Introduction
Apoptosis, a genetically programmed cell death, is
conserved among eukaryotes. It is important during embryonic
development to ensure organogenesis and adulthood for maintenance
of cellular homeostasis (1,2).
Alterations in this process can lead to pathological conditions
such as cancer and degenerative diseases. Apoptosis is controlled
by the BCL-2 family of proteins, this family can be divided into
three different subclasses based on conservation of the BCL-2
homology (BH1-4) domains: multidomain anti-apoptotic proteins
(BCL-2, BCL-XL, MCL-1, BCL-W, and Bfl-1/A1); multidomain
pro-apoptotic proteins (BAX and BAK), and BH3-only pro-apoptotic
proteins (BID, BAD, BIM, PUMA, NOXA and BIK) (3).
BH3-only proteins may function as death sensors that
mediate the activation of the mitochondrial apoptosis pathway in
response to oncogenic stress signals or DNA damage (4). Notably, BH3-only proteins are not
able to kill cells that lack BAX and BAK, indicating that BH3-only
proteins function upstream of these proteins (5). BIK induces apoptosis in a variety of
eukaryotic cells and is non-essential for animal development
(6,7). BIK is a pro-apoptotic tumor
suppressor in several human tissues and its expression in cancers
is prevented by chromosomal deletions of the BIK locus or by
epigenetic silencing (3,8,9).
Several anticancer drugs transcriptionally activate
the BIK gene through transcriptional pathways dependent on
factors such as E2F and p53 (8,10–14).
Bik has also been used as a therapeutic molecule in gene
therapy-based approaches to treat difficult cancers. However, the
relation between BIK and the resistance to TAM is poorly
understood. TAM is widely employed in chemotherapy for breast
cancer. In MCF-7 breast cancer cells, TAM inhibits cell
proliferation and induces oxidative stress (OS) and apoptosis via
mitochondria-dependent mechanisms by estrogen receptor-dependent
modulation of gene expression (14,15).
In the present study, we investigated the relationship between BIK
and treatment with TAM in MCF-7 human breast cancer cells.
Materials and methods
Cell cultures
MCF-7 human breast cancer cells (American Type
Culture Collection, ATCC, Manassas, VA, USA) were maintained in
Dulbecco’s modified Eagle’s medium F:12 (DMEM; Invitrogen,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS)
containing penicillin (100 U/ml) and streptomycin (100
μg/ml) (Life Technologies Inc. BLR, Grand Island, NY,
USA).
The cells were grown in 75-cm2 tissue
culture flasks in (5% CO2) at 37°C and routinely
passaged when confluent. Before each experiment, cells were seeded
in 3.5-cm diameter tissue culture plates (5% CO2).
Half maximal effective concentration
(Ec50) of TAM
TAM (TAM citrate; Sigma Chemical Co., St. Louis, MO,
USA) stock solution was prepared in 2% ethanol. MCF-7 cells were
exposed to different concentrations of TAM (1.0, 2.0, 4.0, 6.0, 8.0
and 10.0 μM) at 37°C for 24 h. Apoptosis of the cells was
measured by flow cytometry by Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) staining (BioLegend, San Diego, CA,
USA).
Suppression of Bik expression with small
interfering RNA interference (siRNAi)
Cells were plated in antibiotic-free DMEM-F12 at a
density of 2.5×105 cells and when 50% confluence was
reached, the cells were transfected with oligofectamine reagent
(Invitrogen) and 100 nmol/l BikRNAi (oligoduplex 5′-AAG
ACCCCUCUCCAGAGACAU-3′,N5′ or AAAUGUCUCUGG AGAGGGGUC-3′) (Labs and
Integrated DNA Technologies, IDT) or control sequence-scrambled
(Silencer Negative Control #3 siRNA, Ambion) composed of a 19 bp
scrambled sequence without significant homology to any known gene
sequences from mouse, rat or human. Briefly, 10 μl
oligofectamine was diluted 7.5-fold in Opti-MEM and incubated at
room temperature for 10 min. In parallel, a separate tube, 5
μl of 50 μmol/l siRNA, was diluted in 425 μl
of Opti-MEM. Diluted oligofectamine (75 μl) was added to the
diluted siRNA and the complex was incubated for 20 min at room
temperature. Cells were washed with 2 ml of Opti-MEM. The siRNA +
oligofectamine complex (500 μl) was added gently to the
dish. The final concentration of siRNA was 100 nmol/l. After 6 h,
1.25 ml of 3X serum medium was added to the dish without removing
the transfection mix. The medium was exchanged for serum-containing
medium after 6 h and the cells were further cultured for 48 h. The
experiments were repeated two to three times.
Western blot analysis
MCF-7 cell groups (non-treated, scrambled, siRNA BIK
and siRNA BIK with TAM) were cultured overnight at room temperature
in 3.5-cm diameter tissue culture plates at a density of
1×105 cells/plate. Cells were transferred into 100
μl of lysis buffer (RIPA-Tris buffer:EGTA 2 mM; NaCl 316 mM;
Na2MoO4 20 mM; NaF 50 mM; Tris-HCl 20 mM;
Na3VO4 100 mM, PMSF 100 mM, and EDTA 100 mM;
0.1% of leupeptin and aprotinin, 0.2% SDS and 2% Triton X-100) and
maintained under constant shaking for 2 h at 4°C. Subsequently, the
sample was centrifuged for 5 min at 20,800 rpm and the supernatant
(30 μg of protein) was denatured in Laemmli sample buffer,
resolved through 12% SDS polyacrylamide gels, and electroblotted
onto polyvinylidene difluoride (PVDF) membranes. Blots were stained
with Ponceau S to confirm that protein loading was identical in all
lanes. Membranes were soaked in PBS to remove the Ponceau S and
incubated for 90 min in Tris-buffered saline (TBS) containing 5%
dried skimmed milk and 0.1% Tween-20 to block the non-specific
protein binding sites. Subsequently, the membranes were incubated
for 14 h at 4°C with the primary antibody 1:1,000: BCL-2; MCl-1;
BAX; BAK; PUMA, and cytochrome c (Cyt C), from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). BIK 1:100 from Abcam was
diluted in 0.1% TBS-Tween-20 including 5% dried skimmed milk, then
washed and incubated with peroxidase-conjugated secondary
antibodies 1:10,000. Protein was detected using an ECL Western blot
detection kit (Millipore). The blots were subjected to densitometry
analysis and data were analyzed using GraphPad Prism5 software
(GraphPad Software, San Diego, CA, USA). Western blot analyses were
repeated three times.
Real-time quantitative RT-PCR
RNA from all samples was amplified by RT-PCR assay
in a rotor gene Real-Time apparatus (Cobbett Research 2004)
utilizing the Superscript III Platinum One-step qRT-PCR kit
(Invitrogen). The 25 μl reaction buffer contained 100 ng of
total RNA, 1X Superscript III Platinum One-step qRT-PCR reaction
mix, and 0.4 μM of each of the primers. The primers were
employed for HPRT gene amplification (13) and BIK gene (forward 5′ GAG
ACA TCT TGA TGG AGA CC3′, reverse 5′ TCT AAG AAC ATC CCT GAT GT3′).
The following thermal profile was used: a single cycle of reverse
transcription (RT) for 15 min at 50°C; another cycle of 2 min at
95°C followed by 45 amplification cycles of 20 sec at 95°C, and 1
min at 57°C. Threshold cycle (TC) value of BIK was normalized to
HPRT (16).
Flow cytometry
Annexin V-FITC/PI double staining was used to detect
the apoptosis index. Briefly, the MCF-7 human breast cancer cells
(1×106 cells/ml) were harvested by trypsinization and
washed twice with cold PBS (0.15 mol/l, pH 7.2). The cells were
centrifuged at 2,500 rpm for 5 min; then, the supernatant was
discarded and the pellet was resuspended in 1X binding buffer (10
mM HEPES/NaOH, pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2),
at a density of 1.0×106 cells/ml, 100 μl of the
sample was transferred into a 5 ml culture tube and incubated in
the dark with 5 μl of FITC-conjugated Annexin V staining
solutions (Biolegend, San Diego, CA, USA) and 5 μl of PI
staining solutions; for 15 min at room temperature. Later, 400
μl of 1X binding buffer was added to each sample tube and
the samples were analyzed by FACSCalibur flow cytometry
(Becton-Dickinson) using Cell Quest Research software
(Becton-Dickinson).
Mitochondrial membrane potential of MCF-7
cells
The mitochondrial membrane potential
(ΔΨm) was measured with JC-1 (5, 56, 6-tetracholoro-1,
1, and 3, 3-tetraethyl-benzimidazolylcarbo cyanine iodide) to
signal the loss of (ΔΨm). MCF-7 cells (non-treated,
scrambled, siRNA BIK, and siRNA BIK with TAM) were harvested; equal
numbers of cells (1×106) were incubated with JC-1 at 2.5
g/ml in 1 ml of PBS for 30 min at 37°C with moderate shaking. Cells
were then centrifuged 1,600 rpm at 4°C for 5 min, washed twice with
ice-cold PBS, resuspended in 200 μl of PBS, and analyzed on
a flow cytometer (Becton-Dickinson). We detected green fluorescence
at excitation/emission wavelengths of 485/530 nm and red
fluorescence at excitation/emission wavelengths of 550/595 nm
utilizing CellQuest Research software.
Caspase assay
Caspases were detected by means of the Vybrant FAM
Poly Caspases assay kit, which detects active caspases by employing
the FAM-VAD-FMK reagent, a fluorescently labeled inhibitor of such
enzymes (FLICA). Briefly, 10 μl aliquots of 30X FLICA
working solution was added to MCF-7 cells in suspension and these
were incubated in the dark for 1 h at 37°C and 5% CO2.
After two washes with Wash Buffer 1X (supplied by the
manufacturer), the samples were fixed by adding 40 μl of 10%
formaldehyde solution (supplied by the manufacturer) for 10 min at
room temperature. Then, MCF-7 cells were again washed and
resuspended in 400 μl of Wash Buffer 1X containing 8
μl of PI for FACS analysis.
Statistical analysis
Results were expressed as the means ± standard error
of the mean (SEM). All data were statistically analyzed using one-
or two-way analysis of variance (ANOVA) for repeated measurements,
followed by the post hoc Tukey’s test. Fisher’s post hoc analysis
was also utilized to analyze differences between groups. Analysis
was performed employing GraphPad Prism5 statistical software
(GraphPad Software). Differences of p<0.05 were considered
statistically significant.
Results
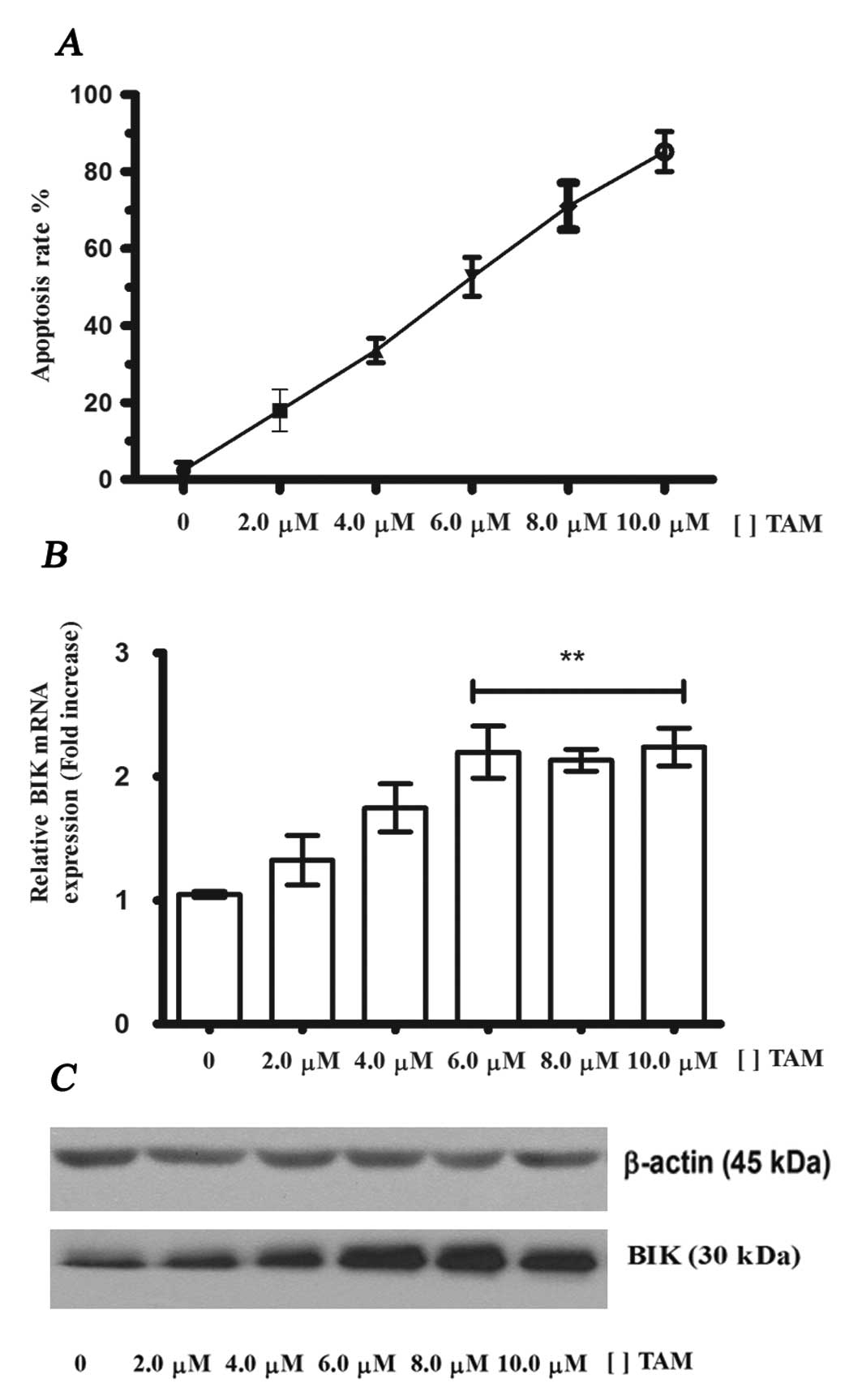
TAM induces apoptosis and increases the
levels of BIK mRNA and its protein in MCF-7 cells
In breast cancer and breast cell lines, TAM-induced
apoptosis is mediated by the estrogen receptor. To identify whether
the expression of the BIK gene and BIK protein in MCF-7
cells are enhanced during TAM induced apoptosis, we incubated these
cells for 24 h at different concentrations of TAM (range, 1–10
μM). Flow cytometry data indicated that TAM increased the
levels of apoptosis (Fig. 1A) and
an EC50 of 6.0 μM was obtained. RT-PCR and
western blot analysis assays revealed that the expression of BIK
mRNA and its protein increases significantly at 6–10 μM
(Fig. 1B and C). These data
indicate that BIK expression is also induced by TAM and suggest the
participation of BIK in TAM-induced apoptosis in MCF-7 cells.
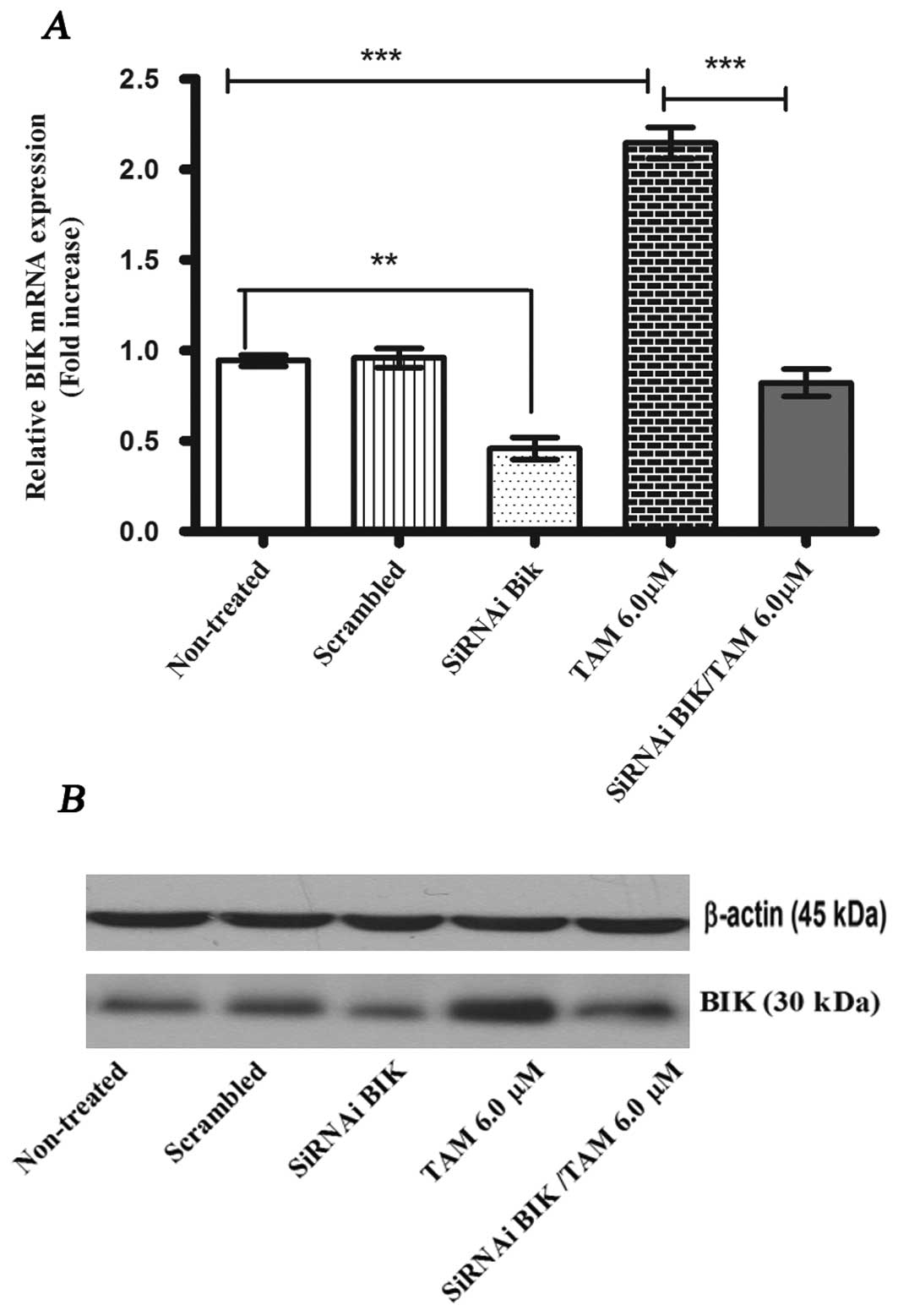
BIK interference protects against
apoptosis
In order to evaluate if BIK expression was blocked
using siRNA, we compared the levels of BIK mRNA and BIK protein
among the following MCF-7 cell groups: non-treated; scrambled;
siRNA BIK, and siRNA BIK with TAM (Fig. 2). The transient transfection of BIK
siRNA reduced the expression levels of BIK mRNA by about 55±0.106%
in MCF-7 cells compared with the controls (non-treated and
scrambled). In the TAM group, mRNA levels increased 2-fold with
respect to control groups. We confirmed these results with the
western blot analysis (Fig.
2).
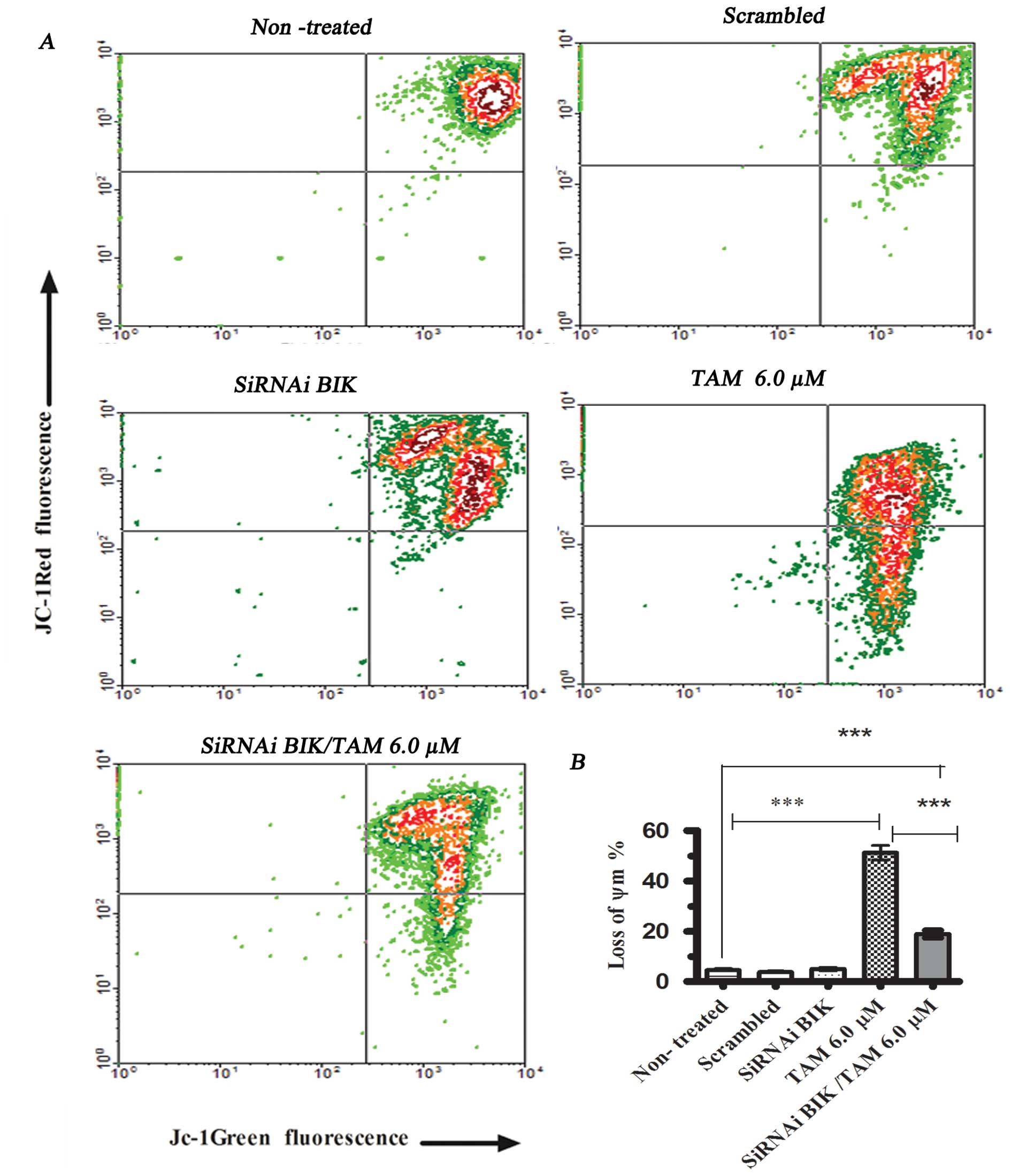
We conducted the ΔΨm assay in different
MCF-7 cell groups for comparison. The groups comprised cells
exposed or not to TAM, after previous transfection or
non-transfected. In cells exposed to TAM 6 μM but not
transfected, the ΔΨm decreased to 52.3±4.56% with
respect to cells not exposed to TAM. We did not find significant
differences between the non-treated cells and cells transfected
with scrambled RNA or with BIK siRNA; however, in cells exposed to
TAM and transfected with BIK siRNA, the ΔΨm decreased
20.2±3.59% (Fig. 3). These results
indicate that BIK could participate in the loss of ΔΨm
modulating anti-apoptotic and pro-apoptotic proteins that regulated
mitochondrial pore formation.
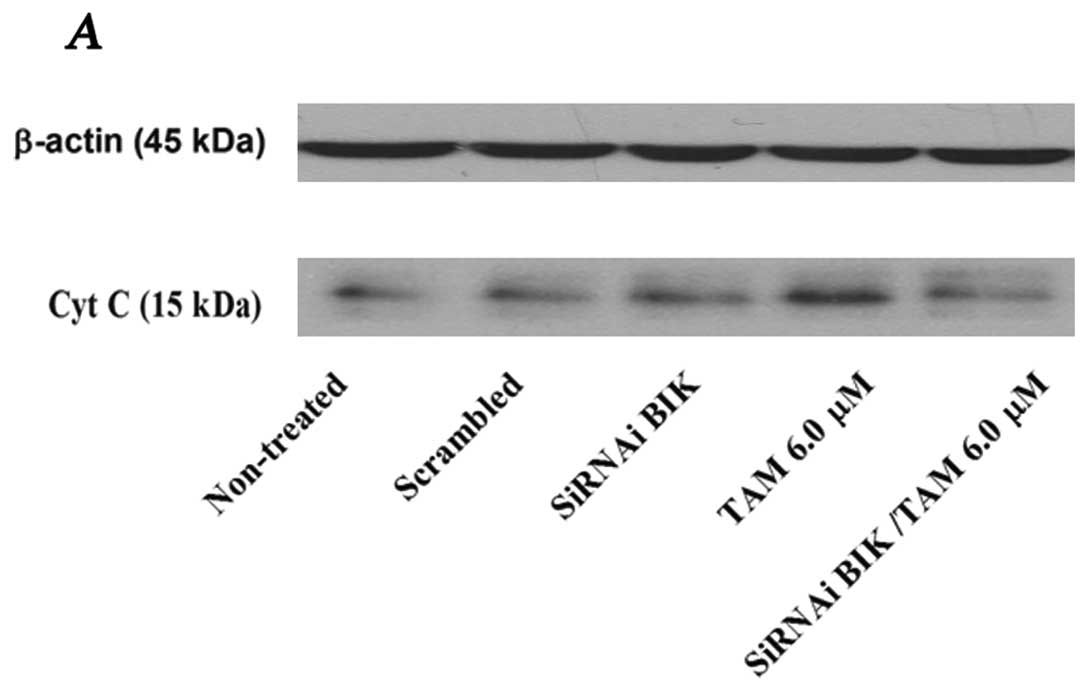
The relation between ΔΨm and apoptotic
initiation is uncertain; however, a change in ΔΨm might
be associated with the release of cytochrome c (Cyt C), and
probably with apoptotic initiation (17). The level of Cyt C protein was
significantly higher in TAM-exposed cells; however, in TAM-exposed
but BIK siRNA-transfected cells, the level was similar to that of
the controls, inhibiting the apoptosis process (Fig. 4). To corroborate these data, we
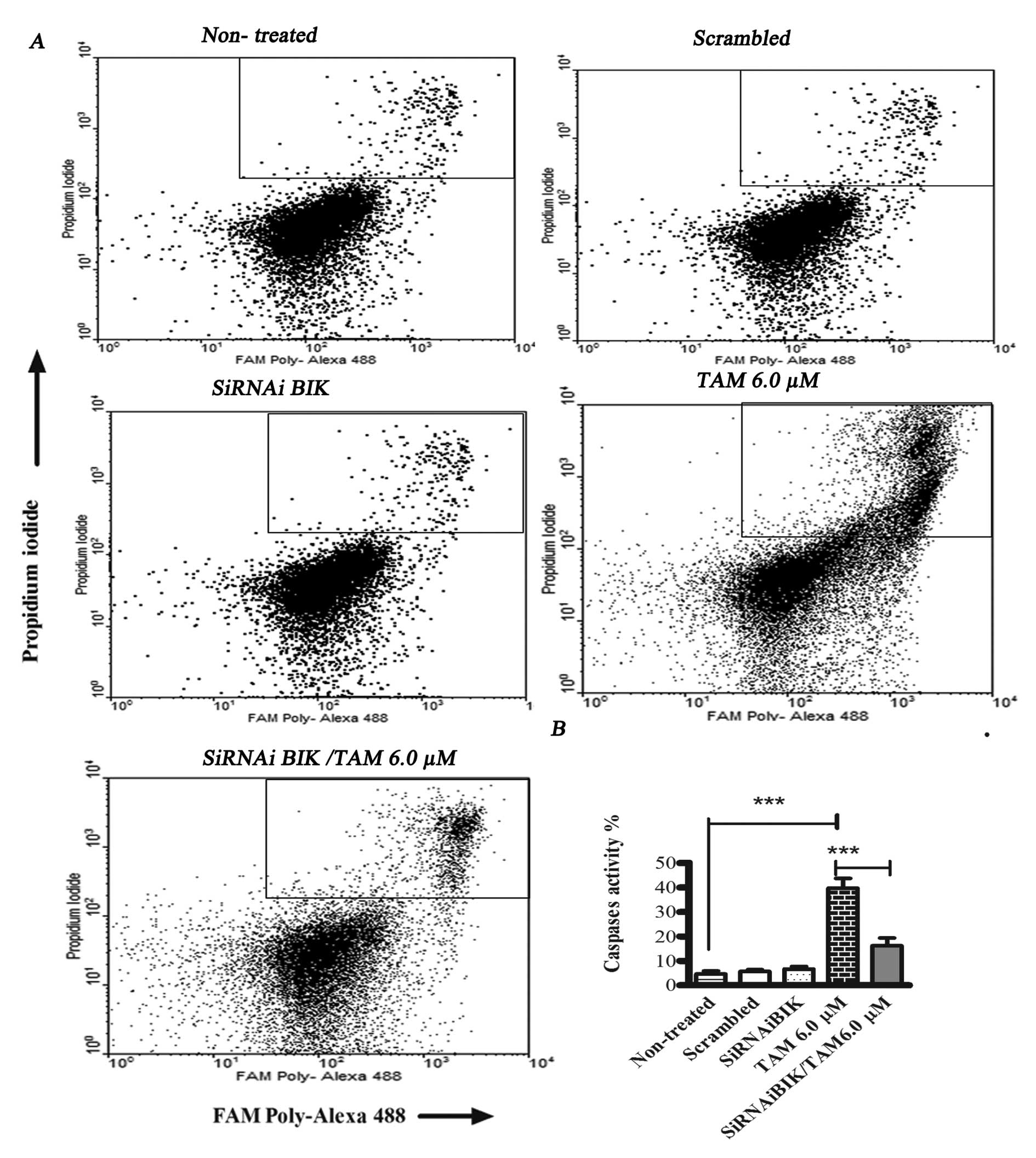
measured total caspase activation.
In TAM-treated MCF-7 cells, total caspase activity
was 50.81±9.17%, while in TAM-infected BIK-exposed MCF-1 cells,
total caspase activity was 20.2±3.59% and in control group showed
no change (Fig. 5). With the aim
of determining the percentage of apoptosis the cells were
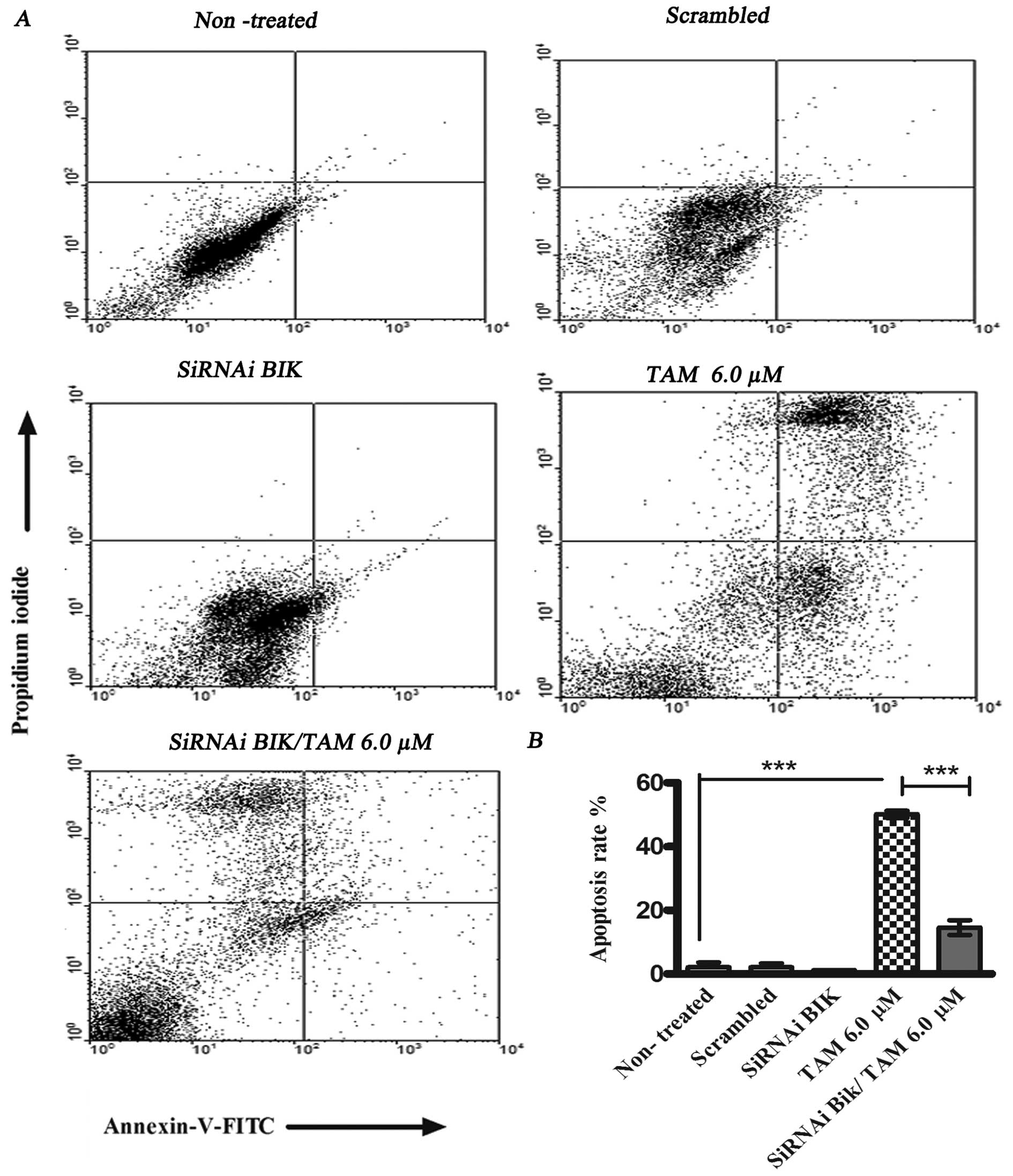
transfected with BIK and treated with TAM. We utilized flow
cytometry staining non-viable cells with PI and Annexin V.
TAM-induced apoptosis was 50.1±6.78% at 24 h, and the percentage of
apoptosis in siRNAi BIK TAM cells was 14.53±3.22%. These data
suggest resistance to TAM-induced apoptosis in BIK
siRNA-transfected cells (Fig.
6).
Low expression of BIK generates
resistance to TAM in MCF-7 regulating pro-apoptotic and
anti-apoptotic family members
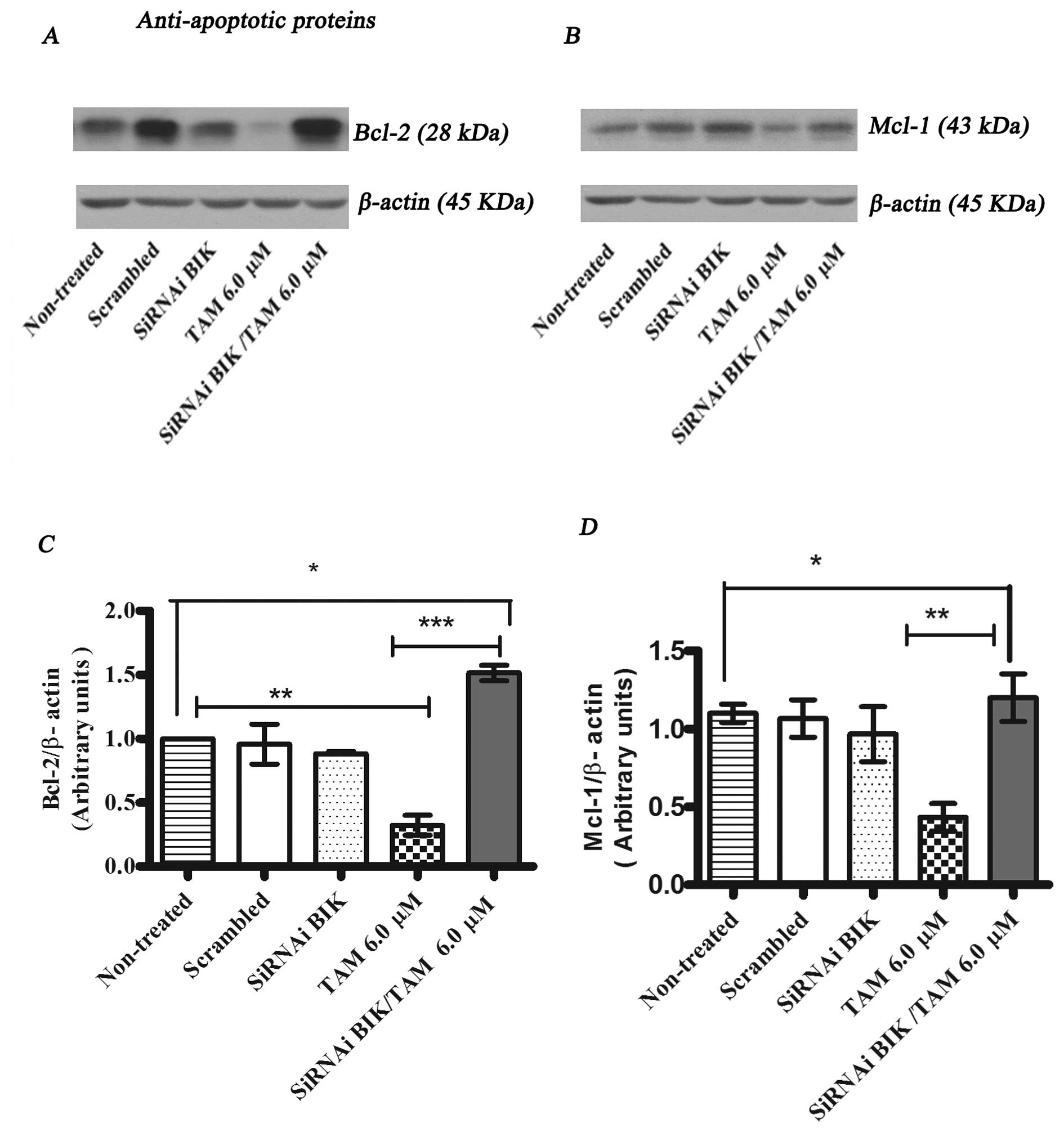
To investigate the molecular mechanisms of the Bik
protein in the process of tamoxifen (TAM)-induced apoptosis, we
determined the protein expression of anti-apoptotic and
pro-apoptotic proteins by western blot analysis. The expression
levels of BCL-2 and MCl-1 in Bik-transfected MCF-7 cells in
response to TAM were higher in comparison with those of TAM
only-treated MCF-7 cells (Fig. 7).
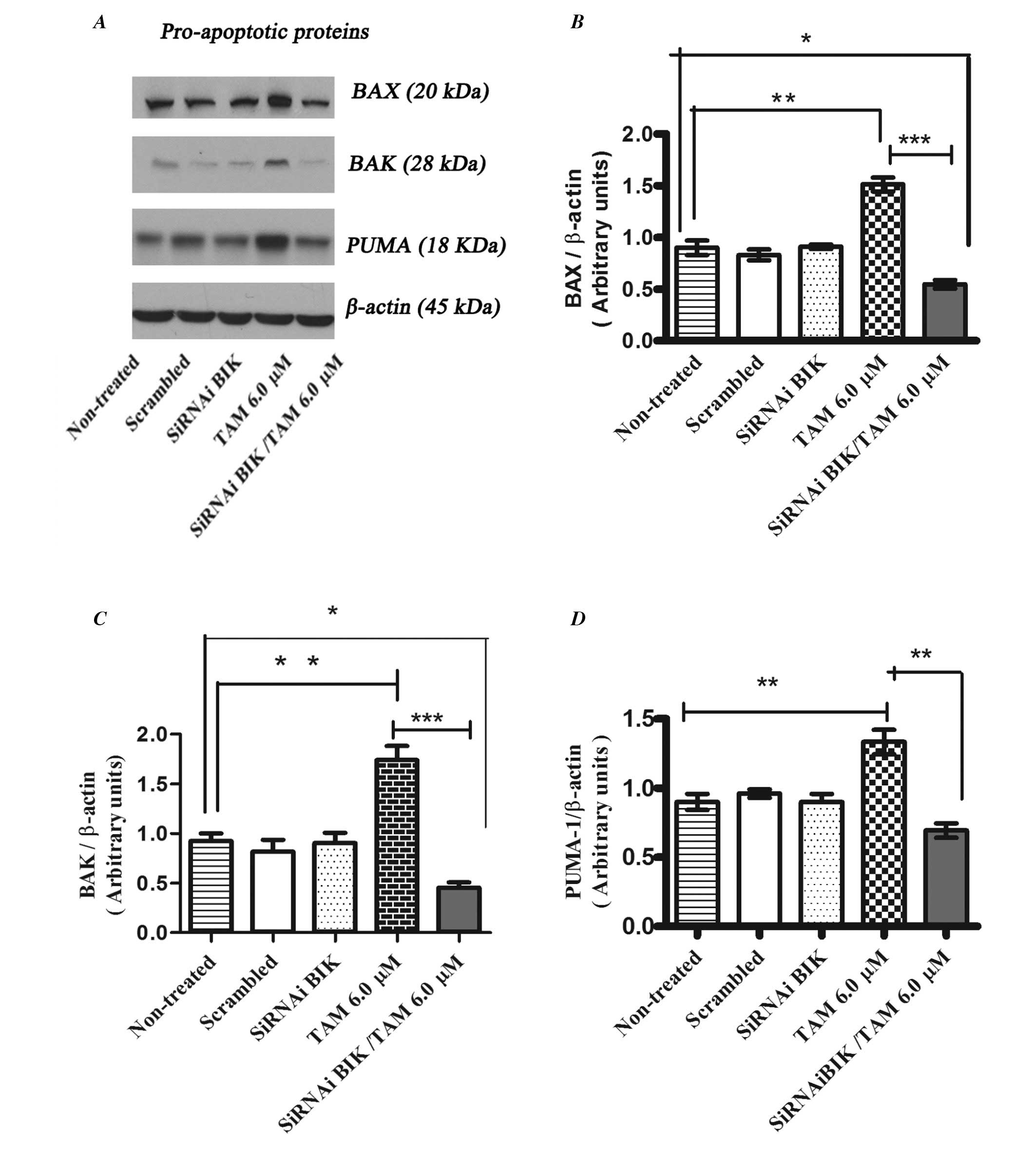
Whereas the levels of pro-apoptotic proteins BAX, BAK and PUMA
increased their expression in TAM treated non-transfected cells in
comparison with TAM-exposed BIK-transfected MCF-7 cells (Fig. 8).
These experiments suggest that low expression of Bik
may generate a process of resistance to apoptosis due to the low
expression of molecules that promote mitochondrial pore formation,
such as BAX and BAK, and induction of the expression of
anti-apoptotic proteins like BCL2 and MCI-1.
Discussion
The Bik gene has been associated with tumor
reversion in different cell lines and was proposed as therapeutic
for inducing apoptosis in cancer, including breast tumors (6,9,18);
however, our group and others have obtained high BIK levels in
breast cancer, non-small cell lung cancer (NSCLC), and
lymphoblastoid cell lines derived from patients with Fanconi anemia
(19,20).
The BH3-only BIK protein, which is inducible by
estrogen starvation and fulvestran treatment, has been suggested to
play a critical role in anti-estrogen-induced apoptosis in breast
cancer cells (18,21). The anti-estrogen TAM is the most
commonly used treatment for estrogen receptor-positive patients
with breast cancer. Although the efficacy of TAM has been
attributed to the induction of tumor cell growth arrest and
apoptosis by inhibition of estrogen receptor signaling (22–25),
the molecular mechanism is not well understood to date. In the
present study, we show that suppression of the BIK gene promotes
resistance to TAM in breast cancer MCF-7 cells.
First, we showed that exposure to different
concentrations of TAM led to the increase of Bik gene
expression possibly by transcriptional pathways. Previous studies
have shown that certain drugs, cytokines and virus infection
affected transcription factors, such as E2F and P53, or removal of
epigenetic marks on the chromatin, which promotes the
transcriptional activation of the BIK gene (11,13).
It is noteworthy that Mathai et al found that BIK expression
in KB human oral epithelial cells depends on P53, but the authors
did not identify functional p53-interacting elements in the BIK
promoter (11). In TAM-treated
MCF-7 cells, we studied the effect of BIK interference. We found
that siRNAi BIK-transfected cells were resistant to apoptosis,
using the Annexin V and PI test, ΔΨm and caspase
activation. Because the relation between ΔΨm and
apoptosis is uncertain, we measured the Cyt C, levels of protein
expression were found to be similar in transfected and control
cells and that TAM treatment increases Cyt C in non-transfected
cells, but not in BIK-siRNA transfected cells. With the aim of
determining the molecular mechanisms of resistance to TAM mediated
by BIK, we evaluated some BCL-2 family proteins. We found low
expression of BAX, BAK and PUMA pro-apoptotic proteins and high
expression of some anti-apoptotic proteins, such as BCL-2 and MCL-1
in BIK siRNA-transfected cells after treatment with TAM, the latter
two proteins have been shown to be involved in the prevention of
Cyt C release (26,27). Our present data demonstrated that
Bik is an important factor in the apoptosis process induced by TAM,
which may regulate mitochondrial integrity by modulation of pro-
and anti-apoptotic proteins; however, it is necessary to conduct
more studies in order to understand BIK-mediated resistance to
TAM-induced apoptosis.
Our results showed that suppression of the
BIK gene exhibited anti-apoptotic effects in TAM-treated
MCF-7 cells. Our data would be useful for future studies to
establish the mechanisms of regulation of TAM resistance in breast
cancer. In women with this neoplasm and with positive estrogen
receptor, it would be important to determine BIK protein levels to
define whether or not TAM would be the appropriate treatment.
Acknowledgements
This study was performed in partial
fulfillment of the requirements for the PhD degree in Biomedical
Sciences of R.V.-R. at the Universidad Nacional Autónoma de México
(UNAM), with a doctoral fellowship provided by CONACyT-México
(grant no. 207148). This study was supported by grants
Salud-2007-785-063 from CONACyT-México.
References
|
1.
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Zhao X, Wang L, Sun Y, et al: The
endoplasmic reticulum (ER)-target protein Bik induces Hep3B cell
apoptosis by the depletion of the ER Ca2+stores. Mol
Cell Biochem. 312:33–38. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lan KL, Yen SH, Liu RS, Shih HL, Tseng FW
and Lan KH: Mutant Bik gene transferred by cationic liposome
inhibits peritoneal disseminated murine colon cancer. Clin Exp
Metastasis. 24:461–470. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Sturm I, Stephan C, Gillissen B, et al:
Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik
is a unifying feature of renal cell carcinoma. Cell Death Differ.
13:619–627. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Gillissen B, Essmann F, Graupner V, et al:
Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is
mediated by an entirely Bax-dependent mitochondrial pathway. EMBO
J. 22:3580–3590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Chinnadurai G, Vijayalingam S and Rashmi
R: BIK, the founding member of the BH3-only family proteins:
mechanisms of cell death and role in cancer and pathogenic
processes. Oncogene. 27(Suppl 1): S20–S29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chinnadurai G, Vijayalingam S and Gibson
SB: BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors
in normal and pathological functions. Oncogene. 27(Suppl 1):
S114–S127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Zou Y, Peng H, Zhou B, et al: Systemic
tumor suppression by the proapoptotic gene bik. Cancer Res.
62:8–12. 2002.PubMed/NCBI
|
|
9.
|
Garcia N, Salamanca F, Astudillo-de la
Vega H, et al: A molecular analysis by gene expression profiling
reveals Bik/NBK overexpression in sporadic breast tumor samples of
Mexican females. BMC Cancer. 5:932005. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Real PJ, Sanz C, Gutierrez O, Pipaon C,
Zubiaga AM and Fernandez-Luna JL: Transcriptional activation of the
proapoptotic bik gene by E2F proteins in cancer cells. FEBS Lett.
580:5905–5909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Mathai JP, Germain M, Marcellus RC and
Shore GC: Induction and endoplasmic reticulum location of BIK/NBK
in response to apoptotic signaling by E1A and p53. Oncogene.
21:2534–2544. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Boyd JM, Gallo GJ, Elangovan B, et al:
Bik, a novel death-inducing protein shares a distinct sequence
motif with Bcl-2 family proteins and interacts with viral and
cellular survival-promoting proteins. Oncogene. 11:1921–1928.
1995.PubMed/NCBI
|
|
13.
|
Hur J, Bell DW, Dean KL, et al: Regulation
of expression of BIK proapoptotic protein in human breast cancer
cells: p53-dependent induction of BIK mRNA by fulvestrant and
proteasomal degradation of BIK protein. Cancer Res. 66:10153–10161.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Nikrad M, Johnson T, Puthalalath H,
Coultas L, Adams J and Kraft AS: The proteasome inhibitor
bortezomib sensitizes cells to killing by death receptor ligand
TRAIL via BH3-only proteins Bik and Bim. Mol Cancer Ther.
4:443–449. 2005.PubMed/NCBI
|
|
15.
|
Monick MM, Powers LS, Butler NS and
Hunninghake GW: Inhibition of Rho family GTPases results in
increased TNF-alpha production after lipopolysaccharide exposure. J
Immunol. 171:2625–2630. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Gottlieb E, Armour SM, Harris MH and
Thompson CB: Mitochondrial membrane potential regulates matrix
configuration and cytochrome c release during apoptosis. Cell Death
Differ. 10:709–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Hur J, Chesnes J, Coser KR, et al: The Bik
BH3-only protein is induced in estrogen-starved and
antiestrogen-exposed breast cancer cells and provokes apoptosis.
Proc Natl Acad Sci USA. 101:2351–2356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Prieto-Remon I, Sanchez-Carrera D,
Lopez-Duarte M, Richard C and Pipaon C: BIK (NBK) is a mediator of
the sensitivity of Fanconi anemia group C lymphoblastoid cell lines
to interstrand DNA cross-linking agents. Biochem J. 448:153–163.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Mebratu YA, Schwalm K, Smith KR, Schuyler
M and Tesfaigzi Y: Cigarette smoke suppresses Bik to cause
epithelial cell hyperplasia and mucous cell metaplasia. Am J Respir
Crit Care Med. 183:1531–1538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Fu Y, Li J and Lee AS: GRP78/BiP inhibits
endoplasmic reticulum BIK and protects human breast cancer cells
against estrogen starvation-induced apoptosis. Cancer Res.
67:3734–3740. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Weng SC, Kashida Y, Kulp SK, et al:
Sensitizing estrogen receptor-negative breast cancer cells to
tamoxifen with OSU-03012, a novel celecoxib-derived
phosphoinositide-dependent protein kinase-1/Akt signaling
inhibitor. Mol Cancer Ther. 7:800–808. 2008. View Article : Google Scholar
|
|
23.
|
Fisher B, Costantino JP, Wickerham DL, et
al: Tamoxifen for prevention of breast cancer: report of the
National Surgical Adjuvant Breast and Bowel Project P-1 Study. J
Natl Cancer Inst. 90:1371–1388. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Mandlekar S and Kong AN: Mechanisms of
tamoxifen-induced apoptosis. Apoptosis. 6:469–477. 2001. View Article : Google Scholar
|
|
26.
|
Simonian PL, Grillot DA and Nunez G: Bak
can accelerate chemotherapy-induced cell death independently of its
heterodimerization with Bcl-XL and Bcl-2. Oncogene. 15:1871–1875.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Zhang H, Nimmer PM, Tahir SK, et al: Bcl-2
family proteins are essential for platelet survival. Cell Death
Differ. 14:943–951. 2007.PubMed/NCBI
|