Introduction
Many new therapeutic maneuvers during the last
decade relates to the introduction of novel biologically targeted
therapeutic agents, improving surgical techniques, and increased
utilization of concomitant chemoradiotherapy for locally advanced
lung cancer (1). Despite this, the
success rates for treatment remain quite dismal, and the limited
options for the management of lung cancer have necessitated the
search for novel preventive approaches for this disease. The
advantages of using a combinatorial chemopreventive approach, in
which 2 or more critical molecules are simultaneously targeted,
include lower dose requirements, reduced incidence of associated
toxicities, and increased likelihood of human acceptability
(2–5).
Non-steroidal anti-inflammatory drugs (NSAIDs) have
attracted substantial attention after the discovery that sulindac
induces the regression of colon adenomatous polyps in cancer
therapy (6). Sulindac, a
structural isoform of indomethacin, exerts antiproliferative and
apoptotic effects, which eventually lead to the regression of
cancer cells (7,8). Many studies have revealed a link
between COX-2 expression and carcinogenesis, suggesting that COX-2
inhibition can prevent cancer growth or progression (9). However, sulindac sulfone, a
metabolite of sulindac that lacks the ability to inhibit COX-2,
reduces the incidence of tumors in breast and colon cancer
(10,11), indicating the involvement of other
mechanisms such as the production of reactive oxygen species (ROS)
and the inhibition of NFκB-mediated signals (12,13).
Statins are a class of drugs that inhibit the
rate-limiting step of the mevalonate pathway, which is catalyzed by
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (14). Besides their lipid-lowering effect,
statins have been studied for their anticarcinogenic properties in
various cancer cell types, including carcinomas of the colon,
prostate, breast, lung and skin (15,16),
which may make them relevant to cancer prevention or treatment.
Many studies have shown that the antiproliferative and proapoptotic
effects of statins are more pronounced in malignant than in
non-malignant cells (17,18). Moreover, low concentrations of
statins have been shown to sensitize cancer cell lines to
cytostatic drugs such as 5-fluorouracil (5-FU), taxol, etoposide,
doxorubicin and cisplatin (19–22).
The relatively high dose required for the observed
chemopreventive effect of NSAIDs in lung cancer patients may
discourage their individual use on a long-term basis for lung
cancer prevention due to the potentially increased risk of serious
gastrointestinal and cardiovascular side-effects (23–25).
However, lower doses of NSAIDs may prove to be more beneficial in
the prevention of lung cancer when administered in combination with
other complementary agents. For example, many reports have
indicated that NSAIDs possess synergistic effects in combination
with other therapeutics such as statins, PPARγ ligands, epidermal
growth factor receptor tyrosine kinase inhibitors (EGFR-TKI) and
TRAIL receptor ligands (26).
Recently, considerable evidence has suggested that
combination treatment of NSAIDs and statin synergistically induced
apoptosis in cancer cells (27,28).
However, the mechanism underlying deregulated survivin by NSAIDs
and statin on human non-small cell lung cancer cells has not been
elucidated. Our results suggest that a mechanism-based approach,
using sulindac at low doses in combination with simvastatin, has
potential in the chemoprevention of lung cancer.
Materials and methods
Materials
RPMI-1640, fetal bovine serum (FBS), and antibiotics
were obtained from Gibco-BRL Co (Grand Island, NY). Sulindac,
simvastatin,
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT), propidium iodide (PI), bicinchoninic acid, dimethyl
sulfoxide (DMSO) and N-acetylcysteine (NAC) were purchased from
Sigma-Aldrich (St. Louis, MO). JC-1 was obtained from Molecular
Probes (Eugene, OR). Primary antibodies against the following
targets were used: caspase-3, -8 and -9; poly(ADP-ribose)
polymerase (PARP); Puma; Bim; Mcl-1; Bcl-XL; and XIAP (Santa Cruz
Biotechnology, Santa Cruz, CA); serine/threonine protein kinase
(Akt); phospho-Akt; JNK; phosphor-JNK; p38; phospho-p38; survivin;
GAPDH (Cell Signaling Technology, Beverly, MA); and cytochrome
c (Pharmingen, San Diego, CA). Anti-rabbit IgG-conjugated
horseradish peroxidase (HRP) antibodies and enhanced
chemiluminescent (ECL) kit were purchased from Amersham Pharmacia
Biotech (Buckinghamshire, UK).
Cell culture and viability test
A549 human lung cancer cells were obtained from the
Korean Cell Line Bank (Seoul, Korea) and grown in RPMI-1640
containing 100 U/ml penicillin, 0.1 mg/ml streptomycin and 10% FBS,
and they were maintained in a humidified atmosphere of 5%
CO2 in air at 37°C and maintained in the log phase. Cell
viability was determined by measuring the mitochondrial conversion
of MTT to a colored product. Cells were treated with the specified
drugs. To determine cell viability, MTT was added to the cell
suspension for 4 h. After 3 washes with phosphate-buffered saline
(PBS; pH 7.4), the insoluble formazan product was dissolved in
DMSO. The optical density (OD) of each well was then measured using
a microplate reader (Titertek Multiskan; Flow Laboratories, North
Ryde, Australia) at 590 nm. The OD resulting from formazan
production in control cells was considered as 100% cell viability,
and all other measurements were expressed as a percentage of the
control cell value.
Annexin V assay for the assessment of
apoptosis
A549 cells undergoing early/late apoptosis were
analyzed by Annexin V-FITC and PI staining. In all,
2.5×105 cells in the exponential growth phase were
seeded in 60-mm2 dishes. Cells were left untreated or
incubated with specified drugs for the indicated times at 37°C.
Both adherent and floating cells were collected and analyzed by the
Annexin V assay, according to the manufacturer’s instructions.
Pelleted cells were briefly washed with PBS and resuspended in an
Annexin binding buffer (BD Pharmingen). Cells were then incubated
with Annexin V-phycoerythrin and propidium iodide for 15 min at
room temperature. After incubation, the stained cells were analyzed
using a FACScan system equipped with Cell Quest software
(Becton-Dickinson, San Jose, CA). Cells with no drug treatment were
used as controls.
Measurement of the mitochondrial membrane
potential (ΔΨm)
Cells were harvested at the indicated treatment time
points, washed with PBS, and then stained with 10 μg/ml JC-1
at 37°C for 30 min. After a brief wash with serum-free medium,
cells were immediately analyzed using a FACScan system equipped
with a Cell Quest software (Becton-Dickinson). At low
concentrations, JC-1 exists mainly in a monomeric form, emitting
green fluorescence (emission maximum at 530 nM), whereas at higher
concentrations it forms aggregates, known as J-aggregates, which
emit orange-red fluorescence (emission maximum at 590 nM).
Measurement of reactive oxygen
species
A549 cells were incubated in the dark with 10
μmol/l 5- (and -6)-carboxy-2′,7′-dichlorodihydrofluorescein
diacetate, carboxy-H2DCFDA (Molecular Probes) for 30
min. Cells were then washed, scraped gently, resuspended in PBS,
and kept on ice for immediate detection by FACScan flow cytometry
using an argon laser (488 nm) for excitation. Green fluorescence
due to intracellularly trapped DCF was collected on the FL1 channel
on a log scale. Data were acquired and analyzed using the Cell
Quest program (Becton-Dickinson).
Western blot analysis
Cells were harvested and lysed using
radioimmunoprecipitation assay buffer [50 mM Tris-Cl (pH 7.4), 1%
NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride,
1 μg/ml each of aprotinin and leupeptin, and 1 mM
Na3VO4]. After centrifugation at 12,000 × g
for 30 min, the supernatant was collected, and the protein
concentration was determined by the Lowry method (29). Equal amounts of protein were
separated on 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) gels under reducing conditions and
subsequently transferred to nitrocellulose membranes. Membranes
were blocked with 5% skim milk in TBS-T [25 mM Tris (pH 7.6), 138
mM NaCl and 0.05% Tween-20] for 1 h and probed with primary
antibodies (at 1:1,000–1:5,000). After a series of washes,
membranes were further incubated with secondary antibody (at
1:2,000–1:10,000) conjugated with HRP. Detection of the
immunoreactive signals was carried out using an ECL detection
system (Amersham Pharmacia Biotech).
Preparation of cytosolic and
mitochondrial fractions
Cytosolic and mitochondrial fractions were prepared
as described previously (30) with
certain modifications. Briefly, A549 cells were harvested, washed
with ice-cold PBS, and then incubated with 500 μM buffer A
[250 mM sucrose; 20 mM HEPES (pH 7.5); 10 mM KCl; 1.5 mM
MgCl2; 1 mM EGTA; 1 mM EDTA; 1 mM DTT; 1 mM PMSF; and 10
μg/ml each of leupeptin, aprotinin and pepstatin A] on ice
for 30 min. Cells were then disrupted by 20 passages through a
26-gauge needle and centrifuged at 750 × g for 10 min. The
supernatant was centrifuged at 10,000 × g for 25 min. After
centrifugation, the cytosolic fraction was frozen at 70°C. The
pellet containing mitochondria was washed with ice-cold buffer A
and then resuspended with cell lysis buffer. The resuspended pellet
was incubated on ice for 30 min and then centrifuged at 10,000 × g
for 25 min. The supernatant thus collected represented the
cytosolic fraction of A549 cells.
Statistical analysis
Each experiment was performed at least 3 times, and
all values are represented as the means ± SD of triplicate samples.
The Student’s t-test was used to determine the statistical
significance of the results. Values of p<0.05 were considered
statistically significant.
Results
Effect of sulindac and simvastatin, alone
and in combination, on the growth of A549 lung cancer cells
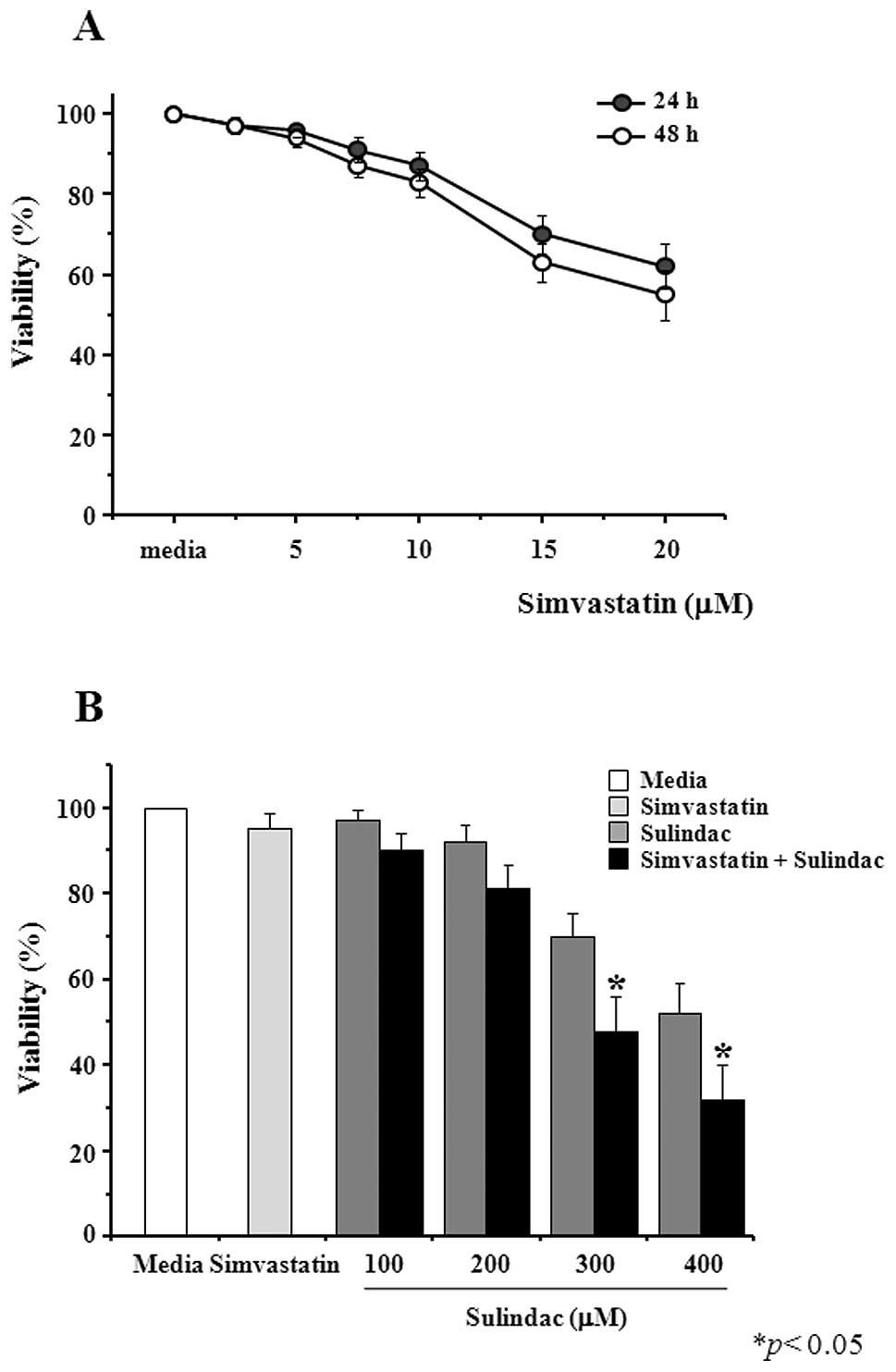
A549 cells were treated with simvastatin after which
their viability was measured by the MTT assay. As shown in Fig. 1A, simvastatin is cytotoxic at
concentrations equal to or greater than 5 μM and up to 48 h
of treatment. We next examined the combined effects of 5 μM
simvastatin and increasing concentrations of sulindac. In the
presence of simvastatin, sulindac-induced cytotoxicity was
significantly enhanced. Incubation of A549 cells with 300 μM
sulindac alone for 48 h decreased cell viability to 70%, whereas
co-treatment with 5 μM simvastatin resulted in 48% viability
(Fig. 1B).
Combination of sulindac and simvastatin
enhances caspase-dependent apoptosis
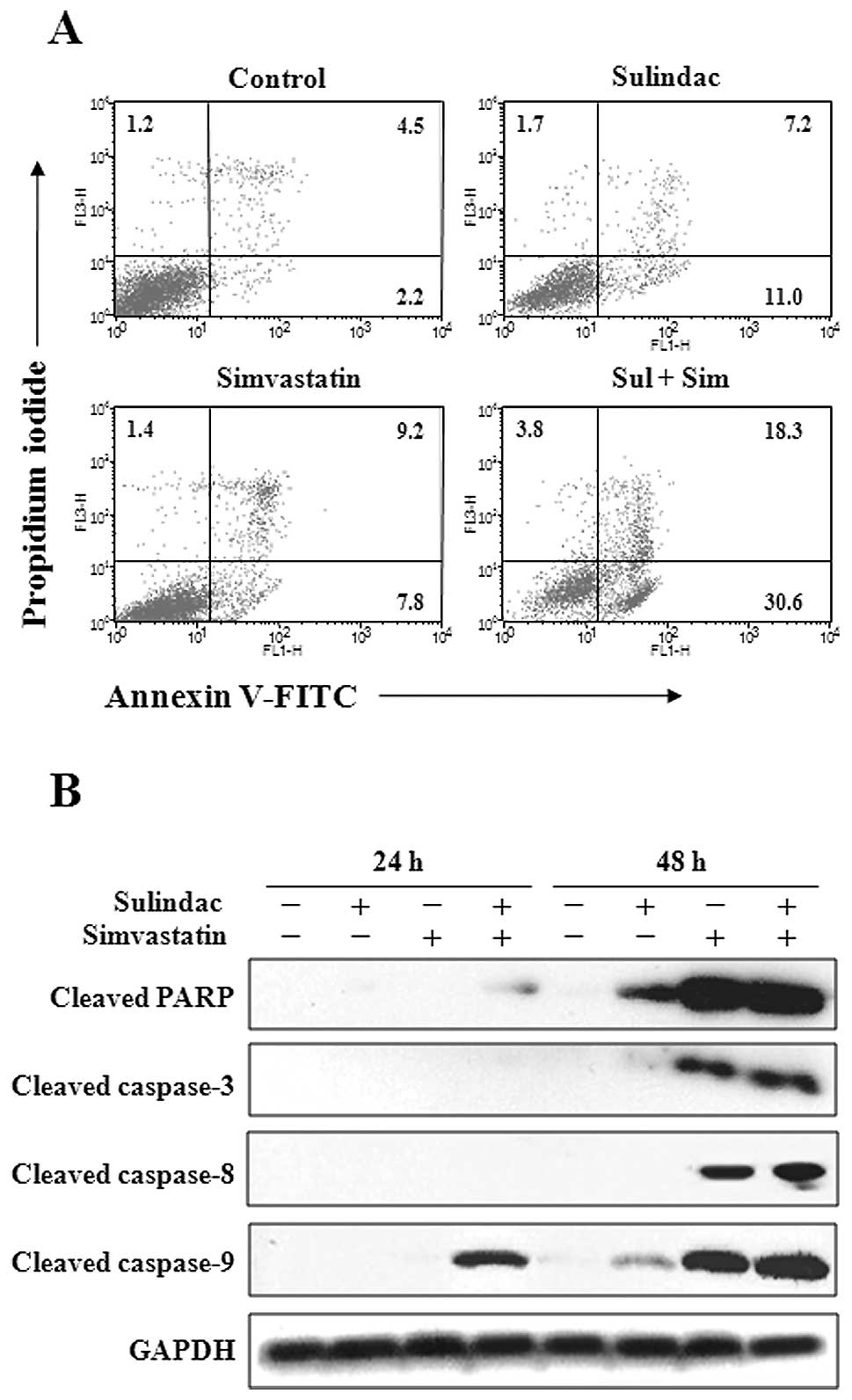
To examine whether the observed growth inhibition
was due to enhanced apoptosis, the proportion of apoptotic cells
was determined using Annexin V-propidium iodide staining, which is
more sensitive for detecting apoptosis than the methods based on
hypodiploid DNA content. Annexin V staining showed that the
combination of sulindac and simvastatin significantly enhanced
apoptosis compared with individual treatment with either drug. As
shown in Fig. 2A, individual
treatment with 300 μM sulindac or 5 μM simvastatin
resulted in apoptosis rates of 18.2 and 17.0%, respectively,
whereas 48.9% Annexin V-positive cells were observed after combined
treatment with both drugs. To further elucidate the mechanism of
apoptosis induced by sulindac and simvastatin, cell lysates were
evaluated by immunoblot analysis (Fig.
2B). Our results showed that the combination of sulindac and
simvastatin enhanced the expression of the processed 85-kDa isoform
of PARP, which is known to play a major role in evading the
apoptosis process. Moreover, combination of sulindac and
simvastatin led to a marked increase in the expression of
caspase-3, -8 and -9. These results indicate that sulindac and
simvastatin play a major role in enhancing caspase-dependent
apoptosis in A549 cells.
Combination of sulindac and simvastatin
leads to mitochondrial dysfunction
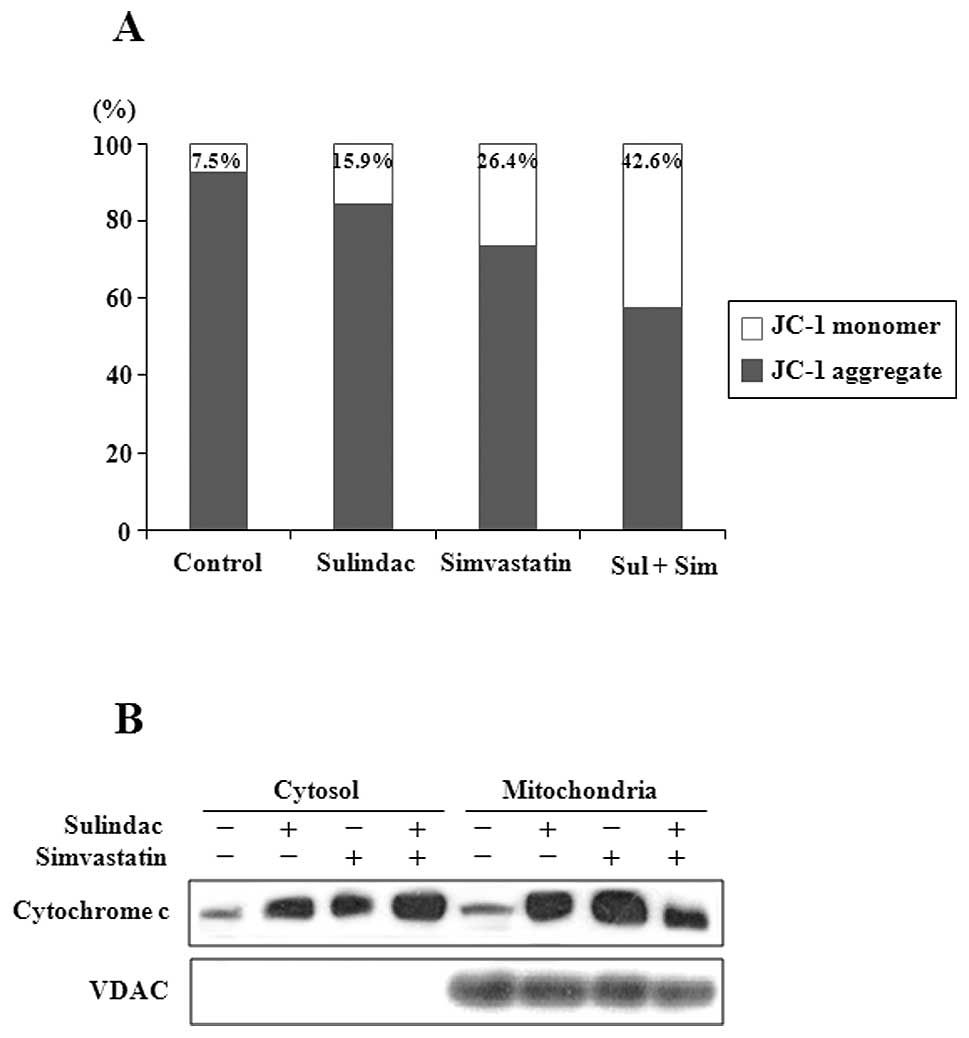
To identify components upstream of caspase-3 in
apoptotic signaling, the status of markers of mitochondrial
dysfunction, including mitochondrial membrane potential transition
(MPT) and cytosolic release of cytochrome c, was evaluated
in cells treated with sulindac and simvastatin. JC-1 is a cationic
dye that exhibits potential-dependent accumulation in mitochondria,
indicated by a fluorescence emission shift from green (525 nm) to
red (590 nm). Therefore, JC-1 has been widely used for the
detection of apoptosis by mitochondrial depolarization, as
indicated by a decrease in the red/green fluorescence intensity
ratio (31). As shown in Fig. 3A, individual treatment with 300
μM sulindac or 5 μM simvastatin resulted in JC-1
monomer levels of 15.9 and 26.4%, respectively, whereas a JC-1
monomer level of 43% was observed in cells treated with the drug
combination. Since the loss of mitochondrial transmembrane
potential (ΔΨm) results in cytochrome c release
into the cytosol, cytochrome c levels were evaluated by
western blot analysis in both mitochondrial and cytosolic fractions
(Fig. 3B). Combination treatment
with sulindac and simvastatin was associated with an increased
cytosolic level of cytochrome c and a corresponding decrease
in its mitochondrial level.
Combination of sulindac and simvastatin
elicits different effects on survival and stress signaling
pathways
To better understand the increased sensitivity of
lung cancer cells to a combination treatment with sulindac and
simvastatin, we next examined the role of a variety of signal
transduction pathways in the modulation of apoptosis. The status of
protein kinase B (Akt), c-Jun NH2-terminal kinase (JNK), and p38
mitogen-activated protein kinase (p38 MAPK) was evaluated in A549
cells treated with sulindac and/or simvastatin for 24 and 48 h
(Fig. 4A). The combination of
sulindac and simvastatin treatment resulted in a significant
time-dependent attenuation of phosphorylated Akt compared to cells
treated with either sulindac or simvastatin alone. In contrast, the
combined drug treatment resulted in enhanced phosphorylated JNK and
p38, compared with single drug treatment. Total protein and GAPDH
levels were unaffected by either treatment type.
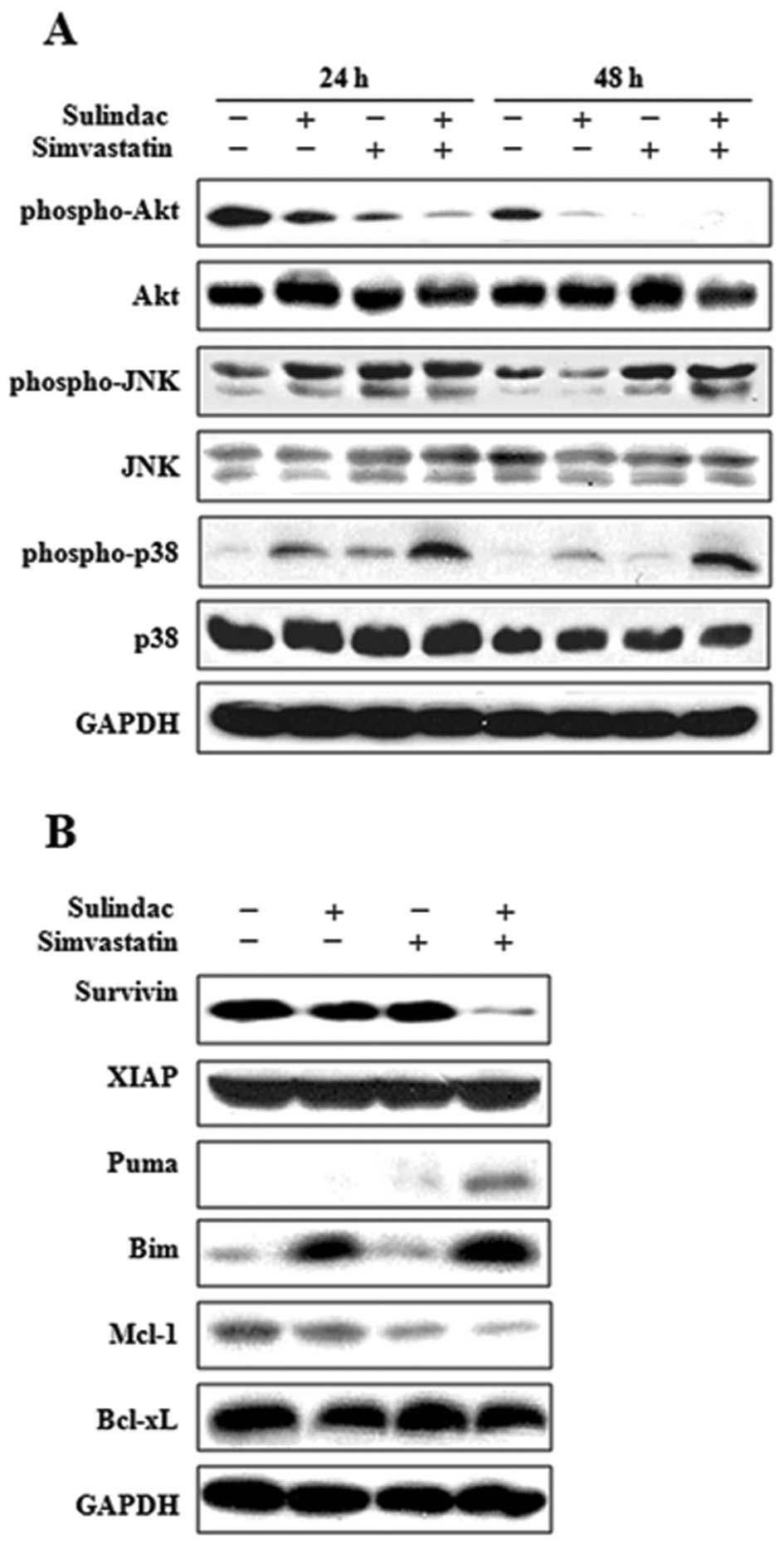 | Figure 4.Involvement of different signal
transduction pathways by combination treatment with sulindac and
simvastatin. (A) Cells were treated with sulindac and simvastatin,
alone and in combination, for 24 and 48 h, and the cell lysate was
subjected to 12% SDS-PAGE to measure the expression of
phosphorylated Akt, JNK and p38. The same membrane used for
anti-phospho antibody staining was stripped and used again with
antibodies for Akt, JNK and p38. (B) Cells were treated with
sulindac and simvastatin, alone and in combination, for 36 h, and
the cell lysate was subjected to 15% SDS-PAGE to measure the
expression of the IAP (survivin and XIAP), proapoptotic (Puma and
Bim), and antiapoptic (Mcl-1, Bcl-xL) Bcl-2 families. Equal protein
loading was confirmed using GAPDH. Immunoblots are representative
of at least 2 independent experiments. |
Combination of sulindac and simvastatin
downregulates survivin and induces changes in Bcl-2 families
Members of the IAP and Bcl-2 families are important
regulators of the mitochondrial apoptotic pathway. To identify the
molecular mechanism underlying apoptosis induced by combined
treatment with sulindac and simvastatin, we examined the expression
level of the IAP (survivin and XIAP), proapoptotic (Puma and Bim),
and anti-apoptotic (Mcl-1 and Bcl-xL) Bcl-2 families, by immunoblot
analysis in A549 cells treated with sulindac and/or simvastatin for
36 h. As shown in Fig. 4B,
treatment of A549 cells with sulindac and simvastatin resulted in a
significant decrease in survivin levels relative to treatment with
either drug alone, but it showed no effect on the expression of
XIAP. In addition, combination treatment with sulindac and
simvastatin increased the expression of the proapoptotic factors
Puma and Bim, whereas it resulted in a decrease in the levels of
the anti-apoptotic factor Mcl-1. The combination treatment had no
effect on the expression level of Bcl-xL.
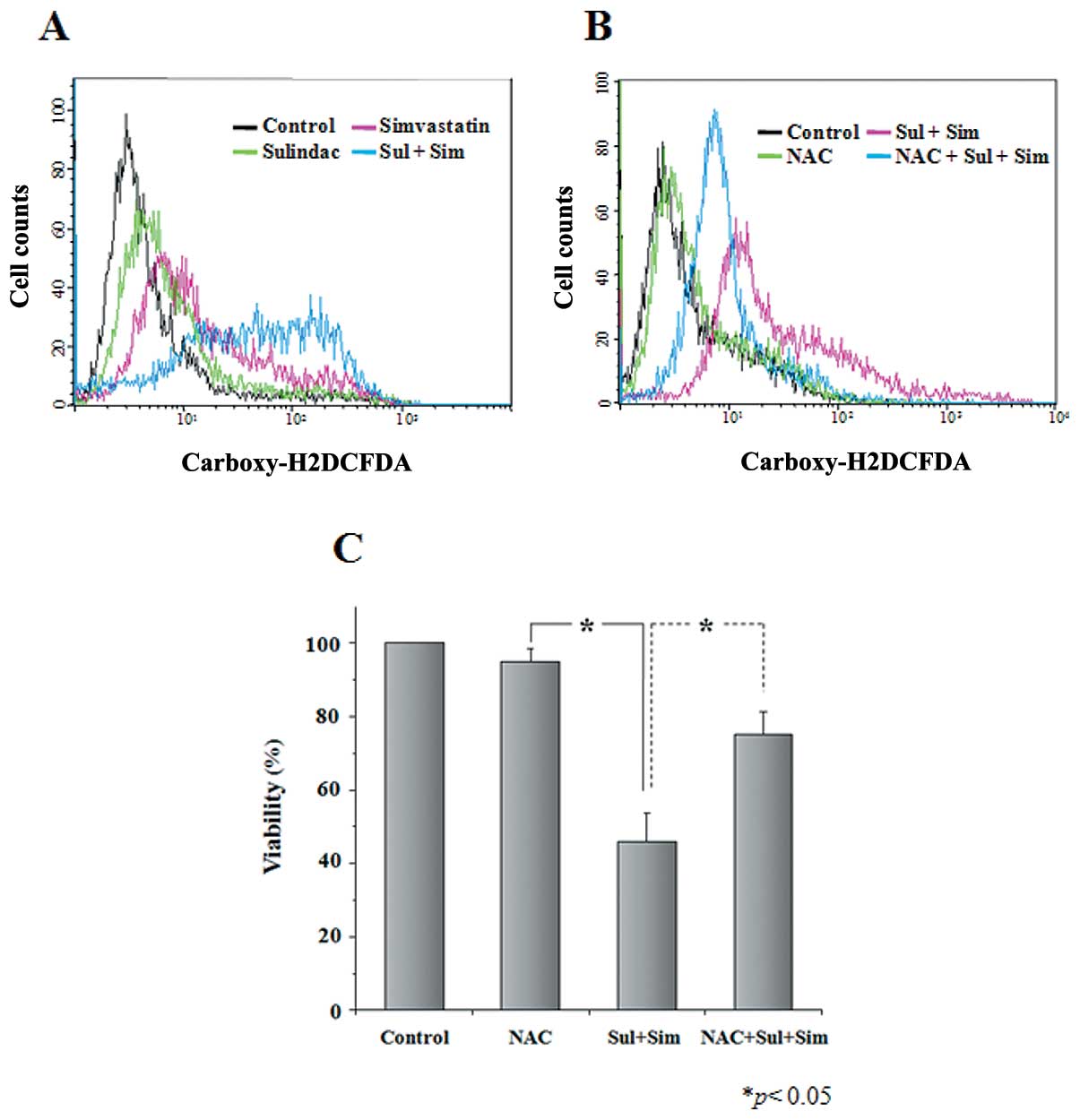
Elevated ROS contributes to anticancer
activity of combination treatment with sulindac and
simvastatin
The generation of intracellular ROS is known to
occur in lung cancer cells after treatment with sulindac or
simvastatin (32). Accordingly, we
examined whether the synergistic cytotoxicity of sulindac and
simvastatin results from the generation of ROS. After 48-h
treatment with 300 μM sulindac alone, 5 μM
simvastatin alone, or a combination of both drugs, cells were
loaded with dichlorofluorescein diacetate, and the resulting
fluorescence was analyzed on a FACSVantage flow cytometer. We
observed a rightward shift of fluorescence signals in cells treated
with both sulindac and simvastatin compared with cells treated with
either sulindac or simvastatin alone (Fig. 5A). We next tested the effect of the
free radical scavenger NAC in sulindac and simvastatin-treated A549
cells. Cells were pretreated with NAC, followed by the addition of
sulindac and simvastatin for 24 h. As shown in Fig. 5B, the enhancement of ROS generation
by combination treatment with sulindac and simvastatin was
abrogated by NAC. Moreover, NAC markedly inhibited combination
therapy-induced anticancer activity, as evaluated by the MTT assay
(Fig. 5C). Our results indicate
that elevated ROS is necessary for the potentiation of cell death
in sulindac plus simvastatin-treated cells.
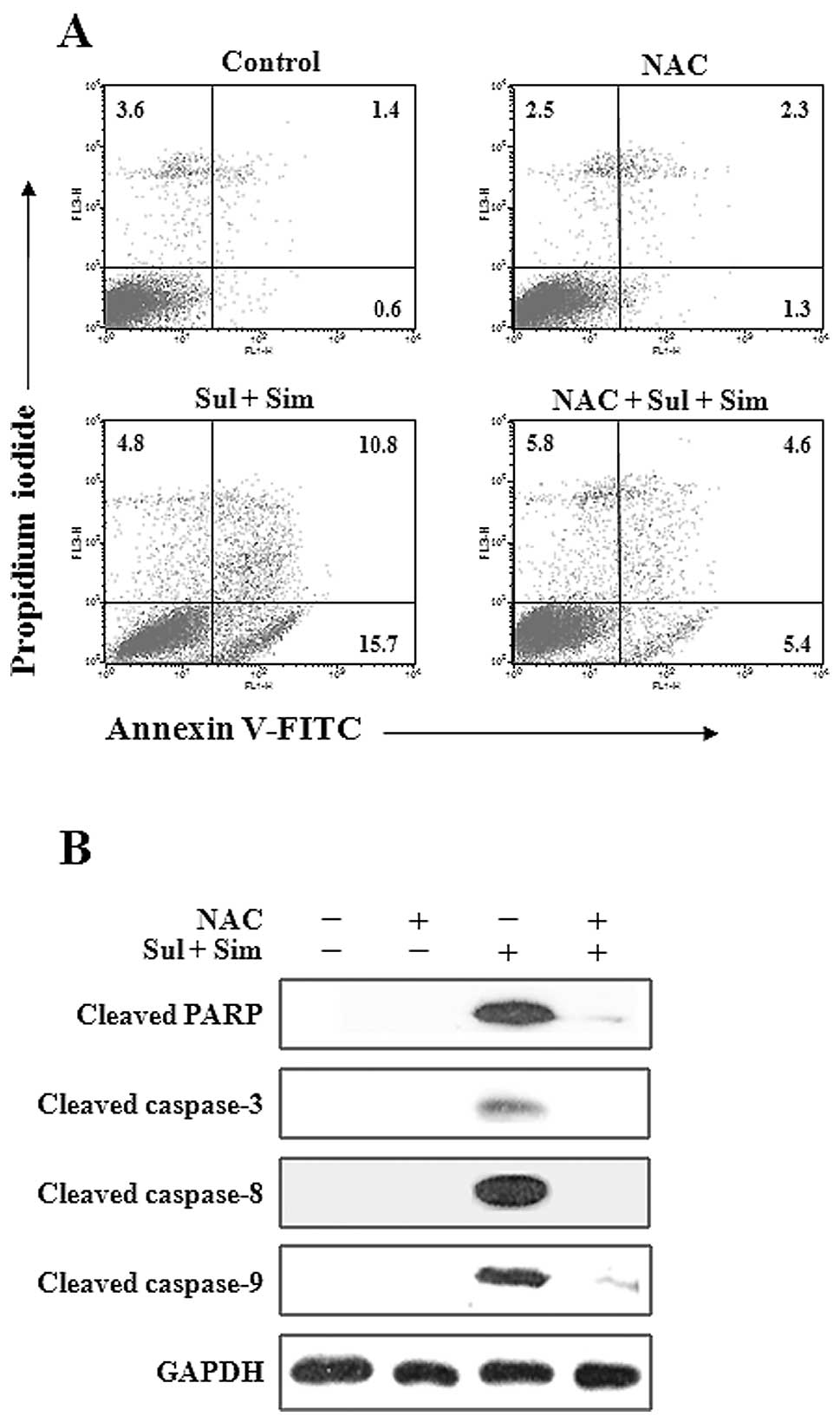
Pretreatment with NAC prevents apoptosis
induced by sulindac and simvastatin
To determine whether elevated ROS participates in
the apoptosis induced by the combination of sulindac and
simvastatin, the proportion of apoptotic cells was determined by
Annexin V-propidium iodide staining (Fig. 6A). While the combination of
sulindac and simvastatin was associated with Annexin V positivity
in approximately 26.5% of cells, pretreatment with NAC markedly
reduced this rate. Moreover, western blot analysis of A549 cell
lysates (Fig. 6B) showed that the
combination of sulindac and simvastatin enhanced the expression of
the 85-kDa form of PARP, and caspase-3, -8 and -9, whereas
pretreatment with NAC blocked this effect. Together, these findings
indicate that ROS generation plays a primary role in apoptosis
induced by sulindac and simvastatin.
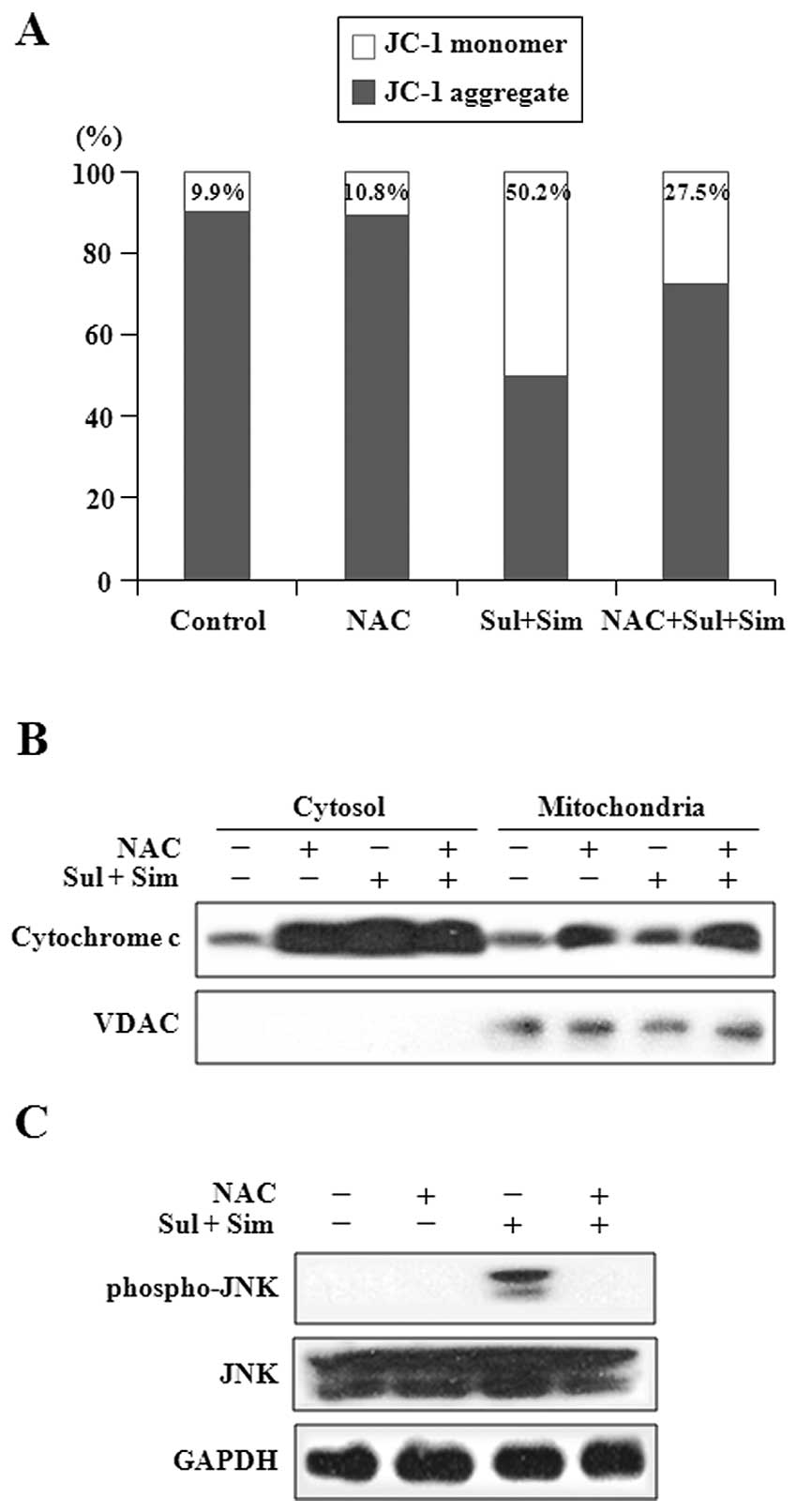
Pretreatment with NAC prevents
mitochondrial dysfunction by sulindac and simvastatin
Elevated ROS has been shown to be involved in the
mitochondrial apoptotic pathway (33). By flow cytometry analysis performed
with the JC-1 fluorescent dye, we investigated the effect of ROS on
the mitochondrial transmembrane potential (ΔΨm) after
combination treatment. As shown in Fig. 7A, we observed JC-1 monomers in
50.2% of cells treated with the drug combination, whereas the loss
of ΔΨm was significantly reduced in cells pretreated
with NAC. Next, we evaluated cytochrome c levels by western
blot analysis of mitochondrial and cytosolic fractions (Fig. 7B). Consistent with its effect in
offsetting the loss of ΔΨm, pretreatment of A549 cells
with NAC abrogated the release of cytochrome c from
mitochondria to the cytosol, which is induced by the combination
treatment with sulindac and simvastatin. Next, we examined the
effect of ROS on the expression of JNK after co-treatment with
sulindac and simvastatin (Fig.
7C). Pretreatment with NAC suppressed the increase in the
levels of phosphorylated JNK induced by the combination treatment
with sulindac and simvastatin. Collectively, these results indicate
that pretreatment with NAC suppresses mitochondrial dysfunction
induced by sulindac and simvastatin co-treatment.
Discussion
In the present study, we demonstrated the
synergistic effect of a combination of sulindac and simvastatin on
apoptosis of A549 lung cancer cells compared to the use of either
agent alone. These findings suggest that a combination of these
agents can potentially be used to kill lung cancer cells more
effectively and with minimal side-effects, thereby affording a
rationale for combining these drugs for the clinical prevention or
treatment of lung cancer.
Mitochondria are dynamic organelles that constantly
undergo fission and fusion to adapt to the changing conditions of
the cell. They play a central role in cellular metabolism and are a
major source of ROS generation in cells (34). Recently, the mitochondrial
megachannel was suggested to be a source of ROS generation induced
by treatment with sulindac and simvastatin (35). Several studies have reported that
mitochondrial morphology changes during apoptosis, resulting in the
appearance of small, round mitochondrial fragments (36,37).
Mitochondria play a major role in many apoptotic responses by
coordinating caspase activation through the release of cytochrome
c (38,39). Moreover, JNK is known to influence
the functions of pro- and anti-apoptotic Bcl-2 family proteins by
various mechanisms in the activation of the intrinsic mitochondrial
apoptotic pathway (40). Our
results demonstrate that the release of cytochrome c into
the cytosol activates caspase-9 and JNK signaling, and subsequently
leads to the activation of caspase-3. Indeed, cleavage of PARP, a
downstream target in this pathway, occurs during sulindac and
simvastatin-induced lung cancer cell apoptosis.
Although ROS are essential to cell survival,
elevated levels of ROS result in slowed growth, cell cycle arrest,
as well as apoptosis or even necrosis (41). Many chemotherapeutic strategies
have been designed to significantly increase cellular ROS levels
with the goal of inducing irreparable tumor cell damage and death.
Intracellular oxidative status has been shown to be important for
simvastatin sensitivity, and sulindac is also known to increase ROS
levels more efficiently than a selective COX-2 inhibitor (42). Increased ROS induces apoptosis by
activating the MAPK and caspase cascades, and/or by disrupting the
mitochondrial membrane potential (43). A challenge for novel treatment
strategies in lung cancer is the fine-tuning of intracellular ROS
signaling for effective therapeutic gain. Accordingly, we
investigated the possibility that ROS plays a role in sulindac and
simvastatin-induced ROS generation in A549 cells. We demonstrated
that, compared to individual treatments, combination treatment with
sulindac and simvastatin increased ROS levels, suggesting that the
combination of these drugs maintains higher ROS levels. If ROS are
indeed involved in apoptosis, ROS quenchers such as antioxidants
would be anticipated to prevent apoptosis. Moreover, ROS is a
possible initiator of JNK activation, which is necessary for
co-treatment-induced ΔΨm change. Indeed, we found that
sulindac and simvastatin-induced apoptosis, mitochondrial
dysfunction, and caspase activation were greatly reduced by
pretreatment with NAC. These results suggest that, in this model
system, ROS generation has a primary role in the induction of
apoptosis by sulindac and simvastatin.
We found that activation of caspases, mitochondrial
cytocyto-chrome c release, and change in ΔΨm
occurred during sulindac and simvastatin-induced apoptosis. JNK and
p38 MAPK were activated in combination with sulindac and
simvastatin, but only JNK activation was necessary for the
co-treatment-induced change in ΔΨm and lung cancer cell
death. Pretreament with NAC reduced co-treatment-induced activation
of caspases and lung cancer cell death. On the basis of our
findings, we suggest that ROS and JNK are involved in sulindac and
simvastatin-induced lung cancer cell apoptosis. Our findings afford
insight into ROS-mediated signaling in tumor cells, as well as
provide a basis for the design of novel improved strategies for the
treatment of lung cancer.
In conclusion, our results indicate that combination
treatment with sulindac and simvastatin augments their apoptotic
potential in lung cancer cells through ROS-dependent mitochondrial
dysfunction. Taken together, these results indicate that sulindac
and simvastatin are clinically promising therapies for the
treatment of lung cancer. Our data elucidate the possible mechanism
of action for sulindac and simvastatin in effecting lung cancer
cell death, and postulate their co-treatment as a chemopreventive
approach in lung cancer.
Acknowledgements
This study was supported by a grant of
the Korean Health Technology R&D Project, Ministry of Health
and Welfare (A120152), Republic of Korea.
References
|
1.
|
Tanaka K, Iwamoto S, Gon G, Nohara T,
Iwamoto M and Tanigawa N: Expression of survivin and its
relationship to loss of apoptosis in breast carcinomas. Clin Cancer
Res. 6:127–134. 2000.PubMed/NCBI
|
|
2.
|
Velmurugan B, Mani A and Nagini S:
Combination of S-allylcysteine and lycopene induces apoptosis by
modulating Bcl-2, Bax, Bim and caspases during experimental gastric
carcinosenesis. Eur J Cancer Prev. 14:387–393. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lin J, Hsiao PW, Chiu TH and Chao JI:
Combination of cyclooxygenase-2 inhibitors and oxaliplatin
increases the growth inhibition and death in human colon cancer
cells. Biochem Pharmacol. 70:658–667. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Banerjee S, Zhang Y, Ali S, Bhuiyan M,
Wang Z, Chiao PJ, Philip PA, Abbruzzese J and Sarkar FH: Molecular
evidence for increased antitumor activity of gemcitabine by
genistein in vitro and in vivo using an orthotopic model of
pancreatic cancer. Cancer Res. 65:9064–9072. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Khor TO, Keum YS, Lin W, Kim JH, Hu R,
Shen G, Xu C, Gopalakrishnan A, Reddy B, Zheng X, Conney AH and
Kong AN: Combined inhibitory effects of curcumin and phenethyl
isothiocyanate on the growth of human PC-3 prostate xenografts in
immunodeficient mice. Cancer Res. 66:613–621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Giardiello FM, Spannhake EW, DuBois RN,
Hylind LM, Robinson CR, Hubbard WC, Hamilton SR and Yang VW:
Prostaglandin levels in human colorectal mucosa: effects sulindac
in patients with familial adenomatous polyposis. Dig Dis Sci.
43:311–316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Shiff SJ, Qiao L, Tsai LL and Rigas B:
Sulindac sulfide, an aspirin-like compound, inhibits proliferation,
causes cell cycle quiescence, and induces apoptosis in HT-29 colon
adenocarcinoma cells. J Clin Invest. 96:491–503. 1995. View Article : Google Scholar
|
|
8.
|
Chan TA, Morin PJ, Vogelstein B and
Kinzler KW: Mechanism underlying nonsteroidal antiinflammatory
drug-mediated apoptosis. Proc Natl Acad Sci USA. 95:681–686. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Williams CS, Mann M and DuBios RN: The
role of cyclooxygenases in inflammation, cancer, and development.
Oncogene. 18:7908–7916. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Thompson HJ, Jiang C, Lu J, Mehta RG,
Piazza GA, Paranka NS, Pamukcu R and Ahnen DJ: Sulfone metabolite
of sulindac inhibits mammary carcinogenesis. Cancer Res.
57:267–271. 1997.PubMed/NCBI
|
|
11.
|
Piazza GA, Rahm AL, Krutzsch M, Sperl G,
Paranka NS, Gross PH, Brendel K, Burt RW, Alberts DS and Pamukcu R:
Antineoplastic drugs sulindac sulfide and sulfone inhibit cell
growth by inducing apoptosis. Cancer Res. 55:3110–3116.
1995.PubMed/NCBI
|
|
12.
|
Tegeder I, Pfeilschifter J and Geisslinger
G: Cyclooxygenase-independent actions of cyclooxygenase inhibitors.
FASEB J. 15:2057–2072. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Seo SK, Lee HC, Woo SH, Jin HO, Yoo DH,
Lee SJ, An S, Choe TB, Park MJ, Hong SI, Park IC and Rhee CH:
Sulindac-derived reactive oxygen species induce apoptosis of human
multiple myeloma cells via p38 mitogen activated protein
kinase-induced mitochondrial dysfunction. Apoptosis. 12:195–209.
2007. View Article : Google Scholar
|
|
14.
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Chan KK, Oza AM and Siu LL: The statins as
anticancer agents. Clin Cancer Res. 9:10–19. 2003.
|
|
16.
|
Demierre MF, Higgins PD, Gruber SB, Hawk E
and Lippman SM: Statins and cancer prevention. Nat Rev Cancer.
5:930–942. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Mantha AJ, Hanson JE, Goss G, Lagarde AE,
Lorimer IA and Dimitroulakos J: Targeting the mevalonate pathway
inhibits the function of the epidermal growth factor receptor. Clin
Cancer Res. 11:2398–2407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Wong WW, Dimitroulakos J, Minden MD and
Penn LZ: HMB-CoA reductase inhibitors and the malignant cell: the
statin family of drugs as triggers of tumor-specific apoptosis.
Leukemia. 16:508–519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Holstein SA and Hohl RJ: Synergistic
interaction of lovastatin and paclitaxel in human cancer cells. Mol
Cancer Ther. 1:141–149. 2001.PubMed/NCBI
|
|
20.
|
Feleszko W, Mlynarczuk I, Olszewska D,
Jalili A, Grzela T, Lasek W, Hoser G, Korczak-Kowalska G and
Jakobisiak M: Lovastatin potentiates antitumor activity of
doxorubicin in murine melanoma via an apoptosis-dependent
mechanism. Int J Cancer. 100:111–118. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Khanzada UK, Pardo OE, Meier C, Downward
J, Seckl MJ and Arcaro A: Potent inhibition of small-cell lung
cancer cell growth by simvastatin reveals selective functions of
Ras isoforms in growth factor signaling. Oncogene. 25:877–887.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kozar K, Kaminski R, Legat M, Kopec M,
Nowis D, Skierski JS, Koronkiewicz M, Jakobisiak M and Golab J:
Cerivastatin demonstrates enhanced antitumor activity against human
breast cancer cell lines when used in combination with doxorubicin
or cisplatin. Int J Oncol. 24:1149–1157. 2004.
|
|
23.
|
Wallace JL: Nonsteroidal anti-inflammatory
drugs and gastroenteropathy: the second hundred years.
Gastroenterology. 112:1000–1016. 1997.PubMed/NCBI
|
|
24.
|
Solomon SD, McMurray JJ, Pfeffer MA,
Wittes J, Fowler R, Finn R, Anderson WF, Zauber A, Hawk E and
Bertagnolli M: Cardiovascular risk associated with celecoxib in a
clinical trial for colorectal adenoma prevention. N Eng J Med.
352:1071–1080. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Psaty BM and Potter JD: Risks and benefits
of celecoxib to prevent recurrent adenomas. N Engl J Med.
355:950–952. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Jalving M, Koorntra JJ, De Jong S, De
Vries EG and Kleibeuker JH: Review article: the potential of
combinational regimen with non-steroidal anti-inflammatory drugs in
the chemoprevention of colorectal cancer. Aliment Pharmacol Ther.
21:321–339. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Xiao H and Yang CS: Combination regimen
with statins and NSAIDs: a promising strategy for cancer
chemoprevention. Int J Cancer. 123:983–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Suh N, Reddy BS, DeCastro A, Paul S, Lee
HJ, Smolarek AK, So JY, Simi B, Wang CX, Janakiram NB, Steele V and
Rao CV: Combination of atorvastatin with sulindac or naproxen
profoundly inhibits colonic adenocarcinomas by suppressing the
p65/β-catenin/cyclin D1 signaling pathway in rats. Cancer Prev Res
(Phila). 4:1895–1902. 2011.
|
|
29.
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
30.
|
Wolf CM and Eastman A: The temporal
relationship between protein phosphatase, mitochondrial cytochrome
c release. Exp Cell Res. 247:505–513. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Smiley ST, Reers M, Mottola-Hartshorn C,
Lin M, Chen A, Smith TW, Steele GD Jr and Chen LB: Intracellular
heterogeneity in mitochondrial membrane potentials revealed by a
J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA.
88:3671–3675. 1991. View Article : Google Scholar
|
|
32.
|
Park JH, Kim EJ, Jang HY, Shim H, Lee KK,
Jo HJ, Kim HJ, Yang SH, Jeong ET and Kim HR: Combination treatment
with arsenic trioxide and sulindac enhances apoptotic cell death in
lung cancer cells via activation of oxidative stress and
mitogen-activated protein kinases. Oncol Rep. 20:379–384. 2008.
|
|
33.
|
Ralph SJ, Rondriguez-Enriquez S, Neuzil J,
Saavedra E and Moreno-Sanchez R: The causes of cancer revisited:
‘mitochondrial malignancy’ and ROS-induced oncogenic transformation
- why mitochondria are targets for cancer therapy. Mol Aspects Med.
31:145–170. 2010.
|
|
34.
|
Copeland WC, Wachsman JT, Johnson FM and
Penta JS: Mitochondrial DNA alterations in cancer. Cancer Invest.
20:557–569. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Seo SK, Lee HC, Woo SH, Jin HO, Yoo DH,
Lee SJ, An S, Choe TB, Park MJ, Hong SI, Park IC and Rhee CH:
Sulindac-derived reactive oxygen species induce apoptosis of human
multiple myeloma cells via p38 mitogen activated protein
kinase-induced mitochondrial dysfunction. Apoptosis. 12:195–209.
2007. View Article : Google Scholar
|
|
36.
|
Frank S, Gaume B, Bergmann-Leitner ES,
Leitner WW, Robert EG, Catez F, Smith CL and Youle RJ: The role of
dynamic-related protein 1, a mediator of mitochondrial fission, in
apoptosis. Dev Cell. 1:515–525. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Karbowski M, Lee YJ, Gaume B, Jeong SY,
Frank S, Nechushtan A, Santel A, Fuller M, Smith CL and Youle RJ:
Spatial and temporal association of Bax with mitochondrial fission
sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 159:931–938.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Li LY, Luo X and Wang X: Endonuclease G is
an apoptotic DNase when released from mitochondria. Nature.
412:95–99. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Tournier C, Hess P, Yang DD, Xu J, Turner
TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA and Davis RJ:
Requrement of JNK for stress-induced activation of the cytochrome
c-mediated death pathway. Science. 288:870–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Burdon RH: Control of cell proliferation
by reactive oxygen species. Biochem Soc Trans. 24:1028–1032.
1996.PubMed/NCBI
|
|
42.
|
Minami T, Adachi M, Kawamura R, Zhang Y,
Shinomura Y and Imai K: Sulindac enhances the proteasome inhibitor
bortezomib-mediated oxidative stress and anticancer activity. Clin
Cancer Res. 11:5248–5256. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Fiers W, Beyaert R, Declercq W and Van den
Abeele P: More than one way to die: apoptosis, necrosis and
reactive oxygen damage. Oncogene. 18:7719–7730. 1999. View Article : Google Scholar : PubMed/NCBI
|