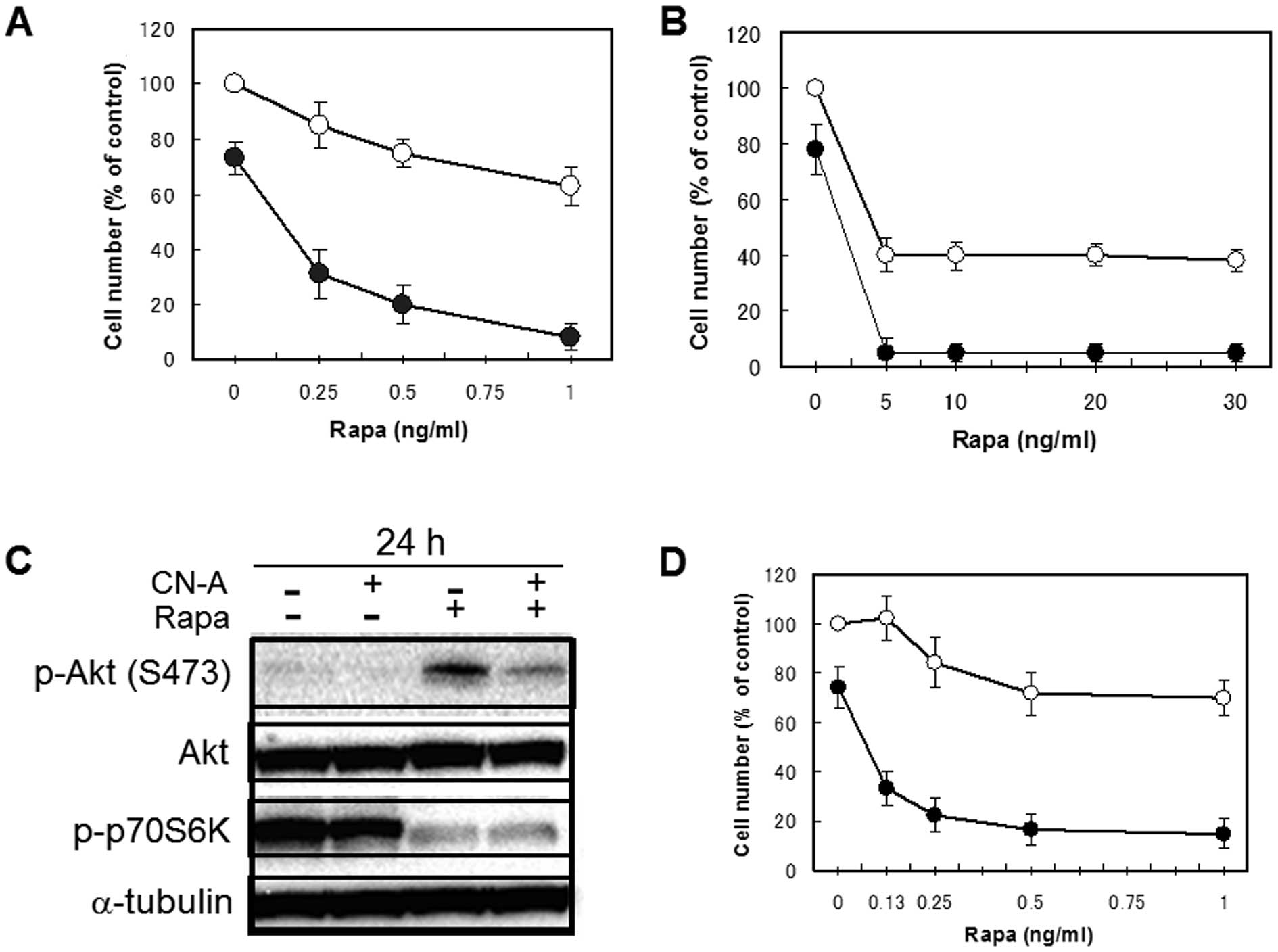
As described in our previous reports (17,19),
CN-A and Rapa synergistically inhibited the proliferation of human
breast cancer MCF-7 cells (Fig. 2A and
B). Although lower doses (0.25–5 ng/ml) of Rapa alone inhibited
the growth of MCF-7 cells dose-dependently to around 40% of
controls, additional higher doses (10–30 ng/ml) of Rapa did not
decrease cell numbers further (Fig.
2B). As previously reported (20,21),
the inhibition of mTOR signaling by Rapa led to an increase in Akt
phosphorylation on Ser473 (Fig.
2C) in MCF-7 cells. Therefore, these blunt effects of Rapa on
the growth of MCF-7 cells may be due to the negative feedback
activation of Akt signaling. We found that CN-A (5 μg/ml)
was able to suppress Rapa-induced phosphorylation of Akt on Ser473
(Fig. 2C). Although CN-A (5
μg/ml) alone slightly inhibited the proliferation of MCF-7
cells, Rapa at 1–30 ng/ml almost completely suppressed the growth
of MCF-7 cells in the presence of CN-A (Fig. 2A and B). Similar results were
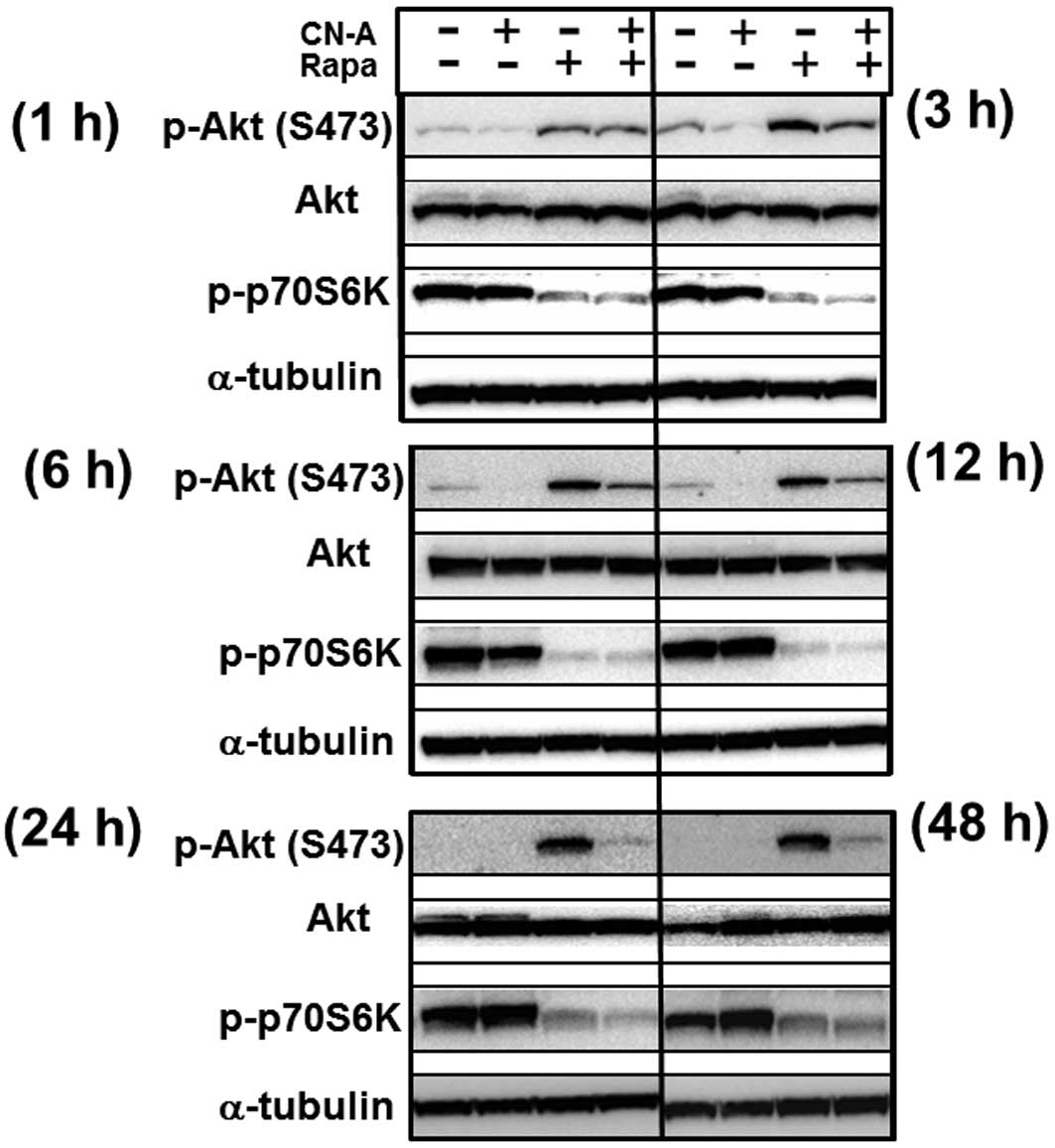
obtained using adriamycin-resistant MCF-7 cells (Fig 2D). Rapa induced phosphorylation of
Akt on Ser473 from 1 h after treatment and maintained higher levels
of phosphorylation of Akt until at least 48 h after treatment,
although Rapa clearly inhibited phosphorylation of p70 S6K on
Thr389 from 1 h after treatment (Fig.
3). CN-A significantly inhibited Rapa-induced phosphorylation
of Akt on Ser473 from 3–48 h after treatment, although CN-A did not
significantly affect the Rapa-induced inhibition of phosphorylation
of p70 S6K on Thr389 (Fig. 3).
Effects of CN-A analogues on the growth
and phosphorylation of Akt (Ser473) in the presence of Rapa
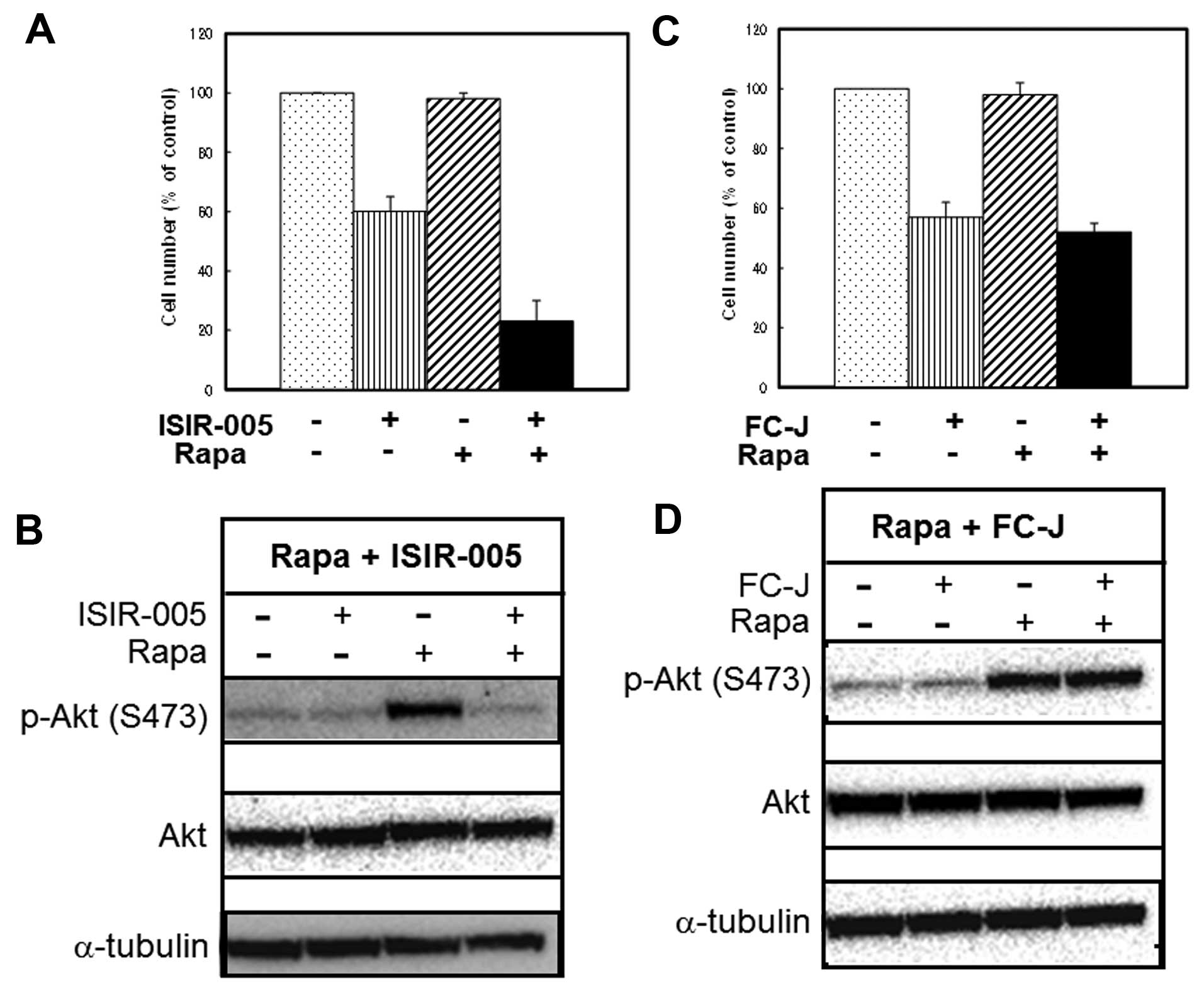
We next examined whether the active CN-A analogue
and Rapa also cooperatively inhibited the growth of MCF-7 cells and
whether the active CN-A analogue could suppress Rapa-induced
phosphorylation of Akt. Although ISIR-005, a synthetic
CN-A-derivative, at 4 μg/ml inhibited the growth of MCF-7
cells to 60% of controls after the 5-day treatment and Rapa at 0.5
ng/ml slightly inhibited the growth of MCF-7 cells, combined
treatment of ISIR-005 plus Rapa inhibited growth to 23% of controls
(Fig. 4A). This combined treatment
of ISIR-005 plus Rapa also clearly inhibited Rapa-induced
phosphorylation of Akt on Ser473 (Fig.
4B). On the contrary, FC-J, a CN-A-related natural product, at
3 μg/ml also inhibited the growth of MCF-7 cells to 57% of
controls after the 5-day treatment (Fig. 4C), but did not enhance Rapa-induced
growth inhibition (Fig. 4C) and
also could not inhibit Rapa-induced phosphorylation of Akt
(Fig. 4D). Furthermore, similar
results were obtained from Rapa treatment plus another inactive
analogue of CN-A (data not shown).
CN-A suppresses Rapa-induced
phosphorylation of Akt and enhances Rapa-induced growth inhibition
of lung cancer A549 cells
We next examined whether CN-A could inhibit
Rapa-induced phosphorylation of Akt and exert synergistic growth
inhibition of cells other than MCF-7 cells when CN-A plus Rapa were
used as the treatment. As we reported previously (17), the sensitivity of human non-small
cell lung carcinoma A549 cells to Rapa was also markedly affected
by CN-A (Fig. 5A). The synergistic
growth inhibitory effects of Rapa and CN-A were also accompanied by
the suppression of Rapa-induced phosphorylation of Akt (Ser473) by
CN-A in A549 cells (Fig. 5B).
These findings indicate that the correlation between the
suppression of Rapa-induced phosphorylation of Akt (Ser473) by CN-A
and their synergistic growth inhibition was not restricted to MCF-7
cells.
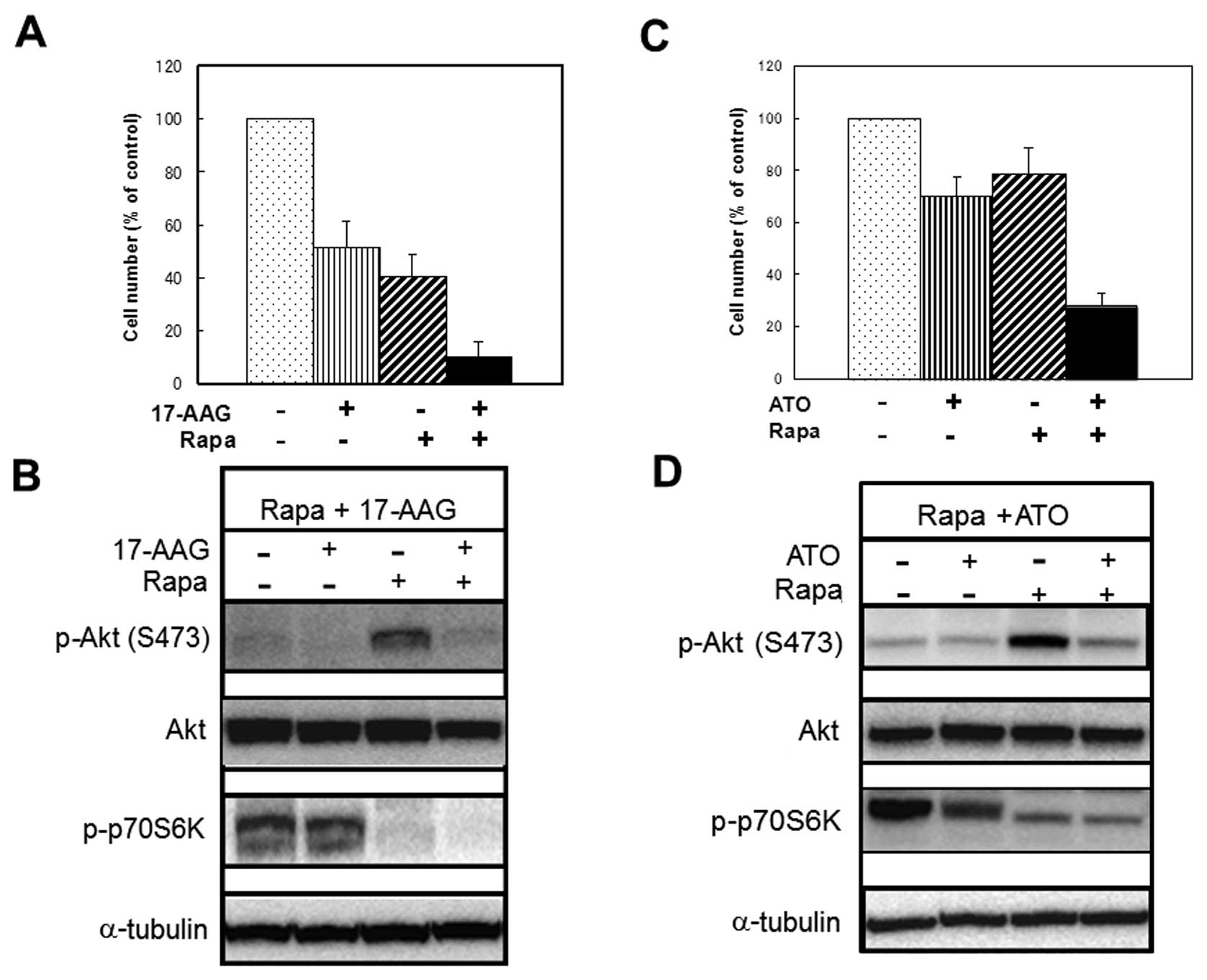
Drugs that inhibit Rapa-induced
phosphorylation of Akt (Ser473) can also enhance Rapa-induced
growth inhibition of MCF-7 cells
We examined whether substances that could inhibit
Rapa-induced phosphorylation of Akt (Ser473) other than CN-A or the
CN-A analogue ISIR-005 could enhance Rapa-induced growth inhibition
of MCF-7 cells. As it was previously reported (22–24)
that the HSP90 inhibitor 17-AAG inhibited phosphorylation of Akt
(Ser473) in several solid tumor cells including MCF-7 cells, we
examined the effects of 17-AAG on Rapa-induced phosphorylation of
Akt (Ser473) and Rapa-induced growth inhibition of MCF-7 cells. As
shown in Fig. 6B, 17-AAG clearly
suppressed Rapa-induced phosphorylation of Akt. Although 17-AAG (30
nM) or Rapa (0.5 ng/ml) alone inhibited cell growth to about 51 or
41% of controls, respectively, combined treatment with 17-AAG and
Rapa inhibited cell growth to 10% of controls (Fig. 6A). Arsenic trioxide (ATO) is also
known to inhibit phosphorylation of Akt in several solid tumor
cells (25–27). Therefore, we examined the effect of
ATO on Rapa-induced phosphorylation of Akt (Ser473) and
Rapa-induced growth inhibition of MCF-7 cells. ATO also suppressed
Rapa-induced phosphorylation of Akt in MCF-7 cells (Fig. 6D). Although ATO (4 μM) or
Rapa (0.2 ng/ml) alone inhibited cell growth to 70 or 79% of
controls, combined treatment with ATO and Rapa inhibited cell
growth to 28% of controls (Fig.
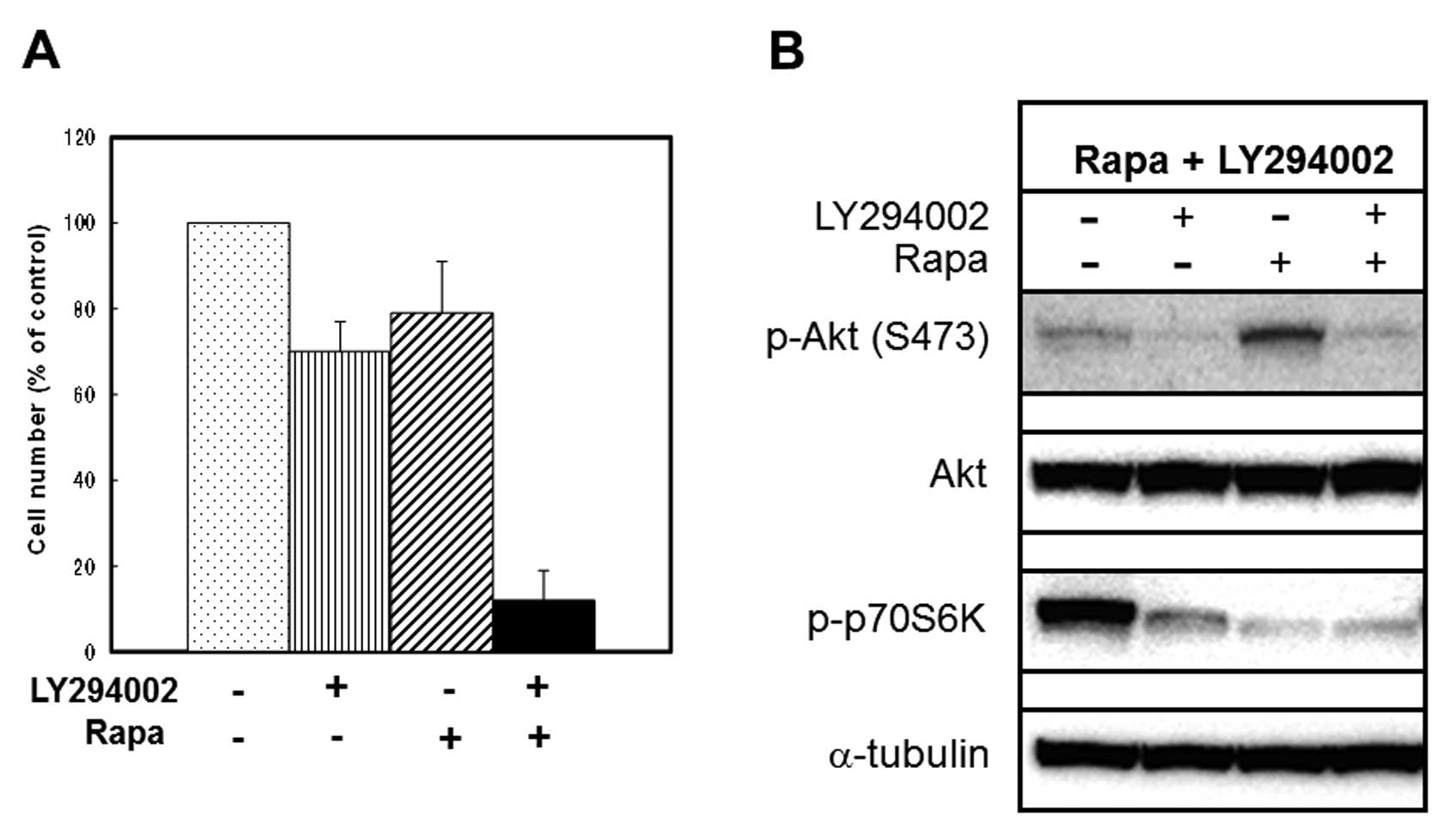
6C). We next examined the effects of a potent PI3K inhibitor
LY294002 on Rapa-induced phosphorylation of Akt (Ser473) and
Rapa-induced growth inhibition of MCF-7 cells. In agreement with
previous reports (28,29), LY294002 (3 μM) alone
significantly inhibited phosphorylation of both Akt (Ser473) and
p70S6K (Fig. 7B). However, this
concentration of LY294002 showed only a 30% inhibition of cell
growth (Fig. 7B). LY294002 (3
μM) also inhibited Rapa-induced phosphorylation of Akt
(Ser473) (Fig. 7B) and could
enhance Rapa-induced growth inhibition (from 21 to 88% inhibition)
(Fig. 7A). These results suggest
that substances that inhibit Rapa-induced phosphorylation of Akt
(Ser473) can enhance Rapa-induced growth inhibition of MCF-7
cells.
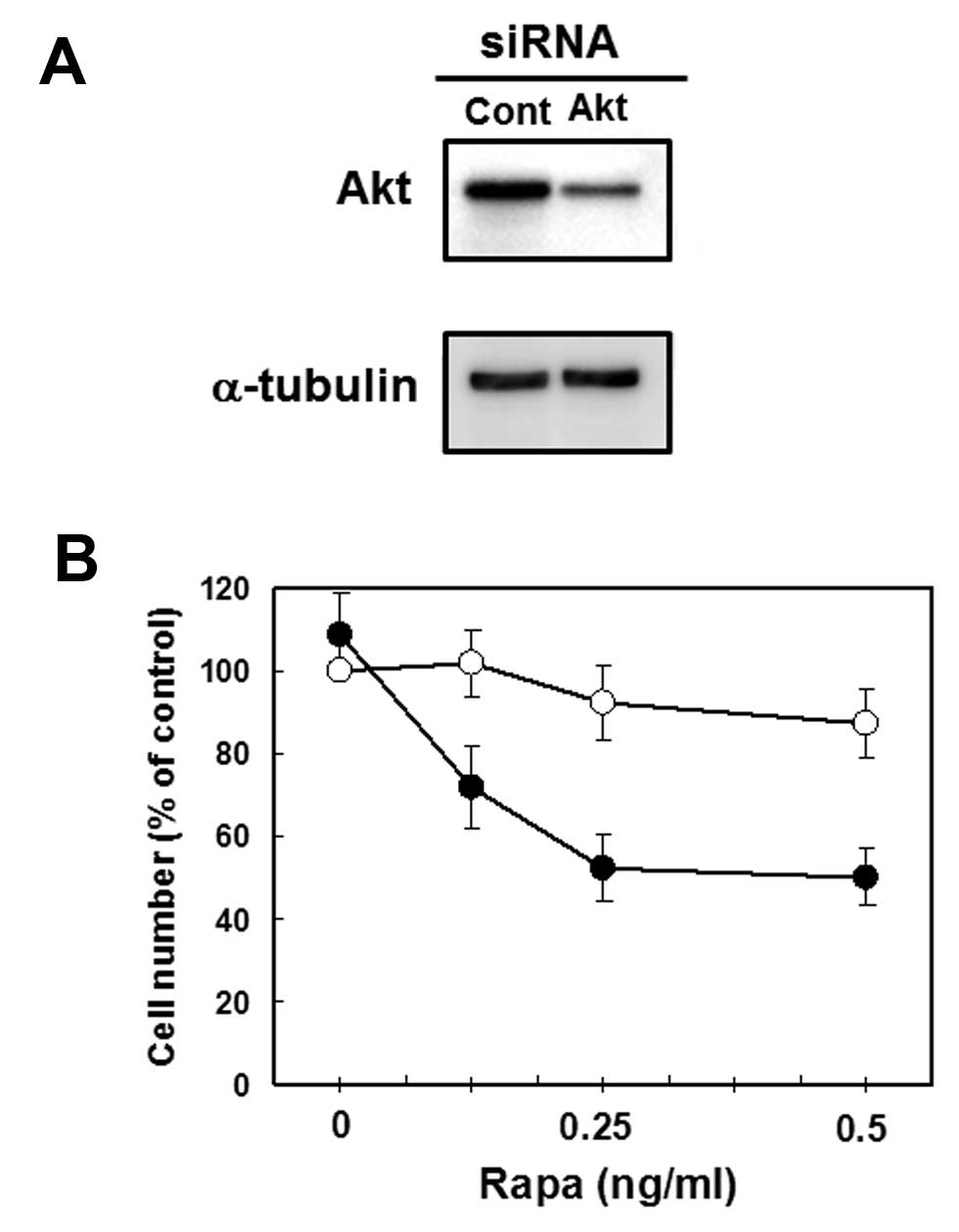
Akt siRNA-transfected MCF-7 cells are
more sensitive to Rapa for growth inhibition
As mentioned above, our results suggest that the
marked effects of combined treatment with CN-A and Rapa on the
growth of cancer cells is due to, at least in part, the inhibition
of Rapa-induced phosphorylation of Akt (Ser473) by CN-A. To
determine the role of Akt protein, we transfected Akt siRNA to
MCF-7 cells and prepared Akt-knockdown MCF-7 cells and then
examined the potency of Rapa to induce growth inhibition in cells.
Akt-specific siRNA (20 nM) reduced Akt protein (60% inhibition)
(Fig. 8A). Although the growth of
control siRNA-transfected MCF-7 cells was slightly inhibited by
Rapa, the growth of Akt siRNA-transfected MCF-7 cells was inhibited
more significantly by Rapa than that of control siRNA-transfected
MCF-7 cells (Fig. 8B). These
results suggest the importance of the down-regulation of Akt
signaling in effective growth inhibition induced by combined
treatment with Rapa plus other substances including CN-A.
Discussion
Activation of the PI3K/Akt/mTOR signaling pathway
contributes to the pathogenesis of many tumor types. The anticancer
drug Rapa is a known mTOR-inhibitor. However, Rapa and rapalogs are
known to induce Akt activation. IGF-I and insulin-dependent
induction of the PI3K/Akt pathway leads to feedback inhibition of
signaling due to mTOR/S6K-mediated phosphorylation and degradation
of IRS-1. Rapa-induced Akt activation has been primarily attributed
to the loss of this negative feedback loop. Akt activation may
limit the antitumor efficacy of Rapa and rapalogs (2–6). We
previously reported that CN-A, which is a potent
differentiation-inducer of leukemia cells, and Rapa, which is also
a differentiation-inducer of leukemia cells, synergistically
inhibited the proliferation of MCF-7 cells in vitro and
cooperatively reduced the growth of MCF-7 cells in
vivo(17,19). This combined treatment with CN-A
and Rapa induced G1 arrest, but not apoptosis, and induced
E-cadherin and senescence-associated β-galactosidase activity in
MCF-7 cells (17). The mechanisms
of these combined effects are largely still unknown, although our
previous COMPARE analysis suggests that CN-A has a unique mode of
action on cancer cells (30). In
this study, we found that CN-A could inhibit Rapa-induced
phosphorylation of Akt in MCF-7 cells. These results suggest that
the inhibition of Rapa-induced Akt phosphorylation by CN-A
correlates with their effective growth inhibition of cancer
cells.
The Hsp90 inhibitor 17-AAG is one of the most
studied inhibitors of Hsp90. 17-AAG destabilizes multiple tyrosine
kinase receptors and other oncoproteins through Hsp90 inhibition,
which results in blocking of proliferation and induction of
apoptosis (22–24). 17-AAG also was reported to impair
Akt stability and activity. For example, 17-AAG downregulated
phosphorylation of Akt (Ser473) in osteosarcoma cell lines (HOS and
KHOS/NP) (22), in several solid
tumor cell lines (RKO colorectal, MCF-7, AGS gastric adenocarcinoma
and U2OS osteosarcoma) (23), and
in ovarian cancer cell lines (SKOV3, OVCAR429 and ES2) (24). We found that 17-AAG could inhibit
Rapa-induced phosphorylation of Akt (Ser473) and could also
significantly enhance Rapa-induced suppression of cell growth in
MCF-7 cells (Fig. 6A and B). ATO
is an effective treatment for patients with acute promyelocytic
leukemia (APL) (31,32). ATO induces differentiation at lower
concentrations and induces apoptosis at higher concentrations in
APL cells. However, ATO has been less successful in other
malignancies at tolerable doses. The doses of ATO required to exert
detectable anticancer effects in solid tumors are much higher than
those required to inhibit hematological malignancies (33). Therefore, new strategies to enhance
the efficacy of ATO, while reducing the dose of ATO in order to
avoid severe side-effects, are essential. Previous reports showed
that ATO decreased not only Akt activity but also total Akt protein
and that the sensitivity to ATO correlated with the degree of Akt
protein reductions in leukemia cells, prostate cancer cells, and
ovarian cancer cells (25–27). In this study, we found that ATO at
lower doses could inhibit Rapa-induced phosphorylation of Akt
(Ser473) in MCF-7 cells, and that ATO and Rapa synergistically
inhibited the proliferation of MCF-7 cells (Fig. 6C and D). These results suggest that
CN-A as well as 17-AAG or ATO is possibly a suitable candidate for
combination therapy with Rapa to inhibit solid tumor cells.
Recently, Loehberg el al(34) reported that anti-malarial
chloroquine could prevent rapalog RAD001-induced Akt activation and
enhance RAD001-induced inhibition of cell proliferation in luminal
MCF-7 cells. However, this combined treatment was not effective in
mesenchymal MDA-MB231 breast cancer cells (34). We previously reported that combined
treatment with Rapa and CN-A was also effective in inhibiting the
growth of MDA-MB231 cells (17).
As with recent reports (28,29),
the potent well-known PI3K inhibitor LY294002 inhibited
Rapa-induced phosphorylation of Akt (Ser473) (Fig. 7B) and was able to enhance the
Rapa-induced growth inhibition of MCF-7 cells in our culture
conditions (Fig. 7A). Moreover,
the growth of Akt siRNA-transfected MCF-7 cells was inhibited more
significantly by Rapa than that of control siRNA-transfected MCF-7
cells by Rapa (Fig. 8B). These
results suggest the importance of downregulating Akt signaling for
the effective growth inhibition induced by combined treatment with
Rapa plus CN-A or Rapa plus other substances.
Cyclin G2 belongs to the ‘G’ family of
unconventional cyclins (35).
Unlike other cyclins that function to promote cell cycle
progression, cyclin G2 inhibits the cell cycle. It is expressed at
modest levels in proliferating cells, peaking during the late
S/early G2-phase, and is significantly upregulated as cells exit
the cell cycle in response to DNA damage and receptor-mediated
negative signaling (36,37). We previously found that CN-A and
Rapa rapidly and markedly induced cyclin G2 gene expression in
several cancer cells including MCF-7 cells (19). Furthermore, we reported that
ectopically inducible cyclin G2 expression potently inhibited the
proliferation of MCF-7 cells and that cyclin G2 knockdown induced
by cyclin G2 small interfering RNA markedly reduced the potency of
CN-A plus Rapa to induce growth inhibition (19). These results suggest that CN-A plus
Rapa induce inhibition of cancer cell growth through, at least in
part, the induction of cyclin G2. FoxO3a is a member of the
Forkhead box class O (FoxO) transcription factor family. FoxO
transcription factors regulate diverse cell functions through the
regulation of genes involved in cell proliferation and apoptosis
(38). In particular, FoxO3a has
been reported to activate genes that induce cell cycle arrest, such
as p27, p21, p15 and p19, and to suppress the expression of cyclin
D2 (38–40). Recently, Fu and Peng (41) reported that several putative
FoxO3a-binding sites were present in the human cyclin G2 promoter
and overexpression of FoxO3a enhanced the cyclin G2 promoter
activity. Since the activity of FoxO3a is mainly regulated at the
level of posttranslational modifications such as phosphorylation
and degradation, activation of the PI3K/Akt signaling pathway in
response to growth stimuli leads to FoxO3a phosphorylation and
subsequent nucleus exclusion (41). In this study, we showed that CN-A
suppressed Rapa-induced phosphorylation of Akt (Ser473). It is
possible that inhibition of Rapa-induced phosphorylation of Akt
(Ser473) by CN-A may induce the accumulation of FoxO3a in the
nucleus and activate FoxO3a. Therefore, it will be interesting to
examine whether CN-A plus Rapa induce inhibition of cancer cell
growth through FoxO3a-mediated induction of cyclin G2.
Meric-Bernstam et al(42) reported that cell lines that are
Rapa-sensitive are more likely to have PIK3CA and/or PTEN mutations
or basal Akt phosphorylation, and that Rapa leads to a greater
increase in Akt phosphorylation in Rapa-sensitive cells. They
suggested that feedback loop activation is an indicator of the Rapa
response. Montero et al(43) showed that mTORC1 and mTORC2 were
constitutively active in ovarian cancer cell lines. Knockdown of
raptor or rictor, proteins required for the functioning of mTORC1
or mTORC2, respectively, resulted in profound inhibition of ovarian
cancer cell proliferation. mTORC2 mainly phosphorylates Akt
(Ser473) as PDK2. They found that the knockdown of raptor had a
more important inhibitory effect than the knockdown of rictor,
indicating mTORC1 had a predominant role over mTORC2 in the control
of ovarian cancer cell proliferation (43). These reports suggest that
Rapa-induced feedback loop activation of Akt is mainly an indicator
of the response to Rapa-induced inhibition of mTORC1-mediated cell
proliferation, since mTORC1 has a predominant role over mTORC2 in
controlling the proliferation of tumor cells. Another study
(44) demonstrated that mTOR
inhibition by the rapalog RAD001 was strongly associated with the
development of drug resistance due to sustained Akt activation in
lung cancer cell lines, and co-targeting mTOR and PI3K/Akt
signaling with separate drugs resulted in blocking Akt
phosphorylation and enhanced antitumor effects. Therefore, since
this Akt activation may limit the antitumor efficacy of Rapa and
rapalogs, approaches to prevent Akt activation are important for
more efficient growth inhibition of Rapa sensitive tumor cells. We
previously reported that treatment with CN-A plus Rapa effectively
inhibited the proliferation of MCF-7 cells in vitro and
in vivo(17). In this
study, we found that CN-A inhibited Rapa-induced phosphorylation of
Akt (Ser473). Our results suggest that the inhibition of
Rapa-induced Akt phosphorylation by CN-A correlates with their
effective growth inhibition of cancer cells. It is also clear that
our present and previous studies (17,19,30,45)
highly support the ability of CN-A to inhibit tumor growth
especially in concert with other drugs. These studies provided
evidence that CN-A represents a potent chemosensitizer in
conjunction with a broad spectrum of drugs including Rapa.
Taken together, our data support the hypothesis that
CN-A exerts at least part of its anticancer effects through the
modification of the PI3K/Akt/mTOR pathway and suggest that CN-A is
a promising combination partner of the mTOR inhibitor Rapa or
rapalogs.
Acknowledgements
This study was supported partly by a
grant from the Ministry of Education, Culture, Sports, Science and
Technology of Japan and Kawano Masanori Memorial Foundation for
Promotion of Pediatrics.
References
|
1
|
Jemar A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Hurvitz SA, Hu Y, O’Brien N and Finn RS:
Current approaches and future directions in the treatment of
HER2-positive breast cancer. Cancer Treat Rev. May 31–2012, (Epub
ahead of print). View Article : Google Scholar
|
|
3
|
Meric-Bernstam FM and Gonzalez-Angulo M:
Targeting the mTOR signaling network for cancer therapy. J Clin
Oncol. 27:2278–2287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Engelman JA: Targeting PI3K signaling in
cancer: opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wong KK, Engelman JA and Cantley LC:
Targeting the PI3K signaling pathway in cancer. Curr Opin Genet
Dev. 20:87–90. 2009. View Article : Google Scholar
|
|
6
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang S, Xiao X, Meng X and Leslie K: A
mechanism for synergy with mTOR and PI3 kinase inhibitors. PLoS
One. 6:e263432011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang X and Sun SY: Enhancing mTOR-targeted
cancer therapy. Expert Opin Ther Targets. 13:1193–1203. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsieh AC, Costa M, Zollo O, et al: Genetic
dissection of the oncogenic mTOR pathway reveals druggable
addiction to translational control via 4EBP-eIF4E. Cancer Cell.
17:249–261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Janes MR, Limon JJ, Chen J, et al:
Effective and selective targeting of leukemia cells using a TOR1/2
kinase inhibitor. Nat Med. 16:205–213. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sassa T, Tojyo T and Munakata K: Isolation
of a new plant growth substance with cytokinin-like activity.
Nature. 227:3791970. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Asahi K, Honma Y and Hazeki K: Cotylenin
A, a plant-growth regulator, induces the differentiation in murine
and human myeloid leukemia cells. Biochem Biophys Res Commun.
238:758–763. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamamoto-Yamaguchi Y, Yamada K, Ishii Y,
Asahi KI, Tomoyasu S and Honma Y: Induction of the monocytic
differentiation of myeloid leukemia cells by cotylenin A, a plant
growth regulator. Br J Haematol. 112:697–705. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamada K, Honma Y, Asahi KI, Sassa T, Hino
KI and Tomoyasu S: Differentiation of human acute myeloid leukemia
cells in primary culture in response to cotylenin A, a plant growth
regulator. Br J Haematol. 114:814–821. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Honma Y: Cotylenin A - a plant growth
regulator as a differentiation-inducing agent against myeloid
leukemia. Leuk Lymphoma. 43:1169–1178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamamoto-Yamaguchi Y, Okabe-Kado J,
Kasukabe T and Honma Y: Induction of differentiation of human
myeloid leukemia cells by immunosuppressant macrolides (rapamycin
and FK506) and calcium/calmodulin-dependent kinase inhibitors. Exp
Hematol. 29:582–588. 2001. View Article : Google Scholar
|
|
17
|
Kasukabe T, Okabe-Kado J, Kato N, Sassa T
and Honma Y: Effects of combined treatment with rapamycin and
cotylenin A, a novel differentiation-inducing agent, on human
breast carcinoma MCF-7 cells and xenografts. Breast Cancer Res.
7:R1097–R1110. 2005. View
Article : Google Scholar
|
|
18
|
Kawakami K, Hattori M, Inoue T, et al: A
novel fusicoccin derivative preferentially targets hypoxic tumor
cells and inhibits tumor growth in xenografts. Anticancer Agents
Med Chem. 12:791–800. 2012. View Article : Google Scholar
|
|
19
|
Kasukabe T, Okabe-Kado J and Honma Y:
Cotylenin A, a new differentiation inducer, and rapamycin
cooperatively inhibit growth of cancer cells through induction of
cyclin G2. Cancer Sci. 99:1693–1698. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun SH, Rosenberg LM, Wang X, et al:
Activation of AKT and eIF4E survival pathways by rapamycin-mediated
mammalian target of rapamycin inhibition. Cancer Res. 65:7052–7058.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Harkavy N, Shen N, Grohar P and
Helman LJ: Rapamycin induces feedback activation of Akt signaling
through an IGF-1R-dependent mechanism. Oncogene. 26:1932–1940.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gazitt T, Kolaparthi V, Moncada K, Thomas
C and Freeman J: Targeted therapy of human osteosarcoma with 17AAG
or rapamycin: characterization of induced apoptosis and inhibition
of mTOR and Akt/MAPK/Wnt pathways. Int J Oncol. 34:551–561.
2009.PubMed/NCBI
|
|
23
|
Vaseva AV, Yallowitz AR, Marchenko ND, Xu
S and Moll UM: Blockade of Hsp90 by 17AAG antagonizes MDMX and
synergizes with Nutlin to induce p53-mediated apoptosis in solid
tumors. Cell Death Disease. 2:e1562011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiao Y, Ou W, Meng F, Zhou H and Wang A:
Targeting HSP90 in ovarian cancers with multiple receptor tyrosine
kinase coactivation. Mol Cancer. 10:1252011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mann KK, Colombo M and Miller WH Jr:
Arsenic trioxide decreases AKT protein in a caspase-dependent
manner. Mol Cancer Ther. 7:1680–1687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan Z, Wang F, Zhao Z, et al:
BIM-mediated AKT phosphorylation is a key modulator of arsenic
trioxide-induced apoptosis is cisplatin-sensitive and - resistant
ovarian cancer cells. PLoS One. 6:e205862011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chiu HW, Chen YA, Ho SY and Wang YJ:
Arsenic trioxide enhances the radiation sensitivity of
androgen-dependent and -independent human prostate cancer cells.
PLoS One. 7:e315792012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ray S, Fry MJ and Darbre PD: Enhanced
sensitivity to rapamycin following long-term oestrogen deprivation
in MCF-7, T-47-D and ZR-75-1 human breast cancer cells. J
Endocrinol. 208:21–29. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Elfiky A, Aziz SA, Conrad PJ, et al:
Characterization and targeting of phosphatidylinositol-3 kinase
(PI3K) and mammalian target of rapamycin (mTOR) in renal cell
cancer. J Trans Med. 9:1332011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Honma Y, Kasukabe T, Yamori T, Kato N and
Sassa T: Antitumor effect of cotylenin A plus interferon-alpha:
possible therapeutic agents against ovary carcinoma. Gynecol Oncol.
99:680–688. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shen Y, Shen ZX, Chen YJ, et al: Studies
on the clinical efficacy and pharmacokinetics of low-dose arsenic
trioxide in the treatment of relapsed acute promyelocytic leukemia:
a comparison with conventional dosage. Leukemia. 15:735–741. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Douer D and Tallman MS: Arsenic trioxide:
new clinical experience with an old medication in hematologic
malignancies. J Clin Oncol. 23:2395–2410. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim KB, Bedikian AY, Camacho LH,
Papadopoulos NE and McCullough C: A phase II trial of arsenic
trioxide in patients with metastatic melanoma. Cancer.
104:1687–1692. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Loehberg CR, Strissel PL, Dittrich R, et
al: Akt and p53 are potential mediators of reduced mammary tumor
growth by Chloroquine and the mTOR inhibitor RAD001. Biochem
Pharmacol. 83:480–488. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Horne MC, Goolsby GL, Donaldson KL, Tran
D, Neubauer M and Wahl AF: Cyclin G1 and cyclin G2 comprise a new
family of cyclins with contrasting tissue-specific and cell
cycle-regulated expressions. J Biol Chem. 271:6050–6061. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Horne MC, Donaldson KL, Goolsby GL, et al:
Cyclin G2 is up-regulated during growth inhibition and B cell
antigen receptor-mediated cell cycle arrest. J Biol Chem.
272:12650–12661. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bates S, Rowan S and Vousden KH:
Characterization of human cyclin G1 and G2: DNA damage inducible
genes. Oncogene. 13:1103–1109. 1996.PubMed/NCBI
|
|
38
|
Fei M, Zhao Y, Wang Y, et al: Low
expression of Foxo3a is associated with poor prognosis in ovarian
cancer patients. Cancer Invest. 27:52–59. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Weidinger C, Krause K, Klagge A, Karger S
and Fuhrer D: Forkhead box-O transcription factor: critical
conductors of cancer’s fate. Endocr Relat Cancer. 15:917–929.
2008.PubMed/NCBI
|
|
40
|
Katayama K, Nakamura A, Sugimoto Y, Tsuruo
T and Fujita N: FOXO transcription factor-dependent p15(INK4b) and
p19(INK4d) expression. Oncogene. 27:1677–1686. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fu G and Peng C: Nodal enhances the
activity of FoxO3a and its synergistic interaction with Smad to
regulate cyclin G2 transcription in ovarian cancer cells. Oncogene.
30:3953–3966. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Meric-Bernstam F, Akcakanat A and Chen H:
PI3CA/PTEN mutations and Akt activation as markers of sensitivity
to allosteric mTOR inhibitors. Clin Cancer Res. 18:1777–1789. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Montero JC, Chen X, Ocana A and Pandiella
A: Predominance of mTORC1 over mTORC2 in the regulation of
proliferation of ovarian cancer cells: therapeutic implications.
Mol Cancer Ther. 11:1342–1352. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu CX, Li Y, Yue P, et al: The combination
of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity
against non-small cell lung cancer in vitro and in vivo. PLoS One.
6:e2208992011.PubMed/NCBI
|
|
45
|
Honma Y, Ishii Y, Yamamoto-Yamaguchi Y,
Sassa T and Asahi K: Cotylenin A, a differentiation-inducing agent,
and IFN-alpha cooperatively induce apoptosis and have an antitumor
effect on human non-small lung carcinoma cells in nude mice. Cancer
Res. 63:3659–3666. 2003.
|