Introduction
Cutaneous melanoma is a tumor arising from epidermal
melanocytes. It particularly affects young patients as it is the
third most frequent cancer in the age range of 20–39 years
(1). Although melanoma accounts
for only 4% of all skin cancers, it is responsible for 80% of
deaths (2). The survival rate at
10 years for patients with metastatic melanoma is <10% (2). During the two last decades, the
incidence of melanoma as well as the associated-mortality have
strongly increased (3),
transforming this disease into a real public health problem.
With the increase in public awareness campaigns, the
diagnosis of melanoma is being diagnosed at an earlier stage when
the disease is still curable by surgery. At this stage, the
survival rate at 5 years is 90% (4). On the other hand, metastatic melanoma
is an incurable disease. Multimodality treatments are necessary.
Historically, chemotherapy (dacarbazine, cisplatin) and
immunotherapy (interferon α-2b and interleukin-2) were for a long
time the mainstay of treatment. Dacarbazine was the only FDA
approved cytotoxic agent even though it produced low response
rates. Interferon and interleukin have showed impressive
antitumoral activity with a number of complete responses but in a
very small subset of patients. The landscape of the treatment of
metastatic melanoma has changed recently with the emergence of new
immuno-modulatory agents such as anti-CTLA-4, anti-PD1, anti-PD-L1
monoclonal antibodies (5,6) and pathway inhibitors such as BRAF and
MEK inhibitors (7–9).
The description of activating BRAF gene mutations in
50–60% of melanomas (10) opened a
new therapeutic perspective. Indeed, treatment with the specific
V600EBRAF inhibitor vemurafenib resulted in spectacular
tumor regressions (11), improved
rates of overall and progression-free survival compared to
dacarbazine (12) and induced
clinical response in >50% of metastatic melanoma patients
harbouring the BRAF mutation (median overall survival was ~16
months) (7). Nevertheless, in
spite of impressive initial responses, selection and/or resistance
developed in many cases due to exogenous growth factors and
cytokines (13), switches between
pathways (14), COT/MAP3K8
activation (15), appearance of
new activation mutations in C121SMEK1 (16), dimerization of aberrantly spliced
V600EBRAF (17) or
prevalence of wild-type cells in the tumors (18).
Besides BRAF, several other mutated or upregulated
proteins involved in major signalling pathways may be considered as
additional potential targets in cutaneous melanoma. First, the
RAS/RAF/MEK/ERK signalling pathway is a major target for therapy as
it is hyper-activated in ~75% of metastatic tumors. Indeed, NRAS
activated mutations are found in 15–25% of cases with the remaining
being BRAF mutated, both mutations being mutually exclusive
(10). Second, PI3K/AKT/mTOR
signalling is another important pathway in melanoma because of its
activation through the loss of the expression of the tumor
suppressor PTEN (30–50%) or the amplification of AKT (60%)
(19). Interestingly, genetic
interaction between NRAS and BRAF mutations and PTEN inactivation
has been reported, (20)
suggesting possible cooperation between both MAPK and PI3K/AKT
activations in melanoma development (21). Finally, STAT3 is a point of
convergence for many tyrosine kinases such as JAK and SRC. It is
constitutively activated in a majority of human melanoma cell lines
and tumors where it plays a key role in growth and survival
(22). Moreover, its activation
has been reported as a compensatory mechanism allowing cell
survival and contributing to resistance to targeted drugs such as
SRC inhibitors (23).
In light of the recent data on targeted therapy and
keeping in mind the identified crosstalk between different
signalling pathways in melanoma, it was evident that more effective
treatments need concomitant inhibition of multiple pathways. The
choice and modalities of such combinations are currently
investigated both in in vitro models and in clinical trials.
Our working hypothesis is based on a particular approach by which a
sustained inhibition of a given signalling pathway renders cells
dependent on other compensatory pathways for their proliferation
and survival. The identification and subsequent targeting of the
latter as well could substantially potentiate cell toxicity. Hence,
we induced changes in signalling profiles in three representative
melanoma cell lines (wild-type or mutated in NRAS or BRAF) by using
a sequential exposure first to non-toxic (<10% toxicity) but
effective (substantial inhibition of targeted kinase)
concentrations of various protein kinase inhibitors targeting MEK,
PI3K, SRC or JAK and second, while maintaining these inhibitions,
to adequate protein kinase inhibitors targeting the activated
compensatory signalling pathways. The results have been
systematically compared to each effector used alone or in
simultaneous combination with the others.
Materials and methods
Inhibitors
Four specific inhibitors of protein kinases has been
used: the SRC family inhibitor PP2 (IC50 = ~5 nM, 50% of
kinase activity inhibition), the MEK1/2 inhibitor U0126
(IC50 = ~0.1 μM), the PI3K inhibitor LY294002 referred
as LY29 (IC50 = ~1 μM) (all from Tocris Bioscience,
Bristol, UK) and the JAK Inhibitor I pyridone 6 referred as PYR
(IC50 = ~5 nM) (Calbiochem, Darmstadt, Germany).
Melanoma cell lines
Human melanoma cell lines were established in our
laboratory from lymph node metastases. HBL cells (LOCE-MM001) are
wild-type for NRAS and BRAF, LOCE-MM057 present the Q61L NRAS
mutation and LOCE-MM074 are bearing the V600E BRAF mutation.
Cell culture
Cells were cultured at 37°C in a humidified 95% air
and 5% carbon dioxide atmosphere. For routine maintenance, cells
were propagated in 175 cm2-flasks containing HAM-F10
medium supplemented with 5% heat-inactivated fetal calf serum and
5% heat-inactivated newborn calf serum and with L-glutamine,
penicillin and streptomycin at standard concentrations (all from
Gibco, Invitrogen, UK). Cells were harvested by trypsinization
(0.05% trypsin - EDTA) (Gibco) and subcultured twice weekly. One
day after seeding, the culture medium was replaced by fresh medium.
All lines are routinely checked for mycoplasma contamination using
MycoAlert™ Mycoplasma Detection kit (Lonza, Basel,
Switzerland).
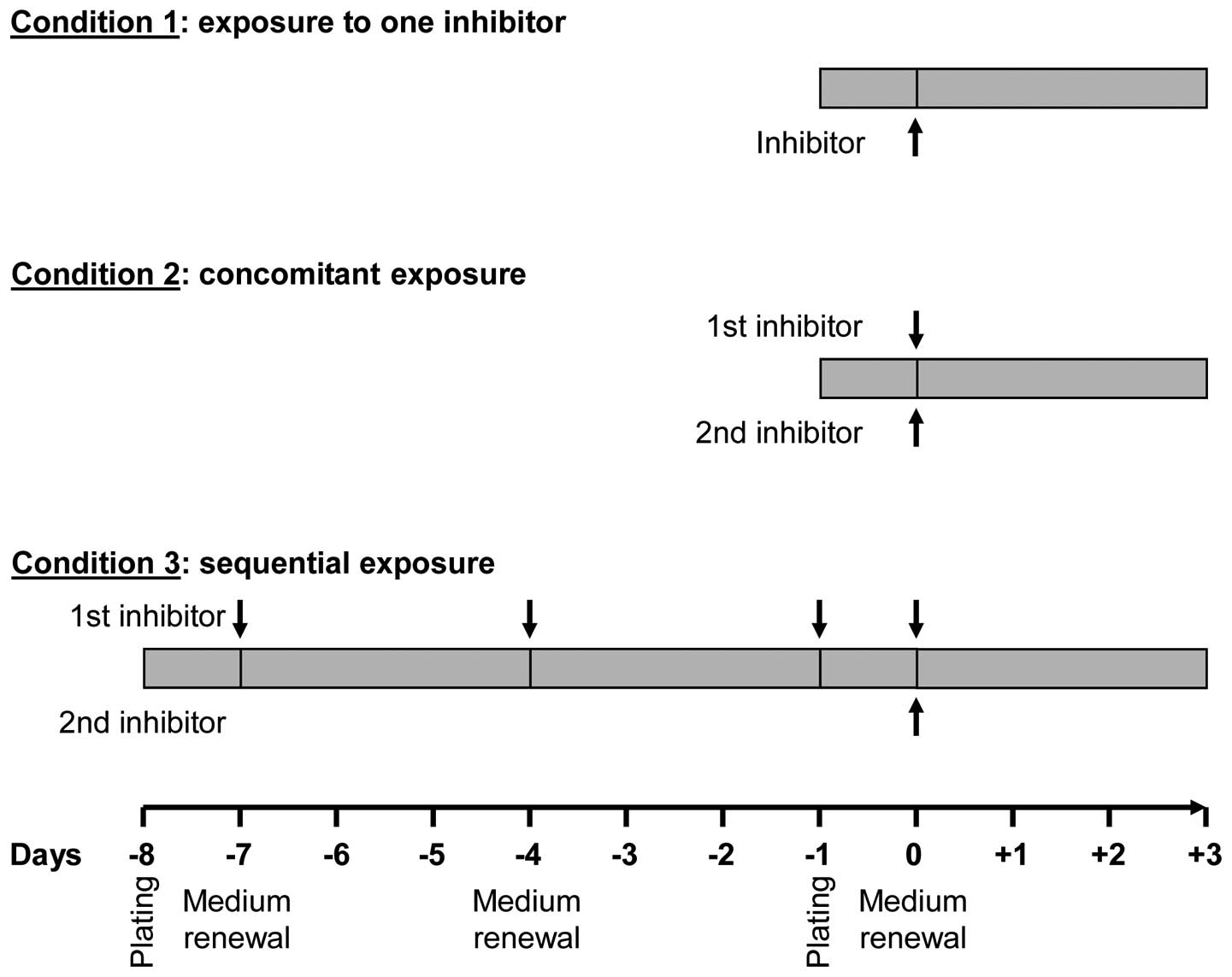
Experimental conditions
Experimental conditions are presented in Fig. 1 as follows: condition 1 (cell
exposure to one single inhibitor), cells were exposed to increasing
concentrations (from 10−12 to 10−4 M) of each
of the protein kinase inhibitors for 3 days; condition 2
(concomitant exposure to two inhibitors), cells were incubated with
a given concentration of an inhibitor and co-treated with
increasing concentrations of another for 3 days; condition 3
(sequential exposure), cells were exposed to each of the inhibitors
for 7 days (medium and inhibitor renewal at days 1, 4 and 7) and,
while maintaining this pre-treatment, cells were incubated for 3
additional days with increasing concentrations of each of the other
inhibitors.
Proliferation assay
All cells were seeded in 96-well plates (8,000
cells/well) containing HAM-F10 medium supplemented or not with
inhibitors. One day after plating, the culture medium was replaced
by a fresh one containing inhibitors depending on experimental
conditions (Fig. 1) and further
cultured for 3 additional days. Cell proliferation was assessed by
crystal violet assay. Briefly, culture medium was removed and cells
were gently rinsed with phosphate-buffered saline (PBS), fixed with
1% glutaraldehyde/PBS for 15 min and stained with 0.1% (w/v in
water) crystal violet for 30 min. Cells were destained under
running tap water and subsequently lysed with 0.2% (v/v in water)
Triton X-100 for 90 min. The absorbance was measured at 570 nm
using a Multiskan EX Microplate Photometer (Thermo Scientific,
Courtaboeuf Cedex, France). On each plate, blank wells containing
medium alone were used for background subtraction and untreated
(control) cells were cultured in parallel to treated cells.
Western blot analysis
Expression and/or phosphorylation levels of key
proteins of targeted signalling pathways were determined by western
blotting in untreated cells and cells exposed to inhibitors for 30
min or 10 days. All cells were plated in Petri dishes
(3×106 cells/dish) containing HAM-F10 medium and
cultured according to the experimental conditions outlined in
Fig. 1. Cells were then lysed
using detergent cocktail (M-PER mammalian extraction buffer)
supplemented with protease inhibitors (Halt protease inhibitor
cocktail) and phosphatase inhibitors (Halt phosphatase inhibitor
cocktail) (all from Pierce, Rockford, IL, USA). Protein
concentrations were determined by the BCA protein assay (Pierce)
using bovine serum albumin as a standard. Equal amounts of cell
proteins (30 μg) were subjected to 10% SDS-PAGE and
electrotransferred onto nitrocellulose membranes using
iBlot® Dry Blotting System (Invitrogen, Life
Technologies, Gent, Belgium). Immunodetection was performed using
antibodies raised against pSRC (Tyr 416) (1/1,000), SRC (1/1,000),
pAKT (Ser 473) (D9E, 1/500), AKT (40D4, 1/2,000), PTEN (138G6,
1/1,000), pSTAT3 (Tyr 705) (D3A7, 1/1,000) (all from Cell Signaling
Technology, Danvers, MA, USA) and pERK (Tyr 204) (E-4, 1/1,000),
ERK2 (C-14, 1/2,000), STAT3 (K-15, 1/200) (all from Santa Cruz
Biotechnology, Santa Cruz, CA, USA). Peroxidase-labeled anti-rabbit
IgG antibody (1/5,000) or peroxidase-labeled anti-mouse IgG
antibody (1/5,000) (both from Amersham Pharmacia Biotech) were used
as secondary reagents to detect corresponding primary antibodies.
Bound peroxidase activity was revealed using the
SuperSignal® West Pico Chemiluminescent Substrate
(Pierce). Immunostaining signals were digitalized with a PC-driven
LAS-3000 CCD camera (Fujifilm, Tokyo, Japan), using a software
specifically designed for image acquisition (Image Reader,
Raytest®, Straubenhardt, Germany).
Statistical analysis
IC10 and IC50 values were
calculated using GraphPad Prism software (GraphPad Software, La
Jolla, CA, USA). Data are reported as means ± SEM and significance
was calculated by Student's t-test using SPSS software (SPSS Inc.,
Paris, France). *p<0.05, **p<0.01,
***p<0.001.
Results
Characteristics of the representative
melanoma cell lines relevant for the study
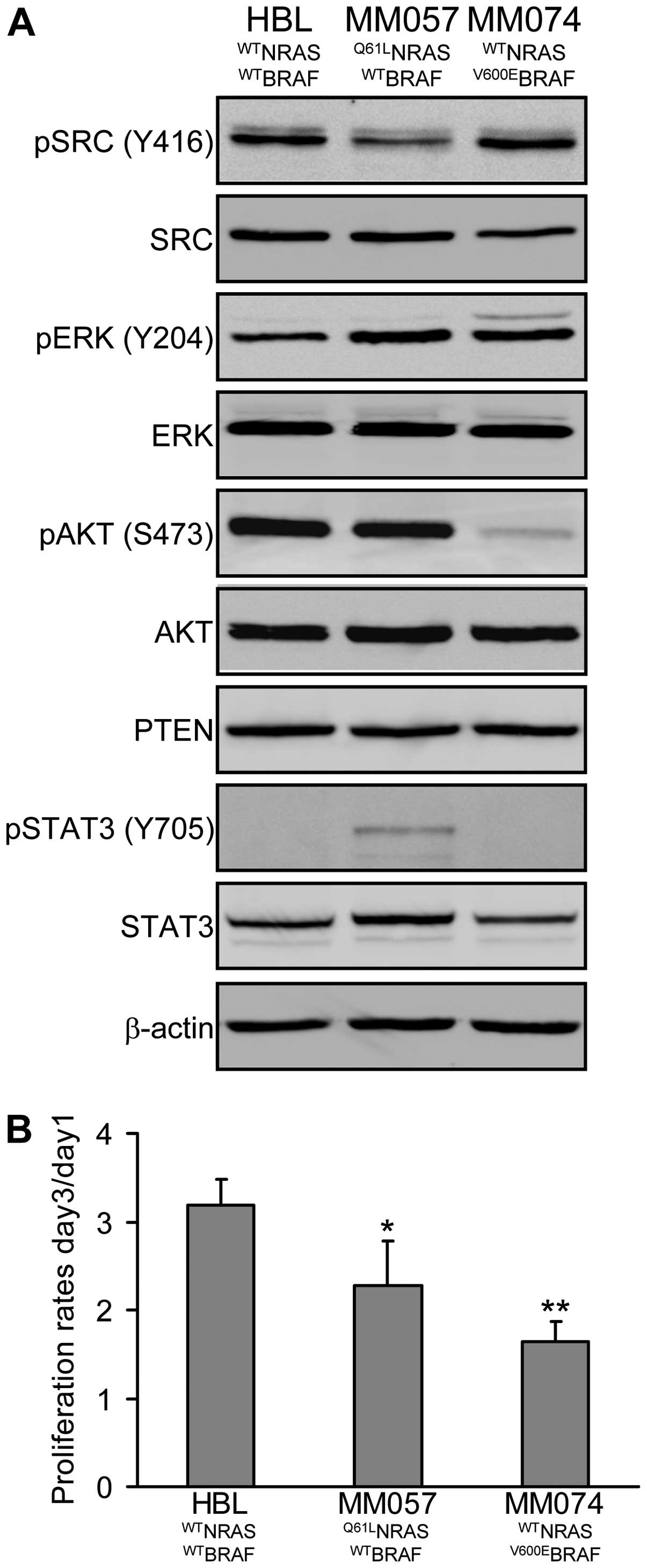
HBL cells are wild-type for NRAS and BRAF, MM057
cells present the Q61L mutation in NRAS and MM074 cells bear the
V600E mutation in BRAF. Analysis of constitutive levels of
phosphorylation/expression of key proteins involved in the master
signalling pathways (SRC, MAPK, PI3K/AKT, JAK/STAT) in melanoma
cells showed that HBL cells exhibit low phosphorylation of ERK and
high phosphorylation of AKT, MM057 cells have high phosphorylation
of both ERK and AKT, while MM074 cells show high phosphorylation of
ERK but low phosphorylation of AKT (Fig. 2A). PTEN expression is not different
among lines. Interestingly, we observed an inverse relation between
the phosphorylations of SRC and STAT3, with high SRC
phosphorylation in HBL and MM074 cells and high STAT3
phosphorylation in MM057 cells. Of note, these phosphorylation
profiles of MM057 and MM074 cells are compatible with the
mutational status of NRAS and BRAF, respectively.
On the other hand, we examined the proliferation
rate of each cell line (day-3/day-1 ratio, crystal violet assay)
and found that HBL cells were highly proliferative, MM057 cells
were moderately, while MM074 cells had the lowest proliferation
rate (2-fold less compared to HBL), suggesting that activating
mutations in NRAS or BRAF are not necessarily associated with a
higher proliferation rate (Fig.
2B) as also suggested by others (22).
Cell exposure to each inhibitor alone
(condition 1)
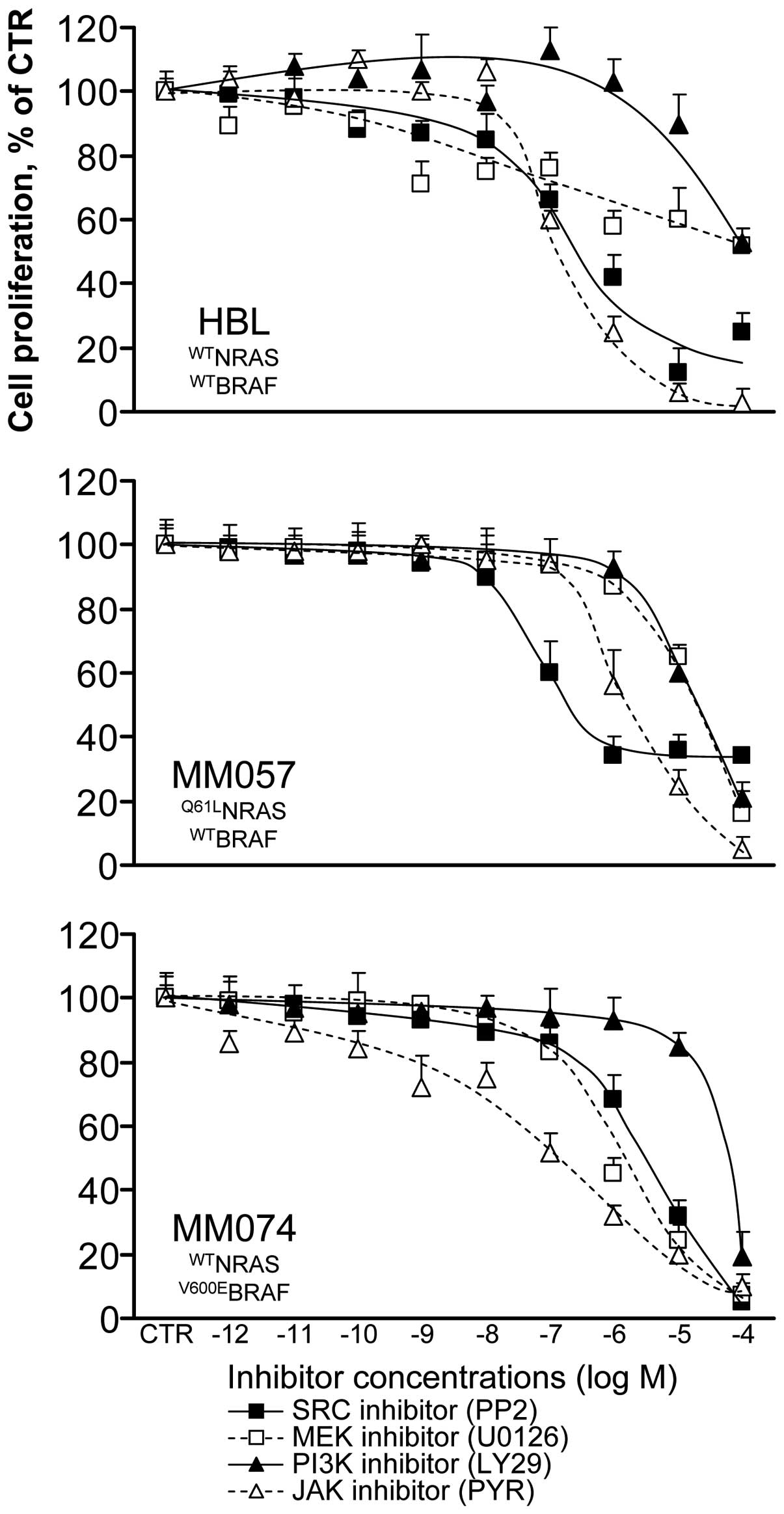
We evaluated the anti-proliferative effect of each
inhibitor in all cell lines after 3 days of treatment (condition 1,
see Materials and methods) (Fig.
3) and we determined the IC10 and IC50 in
all cases (Table I). We found that
wild-type BRAF cells (HBL, MM057) were more sensitive to the SRC
inhibitor PP2 than V600EBRAF cells (MM074), whereas HBL
and MM057 cells had a lower level of phosphorylation of SRC than
MM074 cells (Fig. 2A). As
expected, the V600EBRAF cells were much more responding
to the MEK inhibitor U0126 than the wild-type BRAF ones (especially
HBL cells), in agreement with the hyper-activation of the MAPK
signalling pathway in the former cells. The PI3K inhibitor LY29 had
comparable weak inhibitory effects in the 3 cell lines, while the
PI3K/AKT signalling pathway appeared weakly activated in MM074
cells. Finally, the JAK inhibitor PYR was the less effective in the
Q61LNRAS cells (MM057) which are the only ones to
exhibit an activation of STAT3. Thus, because of probable
activation of alternative signalling pathways in cells under
treatments, inhibitors were not necessary more effective in lines
exhibiting higher activation of their targeted pathways.
 | Table IIC10 and IC50
determination (μM) for HBL (WTNRAS/WTBRAF),
MM057 (Q61LNRAS) and MM074 (V600EBRAF)
exposed to SRC (PP2), MEK (U0126), PI3K (LY29) and JAK (PYR)
inhibitors for 3 days (condition 1). |
Table I
IC10 and IC50
determination (μM) for HBL (WTNRAS/WTBRAF),
MM057 (Q61LNRAS) and MM074 (V600EBRAF)
exposed to SRC (PP2), MEK (U0126), PI3K (LY29) and JAK (PYR)
inhibitors for 3 days (condition 1).
| HBL | MM057 | MM074 |
|---|
|
|
|
|
|---|
| Inhibitors |
IC10 |
IC50 |
IC10 |
IC50 |
IC10 |
IC50 |
|---|
| PP2 | 0.003 | 0.2 | 0.01 | 0.1 | 0.02 | 3 |
| U0126 | <0.001 | 100 | 0.5 | 30 | 0.02 | 0.7 |
| LY29 | 10 | 100 | 2 | 30 | 5 | 100 |
| PYR | 0.02 | 0.5 | 0.1 | 2 | <0.001 | 0.06 |
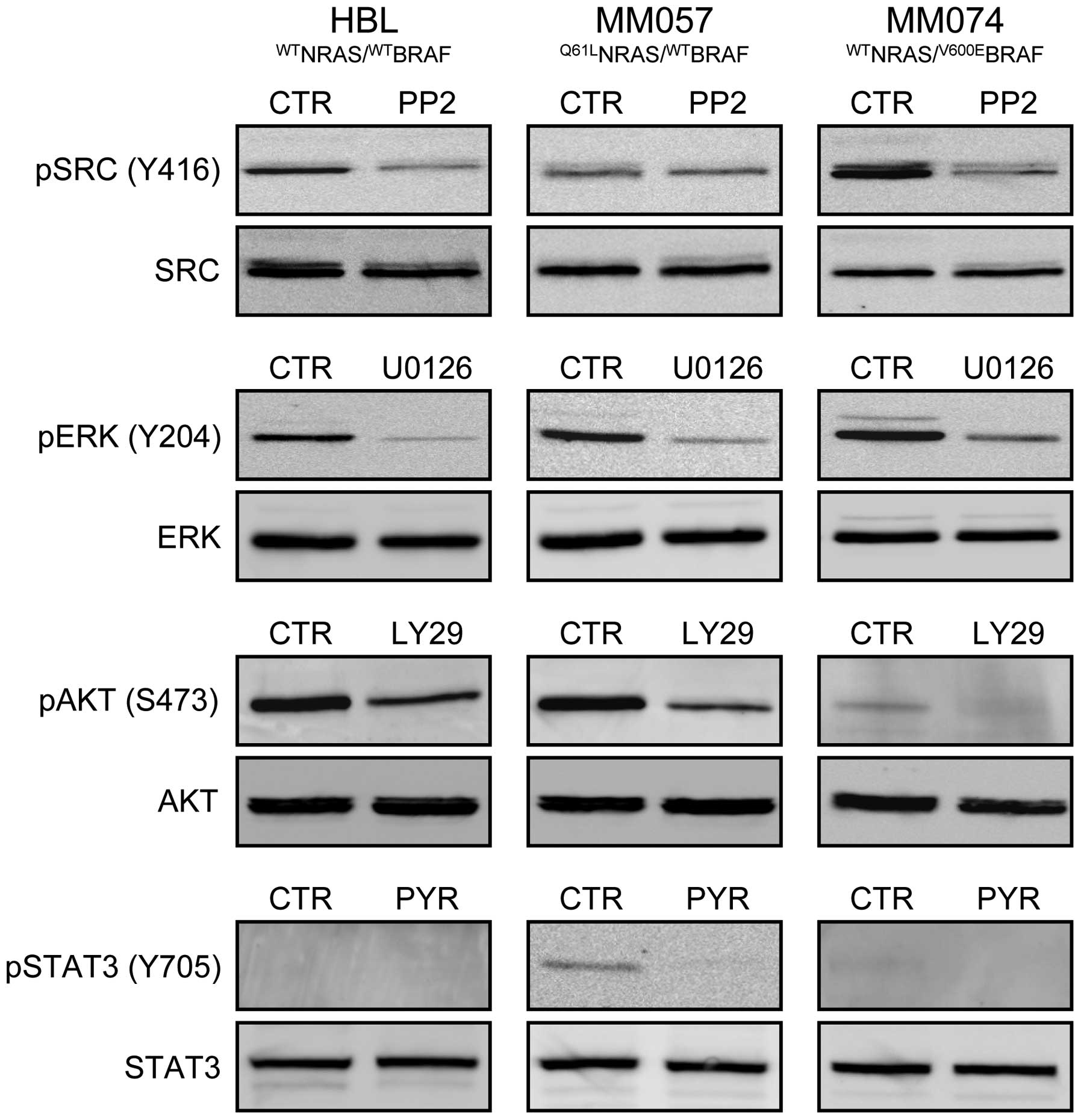
Concomitant cell exposure to two
inhibitors (condition 2)
Based on the effect of each inhibitor in all lines
(Table I), we selected the highest
non-toxic concentrations (0.05 μM PP2, 0.5 μM U0126, 5 μM LY29 and
0.05 μM PYR) after one week exposure to be subsequently used for
inhibitor combination experiments (condition 2, see Materials and
methods). Importantly, using western blot analysis, they were
effective in inhibiting targeted signalling pathways after 30-min
exposure (Fig. 4). Indeed, PP2
inhibited SRC phosphorylation in both HBL and MM074 cells, U0126
affected ERK phosphorylation in all cell lines, LY29 interfered
with AKT phosphorylation in HBL and MM057 lines and PYR decreased
STAT3 phosphorylation in MM057 cells.
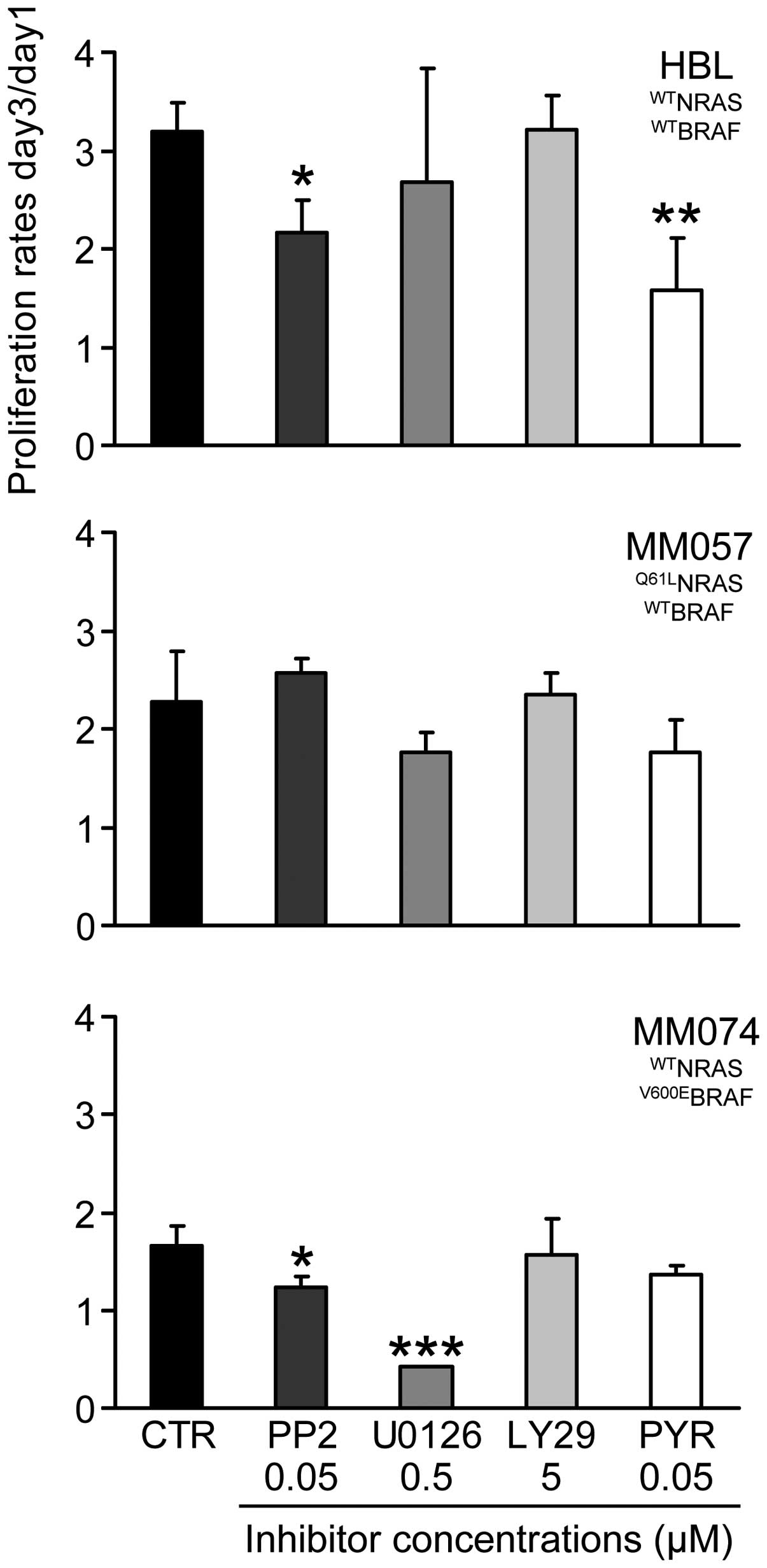
Sequential cell exposure to inhibitors
(condition 3)
We first examined the anti-proliferative effect of
each inhibitor alone at the above mentioned non-toxic
concentrations after 10 days of continuous exposure. As documented
in Fig. 5, we found that all cell
lines were not or were only weakly affected by long-term treatment
with each inhibitor (proliferation rate >1), except in the
V600EBRAF cells (MM074) where 0.5 μM U0126 is highly
toxic (proliferation rate ~0.4).
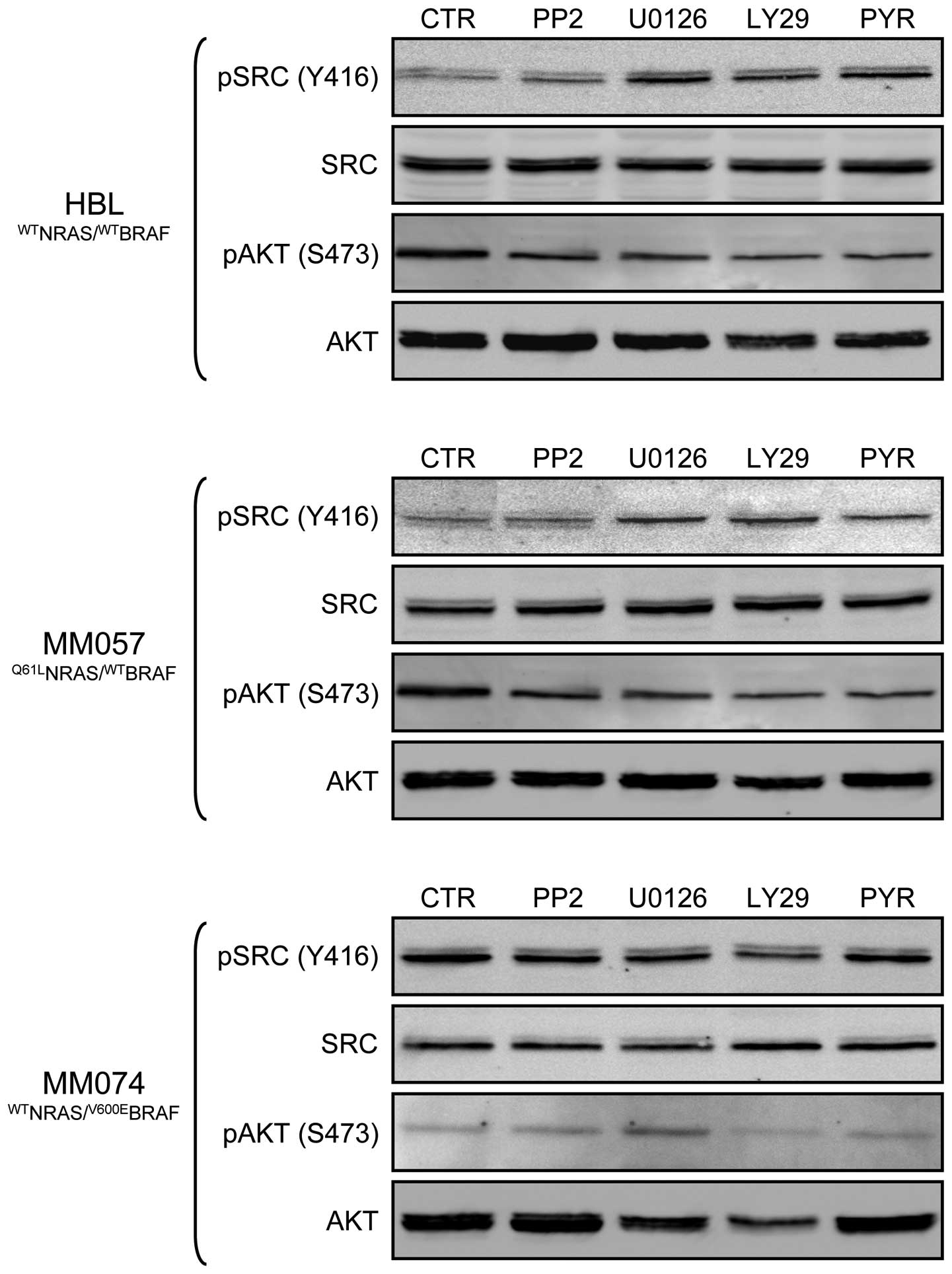
Then, we evaluated whether pre-treatment with a
first inhibitor potentiated the anti-proliferative effect of a
second inhibitor (condition 3), as compared to simultaneous
treatment with both inhibitors (condition 2) and the effect of the
second inhibitor alone (condition 1). By comparing
IC50s, we found that pre-treatment (condition 3) with
MEK, PI3K or JAK inhibitors showed synergistic effects when
specimens were subsequently exposed to an SRC inhibitor (Table II, in italics) in both HBL and
MM057 cell lines. These effects may be explained by the fact that
pre-treatment with U0126, LY29 or PYR increased SRC
expression/phosphorylation in these lines (Fig. 6). In addition, we also observed
synergy between PP2 or U0126 pre-treatment and then exposure to PYR
in HBL cells and between U0126 pre-treatment and PYR in MM074 cells
(Table II, in bold). However,
STAT3 was not necessary more phosphorylated after these
pre-treatments, suggesting that other JAK downstream targets should
have been activated. Furthermore, sequential exposure to U0126 and
then LY29 showed synergy in inhibiting MM074 cell proliferation
(Table II, underlined), in the
line with the AKT higher phosphorylation due to the pre-treatment
with a MEK inhibitor (Fig. 6). By
contrast, some concomitant treatments (condition 2) resulted in
antagonistic effects when SRC and JAK inhibitors are combined
(Table II), as compared to each
inhibitor alone in all three cell lines (Table I).
| Table IIIC50 determination (μM)
for HBL, MM057 and MM074 exposed to concomitant (condition 2) or
sequential (condition 3) combinations of SRC (PP2), MEK (U0126),
PI3K (LY29) and JAK (PYR) inhibitors for 3 days. |
Table II
IC50 determination (μM)
for HBL, MM057 and MM074 exposed to concomitant (condition 2) or
sequential (condition 3) combinations of SRC (PP2), MEK (U0126),
PI3K (LY29) and JAK (PYR) inhibitors for 3 days.
| Inhibitors | HBL | MM057 | MM074 |
|---|
|
|
|
|
|---|
| 1 Fixed conc. | 2 Increasing
conc. | Concomitant
exposure IC50 (μM) | Sequential exposure
IC50 (μM) | Concomitant
exposure IC50 (μM) | Sequential exposure
IC50 (μM) | Concomitant
exposure IC50 (μM) | Sequential exposure
IC50 (μM) |
|---|
| PP2 | U0126 | >100 | >100 | 50 | 20 | 50 | 50 |
| 0.05 μM | LY29 | 30 | 2 | 50 | 20 | 30 | 10 |
| PYR | 50a |
0.002a | >100 | 50 | 50 | 5 |
| U0126 | PP2 | 4a | 0.02a |
>100a | 0.5a | >100 | >100 |
| 0.50 μM | LY29 | 10 | 5 | >100 | 50 | 50a | <0.001a |
| PYR | 20a |
<0.001a | 20 | 50 | 30a |
<0.001a |
| LY29 | PP2 | 0.1a |
<0.001a | 1a |
<0.001a | >100 | >100 |
| 5.00 μM | U0126 | 10 | 0.1 | 3 | 5 | 5 | 30 |
| PYR | 1 | 0.1 | 30 | 10 | 5 | >100 |
| PYR | PP2 | 70a | 0.5a | 20a | 0.03a | >100 | 5 |
| 0.05 μM | U0126 | 2 | 50 | >10 | 50 | 10 | 50 |
| LY29 | 2 | 50 | 2 | 50 | 50 | 50 |
Discussion
Conventional chemotherapy with dacarbazine, approved
by the FDA (Food Drugs Administration) in 1975, was the standard
for treatment of melanoma patients until recently (24). Overall response rate is 15% (mostly
partial responses) but with no evidence of survival benefit.
Immunotherapy with high-dose interleukin-2 or interferon α-2b have
been associated with relatively durable responses but only in a
small subset of patients, with yet no factors predicting which
patients will respond to this therapy (25). Although they showed limited
efficacy, these various therapies allowed considerable progress in
the understanding of melanoma biology and molecular mechanisms
involved in melanomagenesis.
Recent advances in cell biology and the design of
targeted therapies led to unprecedented response rates with the new
targeted agents. A strategy aiming at restoring the immune system
response to disease has been developed, in particular, with
ipilumumab, an antibody raised against CTLA-4 (cytotoxic
T-lymphocyte antigen 4), a protein receptor responsible for immune
tolerance. It improved overall survival in patients within a trial
randomizing patients with previously treated metastatic melanoma
into 3 treatment arms. Moreover, 9 of 15 responders (60%) in the
ipilimumab only group maintained an objective response for ≥2 years
(5). Ipilimumab in combination
with dacarbazine improved overall survival versus dacarbazine plus
placebo in patients with previously untreated metastatic melanoma
(26). A similar immune strategy
with an anti-PD1 or anti-PD-L1 (programmed death 1, ligand)
antibody produced objective responses in ~28% of patients with
melanoma. Interestingly, preliminary data suggest a relationship
between PD-L1 expression on tumor cells and objective responses
(6).
Furthermore, recent clinical data provided a strong
indication for the efficacy of single agent targeted therapy
approaches in melanoma. First, vemurafenib has been reported to
induce clinical responses in >50% of metastatic melanoma
patients bearing V600EBRAF mutated tumors (7). Then, dabrafenib, another
V600EBRAF inhibitor, produced promising tumor shrinkage
in patients with brain metastases, a frequent complication of
metastatic melanoma (8). Lastly,
trametinib, a selective inhibitor of MEK1 and MEK2, downstream
effectors within the MAP kinase pathway, improved progression-free
survival and overall survival as compared to chemotherapy in
metastatic melanoma patients with BRAF V600E/K mutations (9). Importantly, the combination of
dabrafenib and trametinib had an acceptable safety profile with a
lower incidence of MEK inhibitor-related rash and BRAF
inhibitor-induced hyper-proliferative skin lesions compared to the
single agents (27).
Unfortunately, the impressive response rates with these agents are
short-lived, prompting the research for combinatorial strategies
and the discovery of research mechanisms. A discontinuous dosing
regimen with vemurafenib could emerge as a strategy to overcome
resistance because vemurafenib-resistant melanomas become
drug-dependent for their continued proliferation (28). Although many studies both in
vitro and in clinical trials have been devoted to search for
the best targeted drug combinations, encouraging but modest results
have been recorded most probably due to the heterogeneous
biological behaviour of melanoma tumor cells. In a recent study,
pairwise combinations of an array of small-molecule inhibitors on
early-passage melanoma cultures using combinatorial drug screening
revealed several inhibitor combinations effective for melanoma
treatment (29). Such
investigations have to be continued in order to identify the most
effective inhibitor combinations. In our study, we aimed to design
an alternative rationale to combine targeted drugs to potentiate
anti-proliferative effect of protein kinase inhibitors in order to
overcome one of the major resistance mechanism to such drugs in
melanoma. We selected 3 representative cell lines based on their
MAPK mutation status: V600EBRAF cells (MM074),
Q61LNRAS cells (MM057) and wild-type cells (HBL). Of
note, inverse correlations were observed between ERK
phosphorylation and AKT phosphorylation in wild-type HBL cells and
V600EBRAF MM074 cells, confirming crosstalk between MAPK
and PI3K/AKT pathways (21), while
ERK and AKT were highly phosphorylated in the Q61LNRAS
MM057 cells, because NRAS is upstream of both signalling pathways.
Previous studies reported that the consequence of a constitutive
MAPK activation in melanoma includes the increase in cell
proliferation and invasion (30)
and that, whereas V600EBRAF stimulated cell
proliferation in melanoma, it induced senescence in melanocytes
(31–34). However, and in agreement with
others (35), we observed that the
V600EBRAF mutation was associated with significantly
lower proliferation rates in many cell lines that we have examined
up-to-now (data not shown) and in particular is true for MM074
cells used in the present study. Of note, we confirmed that the
BRAF mutation makes cancer cells more dependent on the MAPK pathway
for their survival (36).
We also examined if the phosphorylation level of key
kinases (SRC, ERK, AKT, STAT3) could be predictive of the response
to protein kinase inhibitors regarding cell proliferation. We found
that: (I) although both wild-type (HBL) and V600EBRAF
(MM074) cells exhibited a high phosphorylation level of SRC, the
wild-type cells were 15-fold more sensitive to an SRC inhibitor,
(II) while Q61LNRAS (MM057) and V600EBRAF
(MM074) cells have a high phosphorylation level of ERK,
V600EBRAF cells were 30-fold more sensitive to a MEK
inhibitor, (III) whereas wild-type (HBL) and Q61LNRAS
(MM057) cells showed higher AKT phosphorylation levels than
V600EBRAF (MM074) cells, all cells had similar
sensitivity to PI3K inhibitor and (IV) although Q61LNRAS
cells are the only ones to exhibit STAT3 phosphorylation, they were
the least sensitive to a JAK inhibitor. Altogether, our data
indicate that even if the phosphorylation level of key kinases
correlates with the activation of the corresponding signalling
pathway, it cannot be used alone, as a marker to predict the
response to corresponding up-stream specific inhibitors because
similar level of phosphorylation could be caused by various
alternative mechanisms present in a given cell line, mainly related
to exogenous growth factors and cytokines stimulations, mutation
status, or crosstalk between signalling pathways. For example, ERK
phosphorylation may be induced by growth factor stimulation, RAS or
SRC activation, BRAF mutation and AKT-mediated phosphatase
inhibition.
Many studies have reported the efficacy of multiple
targeted therapies against cancer (37,38)
and suggested a benefit of simultaneous inhibitions of different
signalling pathways (39).
However, little attention has been paid to sequential treatment
that might be more relevant to clinical situation. Our hypothesis
was that a long-term inhibition of a given signalling pathway
renders melanoma cells dependent on other compensatory pathways for
their proliferation and survival that can be different among
subgroups of tumors. This information is crucial to subsequently
target the correct pathway to possibly potentiate the effect
(24). Accordingly, we found that
a 10-day cell exposure to a protein kinase inhibitor affected the
signalling pathway profiles as documented by western blot analyses
of the phosphorylation levels of the master signalling proteins.
For example, in wild-type HBL and Q61LNRAS MM057 cell
lines, MEK, PI3K and JAK inhibitors stimulated the phosphorylation
of SRC, rendering both lines more sensitive to SRC inhibition.
Moreover, in V600EBRAF MM074, MEK inhibitor increased
the phosphorylation of AKT leading to cells more responsive to AKT
inhibition. Thus, we found that long-term cell exposure to a
specific protein kinase inhibitor may enhance the
anti-proliferative effect of another protein kinase inhibitor,
supporting that melanoma cells became dependent on an alternative
pathway for survival after a pre-treatment (40). Most interesting is the fact that
these pre-treatments were done at non-toxic but kinase inhibition
effective concentrations. By contrast, simultaneous use of
inhibitors may surprisingly yield antagonistic effects. Indeed,
this was observed with a concomitant exposure to SRC and JAK
inhibitors, both proteins can affect the STAT pathway (41).
In conclusion, the present study adds new insight
into drug combination strategies by focusing on a sequential use of
kinase targeted inhibitors and on long-term priming/sensitizing
tumor cells with a first effector used at non-toxic but effective
concentrations to potentiate the effect of a second inhibitor. Our
data strongly support a strategy based on the identification of
both mutation status and signalling pathway profiles of a given
tumor to select melanoma patients and propose adequate drug
combinations and most importantly sequential administration
schedules.
Acknowledgements
This study received financial support from ‘MEDIC
Foundation’, ‘Les Amis de l'Institut Bordet’ and ‘Fondation
Lambeau-Marteaux’.
References
|
1
|
Rokuhara S, Saida T, Oguchi M, Matsumoto
K, Murase S and Oguchi S: Number of acquired melanocytic nevi in
patients with melanoma and control subjects in Japan: Nevus count
is a significant risk factor for nonacral melanoma but not for
acral melanoma. J Am Acad Dermatol. 50:695–700. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miller AJ and Mihm MC Jr: Melanoma. N Engl
J Med. 355:51–65. 2006. View Article : Google Scholar
|
|
3
|
Berwick M, Erdei E and Hay J: Melanoma
epidemiology and public health. Dermatol Clin. 27:205–214.
viii2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cummins DL, Cummins JM, Pantle H,
Silverman MA, Leonard AL and Chanmugam A: Cutaneous malignant
melanoma. Mayo Clin Proc. 81:500–507. 2006. View Article : Google Scholar
|
|
5
|
Hodi FS, O'Day SJ, McDermott DF, et al:
Improved survival with ipilimumab in patients with metastatic
melanoma. N Engl J Med. 363:711–723. 2010. View Article : Google Scholar
|
|
6
|
Topalian SL, Hodi FS, Brahmer JR, et al:
Safety, activity, and immune correlates of anti-PD-1 antibody in
cancer. N Engl J Med. 366:2443–2454. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sosman JA, Kim KB, Schuchter L, et al:
Survival in BRAF V600-mutant advanced melanoma treated with
vemurafenib. N Engl J Med. 366:707–714. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Falchook GS, Long GV, Kurzrock R, et al:
Dabrafenib in patients with melanoma, untreated brain metastases,
and other solid tumours: a phase 1 dose-escalation trial. Lancet.
379:1893–1901. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Flaherty KT, Robert C, Hersey P, et al:
Improved survival with MEK inhibition in BRAF-mutated melanoma. N
Engl J Med. 367:107–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davies H, Bignell GR, Cox C, et al:
Mutations of the BRAF gene in human cancer. Nature. 417:949–954.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Flaherty KT, Puzanov I, Kim KB, et al:
Inhibition of mutated, activated BRAF in metastatic melanoma. N
Engl J Med. 363:809–819. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chapman PB, Hauschild A, Robert C, et al:
Improved survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Christensen C and Guldberg P: Growth
factors rescue cutaneous melanoma cells from apoptosis induced by
knockdown of mutated (V 600 E) B-RAF. Oncogene. 24:6292–6302. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Montagut C, Sharma SV, Shioda T, et al:
Elevated CRAF as a potential mechanism of acquired resistance to
BRAF inhibition in melanoma. Cancer Res. 68:4853–4861. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Johannessen CM, Boehm JS, Kim SY, et al:
COT drives resistance to RAF inhibition through MAP kinase pathway
reactivation. Nature. 468:968–972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wagle N, Emery C, Berger MF, et al:
Dissecting therapeutic resistance to RAF inhibition in melanoma by
tumor genomic profiling. J Clin Oncol. 29:3085–3096. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Poulikakos PI, Persaud Y, Janakiraman M,
et al: RAF inhibitor resistance is mediated by dimerization of
aberrantly spliced BRAF(V600E). Nature. 480:387–390. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yancovitz M, Litterman A, Yoon J, et al:
Intra- and inter-tumor heterogeneity of BRAF(V600E) mutations in
primary and metastatic melanoma. PLoS One. 7:e293362012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tsao H, Zhang X, Benoit E and Haluska FG:
Identification of PTEN/MMAC1 alterations in uncultured melanomas
and melanoma cell lines. Oncogene. 16:3397–3402. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsao H, Goel V, Wu H, Yang G and Haluska
FG: Genetic interaction between NRAS and BRAF mutations and
PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol.
122:337–341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheung M, Sharma A, Madhunapantula SV and
Robertson GP: Akt3 and mutant V600E B-Raf cooperate to promote
early melanoma development. Cancer Res. 68:3429–3439. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Niu G, Bowman T, Huang M, et al: Roles of
activated Src and Stat3 signaling in melanoma tumor cell growth.
Oncogene. 21:7001–7010. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Byers LA, Sen B, Saigal B, et al:
Reciprocal regulation of c-Src and STAT3 in non-small cell lung
cancer. Clin Cancer Res. 15:6852–6861. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ji Z, Flaherty KT and Tsao H: Molecular
therapeutic approaches to melanoma. Mol Aspects Med. 31:194–204.
2010. View Article : Google Scholar
|
|
25
|
Yurkovetsky ZR, Kirkwood JM, Edington HD,
et al: Multiplex analysis of serum cytokines in melanoma patients
treated with interferon-alpha2b. Clin Cancer Res. 13:2422–2428.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Robert C, Thomas L, Bondarenko I, et al:
Ipilimumab plus dacarbazine for previously untreated metastatic
melanoma. N Engl J Med. 364:2517–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Flaherty KT, Infante JR, Daud A, et al:
Combined BRAF and MEK inhibition in melanoma with BRAF V600
mutations. N Engl J Med. 367:1694–1703. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Das Thakur M, Salangsang F, Landman AS, et
al: Modelling vemurafenib resistance in melanoma reveals a strategy
to forestall drug resistance. Nature. 494:251–255. 2013.PubMed/NCBI
|
|
29
|
Held MA, Langdon CG, Platt JT, et al:
Genotype-selective combination therapies for melanoma identified by
high-throughput drug screening. Cancer Discov. 3:52–67. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smalley KS: A pivotal role for ERK in the
oncogenic behaviour of malignant melanoma? Int J Cancer.
104:527–532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garnett MJ and Marais R: Guilty as
charged: B-RAF is a human oncogene. Cancer Cell. 6:313–319. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gray-Schopfer VC, Cheong SC, Chong H, et
al: Cellular senescence in naevi and immortalisation in melanoma: a
role for p16? Br J Cancer. 95:496–505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Michaloglou C, Vredeveld LC, Soengas MS,
et al: BRAFE600-associated senescence-like cell cycle arrest of
human naevi. Nature. 436:720–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pollock PM, Harper UL, Hansen KS, et al:
High frequency of BRAF mutations in nevi. Nat Genet. 33:19–20.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dhomen N, Reis-Filho JS, da Rocha Dias S,
et al: Oncogenic Braf induces melanocyte senescence and melanoma in
mice. Cancer Cell. 15:294–303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wellbrock C, Karasarides M and Marais R:
The RAF proteins take centre stage. Nat Rev Mol Cell Biol.
5:875–885. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gopal YN, Deng W, Woodman SE, et al: Basal
and treatment-induced activation of AKT mediates resistance to cell
death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous
melanoma cells. Cancer Res. 70:8736–8747. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Solit DB and Rosen N: Resistance to BRAF
inhibition in melanomas. N Engl J Med. 364:772–774. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Villanueva J, Vultur A, Lee JT, et al:
Acquired resistance to BRAF inhibitors mediated by a RAF kinase
switch in melanoma can be overcome by cotargeting MEK and
IGF-1R/PI3K. Cancer Cell. 18:683–695. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Flaherty KT and Smalley KS: Preclinical
and clinical development of targeted therapy in melanoma: attention
to schedule. Pigment Cell Melanoma Res. 22:529–531. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lui P, Cashin R, Machado M, Hemels M,
Corey-Lisle PK and Einarson TR: Treatments for metastatic melanoma:
synthesis of evidence from randomized trials. Cancer Treat Rev.
33:665–680. 2007. View Article : Google Scholar : PubMed/NCBI
|