Introduction
Choriocarcinoma is a highly malignant trophoblastic
tumor that may occur after miscarriage, abortion, ectopic pregnancy
or even normal pregnancy. In addition, it is characterized
pathologically by the loss of its original structure of
trophoblastic cells and excessive invasion capacity into the
myometrium (1). Choriocarcinoma
can also widely metastasize to other organs or tissues through both
the venous and lymphatic systems. Early hematogenous and lymphatic
metastatic spread can cause rapid death (2).
Although cure rate has been improved because of
progress of chemotherapy, the toxicity and side effects still
remain intolerable. Thus, it is urgent to explore the mechanisms of
proliferation and invasiveness and find new target for drug therapy
(3).
The transforming growth factor-β1 (TGF-β1) is a
multi-functional polypeptide cytokine which belongs to a growth
factor superfamily and it regulates a variety of cellular processes
such as cell differentiation, proliferation, cycle arrest and
extracellular matrix production (4). Our previous studies have demonstrated
that TGF-β1 promotes the invasive capability of choriocarcinoma
JEG-3 cells and TGF/Smad pathway may play a critical role in the
initiation of trophoblastic invasion process (5).
TGF/Smad pathway plays a pivotal role in
intracellular signaling. TGFβ-1 initiates signaling via a cell
surface transmembrane serine/threonine kinase receptor complex
which then activates its downstream Smad protein (6). There are three different functional
groups of Smad proteins: receptor-regulated R-Smads (Smad1, 2, 3, 5
and 8), the co-receptor Smad (Co-Smad) 4 and the inhibitory I-Smads
(Smad6 and 7). In addition, the receptors of the TGF-β superfamily
consist of TβR I and TβR II. TGFβ-1 ligand firstly binds to TβR II,
which then activates and combines to TβR I. After that, receptor
binding induces phosphorylation of its downstream proteins Smad2
and Smad3 (R-Smads) at a C terminal SSXS motif (7). Smad4 (Co-Smad) works as a mediator,
carries phosphorylated R-Smad (Smad2 and Smad3) into the nuclear
where target gene is processed (8).
The function of TGFβ/Smad signaling pathway in
cancer progression is bidirectional. At the early stage of
carcinogenesis, TGF-β works as a tumor suppressor, which induces
growth arrest, but in the later stage or in malignant tumor, it
promotes the growth of tumor cells (9). Furthermore, our previous studies on
the impacts of TGF-β1/Smad signal pathway in JEG-3 cell
proliferation and invasion are in agreement with the above view
(5).
P38 MAPK is a member of the MAPK family that is
responsive to environmental stresses and inflammatory cytokines
(10) and is involved in cell
differentiation, apoptosis and autophagy. It is activated by a
variety of cellular stresses including osmotic shock, inflammatory
cytokines, lipopolysaccharides (LPS), Ultraviolet light and growth
factors (11). As with other MAPK
cascades, the membrane-proximal component is a MAPKKK, typically a
MEKK or a mixed lineage kinase (MLK). The MAPKKK phosphorylates and
then activates MKK3/6, the p38 MAPK kinases. MKK3/6 can also be
activated directly by ASK1, which is stimulated by apoptotic
stimuli (12). P38 MAPK is
involved in regulation of HSP27 and MK2 (MAPKAPK-2), MK3
(MAPKAPK-3) and several transcription factors including ATF-2,
Stat1, the Max/Myc complex, MEF-2, Elk-1 and indirectly CREB via
activation of MSK1 (13,14). Consistent with important function
in tumorigenesis, p38 MAPK signaling is also associated with cancer
in humans.
Recent studies have shown that TGF-β can also
activate other signaling pathways including the p38 MAPK pathway
(15). Studies on pulmonary
epithelial cells suggest that TGF-β activates p38 MAPK within 30
min and inhibition of p38 MAPK can significantly reduce
TGF-β1-dependent gene expression for the extracellular
matrix-related gene fibronectin (16,17).
In addition, blockade of Smad pathway caused by Smad7
overexpression, can result in TGF-β1-induced apoptosis mediated by
p38 MAPK signaling (18).
Moreover, p38 MAPK inhibitors have been shown to
inhibit TGF-β/Smads signaling activity through affecting all levels
of the TGF-β receptor cascade by blockading Smad phosphorylation,
nuclear translocation and target gene activation (4). The study of Dziembowska et al
have shown that the p38 MAPK inhibitor SB202190, decreases the
activity of Smad-dependent promoter induced by TGF-β and the
phosphorylation of Smad2 (19).
According to the study of Tsukada et al, blockade of p38
MAPK signaling with SB203580 inhibited phosphorylation of Smad2 and
Smad3 and the phosphorylation of the TGF-β type I receptor, as
there appears to be similarities in the ATP binding pocket between
TGF-β type I receptor and p38 MAPK (20). Thus, there is an unexpected
interaction between TGF-β and p38 MAPK pathways existing in a
variety of diseases, but the molecular mechanism still remains
unclear. Recently, few studies have been reported on crosstalk
between TGF-β and p38 MAPK in choriocarcinoma (21,22).
Based on the above observations, we investigated
signaling crosstalk between the p38 MAPK pathway and TGF-β pathway
in human choriocarcinoma cells by using the p38 MAPK inhibitor SB
203580 and the TGF-β receptor inhibitor LY 364947.
Materials and methods
Materials
The human placental choriocarcinoma JEG-3 cell line
was obtained from the State Key Laboratory of Reproductive Biology
(SKLRB), Institute of Zoology (IOZ), Chinese Academy of Sciences
(CAS). The study was approved by the ethics committee of the
Natural Science Foundation of Hebei Province and the Education
Department of Hebei province, Hebei, China.
Methods
JEG-3 cell culture
JEG-3 cells were cultured in an incubator with 5%
CO2 at 37°C in RPMI-1640 supplemented with 10% fetal
bovine serum (FBS, Hangzhou Sijiqing Biological Engineering
Materials Co., Ltd., Hangzhou, China), 200 mM glutamine, 100 mM
pyruvic acid Na, 100 μg/ml streptomycin and 100 U/ml
penicillin. When the cells reached ∼70–80%, they were subcultured
with 0.25% trypsin and 0.02% EDTA (5).
Immunofluorescence analysis
JEG-3 cells were seeded on glass coverslips and
cultured in 24-well plate with an initial concentration of
5×104 cells/ml for 48 h prior to staining. Wells were
divided into 8 groups as follows: control group, TGF-β1 group, 1
μM SB203580, 3 μM SB203580, 1 μM LY364947
(Sigma, St. Louis, MO, USA), 3 μM LY364947. The cells were
pretreated in appropriate wells to 80% confluence with different
concentrations of TGF-β1 receptor inhibitor (LY36494, Sigma) and
p38 MAPK inhibitor (SB203580, Sigma) and cultured for 4 h, then 1
μM TGF-β1 (PeproTech Inc., Rocky Hill, NJ, USA) was added
into each well, except control group, continuing incubation for 2
h. After the incubation with TGF-β1, cells were fixed in 4%
paraformaldehyde for 10 min at room temperature, then washed with
PBS 3 times for 3 min each. Then, cells were permeabilized in 0.5
ml of 0.1% Triton X-100 for 15 min at room temperature (23). The cells were washed by PBS 3 times
and followed by blocking in 10% goat serum for 20 min. The primary
antibody was diluted to the concentration of 1:100, then the cells
were incubated in the primary antibody overnight at 4°C. Before
secondary antibody incubation, cells were placed into incubator
with 5% CO2 at 37°C for 45 min, then washed 3 times for
each with PBS (24). The secondary
antibody (Epitomics Inc., USA) was diluted to the concentration of
1:50 and cells were incubated in secondary antibody at 37°C for 30
min, followed by PBS washing 3 times for 5 min each. DAPI, with the
concentration of 1:100, was added to the cell for 5 min at room
temperature in the dark. The primary antibodies were used as
follows: for p-Smad3 (Epitomics); for p38 (Epitomics); and p-p38
(Epitomics). Images were viewed by immunofluorescence microscopy
(Ti-u, Nikon Eclipse, Japan).
Western blot analysis
Cells were incubated in a 6-well plate with an
initial concentration of 5×104 cells/ml for 48 h. Wells
were divided into 6 groups as follows: control group, TGF-β1 group,
1 μM SB203580, 3 μM SB203580, 1 μM LY364947, 3
μM LY364947. When cells reached ∼80%, the same treatments
were given to cells as stated in immunofluorescence analysis. The
cells were collected and lysed on ice with buffer (10% glycerol,
2.3% SDS, 62.5 mM Tris, pH 6.8 150 mM NaCl, 10 mM EDTA, 1 mg/ml
leupeptin, 1 mg/ml pepstatin, 5 mg/ml chymostatin, 1 mg/ml
aprotinin, 1 mM phenyl-methylsulphonyl fluoride) and centrifuged at
12,000 rpm for 5 min at 4°C. The supernatants were collected as a
whole cell protein extract. Protein concentrations were determined
by the bicinchoninic acid assay (25). Then, equal amounts of sample were
denatured at 95°C for 15 min and separated on 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to polyvinylidene difluoride (PVDF) membranes.
Membranes were blocked with 5% non-fat milk in TBS-T (10 mM Tris,
pH 8.0, 150 mM NaCl and 0.1% Tween-20) for 2 h, at room temperature
followed by TBST washing 3 times and incubated with primary
antibody (diluted 1:500) overnight at 4°C (26). After TBST buffer washing, the
membranes were incubated with the secondary antibody (diluted
1:2000) for 1 h at room temperature. β-actin was the internal
control. The primary antibodies were: for Smad3 (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA); for p-Smad3 (Epitomics);
for p38 (Epitomics); and p-p38 (Epitomics). Immunoreactive bands
were detected with Super ECL Plus luminescence fluid (Applygen
Technologies Inc., Beijing, China). The densities of the bands were
scanned and calculated with Quantity One software (Bio-Rad,
Hercules, CA, USA).
Statistical analysis
All data were expressed as means ± standard
deviation (SD). One-way analysis of variance (ANOVA) was used to
compare differences among groups. The SNK-q test was performed to
compare the difference of each two-group. Differences were
considered statistically significant at P<0.05. All statistical
analysis was performed using SPSS 19.0 software (SPSS, Chicago, IL,
USA).
Results
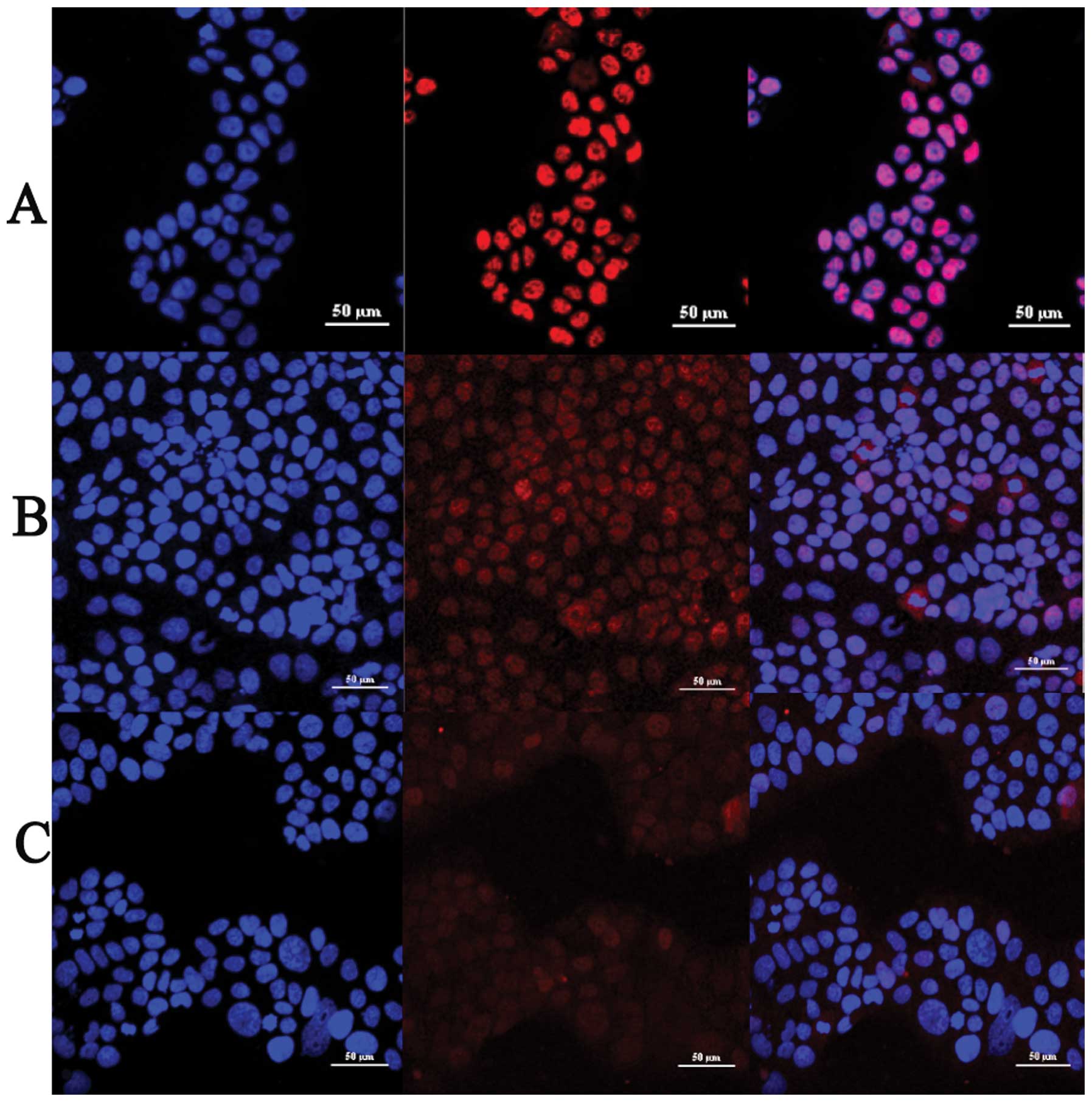
TGF-β1 induces activation and nuclear
translocation of Smad3 in JEG-3 cells
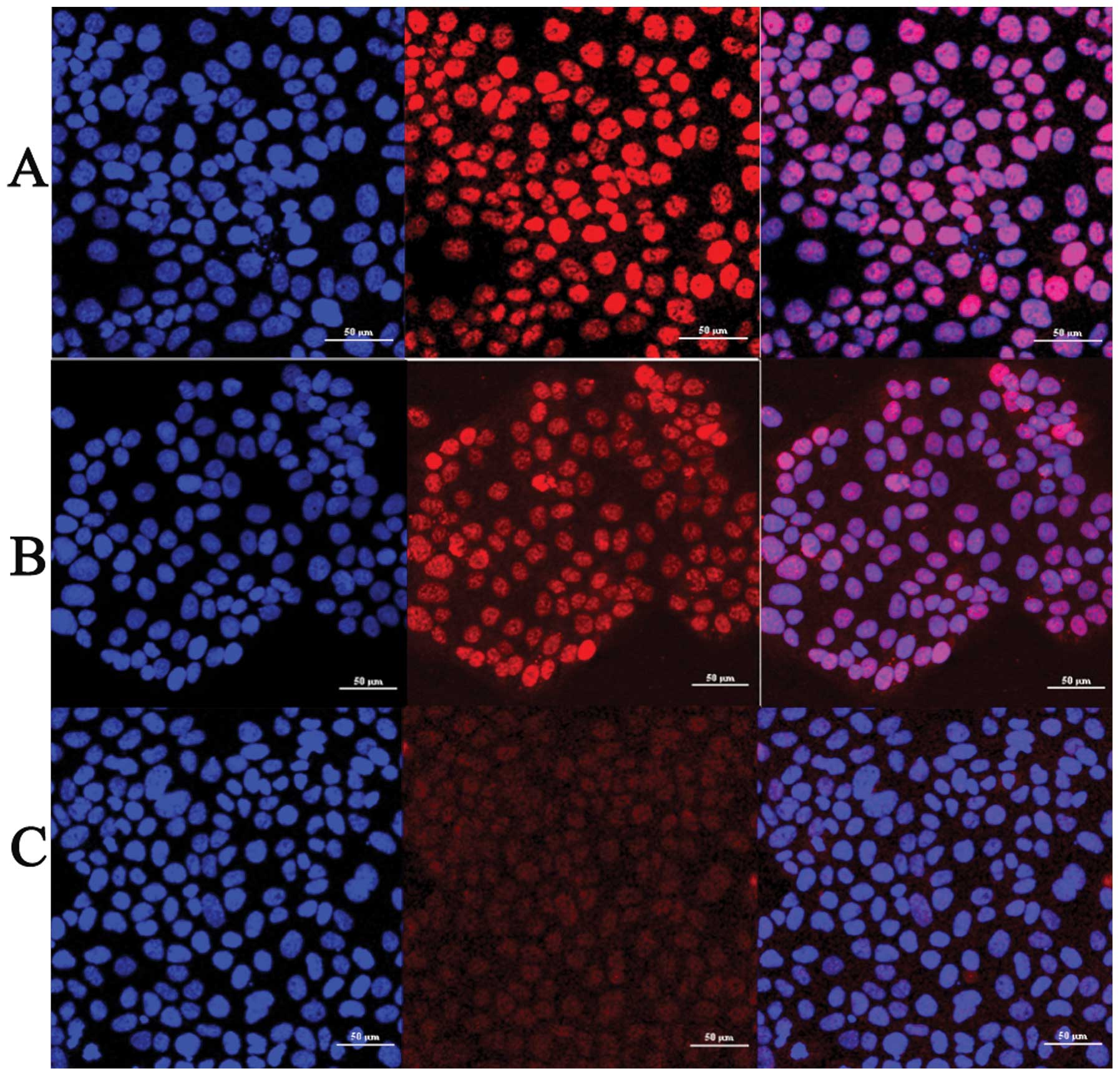
JEG-3 cells were treated by TGF-β1 with the
concentrations of 5 ng/ml. Anti p-Smad3 antibody was employed to
determine the cellular localization of phospho-smad3 in JEG-3 cell
line. We found that TGF-β1 treatment induces phosphorylation of
Smad3 and the intensity of phospho-smad3 immunofluorescence
staining was significantly increased, compared with control group
(Fig. 1A and B). TGF-β1 also
induced Smad3 nuclear translocation. We observed that phospho-smad3
showed a diffuse cytoplasmic staining pattern in control group and
slight staining in the nucleus (Fig.
1A). For the group of 2 h TGF-β1 treatment, phospho-smad3
staining appeared in both the cytoplasm and nucleus (Fig. 1B) whereas in the group of 6 h
TGF-β1 treatment, almost 70% of phospho-smad3 staining was in the
nucleus (Fig. 1C). Thus TGF-β1
activated and induced nuclear translocation of Smad3 in a
time-dependent manner in JEG-3 cells.
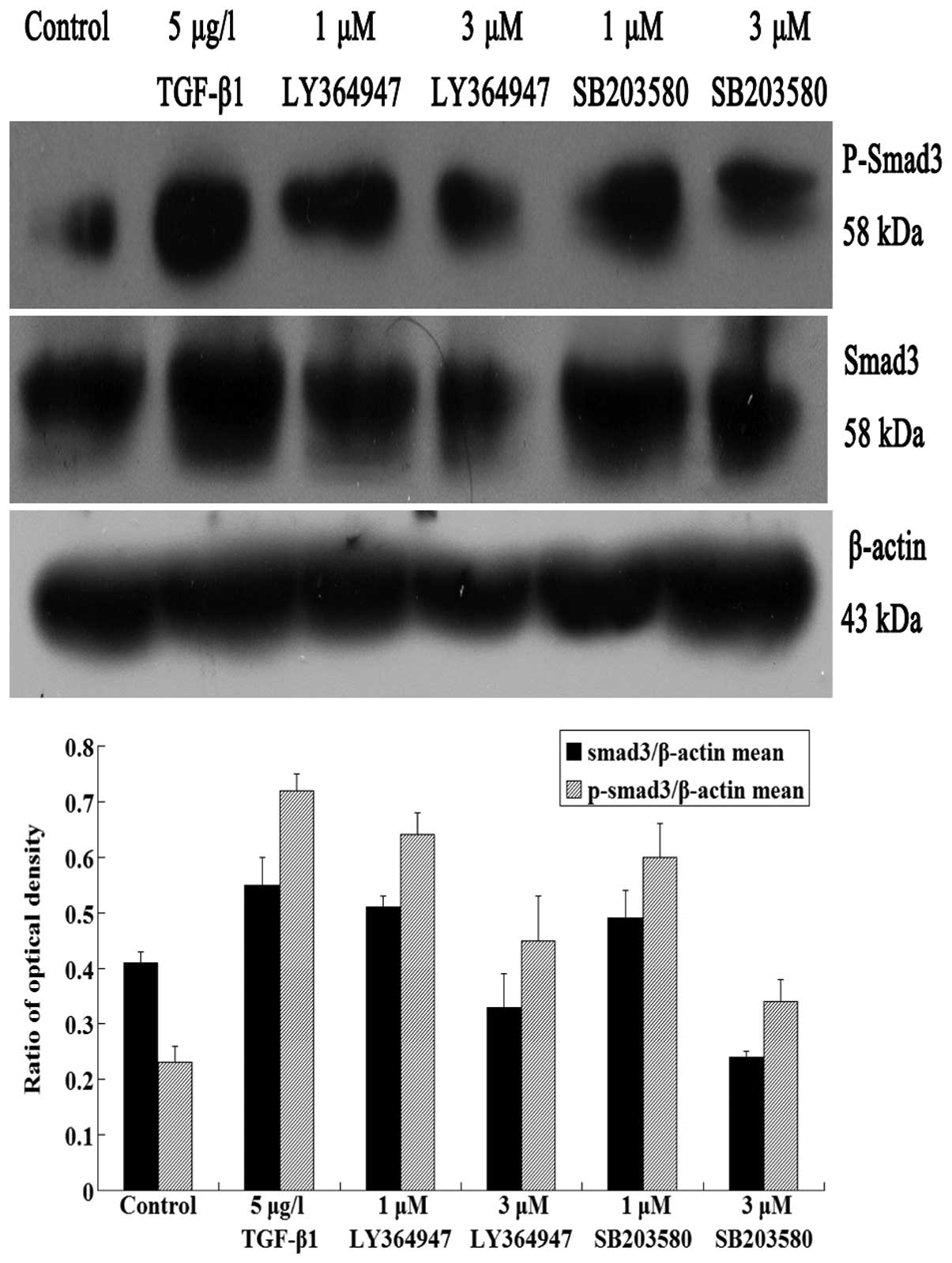
In western blot analysis, JEG-3 cells were
separately pretreated with LY364947, a specific inhibitor of TGF-β1
receptor and SB203580, a specific inhibitor of p38 MAPK, at a
series of concentrations (1 and 3 μM) for 4 h, then
stimulated by TGF-β1 and further cultured for 2 h. However, for
TGF-β1 group, cells were treated with TGF-β1 alone. The protein
expression of Smad3 and phospho-smad3 significantly increased in
TGF-β1 group, comparing with the control group (P<0.05)
(Fig. 4). It also revealed that
TGF-β1 promoted the protein expression of Smad3 and phospho-smad3.
With the increasing concentrations of inhibitors, the Smad3 and
phospho-smad3 protein levels in LY364947 group gradually reduced
compared with the control and TGFβ groups (P<0.05) (Fig. 4). This result demonstrated that
inhibition of TGF-β1 receptor by inhibitor LY364947 blocked the
smad3 activation and nuclear translocation.
TGF-β1 induces activation and nuclear
translocation of p38 in JEG-3 cells
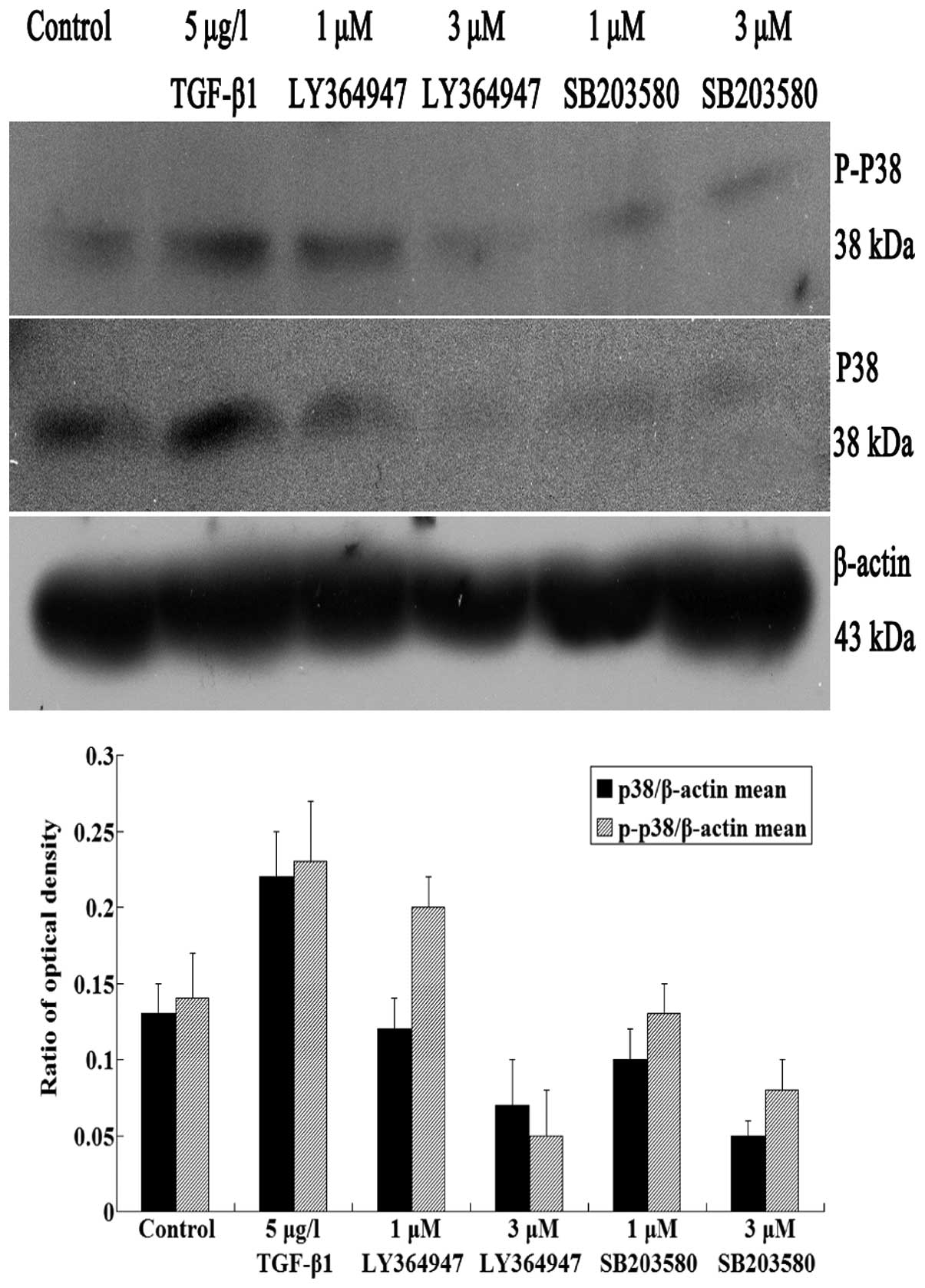
In western blot analysis, protein levels of p38 and
phospho-p38 were obviously increased in TGF-β1 group, which
indicated that TGF-β1 promoted the expression of p38 and
phospho-p38. However, in LY364947 groups, p38 and phospho-p38
protein expression levels decreased as concentrations of LY364947
increased (P<0.05) (Fig.
5).
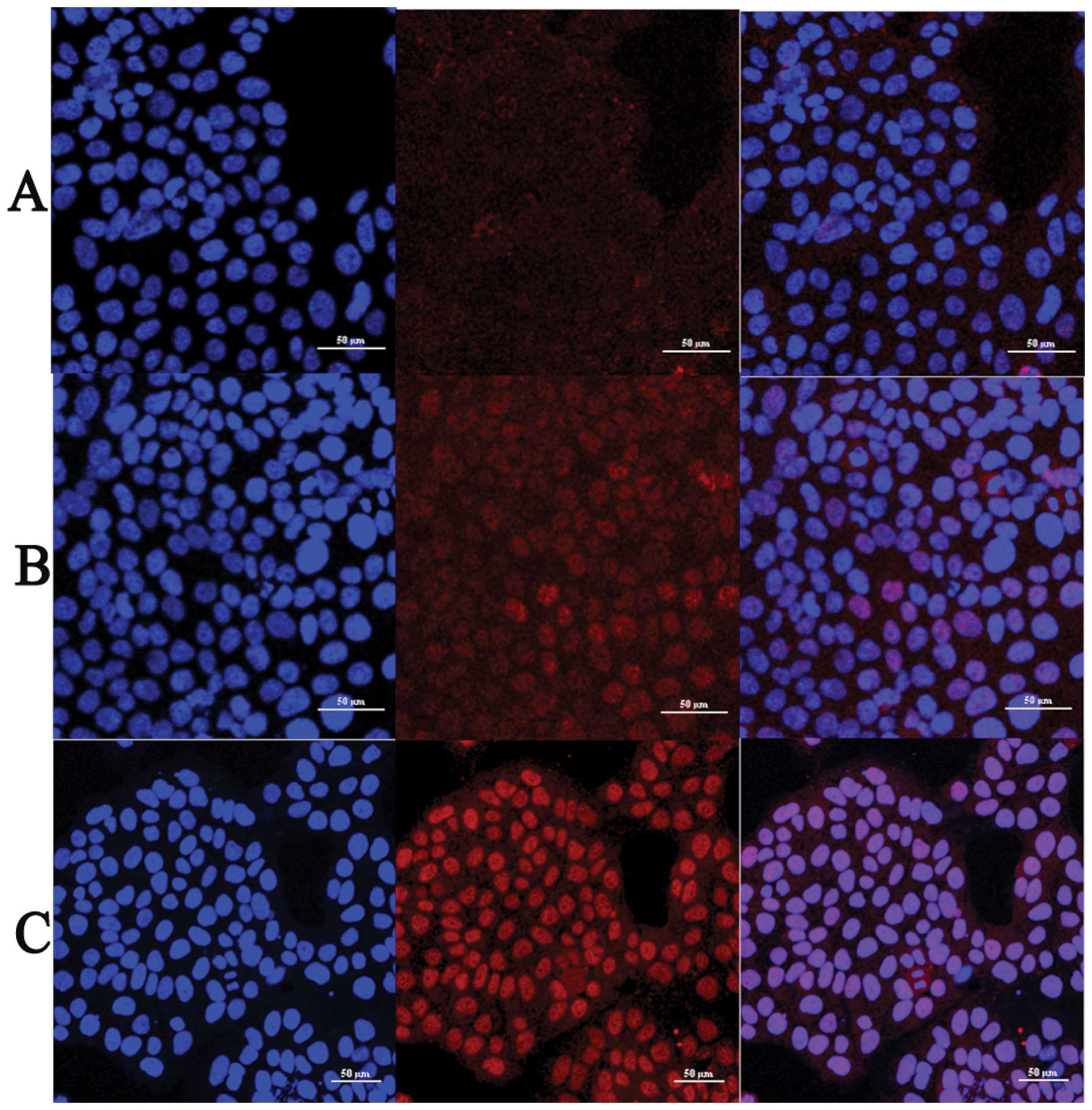
In immunofluorescence analysis, JEG-3 cells were
pretreated with inhibitor of TGF-β1 receptor (LY364947) 1 and 3
μM, respectively, and cultured for 4 h. Then TGF-β1 with the
concentration of 1 μM was added, except for control group,
continuing incubated for 2 h. Phospho-p38 expression was detected
in the cells of LY364947 group. Results showed that compared with
control group, the intensity of phospho-p38 was reduced with
increasing concentrations of LY364947 inhibitor (Fig. 3). Nuclear staining of phospho-p38
also changed into diffuse cytoplasmic staining pattern due to
LY364947 treatment (Fig. 3),
indicating that inhibition of TGF-β1 by inhibitor LY364947 blocked
the activation and nuclear translocation of p38 in a dose-dependent
manner.
Inhibition of p38 MAPK reduces expression
level of smad3 and TGF-β1-induced activation of smad3
SB203580 inhibits p38 in a
dose-dependent manner
In immunofluorescence analysis, the same
concentration of p38 inhibitor (SB203580) was employed and
phospho-p38 expression was detected in the cells of SB203580 group.
For control group, phospho-p38 exhibited nuclear staining in JEG-3
cells (Fig. 2A). However, in
SB203580 groups, phospho-p38 nuclear staining gradually changed
into diffuse cytoplasmic staining pattern as concentrations of the
inhibitor increased. In additon, intensity of the staining was
reduced by ∼50% in the presence of 1 μM SB203580 (Fig. 2B), 70% at 3 μM (Fig. 2C). It indicated that inhibition of
p38 MAPK by inhibitor SB203580 also blocked the activation and
nuclear translocation of p38 in a dose-dependent manner.
SB203580 attenuates TGF-β1-induced
activation of p38 and smad3
In immunofluorescence analysis, anti p-p38 antibody
was employed to detected phospho-p38 expression in JEG-3 cell line.
For control group, phospho-p38 exhibited nuclear staining in JEG-3
cells (Fig. 2A). However in
SB203580 groups, phospho-p38 nuclear staining gradually changed
into diffuse cytoplasmic staining pattern as concentrations of the
inhibitor increased. In additon, intensity of the staining reduced
by ∼50% in the presence of 1 μM SB203580 (Fig. 2B), 70% at 3 μM (Fig. 2C).
In western blot analysis, with the increasing
concentrations of p38 inhibitors, TGF-β1-induced phospho-p38
protein levels were decreased compared with control and TGFβ groups
(P<0.05) (Fig. 4). In additon,
the phospho-smad3 protein levels in SB203580 groups gradually
reduced as the concentrations of p38 inhibitors increased, compared
with control and TGFβ groups (P<0.05) (Fig. 4). Thus SB203580 attenuated
TGF-β1-induced activation of p38 and smad3.
SB203580 decreases the expression
level of Smad3
In western blot analysis, the treatment were as
stated above. Anti-Smad3 antibody was used to detect the expression
level of Smad3. With the increasing concentrations of inhibitors,
the smad3 level in the SB203580 groups gradually reduced compared
with the other two groups (P<0.05) (Fig. 4). This result demonstrated that
SB203580 decreased the expression level of smad3 in a
dose-dependent manner.
Discussion
Choriocarcinoma is a gestational trophoblastic
disease. It is a highly malignant tumor that originates in
developing trophoblasts (27). The
cancer often occurs with a complete hydatidiform mole (28). The abnormal tissue of the mole can
continue to grow even after it is removed. Although choriocarcinoma
is a rare human malignancy which is curable, it is a potentially
fatal disease (29). At present,
availability of different diagnostic aids has turned the prognosis
highly favorable (1), however,
many patients still cannot get effective medical treatment. It is
closely related with cell malignancy of choriocarcinoma and
trophoblastic invasion (27).
TGF-β appears to be a key factor in the development
of choriocarcinoma. Our previous studies have suggested that TGF-β1
can promote JEG-3 cell proliferation and it further enhances the
invasive ability of JEG-3 cells (5). Phosphorylation of smad3 and its
nuclear translocation are critical processes in the whole
TGFβ/Smads pathway. Therefore the process of smad3 activation is
observed by immunofluorescence (30). Our result reveals that TGF-β1 not
only activates Smad3, but also induces smad3 translocation into
nucleus in JEG-3 cell line. The translocation induced by TGF-β1 is
almost complete in 6 h and the nuclear expression of phospho-smad3
is strong positive compared with the control group, whereas, there
is still a small amount of phospho-smad3 expressed in the cytoplasm
of untreated cells, which indicates that TGF-β autocrine mechanism
may exist in JEG-3 cells (31).
After determining the nuclear translocation of
Smad3, we treated the JEG-3 cells, respectively, with the specific
inhibitors LY364947 and SB203580. Then phospho-p38 expression in
the nucleus was detected through immunofluorescence analysis. Not
only in the SB203580 group but also in the LY364947 group, we find
that with increasing concentrations of inhibitors, the intensity of
phospho-p38 in JEG-3 cell nucleus is reduced. It demonstrates that
the inhibitor of TGF-β1 receptor can block activation of p38 MAPK
signaling pathway, indicating that there might be interaction
between TGF-β signaling and p38 MAPK signaling pathways (19).
The development of choriocarcinoma is a complex and
multistep process which includes both cell proliferation and
migration (32). Although the more
exact mechanisms are still controversial, the crosstalk between
TGF-β and p38 MAPK signaling pathway may contribute to solving the
issue. Besides the results of immunofluorescence on p38 nuclear
translocation, our study revealed that p38 and phospho-p38 protein
levels were promoted after TGF-β stimulation and it suggests that
TGF-β can induce the activation of p38 MAPK. An obvious
downregulation of p38 and phospho-p38 protein expression in
TGF-β-induced JEG-3 cells was observed in treatments with LY364947
and SB203580. These results are consistent with those of Bakin
et al (33). The results
presented here also indicate that TGF-β1 receptor inhibition can
suppress activation of p38 MAPK signaling pathway.
Some research on p38 MAPK mediated myofibroblasts in
hepatic stellate cells have manifested that phosphorylation of
Smad3 activated by TGF-β is impaired severely in myofibroblasts
during chronic liver injury, whereas phosphorylation of Smad3 at
the linker region induced by p38 MAPK pathway significantly
increased (34). It indicates that
p38 MAPK pathways can also activate Smad3 in hepatic stellate
cells. However, treatment of SiHa human cervical carcinoma cells
with TGF-β in the presence of p38 MAPK inhibitor does not
significantly inhibit the phosphorylation of Smad2/3 induced by
TGF-β, whereas it reduces phosphorylation of ATF2 (33). Besides the immunofluorescence on
Smad3 nuclear translocation, we have also detected the protein
expression levels of Smad3 and phospho-smad3 with separate
pretreatment of specific p38 MAPK inhibitor (SB203580) and TGF-β
receptor inhibitor (LY364947), following by TGF-β1 stimulation. We
utilized untreated cells as control group and treatment with TGF-β
receptor inhibitor as negative control. Our data indicate that
protein levels of Smad3 and phospho-smad3 are significantly reduced
by using LY364947 and inhibited dose-dependently. Similar results
were observed with the treatment of SB203580, but the inhibitory
effect was slightly lower than the suppression of TGF-β receptor
group. It reveals that the blockade of p38 MAPK pathway can
downregulate protein level of Smad3 and also inhibits the
activation of Smad3. In addition, we believe that p38 MAPK induced
activation of Smad3 has so far appeared to be cell type-dependent,
thus necessitating a careful examination of this mechanism.
In conclusion, TGF-β and p38 MAPK pathways play a
central role in regulating basic cellular processes such as cell
differentiation, proliferation and extracellular matrix production
(35). However, few relative
studies on the interaction of these two signaling pathways in
choriocarcinoma have been reported. The development of
choriocarcinoma has direct relationship with the abnormal cellular
signal transduction pathways. Whether carcinogenic or not, the
active level plays an important role in the process of tumor
development (36). In this study,
p38 and Smad3 protein expression is at the expressive level,
whereas phospho-p38 and phospho-Smad3 protein expression at active
level. Both p38 and phospho-p38 protein expression is reduced
through the use of TGF-β1 receptor inhibitor. In addition, p38 MAPK
inhibitors can also attenuate TGF-β1-induced Smad3 expression and
suppress activation of Smad3. These results suggest that TGF-β and
p38 MAPK signaling are both closely associated with choriocarcinoma
genesis, and progression. Further clarifying the mechanisms of
TGF-β and p38 MAPK pathways in cell models might be informative for
the field of therapeutic approaches.
References
|
1.
|
McGee J and Covens A: Gestational
trophoblastic disease: hydatidiform mole, nonmetastatic and
metastatic gestational trophoblastic tumor: diagnosis and
management. Comprehensive Gynecology. 6th edition. Lentz GM, Lobo
RA, Gershenson DM and Katz VL: Mosby Elsevier; Philadelphia, PA:
chapter 35,. 2012
|
|
2.
|
Braunstein GD: Endocrine changes in
pregnancy. Williams Textbook of Endocrinology. Melmed S, Polonsky
KS, Larsen PR and Kronenberg HM: 12th edition. Saunders Elsevier;
Philadelphia, PA: chapter 21,. 2011, View Article : Google Scholar
|
|
3.
|
Yamamoto E, Ino K, Yamamoto T, Sumigama S,
Nawa A, Nomura S and Kikkawa F: A pure nongestational
choriocarcinoma of the ovary diagnosed with short tandem repeat
analysis: case report and review of the literature. Int J Gynecol
Cancer. 17:254–258. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Fu Y, O’Connor LM, Shepherd TG and
Nachtigal MW: The p38 MAPK inhibitor, PD169316, inhibits
transforming growth factor beta-induced Smad signaling in human
ovarian cancer cells. Biochem Biophys Res Commun. 310:391–397.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Li Y, Xu Q, Zhang Z, Liu S, Shi C and Tan
Y: The impact of TGF-β1 on the mRNA expression of TβR I, TβR II,
Smad4 and the invasiveness of the JEG-3 placental choriocarcinoma
cell line. Oncol Lett. 4:1344–1348. 2012.
|
|
6.
|
Watanabe H, de Caestecker MP and Yamada Y:
Transcriptional cross-talk between Smad, ERK1/2, and p38
mitogen-activated protein kinase pathways regulates transforming
growth factor-β-induced aggrecan gene expression in chondrogenic
ATDC5 cells. J Biol Chem. 276:466–473. 2001.PubMed/NCBI
|
|
7.
|
Ikushima H and Miyazono K: Cellular
context-dependent ‘colors’ of transforming growth factor-β
signaling. Cancer Sci. 101:306–312. 2010.
|
|
8.
|
Nickl-Jockschat T, Arslan F, Doerfelt A,
Bogdahn U, Bosserhoff A and Hau P: An imbalance between Smad and
MAPK pathways is responsible for TGF-β tumor promoting effects in
high-grade gliomas. Int J Oncol. 30:499–507. 2007.PubMed/NCBI
|
|
9.
|
Chapnick DA, Warner L, Bernet J, Rao T and
Liu X: Partners in crime: the TGFβ and MAPK pathways in cancer
progression. Cell Biosci. 1:42–49. 2011.PubMed/NCBI
|
|
10.
|
Coulthard LR, White DE, Jones DL,
McDermott MF and Burchill SA: p38(MAPK): stress responses from
molecular mechanisms to therapeutics. Trends Mol Med. 15:369–379.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Takekawa M, Kubota Y, Nakamura T and
Ichikawa K: Regulation of stress-activated MAP kinase pathways
during cell fate decisions. Nagoya J Med Sci. 73:1–14.
2011.PubMed/NCBI
|
|
12.
|
Cuenda A and Rousseau S: p38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Shiryaev A and Moens U: Mitogen-activated
protein kinase p38 and MK2, MK3 and MK5: ménage à trois or ménage à
quatre? Cell Signal. 22:1185–1192. 2010.PubMed/NCBI
|
|
14.
|
Freund A, Patil CK and Campisi J: p38MAPK
is a novel DNA damage response-independent regulator of the
senescence-associated secretory phenotype. EMBO J. 30:1536–1548.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Li J, Deane JA, Campanale NV, Bertram JF
and Ricardo SD: Blockade of p38 mitogen-activated protein kinase
and TGF-β1/Smad signaling pathway rescues bone marrow-derived
peritubular capillary endothelial cells in adriamycin-induced
nephrosis. J Am Soc Nephrol. 10:2799–2811. 2006.
|
|
16.
|
Gui T, Sun Y, Shimokado A and Muragaki Y:
The roles of mitogen-activated protein kinase pathways in
TGF-β-induced epithelial-mesenchymal transition. J Signal
Transduct. 12:1155–1165. 2012.
|
|
17.
|
Kolosova I, Nethery D and Kern JA: Role of
Smad2/3 and p38 MAP kinase in TGF-β1-induced epithelial-mesenchymal
transition of pulmonary epithelial cells. J Cell Physiol.
226:1248–1254. 2011.
|
|
18.
|
Undevia NS, Dorscheid DR, Marroquin BA, et
al: Smad and p38-MAPK signaling mediates apoptotic effects of
transforming growth factor-β in human airway epithelial cells. Am J
Physiol Lung Cell Mol Physiol. 287:L515–L524. 2004.
|
|
19.
|
Dziembowska M, Danilkiewicz M, Wesolowska
A, et al: Crosstalk between Smad and p38MAPK signalling in
transforming growth factor beta signal transduction in human
glioblastoma cells. Biochem Biophy Res Commun. 354:1101–1106. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Tsukada S, Westwick JK, Ikejima K, et al:
SMAD and p38 MAPK signaling pathways independently regulate α1(I)
collagen gene expression in unstimulated and transforming growth
factor-β-stimulated hepatic stellate cells. J Biol Chem.
280:10055–10064. 2005.
|
|
21.
|
Han YC, Zeng XX, Wang R, Zhao Y, Li BL and
Song M: Correlation of p38 mitogen-activated protein kinase signal
transduction pathway to uPA expression in breast cancer. Ai Zheng.
26:48–53. 2007.PubMed/NCBI
|
|
22.
|
Zhang X-Z, Huang D, Wu F, et al:
Experimental study on role of TGF-β1/p38 mitogen-activated protein
kinase pathway to renal interstitial fibrosis and intervention
mechanisms of Kang Xianling Decoction. China J Traditional Chinese
Medicine Pharmacy. 26:245–248. 2011.
|
|
23.
|
Chen YX, Weng ZH and Zhang SL: Notch3
regulates the activation of hepatic stellate cells. World J
Gastroenterol. 18:1397–1403. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Vyas B, Ishikawa K, Duflo S, Chen X and
Thibeault SL: Inhibitory effects of HGF and IL-6 on TGF-β1 mediated
vocal fibroblast-myofibroblast differentiation. Ann Otol Rhinol
Laryngol. 119:350–357. 2010.
|
|
25.
|
Kashiwagi Y, Horie K, Kanno C, Inomata M,
et al: Trichostatin A-induced TGF-β type II receptor expression in
retinoblastoma cell lines. Invest Ophthalmol Vis Sci. 51:679–685.
2010.PubMed/NCBI
|
|
26.
|
Meng XM, Huang XR, Chung AC, Qin W, Shao
X, et al: Smad2 protects against TGF-β/Smad3-mediated renal
fibrosis. J Am Soc Nephrol. 21:1477–1487. 2010.
|
|
27.
|
Goldstein DP and Berkowitz RS: Gestational
trophoblastic disease. Abeloff’s Clinical Oncology. 4th edition.
Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB and McKenna WG:
Elsevier Churchill Livingstone; Philadelphia, PA: pp. 94–112.
2008
|
|
28.
|
Gerulath AH and Toronto: Gestational
Trophoblastic Disease. Sogc Clinic Practice Guidelines. 114:1–6.
2002.
|
|
29.
|
Denkert C, Darb-Esfahani S, Loibl S,
Anagnostopoulos I and Jöhrens K: Anti-cancer immune response
mechanisms in neoadjuvant and targeted therapy. Semin Immunopathol.
33:341–351. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Giampieri S, Pinner S and Sahai E:
Intravital imaging illuminates transforming growth factor beta
signaling switches during metastasis. Cancer Res. 70:3435–3439.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Tsai S, Hollenbeck ST, Ryer EJ, Edlin R,
Yamanouchi D, Kundi R, Wang C, Liu B and Kent KC: TGF-β through
Smad3 signaling stimulates vascular smooth muscle cell
proliferation and neointimal formation. Am J Physiol Heart Circ
Physiol. 297:H540–H549. 2009.
|
|
32.
|
Javelaud D and Mauviel A: Crosstalk
mechanisms between the mitogen-activated protein kinase pathways
and Smad signaling downstream of TGF-β: implications for
carcinogenesis. Oncogene. 24:5742–5750. 2005.PubMed/NCBI
|
|
33.
|
Bakin AV, Rinehart C, Tomlinson AK and
Arteaga CL: p38 mitogen-activated protein kinase is required for
TGFβ-mediated fibroblastic transdifferentiation and cell migration.
J Cell Sci. 115:3193–3206. 2002.
|
|
34.
|
Furukawa F, Matsuzaki K, Mori S, Tahashi
Y, et al: p38 MAPK mediates fibrogenic signal through Smad3
phosphorylation in rat myofibroblasts. Hepatology. 38:879–889.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Ungefroren H, Groth S, Sebens S, Lehnert
H, Gieseler F and Fändrich F: Differential roles of Smad2 and Smad3
in the regulation of TGF-β1-mediated growth inhibition and cell
migration in pancreatic ductal adenocarcinoma cells: control by
Racl. Mol Cancer. 10:671–679. 2011.
|
|
36.
|
Grzmil M, Morin P Jr, Lino MM, et al: MAP
kinase-interacting kinase 1 regulates SMAD2-dependent TGF-β
signaling pathway in human glioblastoma. Cancer Res. 71:2392–2402.
2011.PubMed/NCBI
|