Introduction
Esophageal cancer is a highly aggressive neoplasm
with a very poor prognosis. The primary treatments of esophageal
cancer include surgery, chemotherapy and radiotherapy (1). In patients with locoregional
esophageal cancer, surgical resection provides the best chance for
a cure. However, in inoperable cases, chemoradiotherapy (CRT) is
extremely important, although the 5-year survival rate remains poor
(1,2). Therefore, the most important issue is
how to improve the therapeutic effects of treatment.
Cyclooxygenase (COX), which consists of two
isoforms, COX-1 and COX-2, is a key enzyme that catalyzes the
transformation of arachidonic acid to prostaglandins (3). It is well known that COX-1 is
expressed in many tissues and is responsible for various
physiological functions (3,4). On
the other hand, recent studies have demonstrated that COX-2 is
overexpressed in various malignancies, including pancreatic,
gastric, prostate, lung, colon, breast, liver, brain and esophageal
cancer (5–10). Recently, it has been reported that
the overexpression of COX-2 induced tumor progression and promotes
resistance to apoptosis activating various factors (11–13).
We previously reported that the overexpression of COX-2 is closely
correlated with resistance to chemoradiotherapy (CRT) for
esophageal squamous cell carcinoma (ESCC) (14). Therefore, reducing the activity of
COX-2 may enhance the antitumor effects of CRT for ESCC (14).
The non-steroidal anti-inflammatory drug (NSAID)
celecoxib (a selective COX-2 inhibitor) belongs to the class of
diaryl heterocycles and constitutes a potent and specific inhibitor
of human COX-2 (15), which is
responsible for resistance to apoptosis, tumor growth, increased
angiogenesis, tumor invasion and metastasis (15–19).
We evaluated the usefulness of the COX-2 inhibitor
celecoxib in combination with treatment including radiation and
chemotherapy for ESCC.
Materials and methods
Cell culture and drugs
The esophageal squamous cell lines TE2 and T.Tn were
plated in culture bottles and cultured in Dulbecco’s modified
Eagle’s medium nutrient mixture (DMEM; Sigma, St. Louis, MO, USA)
with 10% fetal bovine serum (FBS) and 5 mmol/l of L-glutamine, and
100 U/ml of penicillin at 37°C in a 5% CO2
atmosphere.
Celecoxib (a COX-2 inhibitor) kindly provided by
Compound Transfer, Research and Development Operations, Pfizer
Inc., New York, NY, USA, was dissolved in dimethyl sulfoxide
(DMSO). 5-Fluorouracil (5-FU) was purchased from Sigma and
dissolved in DMEM (5% P/S, 10% FBS).
Viability of cells treated with celecoxib
and/or 5-FU
First, the TE2 and T.Tn cell lines were plated in
96-well plates at a density of 3,000 cell/well for 24 h after
seeding. Then, the cell lines were cultured with varying
concentrations of celecoxib and/or 5-FU for 72 h. Following these
treatments, 10 μl/well of the Cell Counting Kit-8 was added,
and the cells were incubated for 2 h. Finally, the optical density
at 450 nm in the multi-mode micro-plate reader was measured in
triplicate. The cell viability was calculated using the following
equation: cell viability (%) = (A experimental group - A control
group)/A control group × 100%.
Drug treatment and irradiation
First, the cells were plated in 96-well plates and
treated with varying concentrations of celecoxib for 72 h. The
cells were cultured after irradiation, combined with celecoxib
treatment for five days. After these treatments, 10 μl/well
of the Cell Counting Kit-8 was added, and the cells were incubated
for 2 h. Finally, the optical density at 450 nm in the multi-mode
micro-plate reader was measured in triplicate.
Observation of apoptosis using flow
cytometry
A total of four groups, including the control and
experimental groups, were evaluated for apoptosis. When the cells
grew to the logarithmic phase, they were treated with 50
μmol/l of 5-FU, or 10 μmol/l of celecoxib, or both
for 48 h. After this treatment, the cells were digested with 0.25%
trypsin to create single cell suspensions. After the cell density
was adjusted to 5×105/100 μl of binding buffer, 5
μl of Annexin V-FITC and 5 μl of propidium iodide
were added and the mixture was gently vortexed followed by
incubation for another 15 min at room temperature in the dark.
After this procedure, a volume of 400 μl of binding buffer
was added to each tube and prepared for an analysis of apoptosis
using flow cytometry within 1 h.
Measurement of the mRNA levels using
real-time reverse transcription polymerase chain reaction (RT-PCR)
for dihydropyrimidine dehydrogenase (DPD) and orotate
phosphoribosyl transferase (OPRT)
The cells were incubated for 48 h with either
celecoxib or 5-FU and/or both. The RNA was extracted using
TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA), and 1
μg of RNA was transcribed to cDNA using a High-Capacity
RNA-to-cDNA kit (Applied Biosystems, Foster City, CA, USA). The
resulting 1 μl of cDNA was amplified via PCR. The sequence
and size of the expected PCR products were as follows: DPD (sense,
5′-AGGACGCAAGGAGG GTTTG-3′; antisense, 5′-GTCCGCCGAGTCCTTACTGA-3′;
118 bp), OPRT (sense, 5′-TAGTGTTTTGGAAACTGTTG AGGTT-3′; antisense,
5′-CTTGCCTCCCTGCTCTCTGT-3′; 91 bp) and β-actin (sense,
5′-TCATGAAGATCCTCAC CGAG-3′; antisense, 5′-TTGCCAATGGTGATGACCTG-3′;
190 bp). The PCR conditions consisted of 40 cycles of 95°C for 30
sec, 95°C for 5 sec, 60°C for 10 sec. This assay was performed in
triplicate.
Cell migration and invasion assay
A membrane invasion culture system (BD BioCoat™
Matrigel™ Invasion Chamber, Tokyo, Japan) was used to quantify the
degree of cell invasion and migration. The cells were incubated for
72 h with either celecoxib or 5-FU or both. A total of
4×104 cells in 500 μl of serum-free medium were
added to the upper chamber of the Transwell-precoated Matrigel
membrane filter inserts with 8-μm pores in 24-well tissue
culture plates. A total of 750 μl of 10% FBS medium was
added to the lower chamber. Thereafter the cells were incubated for
22 h at 37°C. Following incubation, the non-invading cells were
mechanically removed from the upper surface of the membrane.
Subsequently, the cells were fixed and stained with 100% methanol
and 1% Toluidine, blue or 0.25% crystal violet. Next, the membrane
was washed with distilled water to remove excess staining and
dried. The cells on the lower surface were counted manually under a
microscope. This assay was performed in triplicate.
ELISA for prostaglandin E2
The cells were incubated for 24, 48 and 72 h with
celecoxib (0, 10, 20, 30 μM). The PGE2 levels were measured
using an enzyme-linked immunosorbent assay (ELISA) as described by
PGE2 Prostaglandin E2, EIA Kit - Monoclonal (Cayman Chemical
Company, Ann Arbor, MI, USA) according to the manual. The
concentrations were determined following absorbance measurement
with a micro-plate reader (micro-plate manager version 6.0,
Bio-Rad, Tokyo, Japan). This assay was performed in triplicate.
Results
Cytotoxicity of celecoxib in the TE2 and
T.Tn cells
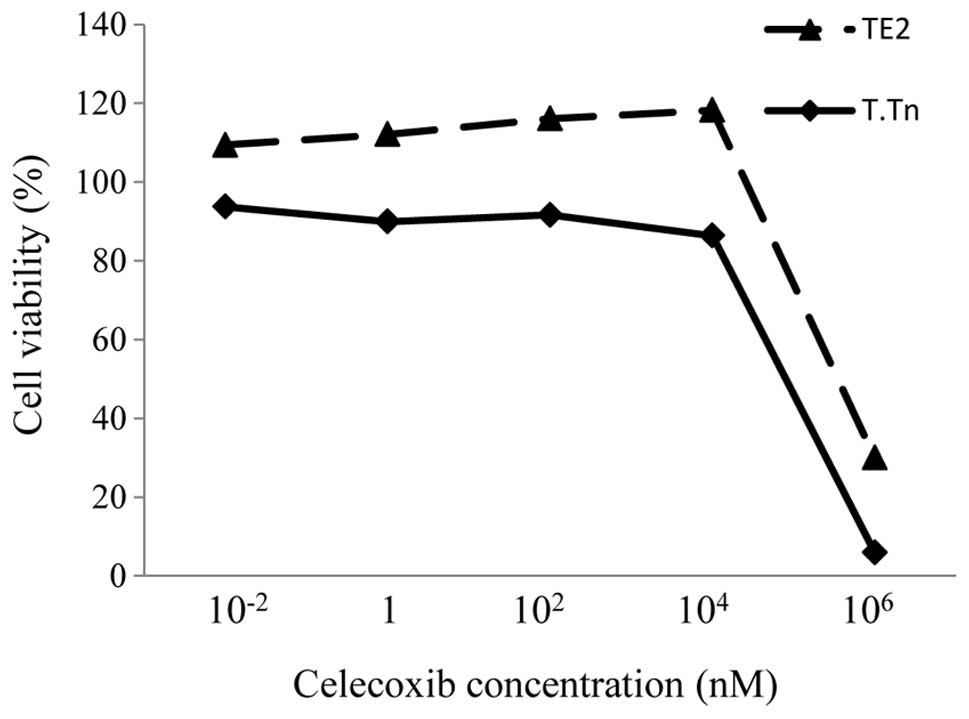
The TE2 and T.Tn cells were treated with different
concentrations of celecoxib for 72 h, and the cell viability was
evaluated. There was no toxicity in the cells treated with
concentrations 10−2 and 104 nM (Fig. 1). However, when the concentration
increased to 106 nM, the cell viability was strongly
reduced. This finding shows that celecoxib itself does not exhibit
any antitumor effects under the normal range of concentrations.
Serum levels of PGE2 in the TE2/T.Tn
cells following treatment with celecoxib
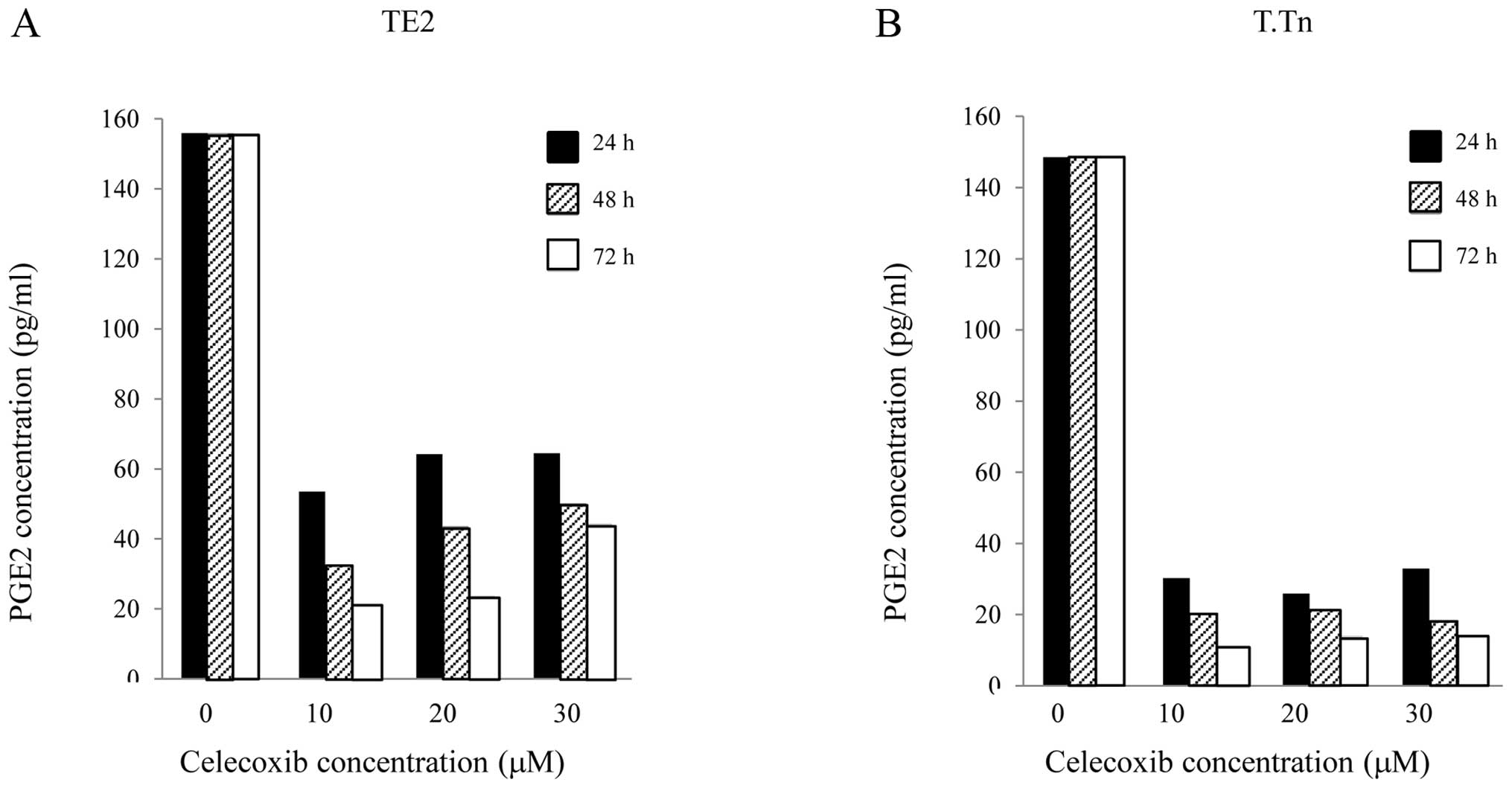
In this study, the PGE2 levels in the TE2 and T.Tn
cells were determined using ELISA. As shown in Fig. 2, celecoxib exhibited obvious
inhibitory effects on PGE2 in the TE2 and T.Tn cells during the
growth process, especially as the drug exposure time was extended,
compared with the normal growth of cancer cells. The production of
PGE2 was also significantly reduced.
Cytotoxicity of TE2 and T.Tn to 5-FU and
celecoxib
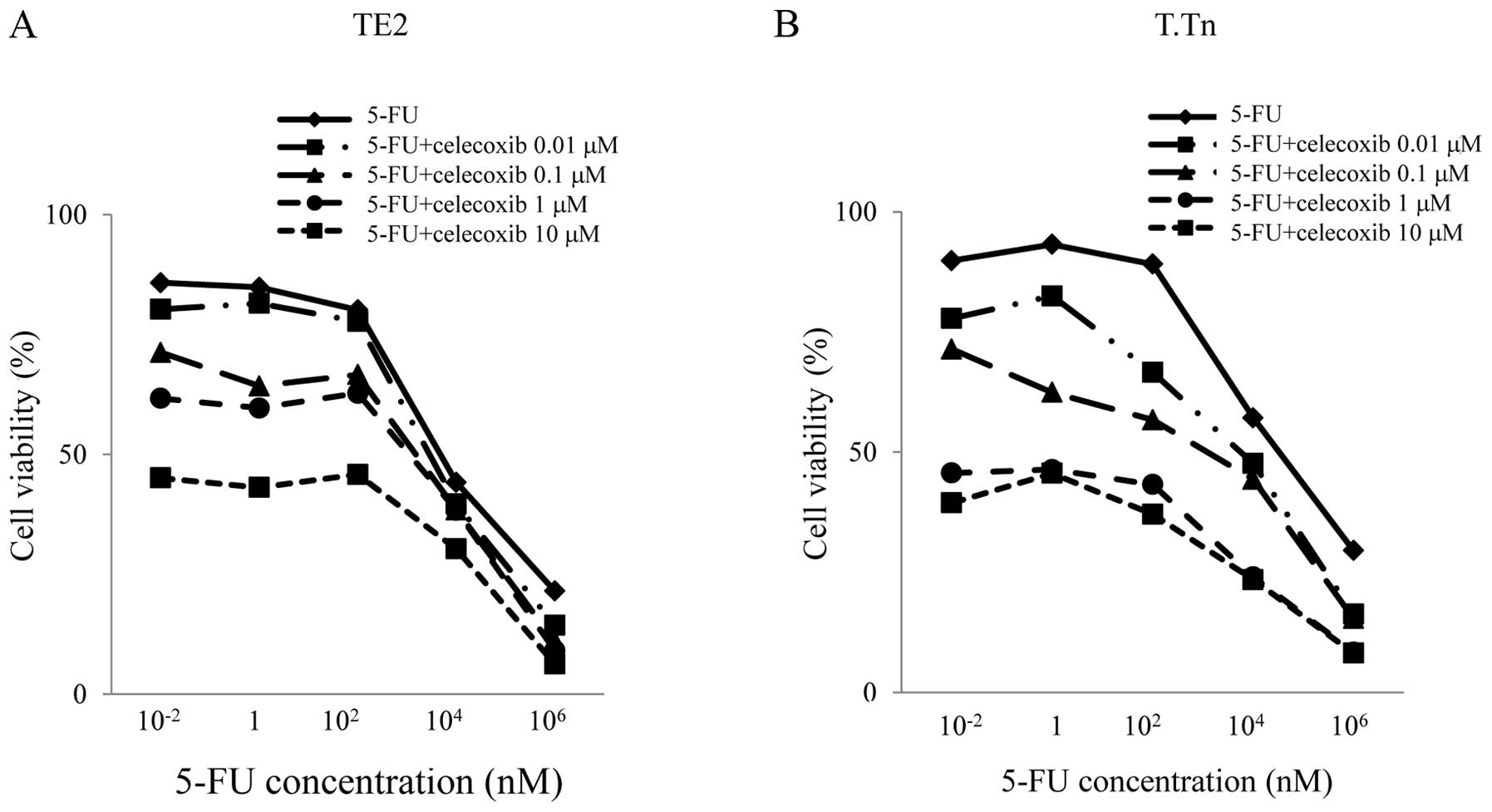
To investigate whether celecoxib can potentiate the
cytotoxic effects of 5-FU in the two cell lines, cytotoxic assays
were conducted. As shown in Fig.
3, in the TE2 cell line, no antitumor effects were observed
between 0 and 100 nM of 5-FU treatment alone. However, when adding
celecoxib, the antitumor effects were enhanced in a celecoxib
concentration-dependent manner. This indicated that celecoxib
enhances the antitumor effects of 5-FU even when the concentration
of 5-FU is low.
Viability of TE2/T.Tn cells treated with
radiation and celecoxib
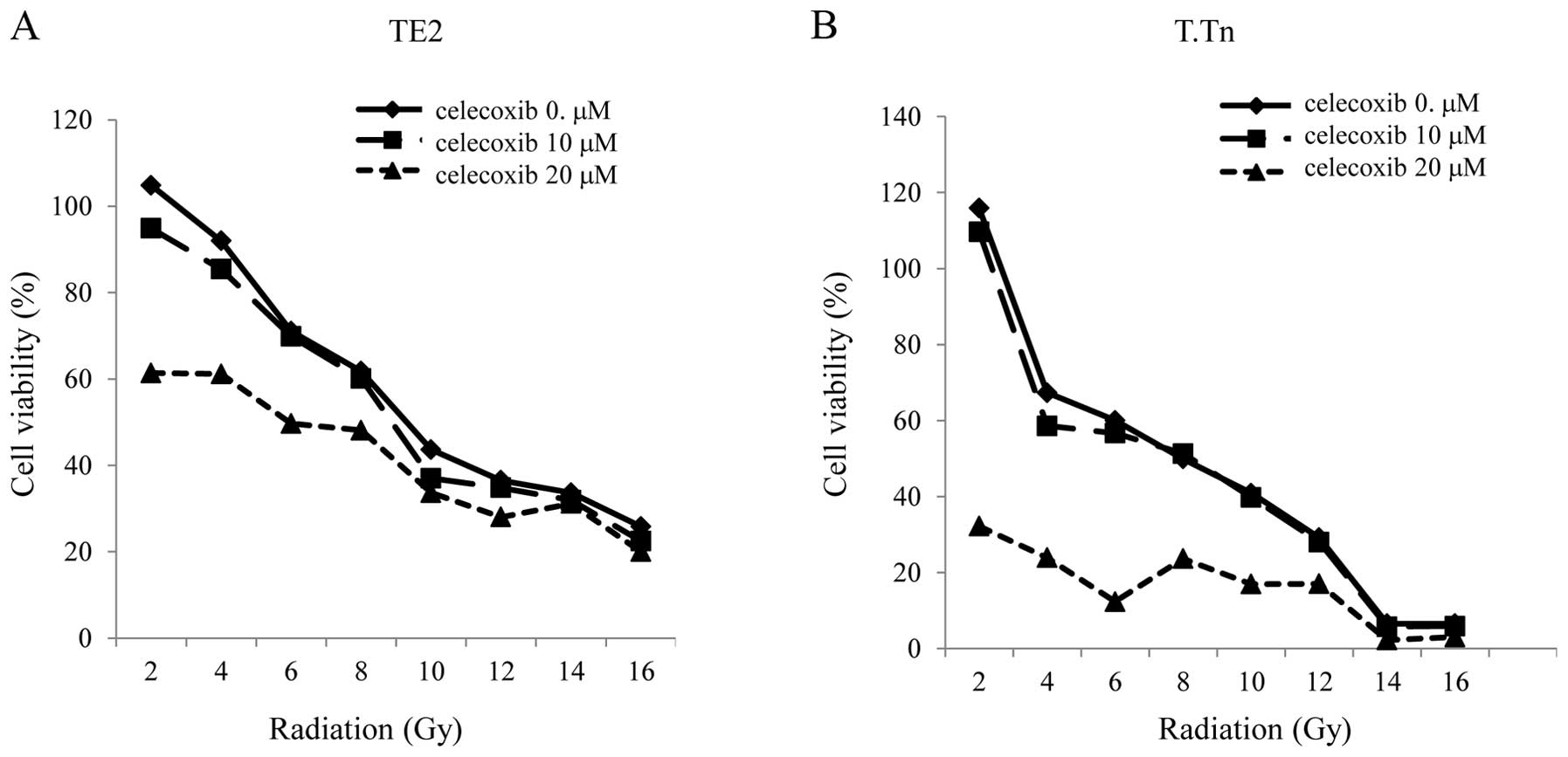
The TE2 and T.Tn cells were cultured with celecoxib
in addition to receiving irradiation, after which the cell
viability was evaluated. There was enhancement of the antitumor
effects in the TE2 cells cultured with the COX-2 inhibitor at 20
μM compared to that observed in the cells exposed to
radiation alone. The discrepancy in cell viability between the
radiation alone group and the radiation plus celecoxib group
increased in the range of lower doses of radiation, especially
after the radiation. The same result was found in the T.Tn cells,
at an even higher level than that observed in the TE2 cells
(Fig. 4).
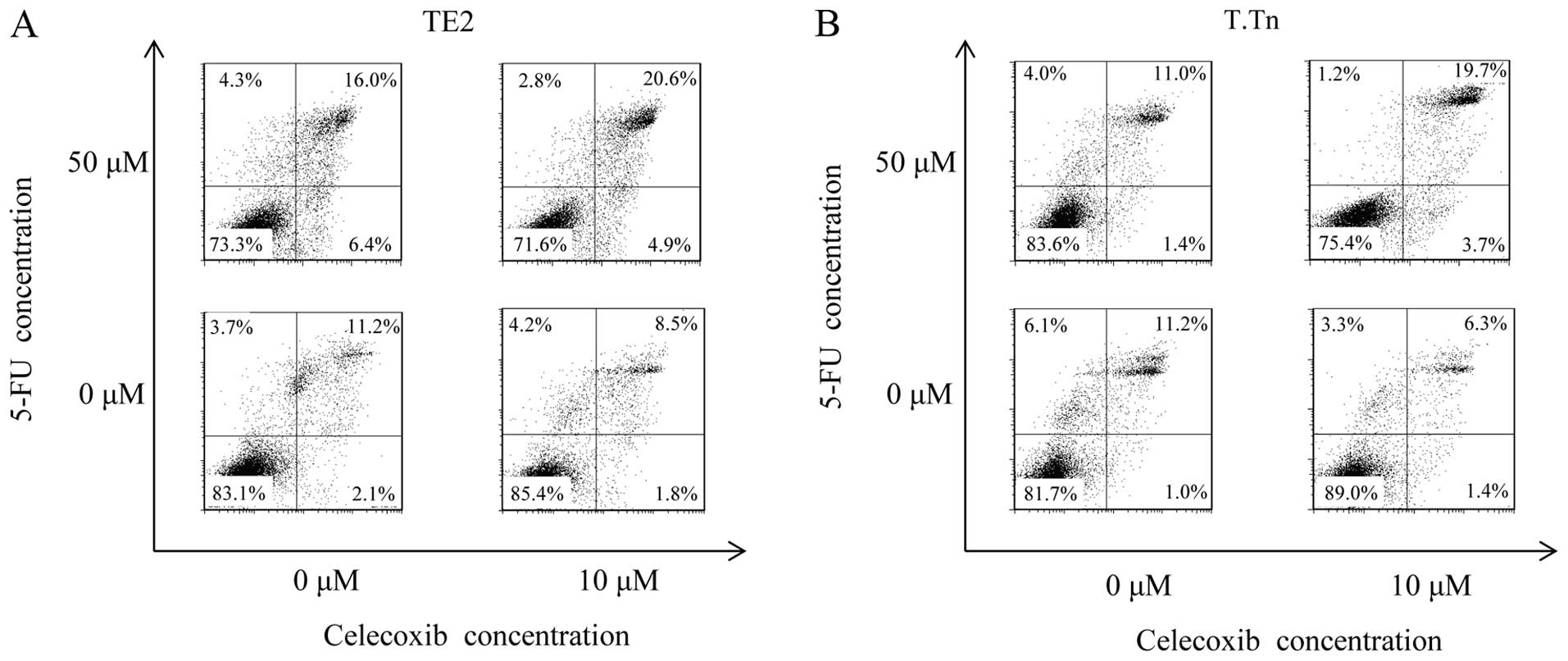
Apoptosis induced by celecoxib and
5-FU
The Annexin V FITC/PI double-labeling method was
performed to detect apoptotic cells. As shown in Fig. 5, the flow cytometry analysis
demonstrated no differences in the Annexin V+PI+ cell population
between the TE2 cells cultured with celecoxib alone (10 μM)
(8.54%) and the control group (11.2%). On the other hand, a
significant increment in the population of apoptotic cells was
observed when the cells were cultured with both celecoxib and 5-FU
(20.6%). The same result was found in the T.Tn cells at an even
higher level than that observed in the TE2 cells.
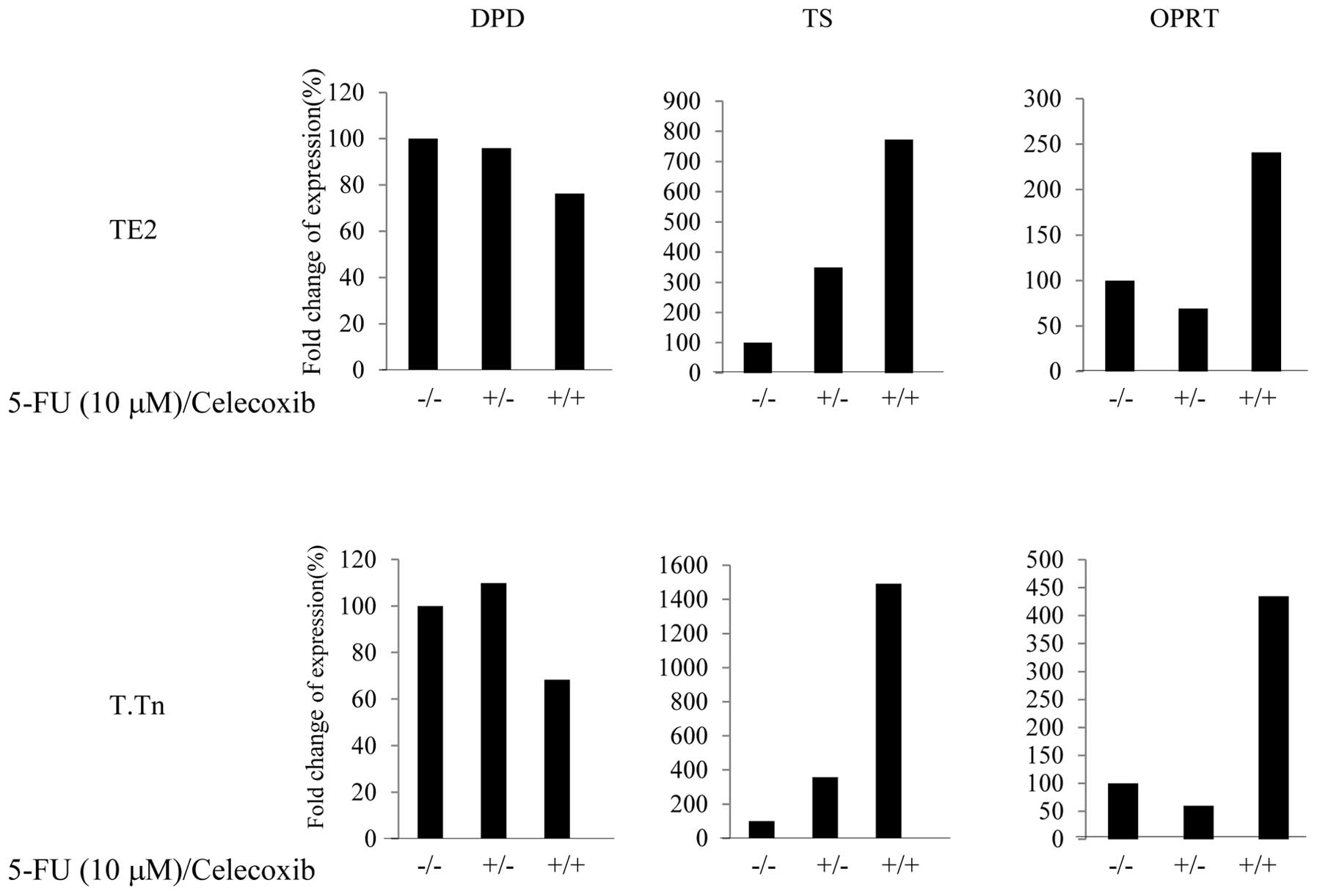
Expressions of OPRT and DPD following
treatment with celecoxib
5-FU was metabolized to the nucleotide level, and
DPD and OPRT are important metabolic enzymes for 5-FU. As shown in
Fig. 6, the expression of DPD, a
degenerative enzyme for 5-FU, was lower (70%) in the TE2 and T.Tn
cells than in the control group when treated with combination of
celecoxib and 5-FU. With regard to the OPRT expression, an
approximately 2.5-fold increase was observed in the TE2 cells and
an approximately 5-fold increase was observed in the T.Tn cells.
Based on these data, celecoxib reduces the degeneration of 5-FU and
increases the metabolism of 5-FU with stronger antitumor effects by
modifying 5-FU-related enzymes, accordingly. This mechanism can be
used to explain the effects of celecoxib in enhancing the antitumor
effects of 5-FU.
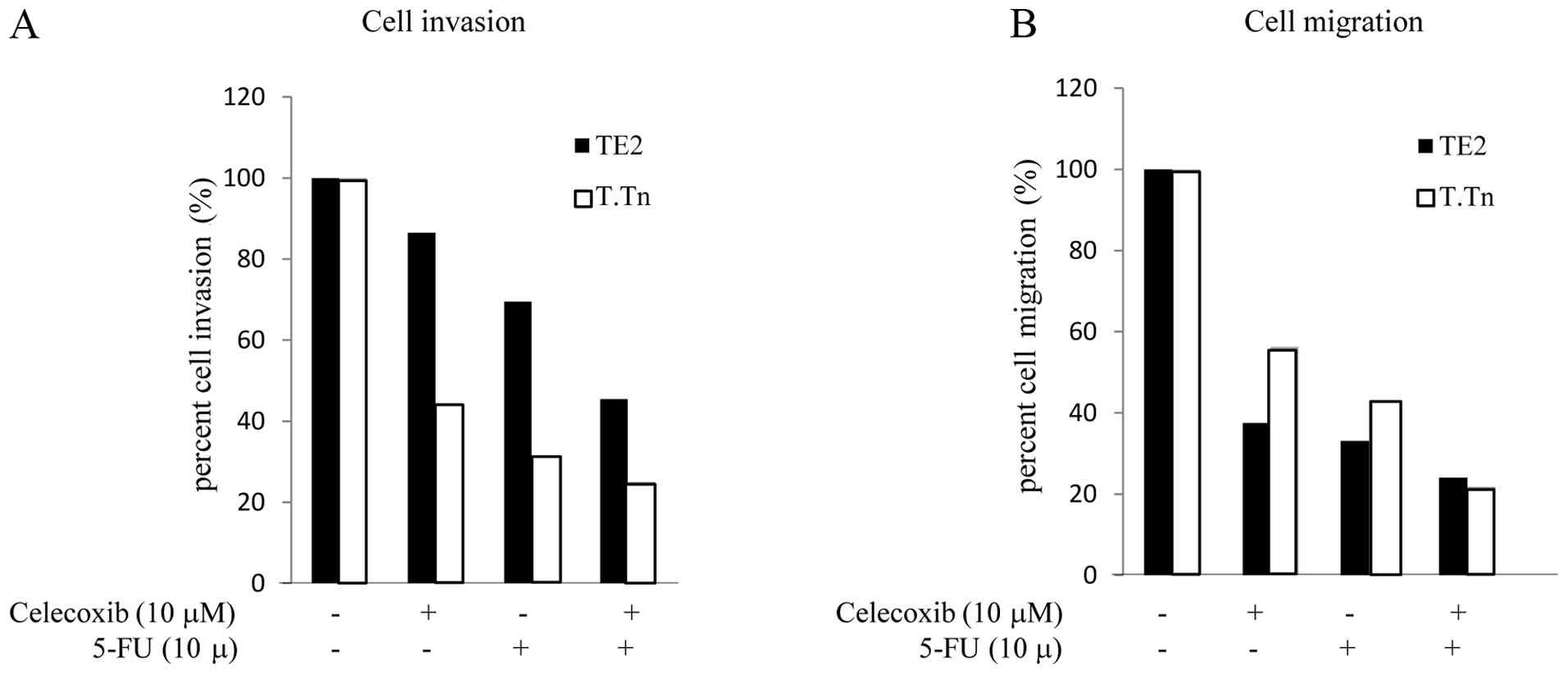
Effects of celecoxib on the cell invasion
and migration potential
The cell migration and invasion activity in the TE2
and T.Tn cells following treatment with 5-FU and celecoxib is shown
in Fig. 7. In the TE2 cells,
treated with celecoxib alone, the cell invasion activity was 86.5%
compared to that observed in the control cells. When cultured with
5-FU alone, the cell invasion activity was reduced to 69.5%
compared to that observed in the controls. However, when cultured
with both celecoxib and 5-FU, the inhibition rate was markedly
reduced to 45.4% (Fig. 7A). This
trend was also observed in the T.Tn cells (celecoxib alone, 44.2%;
5-FU alone, 31.3%; combination group, 25.1%). Next, in order to
quantify the cell migration levels, the cells were treated with
either 5-FU or celecoxib, or both (Fig. 7B). In the T.Tn cells, when treated
with celecoxib alone, the migration activity was reduced to 56.3%.
When treated with 5-FU alone, the invasion rate was 43.1%. However,
when cultured with both celecoxib and 5-FU, the migration activity
was markedly reduced to 21.8% compared to that observed in the
control group. This trend was also observed in the TE2 cells
(celecoxib alone, 37.5%; 5-FU alone, 33.1%; combination group,
24%). These results indicated that celecoxib enhances the
inhibition of both the invasion and migration activity induced
5-FU.
Discussion
The long-term use of non-steroidal anti-inflammatory
drugs (NSAIDs) can reduce the risk of cancer and inhibit the growth
of various cancers, including squamous cell carcinoma (7). The primary target of NSAIDs is COX-2,
which is responsible for resistance to apoptosis, tumor growth,
increased angiogenesis, tumor invasion and metastasis (16,17).
Elevated levels of the COX-2 expression have been found in human
cancers (5–10), and appear to be involved in the
development of cancer via PGE2 production (7,20–23).
In addition, COX-2 overexpression is often associated with a
shorter time to progression and/or overall survival (24). The function of COX-2 in tumor
progression has been recently elucidated. To date, at least five
mechanisms by which COX-2 contributes to tumorigenesis and the
malignant phenotype of tumor cells have been identified, including
inhibition of apoptosis, increased invasiveness, increased
angiogenesis, modulation of inflammation/immune-suppression and
conversion of procarcinogens to carcinogens (5). Research regarding the role of COX-2
in cancer prevention and tumor progression inhibition is ongoing.
With respect to treatment effects, the overexpression of COX-2
leads to tolerance of treatments such as chemotherapy and
chemoradiotherapy, as we previously reported (14).
5-Fluorouracil (5-FU) is one of the most common and
effective clinical chemotherapy medications for the treatment of
digestive tract tumors, with a specific set of effects. The primary
mechanism of action consists of interfering with DNA synthesis and
mRNA translation (25). 5-FU
metabolism is primarily regulated by enzymes, such as DPD and OPRT
(25–27). The rate-limiting enzyme of 5-FU
catabolism is DPD, as more than 80% of administered 5-FU is
catabolized by DPD (26).
Measuring the level of DPD activity can be used as a screening
procedure to identify patients with DPD deficiency, before the
start of treatment with 5-FU (27). The metabolized form of 5-FU is
directly converted by OPRT, and further dephosphorylated to
generate the active metabolite fluorodeoxyuridine monophosphate
(FdUMP) which binds to the nucleotide-binding site of TS and forms
a stable ternary complex with TS and
5,10-methylenetetrahydrofolate(5,10-CH2-THF), leading to DNA damage
(25).
The present study demonstrated that COX-2, was
upregulated in the 5-FU resistant esophageal cancer cell lines.
Although celecoxib, a COX-2 inhibitor, exhibited very slight
anticancer effects in the TE2/T.Tn cells, stronger anticancer
effect was observed following changes in the DPD and OPRT levels in
the 5-FU resistant esophageal cancer cell lines.
In this study, celecoxib inhibited the COX-2
activity, leading to a reduction in the PGE2 levels in the cancer
cells. This indicates that B-cell lymphoma-2 (BCL-2), matrix
metal-loproteinase-2 (MMP-2), epidermal growth factor receptor
(EGFR), endothelial growth factor (VEGF) and other factors are
restrained by COX-2-PGE2-dependent mechanisms (28–31).
However, Elder et al reported no correlations between the
sensitivity of colon cancer cell lines to NS-398 and the COX-2
expression or between the addition of PGE2 and the induction of
apoptosis (32). This discrepancy
may represent differences between the cells analyzed by different
investigators and the types of NSAIDs used. On the other hand,
celecoxib upregulates the OPRT expression and downregulates the DPD
expression, which may increase the antitumor effects of 5-FU and
can inhibit the growth of tumor cells or increase the apoptosis of
cancer cells and inhibit cancer cell migration and invasion.
Consequently, the changes in the reduction of the enzyme activity
observed in experimental systems exhibit good relationships with
the enhancement of the antiproliferative potency of 5-FU. However,
thus far, it is unclear through which factors and pathways COX-2
inhibitors can change the expressions of DPD and OPRT mRNA. In
their in vivo research, Irie et al reported that
celecoxib (a selective COX-2 inhibitor) synergistically potentiates
the antitumor effects of 5-FU in colon cancer cells in an
IFN-γ-dependent manner (33).
In addition, we tested the effects of combination
therapy with celecoxib and radiation in TE2 and T.Tn cells. The
results showed that the effects of combination therapy are stronger
than those of radiotherapy alone. Our experimental results are
supported by those of Kuipers et al (34). Previous studies have revealed that
these effects are due to cell cycle arrest. Radiosensitivity
enhancement using a COX-2 inhibitor is achieved through
COX-2-dependent cell cycle regulation (35) and primarily results from the
inhibition of ionizing radiation-induced G2 arrest (36). It has also been suggested that
these effects are likely due to the inhibition of the
radio-protective effects of prostaglandins (37). Other mechanisms have also been
elucidated. COX-2 inhibitors contribute to the enhancing antitumor
effects via the activation of caspase-3 and caspase-8 or the
inhibition of DNA repair processes (38,39).
In conclusion, resistance to cancer treatment can be
decreased by COX-2 inhibitors. COX-2 inhibitors are useful
enhancers of antitumor drugs and act as radiosensitizers for
radiotherapy for ESCC. Therefore, these drugs may be useful as a
component of combination treatment for cancer.
References
|
1.
|
Pennathur A, Gibson MK, Jobe BA and
Luketich JD: Oesophageal carcinoma. Lancet. 381:400–412. 2013.
View Article : Google Scholar
|
|
2.
|
Siersema PD: Esophageal cancer.
Gastroenterol Clin North Am. 37:943–964. 2008. View Article : Google Scholar
|
|
3.
|
Rizzo MT: Cyclooxygenase-2 in oncogenesis.
Clin Chim Acta. 412:671–687. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Smith WL, DeWitt DL and Garavito RM:
Cyclooxygenases: structural, cellular, and molecular biology. Annu
Rev Biochem. 69:145–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Dempke W, Rie C, Grothey A and Schmoll HJ:
Cyclooxygenase-2: a novel target for cancer chemotherapy? J Cancer
Res Clin Oncol. 127:411–417. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Shamma A, Yamamoto H, Doki Y, et al:
Up-regulation of cyclooxygenase-2 in squamous carcinogenesis of the
esophagus. Clin Cancer Res. 6:1229–1238. 2000.PubMed/NCBI
|
|
7.
|
Hida T, Yatabe Y, Achiwa H, et al:
Increased expression of cyclooxygenase 2 occurs frequently in human
lung cancers, specifically in adenocarcinomas. Cancer Res.
58:3761–3764. 1998.PubMed/NCBI
|
|
8.
|
Dannenberg AJ and Subbaramaiah K:
Targeting cyclooxygenase-2 in human neoplasia: rationale and
promise. Cancer Cell. 4:431–436. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Okamura H, Fujiwara H, Umehara S, et al:
COX-2 overexpression Induced by gene transfer reduces sensitivity
of TE13 esophageal carcinoma cells to 5-fluorouracil and cisplatin.
Anticancer Res. 33:537–542. 2013.
|
|
10.
|
Higashi Y, Kanekura T and Kanzaki T:
Enhanced expression of cyclooxygenase (COX)-2 in human skin
epidermal cancer cells: evidence for growth suppression by
inhibiting COX-2 expression. Int J Cancer. 86:667–671. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Lucci A, Krishnamurthy S, Singh B, et al:
Cyclooxygenase-2 expression in primary breast cancers predicts
dissemination of cancer cells to the bone marrow. Breast Cancer Res
Treat. 117:61–68. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Van Nes JG, de Kruijf EM, Faratian D, et
al: COX2 expression in prognosis and in prediction to endocrine
therapy in early breast cancer patients. Breast Cancer Res Treat.
125:671–685. 2011.PubMed/NCBI
|
|
13.
|
Hashimoto N, Inayama M, Fujishima M and
Shiozaki H: Clinicopathologic significance of expression of
cyclooxygenase-2 in human esophageal squamous cell carcinoma.
Hepatogastroenterology. 54:758–760. 2007.PubMed/NCBI
|
|
14.
|
Akutsu Y, Hanari N, Yusup G, et al: COX2
expression predicts resistance to chemoradiotherapy in esophageal
squamous cell carcinoma. Ann Surg Oncol. 18:2946–2951. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Jendrossek V: Targeting apoptosis pathways
by Celecoxib in cancer. Cancer Lett. 332:313–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Boolbol SK, Dannenberg AJ, Chadburn A, et
al: Cyclooxygenase-2 overexpression and tumor formation are blocked
by sulindac in a murine model of familial adenomatous polyposis.
Cancer Res. 56:2556–2560. 1996.PubMed/NCBI
|
|
17.
|
Tsujii M, Kawano S and DuBois RN:
Cyclooxygenase-2 expression in human colon cancer cells increases
metastatic potential. Proc Natl Acad Sci USA. 94:3336–3340. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kwak YE, Jeon NK, Kim J and Lee EJ: The
cyclooxygenase-2 selective inhibitor celecoxib suppresses
proliferation and invasiveness in the human oral squamous
carcinoma. Ann NY Acad Sci. 1095:99–112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Dai ZJ, Ma XB, Kang HF, et al: Antitumor
activity of the selective cyclooxygenase-2 inhibitor, celecoxib, on
breast cancer in vitro and in vivo. Cancer Cell Int. 12:532012.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Soslow RA, Dannenberg AJ, Rush D, et al:
COX-2 is expressed in human pulmonary, colonic, and mammary tumors.
Cancer. 89:2637–2645. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Gupta S, Srivastava M, Ahmad N, Bostwick
DG and Mukhtar H: Over-expression of cyclooxygenase-2 in human
prostate adenocarcinoma. Prostate. 42:73–78. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Sano H, Kawahito Y, Wilder RL, et al:
Expression of cyclooxygenase-1 and -2 in human colorectal cancer.
Cancer Res. 55:3785–3789. 1995.PubMed/NCBI
|
|
23.
|
Hwang D, Scollard D, Byrne J and Levine E:
Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast
cancer. J Natl Cancer Inst. 90:455–460. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
De Groot DJ, de Vries EG, Groen HJ and de
Jong S: Non-steroidal anti-inflammatory drugs to potentiate
chemotherapy effects: from lab to clinic. Crit Rev Oncol Hematol.
61:52–69. 2007.PubMed/NCBI
|
|
25.
|
Takemura M, Morimura K, Yoshida K and
Fujiwara Y: Four resected cases with basaloid carcinoma of
esophagus - comparison of 5-FU-related enzymes (thymidylate
synthase (TS), dihydropyrimidine dehydrogenase (DPD), orotate
phosphoribosyl transferase (OPRT)) between basaloid carcinoma and
squamous cell carcinoma. Gan To Kagaku Ryoho. 37:2143–2146. 2010.In
Japanese.
|
|
26.
|
Ofverholm A, Arkblad E, Skrtic S,
Albertsson P, Shubbar E and Enerback C: Two cases of 5-fluorouracil
toxicity linked with gene variants in the DPYD gene. Clin Biochem.
43:331–334. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ostapowicz A and Dolegowska B: Review of
methods for determination of dihydropyrimidine dehydrogenase and
possible application in screening previous chemotheraphy with
5-fluorouracil. Przegl Lek. 69:694–697. 2012.In Polish.
|
|
28.
|
Li WZ, Huo QJ, Wang XY and Xu F:
Inhibitive effect of celecoxib on the adhesion and invasion of
human tongue squamous carcinoma cells to extracellular matrix via
down regulation of MMP-2 expression. Prostaglandins Other Lipid
Mediat. 93:113–119. 2010. View Article : Google Scholar
|
|
29.
|
Sheng H, Shao J, Morrow JD, Beauchamp RD
and DuBois RN: Modulation of apoptosis and Bcl-2 expression by
prostaglandin E2 in human colon cancer cells. Cancer Res.
58:362–366. 1998.PubMed/NCBI
|
|
30.
|
Oshima H and Oshima M: The role of
PGE2-associated inflammatory responses in gastric cancer
development. Semin Immunopathol. 35:139–150. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Cianchi F, Cortesini C, Bechi P, et al:
Up-regulation of cyclooxygenase 2 gene expression correlates with
tumor angiogenesis in human colorectal cancer. Gastroenterology.
121:1339–1347. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Elder DJ, Halton DE, Crew TE and Paraskeva
C: Apoptosis induction and cyclooxygenase-2 regulation in human
colorectal adenoma and carcinoma cell lines by the
cyclooxygenase-2-selective non-steroidal anti-inflammatory drug
NS-398. Int J Cancer. 86:553–560. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Irie T, Tsujii M, Tsuji S, et al:
Synergistic antitumor effects of celecoxib with 5-fluorouracil
depend on IFN-gamma. Int J Cancer. 121:878–883. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Kuipers GK, Slotman BJ, Wedekind LE, et
al: Radiosensitization of human glioma cells by cyclooxygenase-2
(COX-2) inhibition: independent on COX-2 expression and dependent
on the COX-2 inhibitor and sequence of administration. Int J Radiat
Biol. 83:677–685. 2007. View Article : Google Scholar
|
|
35.
|
Shin YK, Park JS, Kim HS, et al:
Radiosensitivity enhancement by celecoxib, a cyclooxygenase (COX)-2
selective inhibitor, via COX-2-dependent cell cycle regulation on
human cancer cells expressing differential COX-2 levels. Cancer
Res. 65:9501–9509. 2005. View Article : Google Scholar
|
|
36.
|
Jun HJ, Kim YM, Park SY, et al: Modulation
of ionizing radiation-induced G2 arrest by cyclooxygenase-2 and its
inhibitor celecoxib. Int J Radiat Oncol Biol Phys. 75:225–234.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Davis TW, O’Neal JM, Pagel MD, et al:
Synergy between celecoxib and radiotherapy results from inhibition
of cyclooxygenase-2-derived prostaglandin E2, a survival factor for
tumor and associated vasculature. Cancer Res. 64:279–285. 2004.
View Article : Google Scholar
|
|
38.
|
Kim BM, Won J, Maeng KA, Han YS, Yun YS
and Hong SH: Nimesulide, a selective COX-2 inhibitor, acts
synergistically with ionizing radiation against A549 human lung
cancer cells through the activation of caspase-8 and caspase-3. Int
J Oncol. 34:1467–1473. 2009.
|
|
39.
|
Raju U, Ariga H, Dittmann K, Nakata E, Ang
KK and Milas L: Inhibition of DNA repair as a mechanism of enhanced
radio-response of head and neck carcinoma cells by a selective
cyclooxygenase-2 inhibitor, celecoxib. Int J Radiat Oncol Biol
Phys. 63:520–528. 2005. View Article : Google Scholar : PubMed/NCBI
|