Introduction
Colorectal cancer is a commonly diagnosed cancer and
one of the leading causes of cancer death in the world (1,2).
Metastasis is one of the major causes of deaths of colorectal
cancer (3). Metastasis occurs
through a stepwise process that starts when cancer cells segregate
from a primary tumor, migrate across blood vessel walls then into
the blood stream and invade into tissue (4–6).
Finally, cancer cells disperse throughout new tissues to survive in
the new ectopic sites and generate malignant tumors (1–3,6).
Matrix metalloproteinases (MMPs), a group of
secreted proteinases, play an important role in a variety of
physiological and pathological processing colorectal cancer cell
metastasis (7–10). MMPs degrade extracellular matrix
(ECM) components in the basement membrane, increase cell invasion
and metastasis in colorectal cancer cells (8–12).
Many studies have shown that the functions of various metastatic
molecules are modulated by MMPs (7,13–15).
It has been reported that elevated MMP-2, MMP-7 and MMP-9 protein
levels were detected in colorectal cancer. The correlation of
MMP-2, MMP-7 and MMP-9 expression with invasion and metastasis has
been demonstrated (9,13,14,16,17).
It is well established that receptor tyrosine kinase (RTK)
activates the phosphatidylinositol 3-kinase (PI3K)/AKT and
Ras-Raf-mitogen-activated protein kinase (MAPK) pathways, resulting
in the activation of eIF-4B, eIF-4E, eIF-4G and S6 and NF-κB
transcription factors (18–23).
Those transcription factors are considered to be the regulator of
MMP-2, MMP-7 and MMP-9 translation signaling (24,25).
Quinazolinone derivatives have shown anti-malarial,
anti-inflammatory, anti-bacterial and antitumor activities in many
studies (26,27). Quinazolinone compounds have also
been shown to possess antitumor activity to inhibit cell metastasis
by downregulating the expression and activities of MMP-2 and MMP-9
in many cancer cell lines (28–30).
Our previous study showed that
6-fluoro-(3-fluorophenyl)-4-(3-methoxyanilino)-quinazoline (LJJ-10)
exhibits anti-metastatic effects in human osteosarcoma U-2 OS cells
through targeting the insulin-like growth factor-I receptor
(IGF-IR) (29). The
2-(3-ethoxyphenyl)-6-pyrrolidinylquinazolinone) (MJ-33) exhibits
anti-metastatic effects in human prostate carcinoma DU145 cells via
decreased protein levels of MAPKs (mitogen-activated protein
kinases), AKT, AP-1 and NF-κB, resulting in the inhibition of MMP-2
and MMP-9 (28). Receptor tyrosine
kinases have been indicated as promising molecular targets for
cancer therapy (31–34). The c-Met, a receptor tyrosine
kinase, is overexpressed and/or mutated in a variety of tumor cells
(31,35). Abnormal activation of c-Met
signaling can lead to angiogenesis, proliferation, invasion and
metastasis (36–38). EGFR, epidermal growth factor
receptor, functions with a vital role in colorectal cancer
initiation and progression (39,40).
Although c-Met and EGFR are recognized as important therapeutic
targets for the treatment of malignancies, the inhibitory effect of
c-Met and EGFR in human colorectal cancer cells remains unclear. In
the present study, we investigated the anti-metastatic activity of
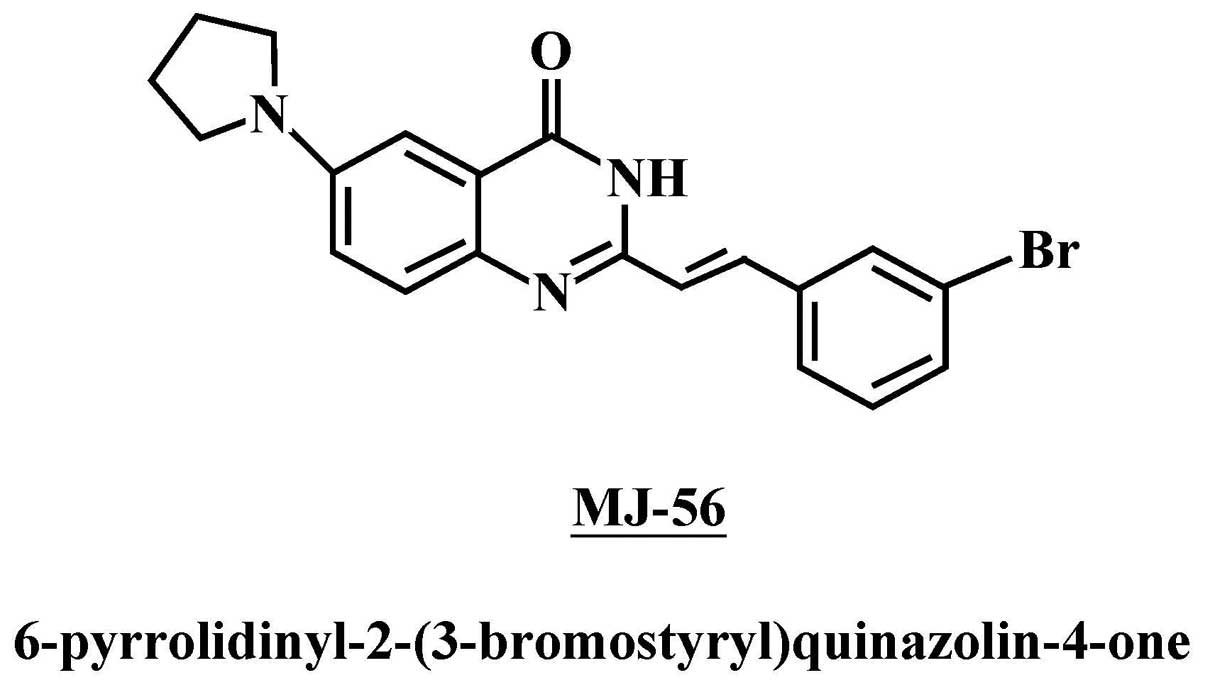
MJ-56 (6-pyrrolidinyl-2-(3-bromostyryl)quinazolin-4-one) (Fig. 1), a novel synthesized quinazolinone
derivate and the anti-metastatic pathways of MJ-56 in the human
colorectal cancer cell line HT29.
Materials and methods
Chemicals and reagents
MJ-56 was designed and synthesized by Mann-Jen Hour
from China Medical University, Taichung, Taiwan (Fig. 1). Antibodies against MMP-1, MMP-2,
MMP-7, MMP-9, MMP-10, TIMP-1, TIMP-2, p85-PI3K, p110-PI3K, p65
NF-κB, PCNA, p38, GAPDH (Santa Cruz Biotechnology, Inc.; Santa
Cruz, CA, USA), EGFR, p-EGFR (Tyr1173), c-Met, p-S6 (Ser240/244),
p-Mnk1 (Thr197/202), p-eIF4E (Ser209), p-eIF4G (Ser1108), p-eIF4B
(Ser422), p-JNK (Thr183/Tyr185), JNK, p-p38 (Thr183/Tyr185), p-ERK
(Thr202/Tyr204), ERK, p-mTOR (Ser2448), mTOR, p-AKT (Ser473), AKT
(Cell Signaling; Danvers, MA, USA) were obtained from the indicated
vendors. Rabbit anti-mouse IgG (HRP) antibody, goat anti-rabbit IgG
(HRP) antibody, donkey anti-goat IgG (HRP) antibody and
FITC-conjugated goat anti-mouse antibody were obtained from Santa
Cruz Biotechnology, Inc. Propidium iodide (PI) was from
Sigma-Aldrich (St. Louis, MO, USA). DMEM medium, fetal bovine serum
(FBS), L-glutamine, penicillin-streptomycin and trypsin-EDTA were
purchased from Invitrogen Life Technologies (Carlsbad, CA,
USA).
Cell culture
Human colorectal cancer cell line, HT29, was
purchased from Food Industry Research and Development Institute
(FIRDI, Hsinchu, Taiwan). HT29 cells were cultured in DMEM medium
supplemented with 10% of fetal bovine serum, 100 U/ml penicillin,
100 μg/ml streptomycin and 2 mM glutamine and incubated at
37°C in a humidified chamber with 5% CO2(24).
Wound healing assay
HT29 cells (cell density: 1×106
cells/well) were seeded 6-well plate and grown to 90% of
confluence. The next day, cells were scratched with a yellow tip
and treated with various concentrations of MJ-56 (0, 5, 10 and 15
μM) in DMEM serum-free medium for 24 h. The cells were
captured and relative cell migration was calculated. All treatments
were in duplicate and three independent experiments were performed
(4,28,30).
Cell invasion assay
The membrane of transwell insert was rinsed with PBS
and coated with Matrigel (BD Matrigel™ Invasion chamber). Cells
were seeded into the chamber of the insert at a density of
2.5×104 cells/ml and incubated with 0.5 ml of complete
DMEM medium in the transwell. HT29 cells were treated with various
concentrations of MJ-56 (0, 5, 10 and 15 μM) for 24 h and
cells inside the chamber were removed. Invaded cells were fixed
with 4% formaldehyde in PBS and stained with 0.1% of hematoxylin
(Sigma-Aldrich), captured and the number of invaded cells were
counted and used for the calculation of inhibitory rate (28,30).
Gelatin zymography analysis
HT29 cells (cell density: 1×106
cells/well) were seeded into 6-well plate and grew for 24 h. Cells
were treated with various concentrations of MJ-56 (0, 5, 10 and 15
μM) in serum-free DMEM medium for 24 h. Culture medium was
collected and spun at 1000 × g for 10 min at 4°C. Supernatant was
collected and 5 μg of total protein was mixed with 2X sample
buffer (0.125 M Tris-HCl, 4% SDS, 20% glycerol, 0.01% bromophenol
blue) and separated in an 8% SDS-polyacrylamide gel with 1%
gelatin. Gel was treated with 2.5% Triton-X-100 at room temperature
for 30 min to remove SDS, incubated in Zymogen developing buffer
(50 mM Tris, pH 7.5, 200 mM NaCl, 5 mM CaCl2, 0.02%
Brij-35; Bio-Rad Laboratories; Hercules, CA, USA) at room
temperature for 30 min, then refreshed with Zymogen developing
buffer and incubated at 37°C for 24 h. Gel was rinsed with water
once and stained with 0.5% of Coomassie blue R-250 (0.5% Coomassie
blue R-250, 50% methanol and 10% acetic acid) for 2 h and
de-stained in de-staining solution (50% methanol and 10% acetic
acid) until clear zones were visualized (28,30).
Preparation of whole cell lysate and
nuclear lysate
HT29 cells were treated with various concentrations
of MJ-56 for the given time and cells were harvested for the
preparation of whole cell lysate using iced-cold RIPA buffer (1%
NP-40, 50 mM Tris-base, 0.1% SDS, 0.5% deoxycholic acid, 150 mM
NaCl, pH 7.5) supplemented with the protease inhibitors including
phenylmethanesulfonyl fluoride (10 mg/ml), leupeptin (17 mg/ml) and
sodium orthovanadate (10 mg/ml). Cells were vortexed briefly and
incubated in ice for 30 min and cell lysate was collected by a spin
at 12,000 × g at 4°C for 10 min. Nuclear extracts were obtained by
using the NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo
Scientific; Rockford, IL, USA). The resulting nuclear pellet was
re-suspended in nuclear extraction buffer (1.5 mM MgCl2,
10 mM HEPES, pH 7.9, 0.1 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM
phenylmethanesulfonyl fluoride, 25% glycerol and 420 mM NaCl) and
incubated in ice for 20 min, then spun at 14,000 × g for 5 min. The
supernatant, the soluble nuclear fraction, was collected and used
for EMSA analysis of NF-κB and western blot analyses of NF-κB and
PCNA (28,30).
Electrophoretic mobility shift assay
(EMSA)
HT29 cells were seeded at a density of
1×106 cells/ml the day before treatment. Cells were then
treated with 15 μM of MJ-56 for 0 and 4 h. Nuclear extracts
were prepared using NE-PER Nuclear and Cytoplasmic Extraction kit
(Thermo Scientific) and soluble nuclear fraction was prepared as
described above. The protein concentrations were determined by
using Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad).
Biotin end-labeled oligonucleotide corresponding to consensus NF-κB
binding site (5′-GATCCAGGGGACTTTCCCTAGC-3′) was prepared with the
LightShift Chemiluminescent EMSA kit (Thermo Scientific) and used
as the probe. The 5 μg of nuclear extract was incubated with biotin
end-labeled duplex DNA, electrophoresed on a 6% polyacrylamide
native gel, transferred to a positive nylon membrane, UV
cross-linked and incubated with streptavidin-HRP and signals were
developed by ECL kit (Millipore) (28,30).
Western blot analysis
Whole cell lysate and nuclear extract were isolated
from treated cells as described above, separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
electro-transferred to a nitrocellulose membrane using the iBot Dry
Blotting System (Invitrogen Life Technologies). The transferred
membranes were incubated in blocking buffer (5% non-fat milk in
Tris-buffered saline/Tween-20) for 1 h and incubated with primary
antibody in blocking buffer at 4°C overnight. Membranes were washed
with Tris-buffered saline/Tween-20 three times for 10 min and
incubated with HRP-conjugated secondary antibody for 1 h. Protein
signals were revealed by using ECL kit (Millipore) and exposed to
Kodak Bio-MAX MR film (Estman Kodak, Rochester, NY, USA) (28,30).
Immuno-fluorescent staining
HT29 cells were seeded onto polylysine-coated slides
(cell density: 1×105 cells/ml) the day before staining.
On the next day, cells were treated with 15 μM of MJ-56 for
4 h, fixed with 4% formaldehyde in PBS, permeabolized with 0.1%
Triton X-100 for 30 min, blocked with 10% normal goat serum for 30
min and probed with primary antibody p65 NF-κB at 4°C overnight.
Then, cells were stained for FITC-conjugated goat anti-mouse
secondary antibodies for 1 h, washed and mounted in Shandan
mounting medium. The images were captured using a fluorescence
microscopy (41).
ERK kinase assay
HT29 cells (cell density: 1×106 cells/ml)
were treated with 15 μM of MJ-56 for 0–4 h and ERK kinase
activity was analyzed using p44/42 MAP Kinase assay kit obtained
from Cell Signaling (#9800). The p44/42 MAP Kinase was
immuno-precipitated from cell lysate of treated cells with
anti-phospho-p44/42 MAPK (Thr202/Tyr204) antibody and mixed with
Elk-1 fusion protein and ATP in kinase buffer (25 mM Tris-HCl, pH
7.5, 5 mM β-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM
Na3VO4, 10 mM MgCl2) and incubated
at 37°C for 30 min. The reaction was stopped by boiling in sample
buffer and subjected to western blot analysis using
anti-phospho-Elk-1 (Ser 383) antibody. The intensity of protein
bands was quantified by NIH ImageJ and the intensity of protein
band from untreated cells was set as 100% and the intensity of
other treatments was calculated accordingly. Shown was the average
data from three independent experiments (42,43).
AKT kinase assay
HT29 cells (cell density: 1×106 cells/ml)
were treated with 15 μM of MJ-56 for 0–4 h and AKT kinase
activity was analyzed using AKT Kinase Assay kit from Cell
Signaling (#9840). AKT was precipitated from cell lysate of treated
cells with anti-AKT antibody and mixed with GSK-3 fusion protein
and ATP in kinase buffer (25 mM Tris-HCl, pH 7.5, 5 mM
β-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM
Na3VO4, 10 mM MgCl2) and incubated
at 30°C for 30 min. The reaction was stopped by boiling in sample
buffer and subjected to western blot analysis using
anti-phospho-GSK3 (Ser219) antibody. The intensity of protein bands
was quantified by NIH ImageJ and the intensity of protein band from
untreated cells was set as 100% and the intensity of other
treatments was calculated accordingly. The average of the data was
shown from three independent experiments (44).
Tyrosine kinase assay
HT29 cells (cell density: 1×106 cells/ml)
were treated with 15 μM of MJ-56 for 0–4 h and Tyrosine
kinase activity was analyzed using Tyrosine Kinase assay kit from
Upstate (#17–315) (EMD Millipore Corp., Billerica, MA, USA). Cell
lysate was incubated with biotinylated poly (Glu4-Tyr) peptide as
the substrate and ATP in a kinase buffer containing
Mn2+/Mg2+ as the co-factors. Tyrosinated
substrate was recognized by anti-phosphotyrosine-HRP and detected
by enzyme-linked immunosorbent assay (ELISA). The kinase activity
from untreated cells was set as 100% and the kinase activity of
other treatments was calculated accordingly. Shown was the average
data from three independent experiments (45).
Statistical analysis
The statistical results were expressed as the mean ±
SEM of triplicate samples. The difference between groups was
analyzed by One-way ANOVA followed by paired two-tailed Student’s
t-test and *P<0.05 was taken as significant.
Results
MJ-56 inhibits migration and invasion of
HT29 cells
Quinazolinone derivatives have been shown to possess
anti-tumor activities (26,30,32).
To obtain more effective drugs for cancer treatment, we have
chemically modified the quinazolinone derivatives. MJ-56, a novel
quinazolinone derivative, was synthesized (Fig. 1) and determined its antitumor
activity by assaying cell migration and invasion in HT29 colorectal
carcinoma cells. HT29 cells were treated with various
concentrations (0, 5, 10 and 15 μM) of MJ-56 for 24 h. The
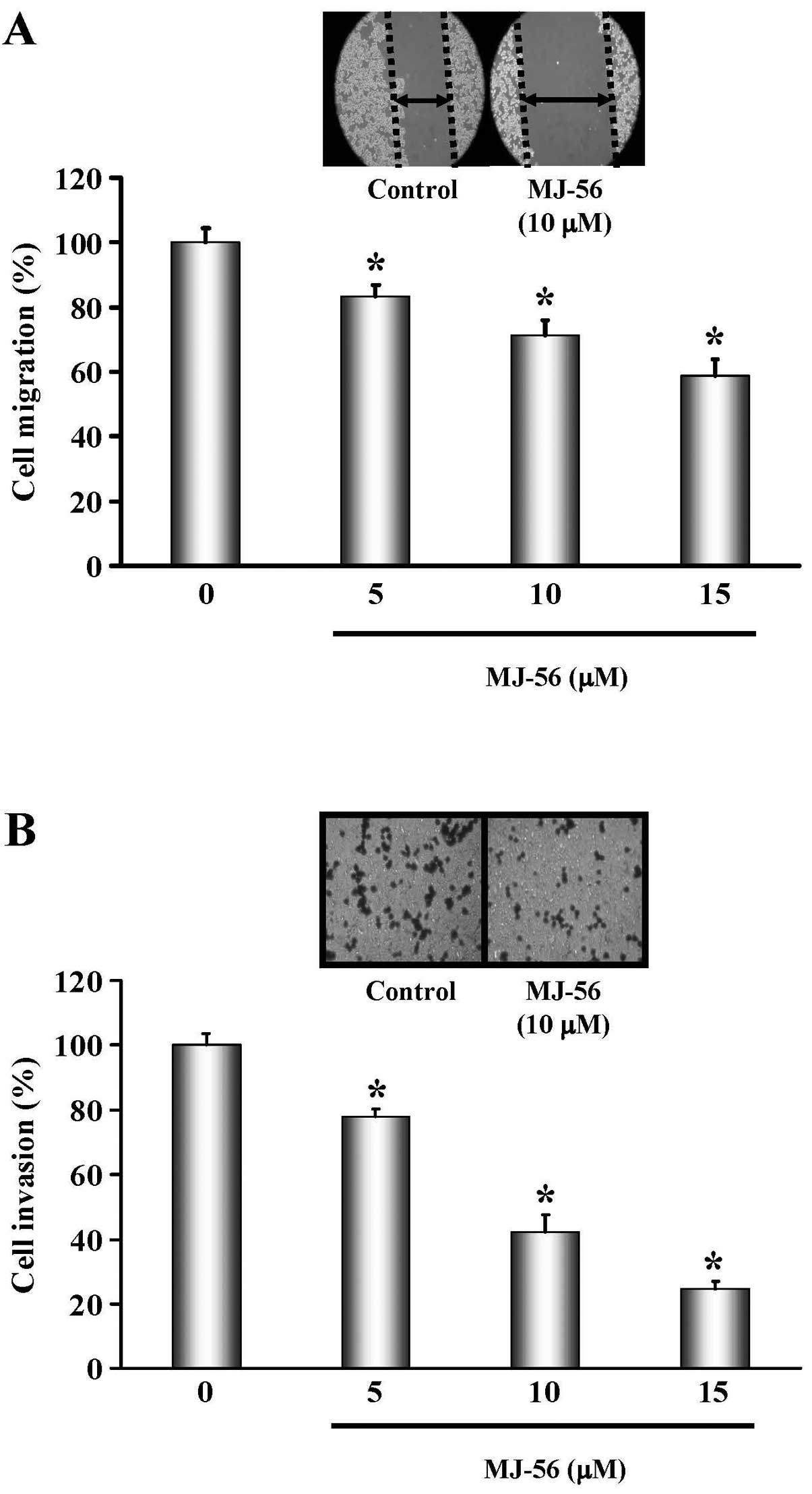
effect of MJ-56 on the cell migration was determined by the scratch
assay. As shown in Fig. 2A,
migration of HT29 cells was gradually impeded with an increase in
the concentrations of MJ-56. Cell invasion is a critical step
during cancer metastasis. To characterize whether MJ-56 inhibits
cell invasion, HT29 cells were treated with various concentrations
(0, 5, 10 and 15 μM) of MJ-56 and the effects of MJ-56 on
cell invasion were examined by the Matrigel-coated transwell assay.
MJ-56 inhibited invasion of HT29 cells in a concentration-dependent
manner. The inhibitory rate was calculated as the percentage of
21.88, 54.24 and 75.58% for 5, 10 and 15 μM of MJ-56-treated
cells, respectively (Fig. 2B).
MJ-56 inhibits the expression and enzyme
activity of matrix metalloproteinases and increases the expression
of metalloproteinase inhibitors TIMP-1 and TIMP-2
During cancer metastasis, matrix metalloproteinases
(MMPs) are indispensible for the degradation of extracellular
matrix, an important step for cancer invasion. Due to the
inhibitory effects of MJ-56 on cell migration and invasion, we
subsequently determined whether MJ-56 may block the expression and
enzymatic activity of MMPs. HT29 ells were treated with various
concentrations of MJ-56 (0, 5, 10 and 15 μM) for 24 h and
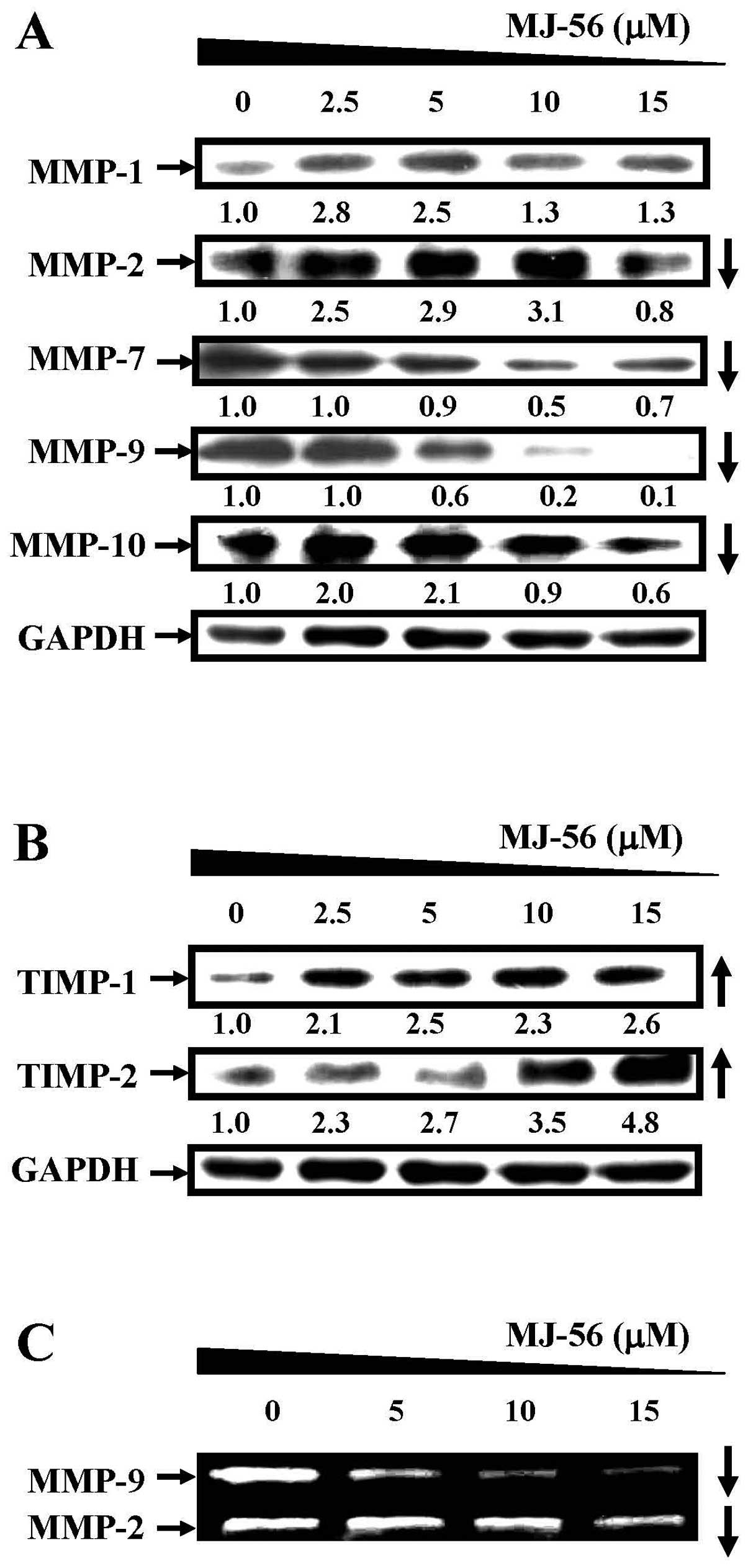
cells were harvested for western blot analyses. MJ-56 suppressed
the expression of matrix metalloproteinases MMP-2, MMP-7, MMP-9 and
MMP-10 in a concentration-dependent manner (Fig. 3A). However, the expression of MMP-1
appeared not to be affected by MJ-56 treatment (top row, Fig. 3A). In contrast, TIMP-1 and TIMP-2
are metalloproteinase inhibitors that can block the enzymatic
activities of MMPs, thereby inhibiting cancer invasion. The
expression of TIMP-1 and TIMP-2 was elevated with the increase in
the concentrations of MJ-56 (0–15 μM) in HT29 cells
(Fig. 3B). Concomitantly, MJ-56
inhibited the enzymatic activities of MMP-2 and MMP-9 by gelatin
zymography analysis (Fig. 3C). The
MMP-9 activity was significantly decreased at 5 μM of MJ-56
and the inhibition of MMP-2 activity was observed at 15 μM
of MJ-56. Our data revealed that MJ-56 may upregulate the
expression of TIMP-1 and TIMP-2, downregulate the expression of
MMP-2, -7, -9, -10 and reduce the enzymatic activities of MMP-2 and
MMP-9 in HT29 cells, which might be responsible for the
concentration-dependent inhibition of cell invasion by MJ-56.
MJ-56 inhibits tyrosine phosphorylation
of EGFR and expression of c-MET
It has been reported that the activation of
autophosphorylation on the receptor tyrosine kinase EGFR can
activate the downstream signaling events to increase the expression
of MMP family proteins such as MMP-2, MMP-7 and MMP-9, contributing
to metastasis and invasion of colorectal cancers (46-48).
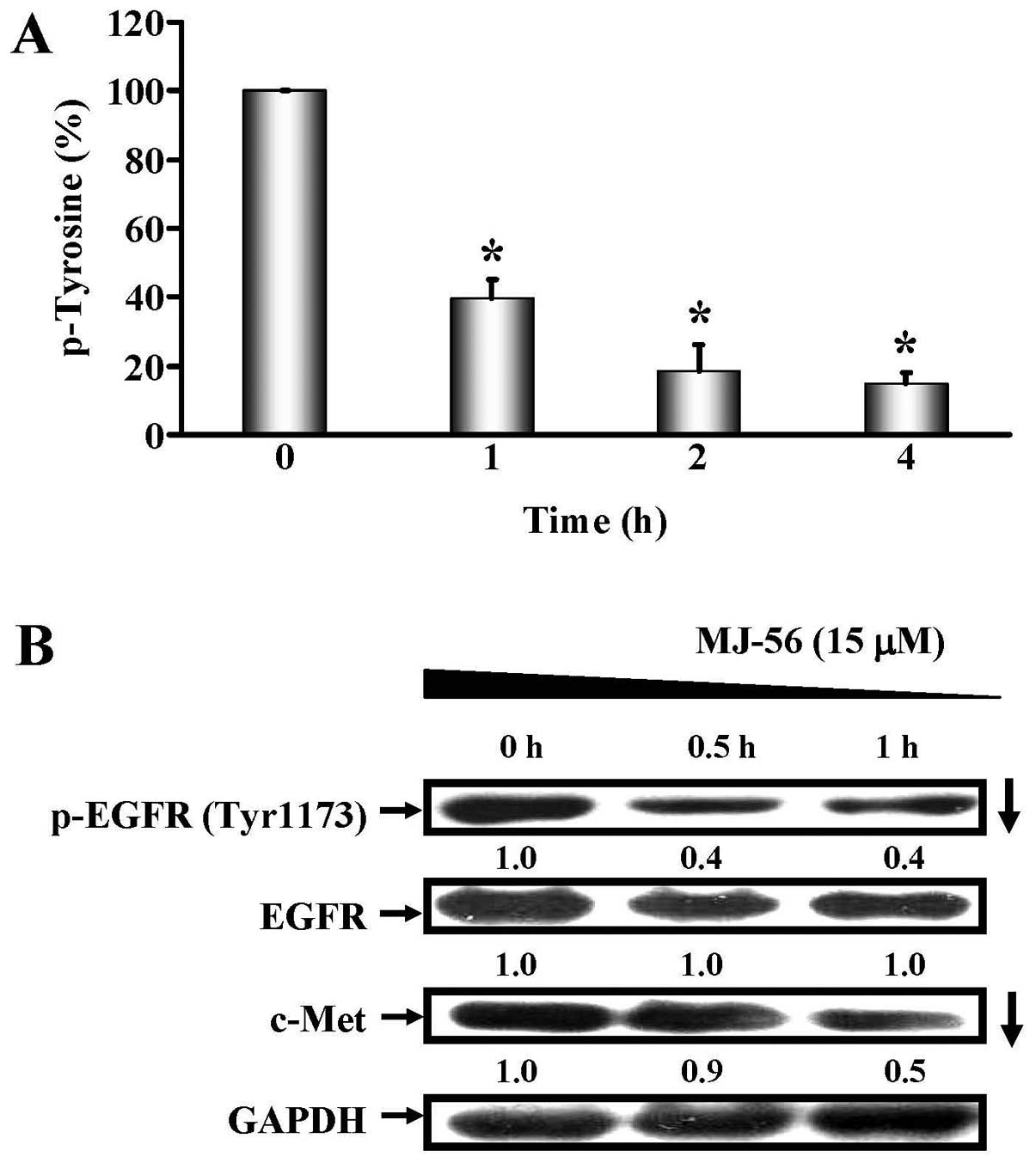
We determined the effects of MJ-56 on the inhibition of tyrosine
kinase activity. HT29 cells were treated with 15 μM of MJ-56
for 0–4 h and tyrosine kinase activity was examined by colorimetric
detection of protein tyrosine kinase activity. As shown in Fig. 4A, MJ-56 reduced the tyrosine kinase
activity of treated cells in a time-dependent manner, with >60%
of inhibition after 1 h of treatment. As tyrosine kinase activity
was inhibited by MJ-56, we then determined whether MJ-56 acts on
the inhibition of receptor tyrosine kinase activity of EGFR, a
receptor protein that undergoes auto-phosphorylation via its
intrinsic tyrosine kinase activity after activation. HT29 cells
were treated with 15 μM of MJ-56 at various time points (0,
0.5 and 1 h) and tyrosine phosphorylation and expression of EGFR
was examined by western blotting. As shown in Fig. 4B, MJ-56 inhibited the tyrosine
phosphorylation of EGFR and c-Met in a time-dependent fashion,
while the protein level of EGFR remained unchanged (second row,
Fig. 4B). Given that MJ-56
inhibited the tyrosine kinase activity of receptor tyrosine kinase
EGFR, we inferred that MJ-56 would be able to inhibit other
receptor tyrosine kinases such as c-Met. MJ-56 also decreased the
expression of receptor tyrosine kinase Met (third row, Fig. 4B). Our results suggest MJ-56 can
inhibit receptor tyrosine kinase by targeting on the tyrosine
kinase activity of EGFR and reducing the expression of c-Met
receptor tyrosine kinase.
MJ-56 inhibits the ERK-mediated MAPK
signaling pathway in HT29 cells
Aforementioned data showed that MJ-56 can inhibit
the tyrosine kinase activity of EGFR and the expression of MMPs,
thus we investigated whether the inhibition in the tyrosine kinase
activity of EGFR and c-Met by MJ-56 results in the inhibition of
downstream signaling, thereby reducing the expression of MMPs. We
determined the effects of MJ-56 on the MAPK signaling pathway
downstream of EGFR, which includes ERK1/2, c-Jun N-terminal kinase
and p38 kinase signaling pathways. HT29 cells were treated with 15
μM of MJ-56 at various time-points (0, 0.5, 1, 3 and 6 h).
The expression and phosphorylation status of JNK, p38 and ERK were
evaluated by western blot analyses. As shown in Fig. 5A, MJ-56 decreased the
phosphorylation of ERK, while the expression of ERK remained
largely unaffected. However, MJ-56 did not affect the activation of
p38 and JNK. Since phosphorylation of ERK is required for the
activation of its kinase activity, we examined whether the
MJ-56-mediated inhibition of ERK phosphorylation will modulate ERK
kinase activity. HT-29 cells were treated with 15 μM of
MJ-56 for 0–4 h and ERK kinase activity was assessed. The results
showed that MJ-56 significantly inhibited ERK kinase activity in a
time-dependent manner (Fig. 5B).
Thus, our data indicated that MJ-56 inhibits the ERK-mediated MAPK
signaling pathway.
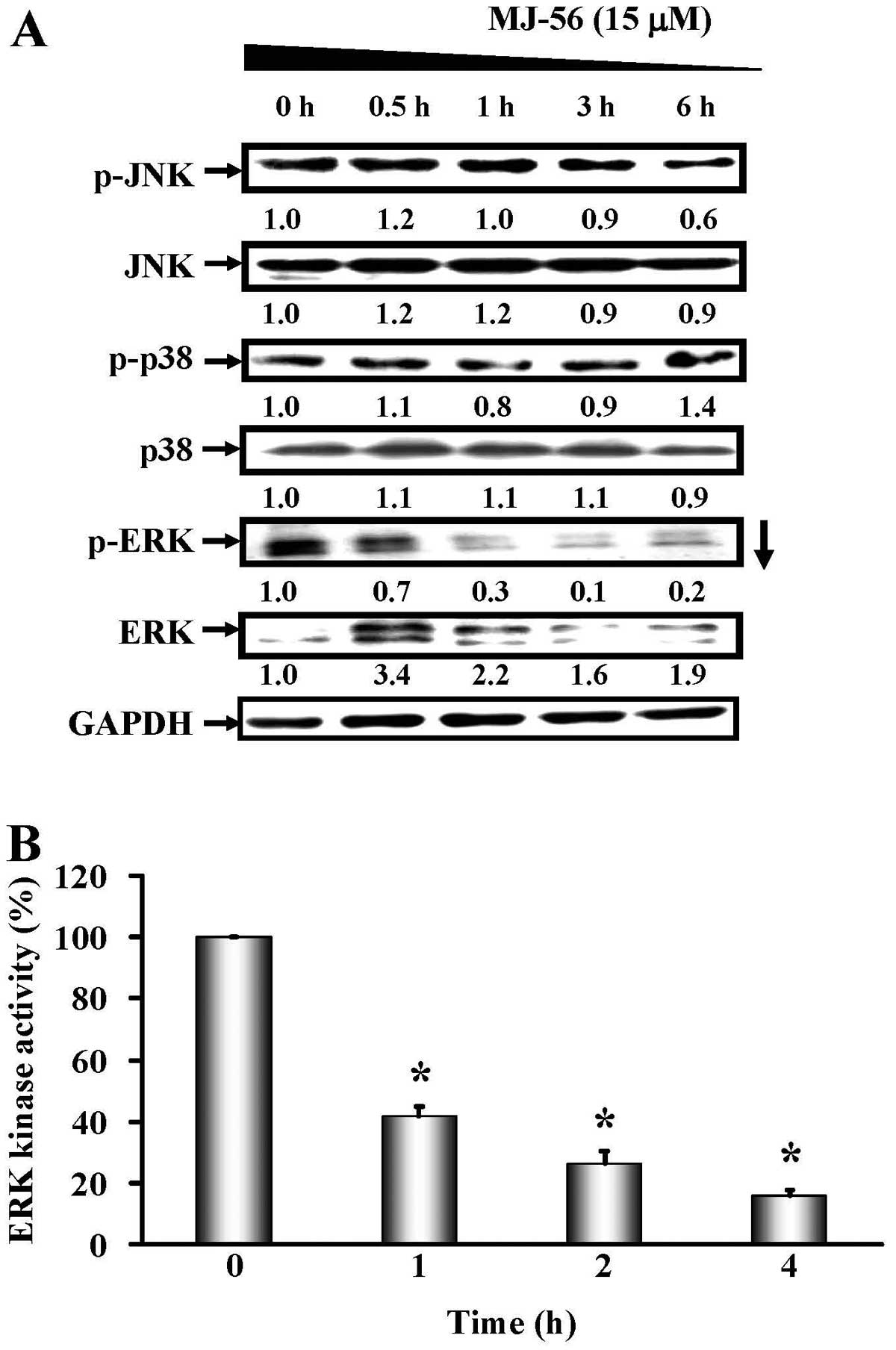 | Figure 5.MJ-56 blocks ERK-mediated MAPK
signaling pathway in HT29 cells. (A) Cells were harvested at 0,
0.5, 1, 3 and 6 h after MJ-56 treatment. The protein levels of
p-JNK, JNK, p-p38, p38, p-ERK and ERK were analyzed by western
blotting. Protein fold changes were normalized to the endogenous
GAPDH control. (B) Cells were harvested at 0, 1, 2 and 4 h after
MJ-56 treatment. Cell lysates were immuno-precipitated with
anti-phospho-p44/42 MAPK (Thr202/Tyr204) antibody and assessed ERK
kinase activity by measuring phosphorylation of Elk-1. ERK kinase
activity in MJ-56 treatment was compared with that in no treatment
control. ERK kinase activity was expressed as mean ± SD of three
independent experiments. *p<0.05, indicates a
significant difference from the control. |
MJ-56 inhibits the
PI3K/AKT/mTOR-dependent signaling pathway in HT29 cells
In addition to MAPK signaling pathway,
PI3K/AKT/mTOR-dependent signaling pathway is also a downstream
signaling pathway of EGFR. To examine the effects of MJ-56 on the
PI3K/AKT/mTOR signaling pathway, HT29 cells were treated with 15
μM of MJ-56 for different time (0–6 h), and western blot
analyses were performed. The results showed that the expression of
PI3K (p85), the regulatory subunit of PI3K, appeared not to be
affected (top row, Fig. 6A). MJ-56
decreased the expression of PI3K (p110), the catalytic subunit of
PI3K kinase (second row, Fig. 6A).
In addition, MJ-56 reduced the phosphorylation of AKT and mTOR,
while the expression of AKT and mTOR seemed not to be altered
(Fig. 6A). As phosphorylation of
AKT is important for its kinase activity, the reduction in
phosphorylation of AKT by MJ-56 implies that the kinase activity of
AKT will be affected. As shown in Fig.
6B, MJ-56 indeed inhibited the enzymatic activity of AKT kinase
in a time-dependent manner. Therefore, our results suggested that
MJ-56 is able to inactivate the PI3K/AKT/mTOR-dependent signaling
pathway in HT29 cells. To evaluate whether MJ-56 inhibits the
downstream proteins of ERK-mediated MAPK and PI3K/AKT/mTOR
signaling pathways, HT29 cells were treated with 15 μM of
MJ-56 for 0 to 6 h and the phosphorylation of S6, Mnk1, eIF-4B,
eIF-4E and eIF-4G was examined by western blot analyses. As shown
in Fig. 6A, MJ-56 treatment led to
a time-dependent inhibition in the phosphorylation of S6, Mnk1,
eIF-4B, eIF-4E and eIF-4G, although MJ-56 treatment resulted in
different inhibition kinetics for each protein. Our data showed
that MJ-56 inhibits the phosphorylation of Mnk1 and elF-4E,
downstream targets of ERK-mediated MAPK signaling pathway and S6,
elF-4B, elF-4G, downstream targets of PI3K/AKT/mTOR signaling
pathway, thereby disturbing translation initiation and protein
synthesis at the ribosomes and hence reduced the expression of
MMPs.
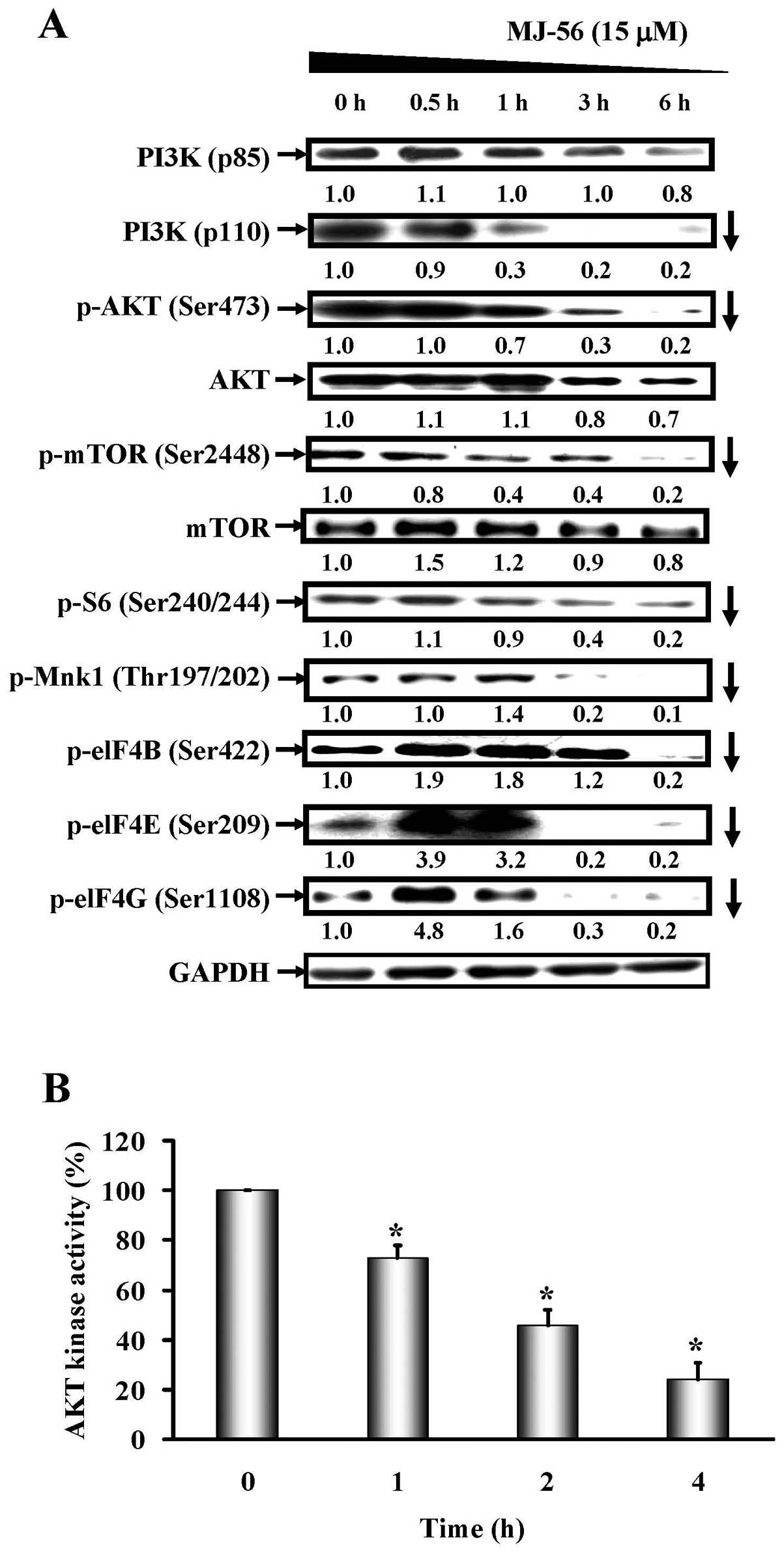 | Figure 6.MJ-56 modulates
PI3K/AKT/mTOR-dependent signaling pathway in HT29 cells. (A) Cells
were harvested at 0, 0.5, 1, 3 and 6 h after MJ-56 treatment. The
protein levels of p110-PI3K, p85-PI3K, p-AKT (Ser473), AKT, p-mTOR
(Ser2448), mTOR, p-S6 (Ser240/244), p-Mnk1 (Thr197/202), p-eIF4B
(Ser422), p-eIF4E (Ser209) and p-eIF4G (Ser1108) were analyzed by
western blotting. Protein fold changes were normalized to the
endogenous GAPDH control. (B) Cells were harvested at 0, 1, 2 and 4
h after MJ-56 treatment. Cell lysates were immunoprecipitated with
anti-AKT antibody and assessed AKT kinase activity by measuring
phosphorylation of GSK-3. AKT kinase activity in MJ-56 treatment
was compared with that in no treatment control. AKT kinase activity
was expressed as mean ± SD of three independent experiments.
*p<0.05, indicates a significant difference from the
control. |
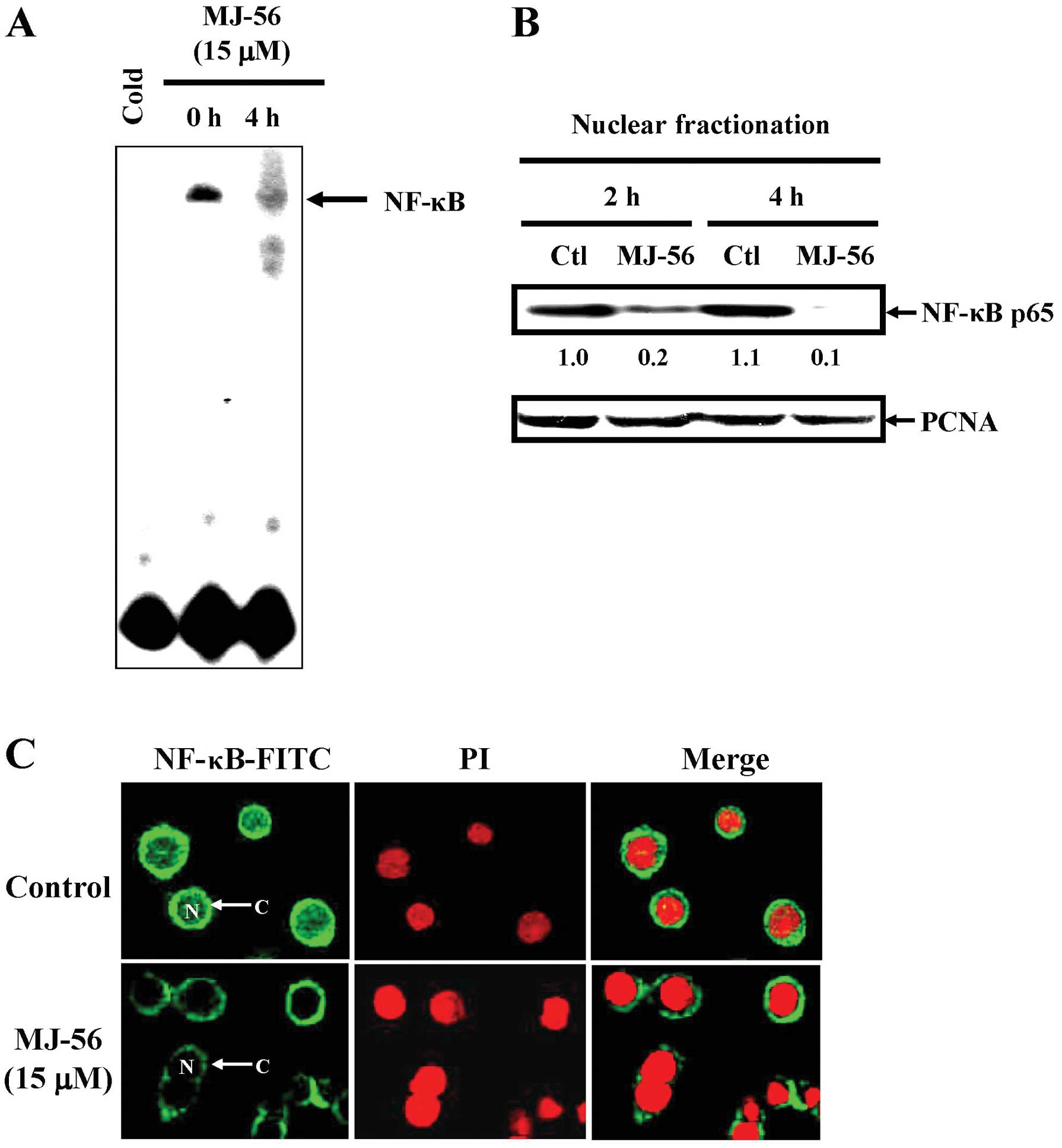
MJ-56 inhibits the NF-κB signaling
pathway in HT29 cells
It is known that PI3K/AKT signaling activates NF-κB.
Activated NF-κB mobilizes to the nucleus, binds to the cognate site
on the target promoters such as MMPs and turns on their gene
expression. To clarify the effects of MJ-56 on the NF-κB signaling
pathway, HT-29 cells were treated with 15 μM of MJ-56 for 0
and 4 h. Translocation of NF-κB into the nucleus resulting in
binding at NF-κB-responsive elements was accessed by
electrophoretic mobility shift assay (EMSA). After HT-29 cells were
treated with 15 μM of MJ-56 for 4 h, the amount of NF-κB
that binds DNA dramatically blocked due to reduced nuclear
translocation of NF-κB (Fig. 7A).
Nuclear extract was obtained from MJ-56-treated cells and assessed
for western blot analyses of NF-κB p65 (Fig. 7B). The results showed that the
mobilization of NF-κB into the nucleus was greatly reduced after 2
h of MJ-56 treatment, as compared to that of control treatment.
Consistently, immuno-fluorescent staining also showed that MJ-56
obstructs the translocation of NF-κB into the nucleus (Fig. 7C). These data suggest that MJ-56
interferes with the NF-κB signaling and therefore NF-κB target
genes such MMPs fail to be expressed.
Discussion
Previous studies demonstrated that quinazolinone
derivatives not only exerts anticancer activity against many cancer
cell lines in vitro and in vivo, but also induced
cell death through apoptosis or autophagy and inhibited cell
metastasis in cancer cells (28,49–52).
In our laboratory, a series of quinazolinone derivatives have been
designed and synthesized and which are established to have
anti-mitotic functions and anticancer activities in colorectal,
lung, ovarian, oral, prostate and breast cancer, as well as in
glioblastoma, osteosarcoma, melanoma and leukemia (28–30,49,50,53–55).
The MJ-56 (6-pyrrolidinyl-2-(3-bromostyryl)quinazolin-4-one), one
of the quinazolinone derivatives, exhibits the most potent
cytotoxicity against colorectal cancer cell lines including HT29,
COLO 205, SW480, SW620 and HCT116 (data not shown). In this study,
our results provide detailed evidence that MJ-56 could modulate
anti-cell migration and invasion effects and trigger MMPs
activities by EGFR and c-Met pathways in HT29 human colorectal
cancer cells. MMPs are known to be an accelerator of colorectal
cancer cell invasion and metastasis and there are associated with
the progression of tumorigenesis (8,9,11,14,24,39,47,48).
Our results demonstrated that MJ-56 can inhibit migration and
invasion of HT29 cells (Fig. 2).
MJ-56 reduced the protein levels of MMP-2, MMP-7, MMP-9, MMP-10
(Fig. 3A) as well as increased the
protein levels of metalloproteinase inhibitors TIMP-1 and TIMP-2 by
western blot analyses (Fig. 3B).
MJ-56 also reduced the enzymatic activities of MMP-2 and MMP-9 by
gelatin zymography assays (Fig.
3C).
Interference with receptor tyrosine kinase provides
a novel approach in cancer therapy agents (56–59).
Activation of the EGFR promotes processes responsible for cancer
cell proliferation, angiogenesis, invasion, metastasis and
inhibition of cell death (60–63).
The agents targeting members of the human epidermal growth factor
receptor (EGFR) have shown hopeful therapeutic efficacy (58,64).
Cetuximab (Erbitux) is a clinical success of selective EGFR
inhibitor. Cetuximab improves the effectiveness of treatment for
metastatic colorectal cancer (64–66).
In this study, we demonstrated that MJ-56 is an EGFR and c-Met
receptor tyrosine kinase inhibition agent using western blot and
kinase assay analyses (Fig. 4).
EGFR and c-Met are phosphorylated, which turn on downstream
intracellular signaling cascades such as MAPK, PI3K/AKT/mTOR and
NF-κB pathways. Our results suggested that inhibition of EGFR and
c-Met receptor tyrosine kinase activity by MJ-56 might be suitable
for novel targeted therapy of colorectal cancer.
MMP promoters have several regulatory motifs
recognized by various proteins such as NF-κB, S6, eIF-4B, eIF-4E
and eIF-4G. The NF-κB protein binding to the MMP-2, MMP-7 and MMP-9
promoter is centrally involved in the induction of MMP-2,
MMP-7 and MMP-9 gene expression associated with cell
invasion (24,28,30,46).
On the other hand, the S6, eIF-4B, eIF-4E and eIF-4G binding to the
MMP-7 promoter are centrally involved in the induction of
MMP-7 gene expression (24). Multiple pathways leading to
activation of those transcription factors in cancer cells may
contribute to MMP transcription and metastatic enhancement
(67–72). MJ-56 reduced the protein levels of
MMPs associated with inactivation of ERK (Fig. 5) and AKT (Fig. 6) as well as displayed inhibitory
effects on NF-κB, S6, eIF-4B, eIF-4E and eIF-4G (Fig. 6). The inhibitory effect on nuclear
entry of NF-κB was consistent with less DNA binding activity of
NF-κB (Figs. 7B and C). ERK is
intricately involved in the expression of the components that are
associated with MMP promoter induction through eIF-4E and its
relation with Mnk1 phosphorylation (73–76).
Activation of mTOR through the PI3K/AKT pathway leads to the
phosphorylation of 4EBP1 and S6K1. The 4EBP1 phosphorylation
inhibits the translation by interrupting the binding of eIF4E with
eIF4G to form an eIF4F translation initiation complex that consists
of eIF4A, 4G and 4E. Activated S6K1 causes phosphorylation of S6
and eIF4B, which, in turn, result in an increase in eIF4A RNA
helicase activity (24,77–82).
We suggested that PI3K/AKT/mTOR and ERK1/2 are the master
regulators of translation initiation in MMPs. Inhibition of the ERK
and PI3K/AKT pathways might have the potential of preventing cancer
cell invasion and migration.
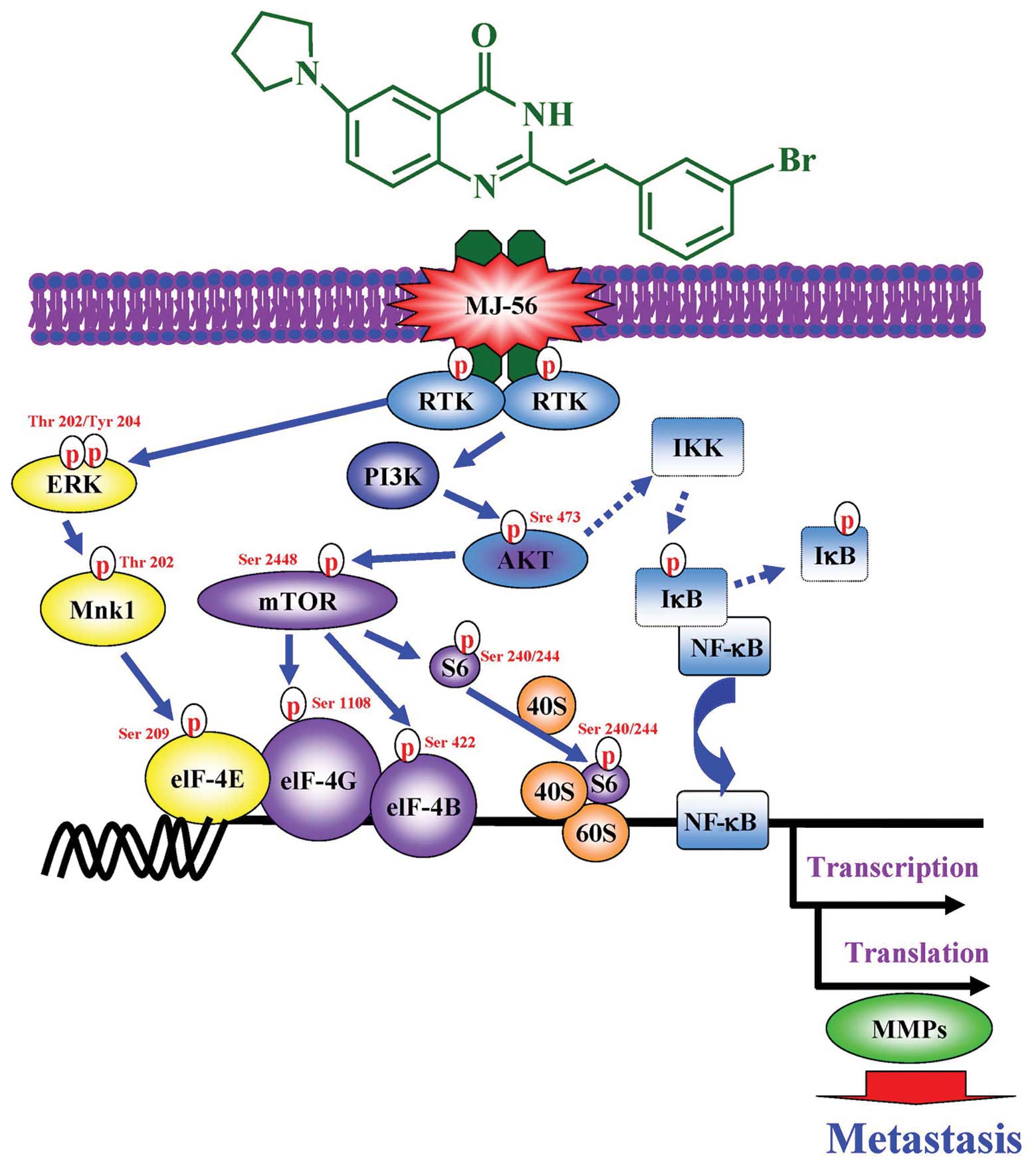
A model was proposed to elucidate molecular
mechanisms by which MJ-56 suppresses cell migration, invasion and
MMP activity in HT-29 human colorectal cancer cells (Fig. 8). In conclusion, MJ-56 might
inhibit invasion and migration of HT29 cells through targeting the
receptor tyrosine kinases, EGFR and c-Met. Thereafter, ERK and
PI3K/AKT/mTOR signaling pathways are inactive. NF-κB signaling
pathway is blocked, which leads to blocking of the transcription
and translation of matrix metalloproteinases.
Acknowledgements
This study was supported by a research
grant from the National Science Council of the Republic of China
(NSC 97-2320-B-039-004-MY3). This study was also supported in part
by grant from China Medical University (CMU101-S-27) awarded to
J.-S. Yang.
References
|
1.
|
Van Cutsem E, Dicato M, Arber N, et al:
Molecular markers and biological targeted therapies in metastatic
colorectal cancer: expert opinion and recommendations derived from
the 11th ESMO/World Congress on Gastrointestinal Cancer, Barcelona,
2009. Ann Oncol. 21(Suppl 6): vi1–vi10. 2010.
|
|
2.
|
Chu E: Colorectal cancer (CRC) continues
to be a major public health problem in the United States and
throughout the world. Cancer J. 16:1952010.PubMed/NCBI
|
|
3.
|
Hoogwater FJ, Nijkamp MW, Smakman N, et
al: Oncogenic K-Ras turns death receptors into metastasis-promoting
receptors in human and mouse colorectal cancer cells.
Gastroenterology. 138:2357–2367. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Vallbohmer D, Kuramochi H, Shimizu D, et
al: Molecular factors of 5-fluorouracil metabolism in colorectal
cancer: analysis of primary tumor and lymph node metastasis. Int J
Oncol. 28:527–533. 2006.PubMed/NCBI
|
|
5.
|
Jin K, Gao W, Lu Y, Lan H, Teng L and Cao
F: Mechanisms regulating colorectal cancer cell metastasis into
liver (Review). Oncol Lett. 3:11–15. 2012.PubMed/NCBI
|
|
6.
|
Ouellette JR, Harboe-Schmidt JE,
Luthringer D, Brackert S and Silberman AW: Colorectal cancer
metastasis presenting as a testicular mass: case report and review
of the literature. Am Surg. 73:79–81. 2007.PubMed/NCBI
|
|
7.
|
Oshima T, Kunisaki C, Yoshihara K, et al:
Clinicopathological significance of the gene expression of matrix
metalloproteinases and reversion-inducing cysteine-rich protein
with Kazal motifs in patients with colorectal cancer: MMP-2 gene
expression is a useful predictor of liver metastasis from
colorectal cancer. Oncol Rep. 19:1285–1291. 2008.
|
|
8.
|
Jensen SA, Vainer B, Bartels A, Brunner N
and Sorensen JB: Expression of matrix metalloproteinase 9 (MMP-9)
and tissue inhibitor of metalloproteinases 1 (TIMP-1) by colorectal
cancer cells and adjacent stroma cells - associations with
histopathology and patients outcome. Eur J Cancer. 46:3233–3242.
2010. View Article : Google Scholar
|
|
9.
|
Gershtein ES, Korotkova EA, Shcherbakov
AM, Prorokov VV, Golovkov DA and Kushlinskii NE: Matrix
metalloproteinases 7 and 9 and their types 1 and 4 tissue
inhibitors in tumors and plasma of patients with colorectal cancer.
Bull Exp Biol Med. 143:459–462. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Mysliwiec AG and Ornstein DL: Matrix
metalloproteinases in colorectal cancer. Clin Colorectal Cancer.
1:208–219. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Mroczko B, Groblewska M, Okulczyk B, Kedra
B and Szmitkowski M: The diagnostic value of matrix
metalloproteinase 9 (MMP-9) and tissue inhibitor of matrix
metalloproteinases 1 (TIMP-1) determination in the sera of
colorectal adenoma and cancer patients. Int J Colorectal Dis.
25:1177–1184. 2010. View Article : Google Scholar
|
|
12.
|
Groblewska M, Mroczko B and Szmitkowski M:
The role of selected matrix metalloproteinases and their inhibitors
in colorectal cancer development. Postepy Hig Med Dosw (Online).
64:22–30. 2010.(In Polish).
|
|
13.
|
Roeb E and Matern S: Matrix
metalloproteinases: promoters of tumor invasion and metastasis - a
review with focus on gastrointestinal tumors. Z Gastroenterol.
39:807–813. 2001.(In German).
|
|
14.
|
Gullu IH, Kurdoglu M and Akalin I: The
relation of gelatinase (MMP-2 and -9) expression with distant site
metastasis and tumour aggressiveness in colorectal cancer. Br J
Cancer. 82:2492000.PubMed/NCBI
|
|
15.
|
Takeha S, Fujiyama Y, Bamba T, Sorsa T,
Nagura H and Ohtani H: Stromal expression of MMP-9 and urokinase
receptor is inversely associated with liver metastasis and with
infiltrating growth in human colorectal cancer: a novel approach
from immune/inflammatory aspect. Jpn J Cancer Res. 88:72–81. 1997.
View Article : Google Scholar
|
|
16.
|
Dziki L, Przybylowska K, Majsterek I,
Trzcinski R, Mik M and Sygut A: A/G polymorphism of the MMP-7 gene
promoter region in colorectal cancer. Pol Przegl Chir. 83:622–626.
2011.PubMed/NCBI
|
|
17.
|
Ichikawa Y, Ishikawa T, Momiyama N, et al:
Function of MMP-7 in colorectal cancer. Nihon Rinsho. 61(Suppl 7):
209–214. 2003.(In Japanese).
|
|
18.
|
Thorp E, Vaisar T, Subramanian M, Mautner
L, Blobel C and Tabas I: Shedding of the Mer tyrosine kinase
receptor is mediated by ADAM17 protein through a pathway involving
reactive oxygen species, protein kinase Cdelta and p38
mitogen-activated protein kinase (MAPK). J Biol Chem.
286:33335–33344. 2011. View Article : Google Scholar
|
|
19.
|
Lou X, Zhou Q, Yin Y, Zhou C and Shen Y:
Inhibition of the met receptor tyrosine kinase signaling enhances
the chemosensitivity of glioma cell lines to CDDP through
activation of p38 MAPK pathway. Mol Cancer Ther. 8:1126–1136. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Narkar V, Hussain T and Lokhandwala M:
Role of tyrosine kinase and p44/42 MAPK in D(2)-like
receptor-mediated stimulation of Na(+), K(+)-ATPase in kidney. Am J
Physiol Renal Physiol. 282:F697–F702. 2002.PubMed/NCBI
|
|
21.
|
Hinohara K, Kobayashi S, Kanauchi H, et
al: ErbB receptor tyrosine kinase/NF-kappaB signaling controls
mammosphere formation in human breast cancer. Proc Natl Acad Sci
USA. 109:6584–6589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lee YJ, Han JY, Byun J, et al: Inhibiting
Mer receptor tyrosine kinase suppresses STAT1, SOCS1/3 and
NF-kappaB activation and enhances inflammatory responses in
lipopolysaccharide-induced acute lung injury. J Leukoc Biol.
91:921–932. 2012. View Article : Google Scholar
|
|
23.
|
Petro JB, Castro I, Lowe J and Khan WN:
Bruton’s tyrosine kinase targets NF-kappaB to the bcl-x promoter
via a mechanism involving phospholipase C-gamma2 following B cell
antigen receptor engagement. FEBS Lett. 532:57–60. 2002.
|
|
24.
|
Kawabata K, Murakami A and Ohigashi H:
Citrus auraptene targets translation of MMP-7 (matrilysin) via
ERK1/2-dependent and mTOR-independent mechanism. FEBS Lett.
580:5288–5294. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Brenneisen P, Wenk J, Wlaschek M, Krieg T
and Scharffetter-Kochanek K: Activation of p70 ribosomal protein S6
kinase is an essential step in the DNA damage-dependent signaling
pathway responsible for the ultraviolet B-mediated increase in
interstitial collagenase (MMP-1) and stromelysin-1 (MMP-3) protein
levels in human dermal fibroblasts. J Biol Chem. 275:4336–4344.
2000.
|
|
26.
|
Mosaad MS, Mohsen KM, Emad KM, Abotaleb N,
Salwa NM and Marwa AF: Novel 6,8-dibromo-4(3H)-quinazolinone
derivatives of promising anti-inflammatory and analgesic
properties. Acta Pol Pharm. 67:159–171. 2010.PubMed/NCBI
|
|
27.
|
Bekhit AA, Habib NS and Park JY: Synthesis
of some thiazolyl and thiadiazolyl derivatives of
4(3H)-quinazolinone as anti-inflammatory-antimicrobial agents. Boll
Chim Farm. 143:34–39. 2004.PubMed/NCBI
|
|
28.
|
Hour MJ, Tsai SC, Wu HC, et al: Antitumor
effects of the novel quinazolinone MJ-33: Inhibition of metastasis
through the MAPK, AKT, NF-κB and AP-1 signaling pathways in DU145
human prostate cancer cells. Int J Oncol. 41:1513–1519.
2012.PubMed/NCBI
|
|
29.
|
Chen KT, Hour MJ, Tsai SC, et al: The
novel synthesized
6-fluoro-(3-fluorophenyl)-4-(3-methoxyanilino)quinazoline (LJJ-10)
compound exhibits anti-metastatic effects in human osteosarcoma U-2
OS cells through targeting insulin-like growth factor-I receptor.
Int J Oncol. 39:611–619. 2011.
|
|
30.
|
Hour MJ, Yang JS, Chen TL, et al: The
synthesized novel fluorinated compound (LJJ-10) induces death
receptor- and mitochondria-dependent apoptotic cell death in the
human osteogenic sarcoma U-2 OS cells. Eur J Med Chem.
46:2709–2721. 2011. View Article : Google Scholar
|
|
31.
|
Wang MH, Padhye SS, Guin S, Ma Q and Zhou
YQ: Potential therapeutics specific to c-MET/RON receptor tyrosine
kinases for molecular targeting in cancer therapy. Acta Pharmacol
Sin. 31:1181–1188. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Uckun FM, Vassilev A and Tibbles H:
Non-receptor tyrosine kinases as molecular targets for patient
tailored cancer therapy. Anticancer Agents Med Chem. 7:5932007.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Gschwind A, Fischer OM and Ullrich A: The
discovery of receptor tyrosine kinases: targets for cancer therapy.
Nat Rev Cancer. 4:361–370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Brunelleschi S, Penengo L, Santoro MM and
Gaudino G: Receptor tyrosine kinases as target for anti-cancer
therapy. Curr Pharm Des. 8:1959–1972. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Rodig SJ and Shapiro GI: Crizotinib, a
small-molecule dual inhibitor of the c-Met and ALK receptor
tyrosine kinases. Curr Opin Investig Drugs. 11:1477–1490.
2010.PubMed/NCBI
|
|
36.
|
You WK, Sennino B, Williamson CW, et al:
VEGF and c-Met blockade amplify angiogenesis inhibition in
pancreatic islet cancer. Cancer Res. 71:4758–4768. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Garouniatis A, Zizi-Sermpetzoglou A, Rizos
S, Kostakis A, Nikiteas N and Papavassiliou AG: FAK, CD44v6, c-Met
and EGFR in colorectal cancer parameters: tumour progression,
metastasis, patient survival and receptor crosstalk. Int J
Colorectal Dis. 28:9–18. 2013. View Article : Google Scholar
|
|
38.
|
Zhao J, Zhang X and Xin Y: Up-regulated
expression of Ezrin and c-Met proteins are related to the
metastasis and prognosis of gastric carcinomas. Histol Histopathol.
26:1111–1120. 2011.PubMed/NCBI
|
|
39.
|
Liska D, Chen CT, Bachleitner-Hofmann T,
Christensen JG and Weiser MR: HGF rescues colorectal cancer cells
from EGFR inhibition via MET activation. Clin Cancer Res.
17:472–482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Cunningham MP, Thomas H, Marks C, Green M,
Fan Z and Modjtahedi H: Co-targeting the EGFR and IGF-IR with
anti-EGFR monoclonal antibody ICR62 and the IGF-IR tyrosine kinase
inhibitor NVP-AEW541 in colorectal cancer cells. Int J Oncol.
33:1107–1113. 2008.PubMed/NCBI
|
|
41.
|
Yang JS, Wu CC, Kuo CL, et al: Solanum
lyratum extracts induce extrinsic and intrinsic pathways of
apoptosis in WEHI-3 murine leukemia cells and inhibit allograft
tumor. Evid Based Complement Alternat Med.
2012:2549602012.PubMed/NCBI
|
|
42.
|
Jia W, Hegde VL, Singh NP, et al:
Delta9-tetrahydrocannabinol-induced apoptosis in Jurkat leukemia T
cells is regulated by translocation of Bad to mitochondria. Mol
Cancer Res. 4:549–562. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
He Z, Cho YY, Ma WY, Choi HS, Bode AM and
Dong Z: Regulation of ultraviolet B-induced phosphorylation of
histone H3 at serine 10 by Fyn kinase. J Biol Chem. 280:2446–2454.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Jost M, Huggett TM, Kari C, Boise LH and
Rodeck U: Epidermal growth factor receptor-dependent control of
keratinocyte survival and Bcl-xL expression through a MEK-dependent
pathway. J Biol Chem. 276:6320–6326. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Abdel-Ghany M, el-Gendy K, Zhang S and
Racker E: Control of src kinase activity by activators, inhibitors
and substrate chaperones. Proc Natl Acad Sci USA. 87:7061–7065.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Spano JP, Milano G and Baselga J:
EGFR/VEGF signalling pathway in colorectal cancer: the way we are!
Bull Cancer. 92:S3–S4. 2005.PubMed/NCBI
|
|
47.
|
Ishikawa T, Uetake H and Sugihara K:
Anti-EGFR antibody therapy for colorectal cancer. Nihon Rinsho.
70:2152–2158. 2012.(In Japanese).
|
|
48.
|
Mimori K, Yamashita K, Ohta M, et al:
Coexpression of matrix metalloproteinase-7 (MMP-7) and epidermal
growth factor (EGF) receptor in colorectal cancer: an EGF receptor
tyrosine kinase inhibitor is effective against MMP-7-expressing
cancer cells. Clin Cancer Res. 10:8243–8249. 2004. View Article : Google Scholar
|
|
49.
|
Yang JS, Hour MJ, Huang WW, Lin KL, Kuo SC
and Chung JG: MJ-29 inhibits tubulin polymerization, induces
mitotic arrest and triggers apoptosis via cyclin-dependent kinase
1-mediated Bcl-2 phosphorylation in human leukemia U937 cells. J
Pharmacol Exp Ther. 334:477–488. 2010. View Article : Google Scholar
|
|
50.
|
Lu CC, Yang JS, Chiang JH, et al:
Inhibition of invasion and migration by newly synthesized
quinazolinone MJ-29 in human oral cancer CAL 27 cells through
suppression of MMP-2/9 expression and combined down-regulation of
MAPK and AKT signaling. Anticancer Res. 32:2895–2903.
2012.PubMed/NCBI
|
|
51.
|
Pospisil P, Korideck H, Wang K, Yang Y,
Iyer LK and Kassis AI: Computational and biological evaluation of
quinazolinone prodrug for targeting pancreatic cancer. Chem Biol
Drug Des. 79:926–934. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Wu YC, Hour MJ, Leung WC, et al:
2-(Naphthalene-1-yl)-6-pyrrolidinyl-4-quinazolinone inhibits skin
cancer M21 cell proliferation through aberrant expression of
microtubules and the cell cycle. J Pharmacol Exp Ther. 338:942–951.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Lu CC, Yang JS, Chiang JH, et al: Novel
quinazolinone MJ-29 triggers endoplasmic reticulum stress and
intrinsic apoptosis in murine leukemia WEHI-3 cells and inhibits
leukemic mice. PLoS One. 7:e368312012. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Chiu YJ, Hour MJ, Lu CC, et al: Novel
quinazoline HMJ-30 induces U-2 OS human osteogenic sarcoma cell
apoptosis through induction of oxidative stress and up-regulation
of ATM/p53 signaling pathway. J Orthop Res. 29:1448–1456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Yang JS, Hour MJ, Kuo SC, Huang LJ and Lee
MR: Selective induction of G2/M arrest and apoptosis in HL-60 by a
potent anticancer agent, HMJ-38. Anticancer Res. 24:1769–1778.
2004.PubMed/NCBI
|
|
56.
|
Garcia ED: Targeted therapy for cancer:
anti-tyrosine kinase receptor agents. An R Acad Nac Med (Madr).
124:171–184. 2007.(In Spanish).
|
|
57.
|
Myers MV, Manning HC, Coffey RJ and
Liebler DC: Protein expression signatures for inhibition of
epidermal growth factor receptor-mediated signaling. Mol Cell
Proteomics. 11:M111 015222,. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Goldwasser F: Treatment of metastatic
colorectal cancer: an illustration of the changes in the cancer
paradigms. Presse Med. 41:46–50. 2012.(In French).
|
|
59.
|
Shaw PH and Adams RA: Where now for
anti-EGF receptor therapies in colorectal cancer? Expert Rev
Anticancer Ther. 11:1543–1553. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Takhar AS, Eremin O and Watson SA: The
role of gastrin in colorectal carcinogenesis. Surgeon. 2:251–257.
2004. View Article : Google Scholar
|
|
61.
|
Cohen RB: Epidermal growth factor receptor
as a therapeutic target in colorectal cancer. Clin Colorectal
Cancer. 2:246–251. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Yokozaki H and Tahara E:
Metastasis-related genes. Gan To Kagaku Ryoho. 21:2541–2548.
1994.(In Japanese).
|
|
63.
|
Wilson KJ, Gilmore JL, Foley J, Lemmon MA
and Riese DJ II: Functional selectivity of EGF family peptide
growth factors: implications for cancer. Pharmacol Ther. 122:1–8.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Broadbridge VT, Karapetis CS and Price TJ:
Cetuximab in metastatic colorectal cancer. Expert Rev Anticancer
Ther. 12:555–565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Harding J and Burtness B: Cetuximab: an
epidermal growth factor receptor chemeric human-murine monoclonal
antibody. Drugs Today (Barc). 41:107–127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Takayama T, Goji T, Taniguchi T and Inoue
A: Chemoprevention of colorectal cancer-experimental and clinical
aspects. J Med Invest. 56:1–5. 2009. View Article : Google Scholar
|
|
67.
|
Ho BY, Wu YM, Chang KJ and Pan TM:
Dimerumic acid inhibits SW620 cell invasion by attenuating
H(2)O(2)-mediated MMP-7 expression via JNK/C-Jun and ERK/C-Fos
activation in an AP-1-dependent manner. Int J Biol Sci. 7:869–880.
2011.PubMed/NCBI
|
|
68.
|
Zugowski C, Lieder F, Muller A, et al:
STAT3 controls matrix metalloproteinase-1 expression in colon
carcinoma cells by both direct and AP-1-mediated interaction with
the MMP-1 promoter. Biol Chem. 392:449–459. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Fang YJ, Lu ZH, Wang GQ, et al: Elevated
expressions of MMP7, TROP2 and survivin are associated with
survival, disease recurrence and liver metastasis of colon cancer.
Int J Colorectal Dis. 24:875–884. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Shin JE, Jung SA, Kim SE, et al:
Expression of MMP-2, HIF-1alpha and VEGF in colon adenoma and colon
cancer. Korean J Gastroenterol. 50:9–18. 2007.(In Korean).
|
|
71.
|
Zinzindohoue F, Lecomte T, Ferraz JM, et
al: Prognostic significance of MMP-1 and MMP-3 functional promoter
polymorphisms in colorectal cancer. Clin Cancer Res. 11:594–599.
2005.PubMed/NCBI
|
|
72.
|
Yamamoto H, Itoh F, Senota A, et al:
Expression of matrix metalloproteinase matrilysin (MMP-7) was
induced by activated Ki-ras via AP-1 activation in SW1417 colon
cancer cells. J Clin Lab Anal. 9:297–301. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Ueda T, Sasaki M, Elia AJ, et al: Combined
deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and
Mnk2) delays tumor development. Proc Natl Acad Sci USA.
107:13984–13990. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Joshi S, Kaur S, Redig AJ, et al: Type I
interferon (IFN)-dependent activation of Mnk1 and its role in the
generation of growth inhibitory responses. Proc Natl Acad Sci USA.
106:12097–12102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Zhang Y, Li Y and Yang DQ: Phosphorylation
of eIF-4E positively regulates formation of the eIF-4F translation
initiation complex following DNA damage. Biochem Biophys Res
Commun. 367:54–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Duncan RF, Peterson H and Sevanian A:
Signal transduction pathways leading to increased eIF4E
phosphorylation caused by oxidative stress. Free Radic Biol Med.
38:631–643. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Populo H, Lopes JM and Soares P: The mTOR
signalling pathway in human cancer. Int J Mol Sci. 13:1886–1918.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Hong S, Mannan AM and Inoki K: Evaluation
of the nutrient-sensing mTOR pathway. Methods Mol Biol. 821:29–44.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Nyfeler B, Bergman P, Triantafellow E, et
al: Relieving autophagy and 4EBP1 from rapamycin resistance. Mol
Cell Biol. 31:2867–2876. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Ha SH, Kim DH, Kim IS, et al: PLD2 forms a
functional complex with mTOR/raptor to transduce mitogenic signals.
Cell Signal. 18:2283–2291. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
81.
|
Arvisais EW, Romanelli A, Hou X and Davis
JS: AKT-independent phosphorylation of TSC2 and activation of mTOR
and ribosomal protein S6 kinase signaling by prostaglandin F2alpha.
J Biol Chem. 281:26904–26913. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
82.
|
Hannan KM, Brandenburger Y, Jenkins A, et
al: mTOR-dependent regulation of ribosomal gene transcription
requires S6K1 and is mediated by phosphorylation of the
carboxy-terminal activation domain of the nucleolar transcription
factor UBF. Mol Cell Biol. 23:8862–8877. 2003. View Article : Google Scholar
|