Introduction
African-American men have a two-fold risk of
mortality from prostate cancer, and currently one of the most
unsettled debates is whether this high risk is driven by behavior,
biology or both (1,2). Pathological features, such as larger
prostate sizes, greater tumor volumes and tumor grades of African
Americans, in contrast to Caucasians strongly implicate biological
differences for this disparity (3–5). In
contrast, the disparity in incidence of the disease between second
generation Asian migrants and their prostate cancer-free pedigrees
strongly implicate environmental or behavioral/lifestyle variables
as risk factors in the etiology or prognosis of the disease
(6). Adoption of a Western
lifestyle characterized by less physical activity, increased intake
of red meat and dairy products high in saturated fats and
cholesterol has been associated with increased risk and poor
prognosis of the disease (6–8).
Regardless, epidemiological and population studies reveal that
lifestyle or environmental factors alone seldom account for the
disparity in cancer risk among different populations (9). Current evidence rather suggests that,
the interaction of specific genetic and lifestyle factors
predispose populations to most cancers (10–12).
The suggestion that some diets increase the burden
of prostate cancer has encouraged the increased scrutiny of
cholesterol-rich, Western-style diets as risk factors for the
disease. Overwhelming evidence suggests that all direct human
ancestors were largely herbivorous, and that the shift to meat or
cholesterol-rich diets in our immediate human ancestors selected
for genes that modulate the extra cholesterol burden (13). One meat-adaptive gene that
critically mediates cholesterol and lipid uptake by cells
throughout the body is apolipoprotein (Apo) E (13,14).
ApoE displays genetic polymorphism with three common alleles
namely, ε2, ε3, and ε4 in a single gene-locus in chromosome 19,
giving rise to 3 homo-zygous (apoε2/ε2, apoε3/ε3, apoε4/ε4) and 3
heterozygous genotypes (Apoε2/ε3, Apoε2/ε4, Apoε3/ε4) (15). Variants of this gene account for
more genetic differences in cholesterol metabolism than any other
gene (16,17). Apoε3 genotype is reportedly
selected for its positive effects in cholesterol control and for
reducing the risk of various diseases (13). Yet again for cholesterol control,
the ε2 and ε4 alleles are regarded as dysfunctional forms of the ε3
alleles (18), and are considered
to enhance the susceptibility to certain cancers (19). The ApoE3 gene is the most prevalent
in all human populations, and is considered to have evolved from
ancestral ApoE 4-like gene (13).
Based on the different binding affinities of apoE4 and apoE2 to the
LDL receptor and the LDL-related protein receptor, rates of
postprandial clearance of remnant lipoproteins are reportedly low
in subjects with the ApoE2/E2 phenotype, and high in those with the
apoE4/E4 phenotype (20). Overall,
carriers of the ε2 allele have lower serum LDL-cholesterol
concentrations relative to carriers of the ε3 and ε4 alleles
(21).
The ApoE gene also regulates reverse cholesterol
transport by transporting the excess cholesterol in peripheral
tissues (including the prostate) to the liver for excretion
(22). Efficient cellular
cholesterol efflux depends on ATP-binding cassette transporters
(ABCA1 and ABCG1), and uses poor ApoAI and ApoE-containing
particles (γ-LpE) from ApoE3/E3 subjects as avid cholesterol
acceptors (22). In some cells,
the transcription of ApoE is induced by exposure to cholesterol,
and its release into the extracellular medium is by classical
secretory pathway (23). There is
evidence that secreted ApoE also removes cholesterol in an
ABCA1-independent manner (24).
This pathway requires the intracellular synthesis and transport of
ApoE through internal membranes before its secretion (24). Regardless of the pathway used for
cholesterol efflux, theApoE isoforms differ in their ability to
deliver cholesterol to cells, and in their ability to mediate
cholesterol efflux (25,26). This is consistent with the
plausible differences in isoform-specific build-up in intracellular
cholesterol. Nascent cellular cholesterol first appears in rafts or
caveolae domains of plasma membranes, where it forms a complex with
caveolin-1 (cav-1), a cholesterol-binding protein (27,28).
The caveolae provide scaffolds for the aggregation of signaling
molecules that increase biological activities for cell survival,
and cell proliferation (29). Also
stored cholesterol is vital for mammalian cell growth, as growth
requires cholesterol for membrane biogenesis (28). Since cav-1 is a growth signaling
molecule, it may serve as a surrogate for cell proliferation, which
is the major feature of prostate tumor progression. The aim of this
study is to explore whether prostate cancer cells with apoE2/E4
phenotypes accumulate higher prostatic tissue cholesterol, and
whether cholesterol storage in such cell lines is associated with
aggressive cell phenotypes.
Materials and methods
Materials
Human prostatic adenocarcinoma cells LNCaP, PC3,
DU145 and MDA PCa 2b were obtained from American Type Culture
Collection (ATCC, Manassas, VA, USA). Cell culture media, MEM and
RPMI-1640 were purchased from Sigma-Aldrich (St. Louis, MO, USA),
while HPC1 serum free medium and FNC coating mix were obtained from
Athena Enzyme Systems (Baltimore, MD, USA). Sigma-Aldrich supplied
all the cell culture supplements, adenosine 3′,5′ cyclic
monophosphate (cAMP), acyl-CoA cholesterol acyltransferase (ACAT)
inhibitor (Sandoz 58-035), cholic acid, enhanced avian reverse HS
RT-PCR kit, and (2-hydroxypropyl)-β-cyclodextrin.
BODIPY-cholesterol was purchased from Avanti Polar Lipids, Inc.
(Alabaster, AL, USA), while Life Technologies (Grand Island, NY,
USA) supplied fetal bovine serum (FBS). Human ApoAI was purchased
from BioVision Inc. (Milpitas, CA, USA). Gene ruler ultra-low range
DNA ladder and Fast digest HhaI restriction enzyme kit was
supplied by Fermentas Inc. (Glen Burnie, MD, USA), while Novex TBE
running buffer, Hi-density sample buffer and polyacrylamide 8% TBE
gels were supplied by Invitrogen (Carlsbad, CA, USA). QIAmp DNA
mini kit and RNeasy kit were supplied by Qiagen (Valencia, CA,
USA).
Cell cultures
LNCaP cells were grown in RPMI-1640 medium
supplemented with 10% FBS, 1% L-glutamine, 0.5 ml fungizone, 5 ml
100 mM sodium pyruvate, 1% penicillin-streptomycin and buffered
with 0.75% HEPES. PC3 and DU145 cells were cultured in MEM with 10%
FBS 1% penicillin/streptomycin, 1% glutamine, 1% non-essential
amino acids, 0.1% gentamicin and fungizone, and buffered with 0.75%
HEPES. MDA PCa 2b cells were grown in HPC1 medium in FNC-precoated
6-well plates. The cells were incubated at 37°C in 5%
CO2.
Amplification of ApoE sequences from
genomic DNA
Genomic DNA was extracted as described in QIAMP DNA
Mini and blood Mini handbook (Qiagen) from confluent prostate
cancer cells. ApoE sequences were amplified from 0.5 μg of
genomic DNA using Jump Start AccuTaq DNA polymerase
(Sigma-Aldrich), and oligonucleotide primers F4
(5′-ACAGAATTCGCCCCGGCCTGGTACAC-3′) and F6
(5′-TAAGGTTGGCACGGCTGTCCAAGGA-3′) in a Master cycler (Eppendorf) as
previously described (30). The
ApoE sequences were amplified using the enhanced avian reverse
transcriptase-PCR kit procedure (Sigma-Aldrich). The reaction was
carried out in a reaction mixture volume of 50 μl. Reaction
conditions were: 1 cycle of 95°C for 5 min. This was followed by 30
cycles of heating (denaturation) at 95°C for 1 min and annealing at
60°C for 1 min. Extension was at 70°C for 2 min, and final
extension was at 70°C for 2 min. To confirm the amplification of
the ApoE sequences, the PCR products were analyzed on 1.5% agarose
gel and visualized by ethidium bromide staining methods using the
Gel Imaging Kodak MI standard Logic. The concentration of PCR
products was then measured using NanoDrop 1000 spectrophotometer
(Thermo Scientific, Wilmington, DE, USA).
Restriction isotyping of amplified ApoE
sequences with HhaI and gel analysis
After PCR amplification 3 μl of FastDigest
HhaI enzyme (Fermentas) was added directly to each reaction
mixture comprised of 30 μl of PCR amplification product, 51
μl of nuclease free water and 6 μl of 10X FastDigest
green buffer. The reaction mixtures were mixed gently and incubated
at 37°C in a water bath for 5 min, and then loaded onto 8%
non-denaturing pre-cast Novex polyacrylamide TBE gels (1.5-mm thick
and 8-cm long) and electrophoresed with 5X Novex TBE running buffer
for 20 min. The gel was run in an XCell SureLock Mini-Cell
electrophoresis tank at 200 constant voltage and stopped when the
bromophenol tracking dye was approximately two inches from the
bottom of the tank to enable visibility of the 20- and 25-bp bands.
After electrophoresis, the gel was treated with ethidium bromide (6
μl of 30%) for 10 min, washed three times in distilled water
and visualized by UV using the Gel Imaging Kodak MI standard Logic.
The sizes of the HhaI fragments were estimated by comparison
with Ultra low range DNA gene ruler (Fermentas Inc.).
Cav-1 gene expression analysis by
RT-PCR
After seeding all the prostate cancer cell lines to
confluence as described above, they were washed with PBS,
trypsinized and harvested on ice. RNA was extracted from them
according to the procedure described in the RNeasy isolation kit
(Qiagen). From each sample, 0.25 μg of total RNA was
subjected to first-strand cDNA synthesis using the enhanced avian
reverse transcriptase-PCR kit procedure (Sigma-Aldrich). The same
amount of each cDNA was used to amplify fragments of cav-1 and
GAPDH genes in the presence of Taq DNA polymerase (Sigma-Aldrich).
Specific primers for PCR amplification of the two genes were: i)
cav-1: sense 5′-CAACAAGGCCATGGCA GACGAGCT-3′ and antisense
5′-CATGGTACAACTGCC CAGATG-3′; (b) GAPDH: sense 5′-AGGTCGGAGTCAACG
GATTG-3′ and antisense 5′-GTGATGGCATGGACTGT GGT-3′. Cycling
conditions were: one initial denaturation cycle of 95°C for 5 min.
This was followed by 38 cycles of denaturation at 95°C for 2 min
(for cav-1) or 1 min (for GAPDH), and annealing at 55°C for 2 min
(for cav-1) or 57°C for 1 min (for GAPDH). Extension was at 72°C
for 2 min and final extension was for 1 cycle at 72°C for 5 min.
The amplified products were analyzed on 1.5% agarose gel and
visualized by ethidium bromide staining method. The intensity of
the bands was quantified with the Gel Imaging Kodak MI standard
Logic. All the reactions were normalized with GAPDH mRNA
expression.
Treatment of cells with
BODIPY-cholesterol
After growing the cells to confluence in their
respective media, they were washed with phosphate buffered saline
(PBS), and labeled with a complex mixture of 2 μg/ml ACAT
inhibitor, unlabeled cholesterol, BODIPY-cholesterol and
2-hydroxypropyl-β-cyclodextrin (2HβCD), prepared as previously
described (31). All the cells
were incubated in 1 ml of the labeling medium for 1 h, and then
washed with their respective HEPES-buffered growth media. The cells
were then equilibrated with these growth media containing 0.2% BSA,
ACAT inhibitor, and cAMP (0.3 mmol/l) for 18 h. After the
equilibration period, the cells were washed with the HEPES-buffered
media and triplicates of the experimental groups were incubated for
12 h in the media having cholesterol acceptors (10 μg/ml of
ApoA-I), while the control group received no such treatment.
Analysis of efflux and intracellular
BODIPY-cholesterol
At the end of the incubation period, the efflux
media in the control and cholesterol acceptor groups were removed,
filtered through a 0.45-μm filter, and the fluorescence
intensity measured with Infinite M200 microplate reader (Tecan) at
482-nm excitation and 515-nm emission. To analyze the intracellular
concentration of BODIPY-cholesterol, all the cells in the control
and cholesterol acceptor groups were solubilized with 1% cholic
acid and thoroughly mixed by shaking on a plate shaker for 4 h at
room temperature. The fluorescence was again measured as
described.
Microscopy and image analysis
After labeling the cells with BODIPY-cholesterol
mixture and subsequent equilibration in the respective growth
media, the cells were washed with PBS and treated with cholesterol
acceptors as described above. They were then split into two groups;
one group was incubated for 12 h in cholesterol acceptors (ApoAI),
and the second group had no acceptors. In both groups, the labeled
BODIPY-cholesterol was visualized within the living cells using an
IX70 inverted microscope (Olympus) equipped with a polychrome IV
monochromator (TILL Photonics) with appropriate filters. BODIPY
fluorescence intensity in the plasma membrane was then analyzed
with Image-Pro Plus software (Cybernetics).
Statistical analysis
Statistical analyses were performed using Minitab 16
(Minitab Inc., University Park, PA, USA) and SigmaPlot 10.0 (Systat
Software Inc., San Jose, CA, USA). Data are presented as the means
± SD, n=3. Differences between the groups were analyzed by a
one-way analysis of variance (ANOVA) and by Tukey’s post-hoc
test.
Results
ApoE genotypes of the prostate cancer
cell lines
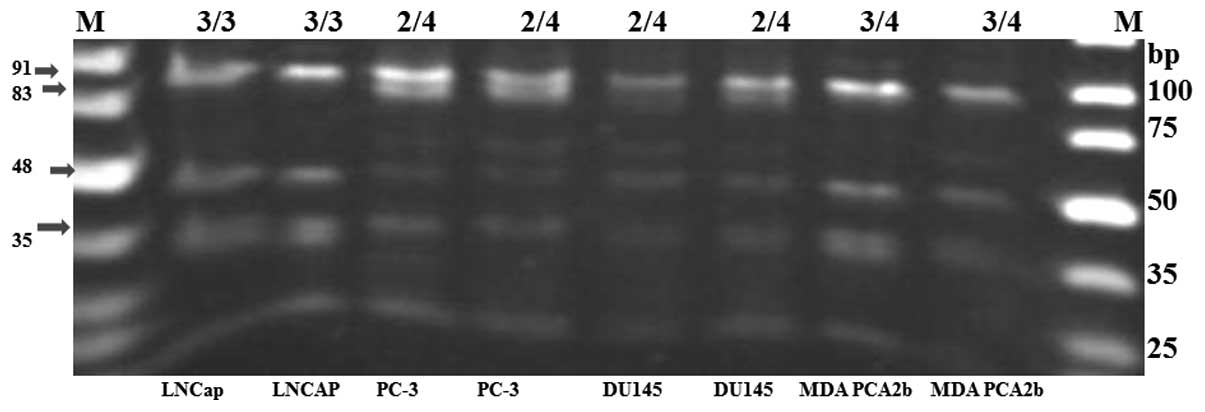
In this study, the unique restriction fragments from
the Apoε2 genotypes were detected as the 91- and 83-bp bands, while
the fragments for the Apoε3 genotypes aligned with the 91-, 48- and
35-bp bands of the DNA marker. The restriction fragments of the
Apoε4 genotypes were identified by their 48- and 35-bp fragments as
well as their unique 72-bp fragment. The intensities of the
overlapping 35/38-bp bands were lower for all the Apoε2/ε4
restriction fragments because of the absence of the 35 bp fragment
in all Apoε2 genotypes. Fig. 1
shows the polyacrylamide gel separation of ApoE isoforms from
genomic DNA of prostate cancer cells, after the DNA amplification
by PCR, and digestion with the restriction enzyme HhaI. The
digested fragments revealed the presence of homozygous and
heterozygous combinations of Apoε alleles. From the HhaI
cleavage signature fragments, the LNCaP cell line carried
homozygous Apoε3/ε3 alleles, while PC3 and DU145 were both
heterozygous for the Apoε2/ε4 alleles. Another heterozygous allele
combination (Apoε3/ε4) was found in MDA PCa 2b cell line.
Relative expression of cav-1 gene in
aggressive and non-aggressive prostate cancer cell lines
Endogenous ApoE colocalizes with cav-1 at the plasma
membrane to maintain lipid flux (32), thus we investigated the relative
expression of cav-1 mRNA in the prostate cancer cell lines in order
to validate its relationship with variants of ApoE gene and
cholesterol balance. Our results showed a relationship between
specific ApoE isoforms and the level of expression of cav-1 mRNA in
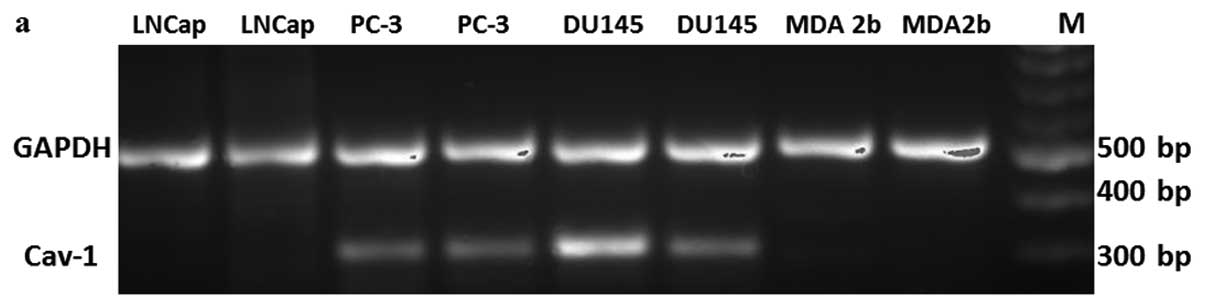
prostate cancer cell lines. According to the agarose gel
electrophoresis separation image shown in Fig. 2a, cav-1 mRNA expressed markedly in
the aggressive cell lines (PC3 and DU145). By contrast, cav-1 mRNA
was undetectable in the non-aggressive cell lines (LNCaP and MDA
PCa 2a). Semi-quantitative evaluation of cav-1 expression by RT-PCR
showed almost a two-fold increase in cav-1 expression in the
aggressive cell lines as compared to the non-aggressive cell lines
(Fig. 2b). The difference between
cav-1 gene expression in the aggressive (PC3 and DU145) and
non-aggressive (LNCaP and MDA PCa 2b) cell lines was statistically
significant (p<0.05).
Analysis of intracellular
BODIPY-cholesterol efflux
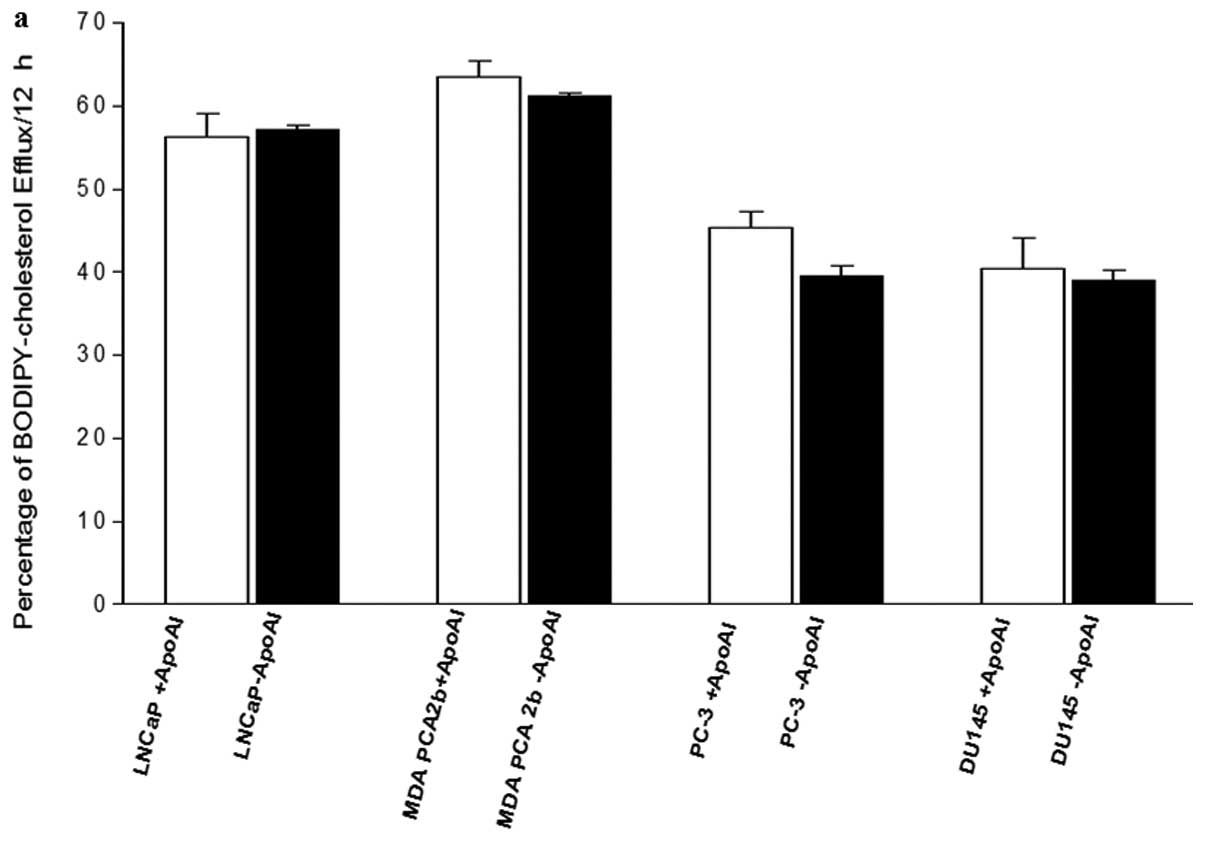
Examination of the relationship between ApoE
phenotypes and its cholesterol regulatory mechanism revealed a
significantly (p<0.001) greater efflux of BODIPY-cholesterol
from the non-aggressive cell lines compared to the aggressive cell
lines (Fig. 3a). The statistical
summary of the reverse cholesterol transport process showed a
significant difference (p<0.001) in cholesterol efflux across
all cell lines. Specifically, interval plots illustrating the
differences in cholesterol efflux between aggressive and
non-aggressive prostate cancer cell lines assessed with Tukey’s
test (Fig. 3b) revealed a
significantly higher cholesterol efflux in the non-aggressive
prostate cancer cell lines (LNCap, 56.32±3.33% and MDA PCa 2b,
63.49±2.28%) than from the aggressive cell lines (PC-3, 45.29±2.40%
and DU-145, 40.46±4.49%). It has been established that cells
incubated in the presence of cholesterol acceptors such as ApoA-I
release more cholesterol to the medium than those incubated without
the acceptors. This is consistent with our data showing a
statistically significant (p<0.05) difference between the mean
values of cholesterol efflux from cells cultured in the presence
and absence of cholesterol acceptors. There was no interaction
(p>0.17) between prostate cancer cell lines and the presence or
absence of cholesterol acceptors.
Analysis of retained
BODIPY-cholesterol
Consistent with the expected relationship between
apoE phenotypes and intra-cellular cholesterol regulation, we
observed a significantly higher (p<0.001) retention of
cholesterol by the aggressive cell lines, compared to the
non-aggressive cell lines (Fig.
3c) Likewise, the difference between cholesterol retention in
the presence and absence of efflux acceptors in some of the cell
lines was statistically significant (p<0.001).
Microscopic analysis of membrane
localized BODIPY-cholesterol
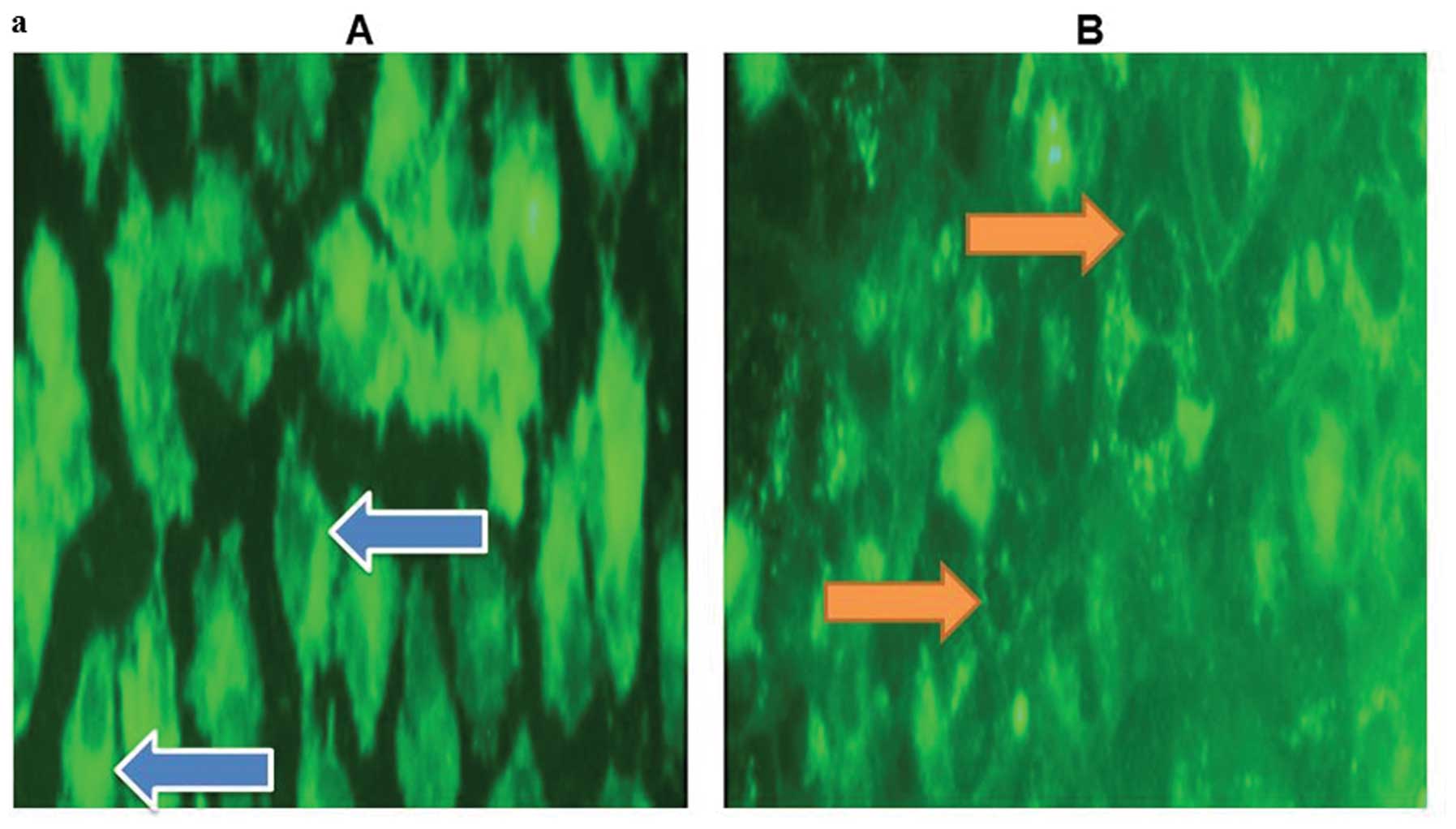
To probe the actual localization of the retained
BODIPY-cholesterol using fluorescence microscopy, we observed
higher membrane localization in of BODIPY-cholesterol in cell
cultures lacking cholesterol acceptors (Fig. 4a). Finally, the fluorescence
microscopy images showed a higher localization of
BODIPY-cholesterol in membranes of aggressive prostate cancer cell
lines, than those of non-aggressive ones (Fig. 4b).
Discussion
In this study, we explored the association between
ApoE phenotypes of prostate cancer cell lines and the risk of
aggressive prostate cancer. We investigated PC3 and DU145, the two
established androgen-insensitive cell lines that are highly
invasive and tumorigenic in athymic nude mice, and uniquely express
PROS1, a crucial protein mediator of cancer progression, thus
justifying their consistently undisputed characterization as
hallmarks of aggressiveness (33,34).
In contrast, LNCaP and MDA PCa 2b, which are androgen
receptor-expressing, androgen-refractory, poorly migratory and less
invasive cell lines that universally characterize
non-aggressiveness were also investigated (33,35–39).
Accordingly, we classified tumor aggressiveness by regarding the
highly tumorigenic and the moderately tumorigenic prostate cancer
cell lines (PC3 and DU145, respectively) as ‘aggressive’; while the
weakly tumorigenic cell lines (LNCaP and MDAPCa 2b) (33) were regarded as ‘non-aggressive’. We
observed that the aggressive cell lines were heterozygous for the
Apoε2/ε4 genotype, while the non-aggressive cell lines were
heterozygous carriers of at least one Apoε3 genotype. The
complementary alleles of the non-aggressive cell lines were Apoε3
(PC3) and Apoε4 (MDAPCa 2b). Thus, PC3 was homozygous for the
ApoE3/E3 phenotype, while MDAPCa 2b was heterozygous for the
ApoE3/E4 phenotype.
To the best of our knowledge, the genetic
polymorphism of ApoE in prostate cancer cell lines has not been
previously documented. Rather, an earlier investigation focused on
the linear relationship between the expression of ApoE mRNA and the
aggressiveness of prostate cancer cell lines (40). Remarkably, a number of previous
investigations had investigated the relationship between genetic
polymorphism of ApoE and susceptibility to breast (41) and prostate tumors (42). While these studies were generally
focused on the transcriptional regulation of tumor aggressiveness
by ApoE, the present study exclusively investigated the
relationship between ApoE variants and acknowledged aggressive
prostate cancer cell lines. Accordingly, we observed that the
aggressive prostate cancer cell lines carried the ApoE2/E4
phenotype, while the non-aggressive types carried the ApoE3/E3 or
E3/ E4 phenotypes. Although there is scarcely any data on the
allelic variation of prostate cancer cell lines, an earlier study
in human subjects revealed overexpressed Apoε2/ε4 alleles in
hormone-refractory, locally recurrent prostate cancer patients as
compared to control subjects (43). The observed carriage of the
Apoε2/ε4 alleles in these patients and our demonstration of its
frequency in aggressive prostate cancer cell lines highlight its
probable role in aggressive disease, since hormone-refractory and
recurrent prostate carcinomas are regarded as clinically aggressive
(44).
Currently, there are no clearly defined mechanisms
to explain the relationship between the ApoE isoforms, especially
the ApoE2/E4 phenotype and aggressive prostate cancer. However, a
possible link could be inferred from our observed accumulation of
intracellular cholesterol in cells carrying certain ApoE
phenotypes. A preponderance of evidence suggests that intracellular
cholesterol overload supports the progression of prostate cancer to
advanced disease (45). In this
context, the role of ApoE in clearing circulating cholesterol is
the regulation of its influx to cells (18), and its efflux by reverse
cholesterol transport, involving its extraction from peripheral
tissues to the liver for excretion (46). Phenotypic differences in the
regulation of cholesterol efflux from cells by ApoE (46,47),
may govern cholesterol accumulation in macrophages and RAW 264.7
cell lines (47,48). Although we are not aware of such a
relationship in prostate cancer cell lines, the concept is
consistent with our findings that the three ApoE phenotypes in our
prostate cancer cell lines have distinct abilities to promote
cholesterol efflux, and to inversely accumulate cholesterol.
The Apoε3 allele-carrying non-aggressive cell lines,
or precisely the LNCaP which carries the ApoE3/E3 phenotype, and
the MDA PCa 2b, which carries the ApoE3/E4 phenotype displayed
higher BODIPY-cholesterol efflux, while the aggressive cell lines
carrying the ApoE2/E4 phenotype (PC3 and DU145) displayed lower
BODIPY-cholesterol efflux. There was no significant difference in
the BODIPY-cholesterol efflux of the poorly aggressive cell lines,
and neither was there any difference in that of the aggressive cell
lines. To support the isoform-mediated differences in cellular
cholesterollevels, an assessment of BODIPY-cholesterol accumulation
by fluorescence microscopy revealed that the non-aggressive
prostate cancer cell lines, which carry at least one Apoε3 allele
retained less membrane cholesterol, in contrast to the Apoε3
allele-deficient aggressive cell lines. These data appear
consistent with the high expression of key effectors of cholesterol
production and downregulation of the expression of major
cholesterol exporters in prostate cancer cells, thus supporting the
reprogramming of their cholesterol metabolism to favor its
increased production and rapid cell growth (48). Our data revealed a higher
BODIPY-cholesterol within membranes of the aggressive prostate
cancer cell lines, which carry the ApoE2/E4 phenotype, while a
lower amount was found in membranes of the non-aggressive prostate
cancer cell lines, which carry at least one Apoε3 allele.
Taken together, these exploratory findings support
the premise that in prostate cancer cell lines, efficient
cholesterol efflux and its resultant depletion is associated with
certain ApoE isoforms. To the contrary, our data imply that other
ApoE isoforms are associated with deregulated cholesterol efflux,
and membrane accumulation. Consistent with this, our results
suggest that aggressive prostate cancer cell lines, carrying the
ApoE2/E4 phenotype, exhibited low BODIPY-cholesterol efflux and
accumulated BODIPY-cholesterol in the membranes.
To our knowledge there is paucity of data on how the
different ApoE isoforms influence cholesterol efflux in prostate
cancer cell lines. Nevertheless, studies from human intact
fibroblasts showed that lipoprotein-containing ApoE particles
(γ-LpE) from ApoE3/E3 individuals stimulated 7- to 13-fold more
cholesterol efflux than from ApoE2/E2 or ApoE4/E4 individuals
(46). Earlier studies using RAW
264.7 mouse macrophage cells did not find any difference in
cholesterol efflux from cells with the three ApoE isoforms
(49). However, all the clonal
macrophage cell lines used in that study carried three different
homozygous ApoE-isoforms, whereas our investigated prostate cancer
cell lines had a combination of heterozygous and homozygous ApoE
isoforms. Again, the differences in the ApoE isoform-dependent
cholesterol efflux in RAW 264.7 cells may have been annulled by the
presence in the culture of a cAMP analogue, which induces a
cellular ApoE receptor-mediated transfer of cholesterol to all
apolipoproteins, followed by the subsequent release of the
lipoprotein particles (49).
ApoE influences cholesterol efflux from cells by
reverse cholesterol transport, and rids cells of excess
cholesterol, which is transported to the liver for excretion. To
influence this cholesterol efflux, ApoE is generally synthesized in
various cell types including the prostate, and then transported to
the plasma membrane for secretion (23). In cholesterol-rich cells, this
secreted ApoE promotes cholesterol efflux even in the absence of
cholesterol acceptors (23). The
exact mechanism by which ApoE mediates cellular cholesterol efflux
and whether this cholesterol efflux is isoform-specific is still
unknown. The most consistent explanation for ApoE-mediated
cholesterol efflux had relied on the intracellular association of
ApoE with cAMP-induced ABCA1, which induces increased secretion of
ApoE and catalysis of an initial transfer of cholesterol to the
lipid poor ApoE (16). Doubt has
been cast on the sole regulation of cholesterol efflux by ABCA1,
leading to the recognition of ABCG1 as finalizing the full transfer
of this cholesterol to the apolipoprotein (16). One reason cited for the
plausibility of the ApoE isoform-dependent cholesterol efflux was
the low affinity of the apoE2 isoform versus the ApoE3 or ApoE4
isoforms for heparan sulfate proteoglycans (HSPGs), which modulate
cholesterol depleting ability of ApoE (50). The higher cholesterol efflux of
ApoE2-carrying cells was explained by several demonstrations that
the higher affinity of ApoE3 and ApoE4 for HSPGs caused the
sequestration of the latter isoforms into the pericellular
proteoglycan matrix, leading to their eventual cellular degradation
(47,50). Thus, ApoE2 is the most effective
for cholesterol efflux, followed by ApoE3, while ApoE4 is least
effective. The effective cholesterol efflux of the ApoE2/E2
phenotype-carrying cells is consistent with an earlier observation
that cholesterol loaded cells redirect this ApoE isoform from the
degradatory to the secretory pathway (47). This study also revealed that cells
carrying the ApoE3/E3 phenotype secreted nearly 77% of ApoE
proteins, leading to reduced intracellular cholesterol accumulation
(47). These investigations
concluded that ApoE4/ E4-carrying cells secreted the most ApoE
apolipoproteins, but lacked effective net cholesterol efflux
because of greater HSPGs affinity and re-uptake of cholesterol
particles (47,50).
Our observed reduction in cholesterol efflux and
increased cholesterol retention by the ApoE2/E4 phenotype-carrying
prostate cancer cell lines highlights the dominance of the ε4
allele over the ε2 allele in heterozygosity. Elucidating how the ε4
allele dominates the ε2 allele in cholesterol transport will
significantly improve our understanding of the isoform-dependent
regulation of cholesterol efflux and retention. The preceding
discussion is consistent with previous evidence showing that
reduced cholesterol efflux by ApoE contributes to membrane
cholesterol accumulation (51).
The current paradigm in cholesterol homeostasis holds that the
increased prevalence of lipid rafts and caveolae in cells is
attributable to membrane cholesterol accumulation. Cav-1 is the
most celebrated structural protein of the caveolae that binds
cholesterol, and its absence impairs cholesterol homeostasis
(52). Additionally, membrane
cholesterol enrichment has been associated with increased cav-1
mRNA expression (53). This is
consistent with our studies showing that
BODIPY-cholesterol-accumulating and aggressive prostate cancer cell
lines overexpress cav-1 mRNA, whereas BODIPY-cholesterol poor and
non-aggressive prostate cancer cell lines express low or no cav-1
mRNA.
It has been previously demonstrated that cav-1
expression is highly dependent on the availability of cholesterol
(54), and its depletion
diminishes the caveolae by removing cav-1 from the membrane
(55). We observed a higher
retention of the fluorescent cholesterol analogue in the aggressive
prostate cancer cell lines, which concurrently overexpressed cav-1
mRNA, further strengthening the relationship between cav-1
expression and cell aggression. This relationship supports earlier
demon stration that cav-1 is a potential biomarker of aggressive
prostate cancer (56). To the
contrary, we found no expression of cav-1 mRNA in the
non-aggressive prostate cancer cell lines. This finding is
consistent with previous studies indicating the overexpression of
cav-1 in mouse and human metastatic prostate cancer cells (57). This has been corroborated by a
recent report indicating that the absence of cav-1 significantly
inhibited the progression of prostate cancer to highly invasive and
metastatic disease (58). Overall,
molecular approaches highlight the plausible relationship between
intracellular cholesterol accumulation and prostate cancer.
Pathological studies of prostate tumor samples had provided
evidence for such relationship by demonstrating a positive
correlation between Gleason grade and the levels of expression of
this cholesterol-binding protein or cav-1 (55).
Although the role of caveolae in the progression of
solid tumors is not fully understood, the contribution of cav-1 to
signaling processes that initiate prostate cancer has been
extensively investigated. The initiation and progression of cancer
by cav-1 is linked to its tendency to form platforms that aggregate
membrane proteins for cell proliferation signaling. This function
of cav-1 is partly related to its possession of an amino acid
domain that binds a variety of signaling proteins (49), which recently includes ABCA1 and
ApoE (32,59). Binding of these proteins to cav-1
is believed to inactivate downstream growth signals (59). An interaction between cav-1, ABCA1
and ApoE is appropriate as it suggests crosstalk among them
favoring cholesterol balance, reduced cholesterol overload and
inhibition of cell proliferation. However, the effect of cav-1
expression on cholesterol efflux is still controversial and has
been attributed to differences in the cell types used for such
studies (60). The molecular
mechanism by which these membrane-anchored proteins maintain the
correct intracellular cholesterol balance and the particular ApoE
isoforms that perturb such equilibrium has not been elucidated. Our
results highlight the need for further experiments to confirm
whether the ApoE2/E4 is the dysfunctional phenotype that inhibits
cholesterol efflux, leading to prostate cancer cell
proliferation.
In summary, our data suggest the possibility of the
relationship between the ApoE2/E4 phenotype, the intracellular
cholesterol efflux and cholesterol content of prostate cancer
cells. This is consistent with our observed overexpression of
cav-1, the sentinel gene for cholesterol overload exclusively by
the ApoE2/E4-carrying aggressive prostate cancer cell lines.
We conclude that the overexpression of cav-1 and
cholesterol overload in aggressive prostate cancer cell lines,
suggests that cav-1 may confer survival advantage on prostate
cancer cells leading to disease progression. The less than
anticipated aggressiveness of the African American cancer cell line
(MDA PCa 2b) suggests that the Apoε3 allele masks the deregulated
cholesterol balance associated with the ancestral Apoε4 allele. The
low cholesterol efflux and higher cholesterol retention in ApoE2/E4
phenotype-carrying aggressive prostate cancer cells justifies
further investigation of ApoE2/E4 phenotype as a biomarker of
aggressive disease. While the evidence amassed so far and elsewhere
clearly indicates that genetic factors play a key role in
determining the susceptibility to aggressive prostate cancer, the
sample size in our study imposes limited statistical power and
restrains definitive conclusions. Unraveling the mechanism by which
the dysfunctional apoE isoforms transforms the prostate cancer cell
lines to aggressive phenotypes could be a daunting task, which
however could be overcome by genetic manipulation under varying
physiological conditions, and may provide new insights into the
pathogenesis and therapeutic targets of the disease.
Acknowledgements
Funding was provided by the National
Cancer Institute (NCI) Cancer Prevention and Control Training
Program Grant R25 CA04788 (G.O.I.).
References
|
1.
|
Williams SD, Cook ED, Anderson KB and
Hamilton SJ: Bridging the gap between populations: the challenge of
reducing cancer disparities among African-American and other ethnic
minority populations. US Oncol. 4:72–75. 2008.
|
|
2.
|
Ifere GO, Abebe F and Ananaba GA: Emergent
trends in the reported incidence of prostate cancer in Nigeria.
Clin Epidemiol. 4:19–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Sakr WA: Prostatic intraepithelial
neoplasia: A marker for high-risk groups and a potential target for
chemoprevention. Eur Urol. 35:474–478. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Bock CH, Powell I, Kittles RA, Hsing AW
and Carpten J: Racial disparity in prostate cancer incidence,
biochemical recurrence, and mortality. Prostate Cancer. 716178:1–2.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bigler SA, Pound CR and Zhou X: A
retrospective study on pathological features and racial disparities
in prostate cancer. Prostate Cancer. 2011:2394602011. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lee J, Demissie K, Lu SE and Rhoads GG:
Cancer incidence among Korean-American immigrants in the United
States and native Koreans of South Korea. Cancer Control. 14:78–85.
2007.PubMed/NCBI
|
|
7.
|
Ma RW and Chapman K: A systematic review
of the effect of diet in prostate cancer prevention and treatment.
J Hum Nutr Diet. 22:187–199. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Michaud DS, Augustsson K, Rimm EB,
Stampfer MJ, Willet WC and Giovanucci E: A prospective study on
intake of animal products and risk of prostate cancer. Cancer
Causes Control. 12:557–567. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Waddell WJ: Epidemiological studies and
effects of environmental estrogens. Int J Toxicol. 17:173–191.
1998. View Article : Google Scholar
|
|
10.
|
Le Marchand L and Wilkens LR: Design
considerations for genomic association studies: importance of
gene-environment interactions. Cancer Epidemiol Biomarkers Prev.
17:263–267. 2008.PubMed/NCBI
|
|
11.
|
Nothlings U, Yamamoto JF, Wilkens LR,
Murphy SP, Park SY, Henderson BE, Kolonel LN and Le Marchand L:
Meat and heterocyclic amine intake, smoking, NAT1 and NAT2
polymorphisms, and colorectal cancer risk in the multiethnic cohort
study. Cancer Epidemiol Biomarkers Prev. 18:2097–2106. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Wünsch Filho V and Zago MA: Modern cancer
epidemiology research: genetic polymorphism and environment. Rev
Saude Publica. 39:490–497. 2005.
|
|
13.
|
Finch CE and Stanford CB: Meat-adaptive
genes and the evolution of slower aging in humans. Q Rev Biol.
79:3–50. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Mahley RW: Apolipoprotein E: cholesterol
transport protein with expanding role in cell biology. Science.
240:622–630. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Tziakas DN, Chalikias GK, Antonoglou CO,
Veletza S, Tentes IK, Kortsaris AX, Hatseras DI and Kaski JC:
Apolipoprotein E genotype and circulating interleukin-10 levels in
patients with stable and unstable coronary artery disease. J Am
Coll Cardiol. 48:2471–2481. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Leduc V, Domenger D, De Beaumont L,
Lalonde D, Bélanger-Jasmin S and Poirier J: Function and
comorbidities of apolipoprotein E in Alzheimer’s disease. Int J
Alzheimers Dis. 2011:1–22. 2011.
|
|
17.
|
Eichner JE, Dunn ST, Perveen G, Thompson
DM, Stewart KE and Stroehla BC: Apolipoprotein E polymorphism and
cardiovascular disease: a HuGE review. Am J Epidemiol. 155:487–495.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Mahley RW and Rall SC Jr: Apolipoprotein
E: far more than a lipid transport protein. Annu Rev Genomics Hum
Genet. 1:507–537. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Moore RJ, Chamberlain RM and Khuri FR:
Apolipoprotein E and the risk of breast cancer in African-American
and non-Hispanic white women. A review. Oncology. 66:79–93. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kallio MJ, Salmenperä L, Siimes MA,
Perheentupa J, Gylling H and Miettinen TA: Apolipoprotein E
phenotype determines serum cholesterol in infants during both
high-cholesterol breast feeding and low-cholesterol formula
feeding. J Lipid Res. 38:759–764. 1997.PubMed/NCBI
|
|
21.
|
Berglund L: The APOE gene and diets - food
(and drink) for thought. Am J Clin Nutr. 73:669–670.
2001.PubMed/NCBI
|
|
22.
|
Papaioannou I, Simmons JP and Owen JS:
Targeted in situ gene correction of dysfunctional APOE alleles to
produce atheroprotective plasma ApoE3 protein. Cardiol Res Pract.
2012:1487962012.PubMed/NCBI
|
|
23.
|
Kockx M, Jessup W and Kritharides L:
Regulation of endogenous apolipoprotein E secretion by macrophages.
Arterioscler Thromb Vasc Biol. 28:1060–1067. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Huang ZH, Fitzgerald ML and Mazzone T:
Distinct cellular loci for the ABCA1-dependent and
ABCA1-independent lipid efflux mediated by endogenous
apolipoprotein E expression. Arterioscler Thromb Vasc Biol.
26:157–162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Huang Y, von Eckardstein A, Wu S and
Assmann G: Effects of the apolipoprotein E polymorphism on uptake
and transfer of cell derived cholesterol in plasma. J Clin Invest.
96:2693–2701. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lane RM and Farlow MR: Lipid homeostasis
and apolipoprotein E in the development and progression of
Alzheimer’s disease. J Lipid Res. 46:949–968. 2005.
|
|
27.
|
Uittenbogaard A, Ying Y and Smart EJ:
Characterization of a cytosolic heat-shock protein-caveolin
chaperone complex. Involvement in cholesterol trafficking. J Biol
Chem. 273:6525–6532. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Liscum L and Munn NJ: Intracellular
cholesterol transport. Biochim Biophys Acta. 1438:19–37. 1999.
View Article : Google Scholar
|
|
29.
|
Li YC, Park MJ, Ye SK, Kim CW and Kim YN:
Elevated levels of cholesterol-rich lipid rafts in cancer cells are
correlated with apoptosis sensitivity induced by
cholesterol-depleting agents. Am J Pathol. 168:1107–1118. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Hixson JE and Vernier DT: Restriction
isotyping of human apolipoprotein E by gene amplification and
cleavage with Hha I. J Lipid Res. 31:545–548.
1990.PubMed/NCBI
|
|
31.
|
Sankaranarayanan S, Kellner-Weibel G, de
la Llera-Moya M, Phillips MC, Asztalos BF, Bittmam R and Rothblat
GH: A sensitive assay for ABCA1-mediated cholesterol efflux using
BODIPY-cholesterol. J Lipid Res. 52:2332–2340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Yue L and Mazzone T: Endogenous adipocyte
apolipoprotein E is colocalized with caveolin at the adipocyte
plasma membrane. J Lipid Res. 52:489–498. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Nair HK, Rao KV, Aalinkeel R, Mahajan S,
Chawda R and Schwartz SA: Inhibition of prostate cancer cell colony
formation by the flavonoid quercetin correlates with modulation of
specific regulatory genes. Clin Diagn Lab Immunol. 11:63–69.
2004.PubMed/NCBI
|
|
34.
|
Saraon P, Musrap N, Cretu D, Karagiannis
GS, Batruch I, Smith C, Drabovich AP, Trudel D, van der Kwast T,
Morrissey C, Jarvi KA and Diamandis EP: Proteomic profiling of
androgen-independent prostate cancer cell lines reveals a role for
protein S during the development of high grade and
castration-resistant prostate cancer. J Biol Chem. 287:34019–34031.
2012. View Article : Google Scholar
|
|
35.
|
Wang B, Yang Q, Ceniccola K, Bianco F,
Andrawis R, Jarret T, Frazier H, Patierno SR and Lee NH: Androgen
receptor-target gene in African American prostate cancer
disparities. Prostate Cancer. 2013:7635692013. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Kim DH and Wirtz D: Recapitulating cancer
cell invasion in vitro. Proc Natl Acad Sci USA. 108:6693–6694.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Hoosein NM: Neuroendocrine and immune
mediators in prostate cancer progression. Front Biosci.
3:D1274–D1279. 1998.PubMed/NCBI
|
|
38.
|
Hara T, Miyazaki H, Lee A, Tran CP and
Reiter RE: Androgen receptor and invasion in prostate cancer.
Cancer Res. 68:1128–1135. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Nomura DK, Lombardi DP, Chang JW, Niessen
S, Ward AM, Long JZ, Hoover HH and Cravatt BF: Monoacylglycerol
lipase exerts dual control over endocannabinoid and fatty acid
pathways to support prostate cancer. Chem Biol. 18:846–856. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Venanzoni MC, Giunta S, Murara GB, Storari
L, Crescini C, Mazzucchelli R, Montironi R and Seth A:
Apolipoprotein E expression in localized prostate cancers. Int J
Oncol. 22:779–786. 2003.PubMed/NCBI
|
|
41.
|
Saadat M: Apolipoprotein E (ApoE)
polymorphism and susceptibility to breast cancer: a meta-analysis.
Cancer Res Treat. 44:121–126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Niemi M, Kervinen K, Kiviniemi H,
Lukkarinen O, Kyllönen AP, Apaja-Sarkkinen M, Savolainen MJ,
Kairaluoma MI and Kesäniemi YA: Apolipoprotein E phenotype,
cholesterol and breast and prostate cancer. J Epidemiol Community
Health. 54:938–939. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Haapla K, Lehtimäki T, Ilveskoski E and
Koivisto PA: Apolipoprotein E is not linked to locally recurrent
hormone-refractory prostate cancer. Prostate Cancer Prostatic Dis.
3:107–109. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Koivisto P, Visakorpi T, Rantala I and
Isola J: Increased cell proliferation activity and decreased cell
death are associated with the emergence of hormone-refractory
recurrent prostate cancer. J Pathol. 183:51–56. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Mostaghel EA, Solomon KR, Pelton K,
Freeman MR and Montgomery RB: Impact of circulating cholesterol
levels on growth and intratumoral androgen concentration of
prostate tumors. PLoS One. 7:e300622012. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Krimbou L, Denis M, Haidar B, Carrier M,
Marci M and Genest J Jr: Molecular interactions between apoE and
ABCA1: impact on apoE lipidation. J Lipid Res. 45:839–848. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Cullen P, Cignarella A, Brennhausen B,
Mohr S, Assmann G and Von Eckardstein A: Phenotype-dependent
differences in apolipoprotein E metabolism and in cholesterol
homeostasis in human monocyte-derived macrophages. J Clin Invest.
101:1670–1677. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Murtola TJ, Syvälä H, Pennanen P, Bläuer
M, Solakivi T, Ylikomi T and Tammela TL: The importance of LDL and
cholesterol metabolism for prostate epithelial cell growth. PLoS
One. 7:e394452012. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Smith JD, Miyata M, Ginsberg M, Grigaux C,
Shmookler E and Plump AS: Cyclic AMP induces apolipoprotein E
binding activity and promotes cholesterol efflux from a macrophage
cell line to apolipoprotein acceptors. J Biol Chem.
271:30647–30655. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Hara M, Matsushima T, Satoh H, Iso-o N,
Noto H, Togo M, Kimura S, Hashimoto Y and Tsukamoto K:
Isoform-dependent cholesterol efflux from macrophages by
apolipoprotein E is modulated by cell surface proteoglycan.
Arterioscler Thromb Vasc Biol. 23:269–274. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Karasinska JM and Hayden MR: Cholesterol
metabolism in Huntington disease. Nat Rev Neurol. 7:561–572. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Fernández-Hernando C, Yu J, Dávalos A,
Prendergast J and Sessa WC: Endothelial-specific overexpression of
caveolin-1 accelerates atherosclerosis in apolipoprotein
E-deficient mice. Am J Pathol. 177:998–1003. 2010.PubMed/NCBI
|
|
53.
|
Cohen AW, Hnaska R, Schubert W and Lisanti
MP: Role of caveolae and caveolins in health and disease. Physiol
Rev. 84:1341–1379. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Frank PG, Cheung MW, Pavlides S, Llaverias
G, Park DS and Lisanti MP: Caveolin-1 and regulation of cellular
cholesterol homeostasis. Am J Physiol Heart Circ Physiol.
291:H677–H686. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Daniel EE, El-Yazbi A and Cho WJ: Caveolae
and calcium handling, a review and a hypothesis. J Cell Mol Med.
10:529–544. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Yang G, Truong LD, Wheeler TM and Thompson
TC: Caveolin-1 expression in clinically confined human prostate
cancer: a novel prognostic marker. Cancer Res. 59:5719–5723.
1999.PubMed/NCBI
|
|
57.
|
Thompson TC: Metastasis-related genes in
prostate cancer: the role of caveolin-1. Cancer Metastasis Rev.
17:439–442. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Williams TM, Hassan GS, Li J, Cohen AW,
Medina F, Frank PG, Pestell RG, Di Vizio D, Loda M and Lisanti MP:
Caveolin-1 promotes tumor progression in an autochthonous mouse
model of prostate cancer. J Biol Chem. 280:25134–25145. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Lin YC, Ma C, Hsu WC, Lo HF and Yang VC:
Molecular interaction between caveolin-1 and ABCA1 on high-density
lipoprotein-mediated cholesterol efflux in aortic endothelial
cells. Cardiovasc Res. 75:575–583. 2007. View Article : Google Scholar
|
|
60.
|
Le Lay S, Rodriguez M, Jessup W, Rentero
C, Li Q, Cartland S, Grewal T and Gaus K: Caveolin-1-mediated
apolipoprotein A-I membrane binding sites are not required for
cholesterol efflux. PLoS One. 6:e233532011.PubMed/NCBI
|